

Abstract
Previous studies demonstrated that a high fat/high cholesterol (HFC) diet results in a loss of working memory in mice correlated with neuroinflammatory changes and increased AβPP processing (Thirumangalakudi et al. J. Neurochem. 106:475–485; 2008). To further explore the nature of the molecular correlates of cognitive impairment, in this study, we examined changes in tau phosphorylation, insulin/IGF-1 signaling including GSK3 and levels of specific synaptic proteins. Immunoblot analysis of hippocampal tissue from C57BL/6 mice fed HFC for 2 months with anti-phospho-tau (i.e., PHF1 and phospho-Thr-231 tau) antibodies demonstrated the presence of hyperphosphorylated tau. The tau phosphorylation correlated with activated GSK3, a prominent tau kinase normally kept inactive under the control of insulin/IGF signaling (IIS). That IIS itself was impaired due to the hyperlipidemic diet was confirmed by a down-regulation of insulin receptor substrate-1 and phospho-Akt and levels. Although no significant changes in the levels of the pre-synaptic protein i.e., synaptophysin in response to HFC were apparent in immunoblot analysis, there was a clear down-regulation of the post-synaptic protein, PSD95 and drebrin, a dendritic spine-specific protein, indicative of altered synaptic plasticity. The results, in concert with previous findings with the same model, suggest that high dietary fat/cholesterol elicits brain insulin resistance and altered IIS leading to Alzheimer’s disease (AD)-like cognitive impairment in ‘normal’ mice.
Keywords: Dietary fat, tau phosphorylation, Insulin/IGF signaling, Brain insulin resistance, Hippocampus, Synaptic plasticity
INTRODUCTION
It is becoming clear that genetic, metabolic and environmental factors play interactive roles in the etiology of a majority of sporadic cases of Alzheimer’s disease (AD). The risk of AD seems to increase in subjects with cardiovascular diseases including atherosclerosis, hypercholesterolemia and type-2 diabetes (T2DM) [1–5] and by extension, with ‘metabolic syndrome’, a cluster of related conditions including obesity, hypertension, hyperlipidemia and hyperglycemia [6, 7]. Epidemiological and clinical studies also demonstrate that the pathogenesis of metabolic syndrome is largely attributable to dietary factors. Thus, unhealthy dietary life-styles in particular, the consumption of food rich in saturated fat and cholesterol represented by the so-called ‘Western diet’ are now commonly associated with the development of most metabolic disorders [8, 9]. Experimental and diet-induced animal models of these disorders have helped identify certain convergent mechanisms underlying abnormal metabolic changes including hyperglycemia, hyperinsulinemia, oxidative stress and atherogenesis. These mechanisms prominently include ‘metabolic inflammation’ and resulting insulin resistance, a consequence of impaired insulin/IGF signaling (IIS) [10, 11].
There is accumulating evidence, especially from animal studies, that diets rich in fat (primarily diabetogenic) and cholesterol (primarily atherogenic) can induce brain dysfunction and loss of memory [8, 12]. Thus, in early studies, Greenwood and Winocur [13] showed that rats fed a high fat diet for 3 months experience severely impaired cognitive function. [14] noted that the effect of saturated fat diet on cognitive function was related to altered synaptic plasticity and a down-regulation of brain-derived neurotrophic factor (BDNF). High fat diet-induced cognitive impairment in normal rats tied to hippocampal plasticity has been confirmed in recent studies [15]. A connection between high circulating cholesterol and brain amyloid accumulation, a hallmark of AD pathology, was first made using New Zealand white rabbits [16]. Since then, there have been several studies using transgenic mouse models of AD demonstrating that feeding of diets rich in fat/cholesterol can cause increased amyloidosis, tau phosphorylation and behavioral deficits, essential features associated with Alzheimer pathology [17–21]. Although the underlying mechanisms are obviously complex, there are indications that altered insulin signaling may play a key role in diet-induced AD-like changes [8]. The tau pathology in particular, is thought to result from defective IIS which causes an activation of glycogen synthase kinase3 (GSK3), a predominant tau kinase. There is evidence for reduced levels of insulin/IGF receptors and related signaling proteins along with an activation of GSK3 in postmortem AD brain suggesting that the neurons that degenerate in this disease may be defective in IIS [22–25]. It is also thought that cognitive decline and dementia often observed in patients with diabetes may be the result of insulin/IGF-I resistance and/or deficiency [26, 27]. In support of this, rodent models of spontaneous and experimental diabetes show AD-like changes such as amyloidosis, tau hyperphosphorylation, neurite degeneration and neuronal loss [28–30]. The changes are more severe in the T2DM model and appear to be associated with insulin resistance and possibly hypercholesterolemia.
We previously showed that a high fat/cholesterol (HFC) diet induces neuroinflammatory changes in association with increased Aβ precursor protein (AβPP) processing and a loss of working memory in normal mice [31]. In the present study, we investigated the changes in the molecular indices of synaptic/cognitive function in this model. Our findings show that HFC intake results in altered IIS (i.e., IRS-1 and Akt down-regulation and GSK3 activation) accompanied by increased tau phosphorylation and reduced levesls of post-synaptic proteins. Likely, these alterations contribute to behavioral abnormalities seen in this model.
MATERIALS AND METHODS
Animals, diet and sample collection
Four-month old C57BL/6 mice (Jackson labs) were divided into two groups: A control group fed a standard chow (5% fat with no added cholesterol) and a high fat/high cholesterol group fed a custom (Teklad, Harlan Labs) chow (21% fat and 1.25 % cholesterol) as before [31]. After a period of 2 months the mice were anesthetized with an intraperitoneal (IP) injection of sodium pentobarbital (50mg/kg), transcardially perfused with phosphate-buffered saline (PBS, 4°C) and sacrificed. The brains were removed and divided sagittally. One hemibrain was post-fixed in phosphate-buffered 4% paraformaldehyde, pH 7.4 at 4°C for 48 hours for sectioning; the other was rapidly dissected for hippocampus and cortical tissues, snap-frozen and stored at -70°C for biochemical analyses. All animal procedures were carried out in accordance with the USPHS policy on the Humane Care and Use of Laboratory Animals.
Glucose tolerance test
The two groups of mice i.e., the control fed the normal diet and that fed HFC for 2 months were fasted for 3.5h and blood samples (via tail prick) from each mouse tested for glucose levels using a Contour glucometer and test strips. Then they were injected with glucose (2g/kg body wt, ip) and after regular intervals (i.e., every 30 min for 3h), blood samples drawn were tested for glucose levels.
Western blot analysis
Proteins were extracted from the hippocampi (from 5 mice/group) using 1% Triton-X100 and 0.1% SDS in 10mM Tris pH 7.4 containing protease inhibitors. The proteins were resolved in 4–20% SDS-PAGE and transferred to PVDF membrane. The membranes were then blocked in TBST containing 5% nonfat dry milk and subsequently incubated with primary antibodies including insulin receptor (IR), IGF-IR (Santa Cruz Biotech), IRS-1, phospho-IRS-1(Ser 612), pAkt (Ser473), p-GSK3(pSer9) (Cell Signaling Tech), p-tau (PHF1, kindly provided by Dr. Peter Davies), p-tau Thr-231, pGSK3(pY279/pY216) (Millipore), drebrin (Novus), PSD95 (Chemicon) and synaptophysin (Epitomics). Bound antibody was detected with anti-rabbit/mouse horseradish peroxidase conjugate and developed using chemiluminescence (Pearce). Beta-actin or GAPDH was used as a loading control.
RESULTS
Previously, we showed that feeding of C57BL/6 mice with HFC diet increased the blood levels of LDL cholesterol and triglycerides indicating a state of hyperlipidemia/cholesterolemia that was correlated with AD-like biochemical and behavioral changes [31]. To test if such metabolic changes were also accompanied by insulin resistance, we carried out the glucose tolerance test as described under Methods. As shown in Fig 1, the mice fed the HFC diet showed glucose intolerance indicative of peripheral insulin resistance. There was only a slight increase in the level of insulin (2.2 ±0.14 ng/ml ND vs. 2.6 + ± 0.13 ng/ml HFC), however.
Fig 1.

HFC-induced glucose intolerance. Groups of mice fed either the standard chow or HFC for 2 months were subjected to glucose tolerance test as described under Methods. Black line, ND; Red line, HFC.
Increased tau phosphorylation and GSK3 activation in response to dietary HFC
We also made the observation previously that increased neuroinflammation and AβPP processing, among other factors, might have contributed to diet-induced cognitive impairment [31]. In the present study, we examined changes in additional and more specific indices of cognitive function. Tau is a microtubule binding protein with important functions in maintaining neuronal cytoskeletal dynamics and normal axonal transport. Its phosphorylation makes it soluble and causes microtubule disassembly. In extreme situations as in AD, hyperphsophorylation of tau leads to the formation of neurofibrillary tangles (NFT). To determine tau phosphorylation status in HFC-fed mice, Western blots of hippocampal extracts were probed with anti-phospho-tau antibodies i.e., anti-PHF1 (A, C) and anti-phospho tau-Thr-231 (B, D) that identify Alzheimer’s type phospho-tau. The data shown in Fig 2 confirm an increased level of hyperphosphorylated tau in the hippocampi of HFC fed mice relative to the controls.
Fig 2.


A. HFC-induced tau phosphorylation. Hippocampal protein samples from mice fed normal chow and HFC were analyzed by Western blot using anti-PHF1 (A) and anti-Thr-231 phospho-tau (B) antibodies for the detection of hyperphosphorylated tau. C, D. Quantitative densitometric analysis of blots A and B shows increased levels of phospho-tau in HFC-fed mice. Please note: The results shown in this and other figures in the manuscript are representative of at least 3 separate experiments using groups (n = 5 or 6) of HFC- or standard chow-fed mice. * p < 0.05 vs. the normally (basal diet) fed control group.
Of the several protein kinases that potentially phosphorylate tau, GSK3 in particular, is considered to play a predominant role. Its activity itself is modulated by differential phosphorylations by upstream kinases and by autophosphorylation. Akt, a key mediator of IIS phosphorylates Ser9 residue of GSK3 and keeps it inactive. In contrast, dual Tyr phosphorylation of GSK3 at residues 279 and 216 (either by autophosphorylation or by an unknown kinase) increases the kinase activity. We determined the phosphorylation status of GSK3 using antibodies that specifically identify the two types of phosphoryations. As shown in Fig 3A, there was a decrease in the inactivating phosphorylation i.e., (pSer-9)-GSK3β of GSK3 and an increase in the activating phosphorylation i.e., (pY279/pY216)-GSK3α/β, indicating the presence of an active form of GSK3 in the hippocampus of HFC-fed mice. Fig 3B shows densitometric analysis of the Western blots for the two phospho forms of GSK3.
Fig 3.

HFC-induced GSK3 activation. The Western blots of hippocampal tissue extracts from mice fed normal chow and HFC were probed with two separate phospho-specific anti-GSK3 antibodies i.e., anti-phospho(p-Ser9)-GSK3β (A, top panel) and anti-phospho(pY279/pY216)-GSK3αβ (A, bottom panel) antibodies. Note a reduction in p-Ser9, an inactivating phosphorylation and an increase in pY279/pY216, an activating phosphorylation implicating an activation of GSK3 enzyme activity in HFC-fed mice. B. Densitometric analyses of p-Ser9 GSK3β and pY279/pY216 GSK3αβ. * p < 0.05 vs. the normally (basal diet) fed control group.
HFC-induced down-regulation of IIS: changes in IRS-1 and p-Akt
As noted above, being a substrate for Akt activity, GSK3 represents a target of IIS pathway. Normally, insulinR/IGF-IR-mediated signal transduction following ligand binding proceeds through Tyr phosphorylation of insulin receptor substrates (IRS1/2) followed by an activation of phosphatidylinositol 3-kinase (PI3K)-Akt and MAPK cascades. While the MAPK pathway is involved in growth and differentiation-related gene expression, the PI3K pathway is largely responsible for insulin/IGF action on glucose uptake, suppression of gluconeogenesis and inhibition of GSK3. Phosphorylation of IRS on critical Ser sites by proinflammatory signaling kinases, as opposed to Tyr by insulin/IGF-1R activation, renders IRS inactive and prone to degradation resulting in a blockade of IIS [32, 33]. To see if activated GSK3 and increased phosphorylation of its target i.e., tau can be accounted for by a down-regulation of IIS and Akt, we determined the expression levels of IRS-1, a key intermediate in IIS whose loss represents a signature of insulin resistance, and phospho-Akt. As shown in Fig 4, there was a down-regulation of IRS-1 (A), which was correlated with decreased levels of phospho-Akt (C) (quantitation for both given in Fig D), although we did not see significant changes in the levels of either IGF1R or IR (data not shown). Probing of Ser phosphorylation of IRS-1 however, did not yield consistant results (-an example given in Fig 4B) perhaps due to the susceptibility of phosphorylated species to degradation, as mentioned above.
Fig 4.

Down regulation of IRS-1 and p-Akt levels in response to HFC diet. The samples from mice fed normal chow and HFC diet were analyzed by Western blot for IRS-1 (A), phospho-IRS-1(Ser612) and phospho-Akt (C) levels using specific antibodies. The bands (A and C) were analyzed by densitometry relative to β-actin (C). * p < 0.05 versus the normally (basal diet) fed control group.
Effects of HFC diet on synaptic protein levels
Lastly, we determined changes in the levels of representative pre- and post-synaptic proteins i.e., synaptophysin and PSD-95 respectively as well as a marker of dendritic spine i.e., drebrin. As shown in Fig 5 (A & D), other than a down-ward trend, there was no significant change in the levles of synaptophysin in the hippocampi from HFC-fed mice relative to the control tissue from normally fed mice. However, HFC feeding resulted in a significant decrease in the expression level of PSD95 (B), the post-synaptic protein and drebrin (C), an actin-binding spine-specific protein intimately associated with cytoskeletal dynamics and synaptic plasticity.
Fig 5.
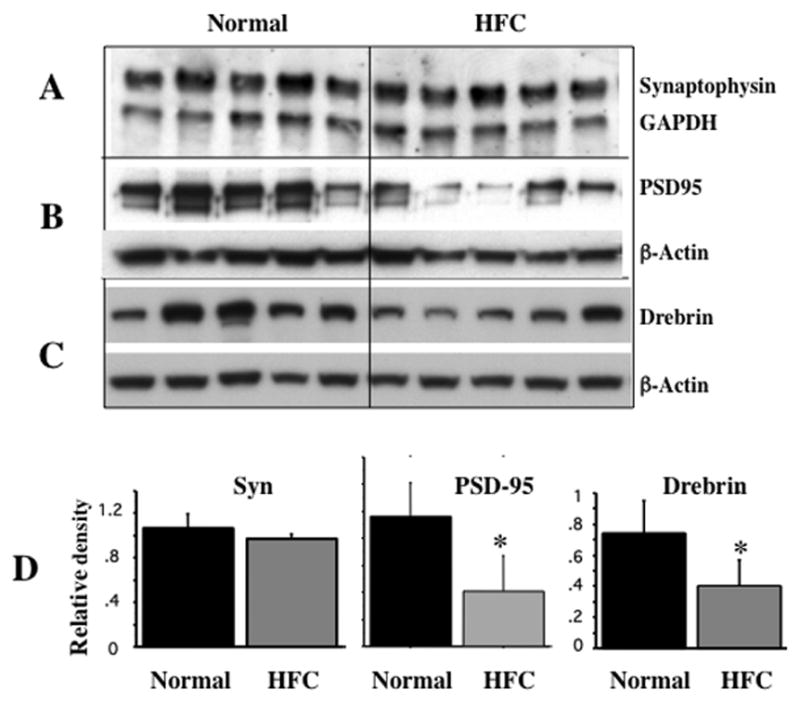
HFC-induced changes in synaptic proteins. Hippocampal extracts from mice fed normal chow and HFC were analyzed by Western blot for synaptophysin (A), PSD95 (B) and drebrin (C) levels and quantitated by densitometry (D) relative to GAPDH or β-actin using NIH ImageJ software. * p < 0.05 versus the normally (basal diet) fed control group.
DISCUSSION
In this study we analyzed the effects of an atherogenic diet in normal C57BL/6 mice on the expression/activity of molecules associated with neuronal and synaptic structure-function relevant to cognitive impairment that we had observed previously with this model [31]. We found that the HFC diet resulted in increased levels of hyperphosphorylated tau in the hippocampus and that the increased presence of phospho-tau correlated with the active form of its kinase i.e., GSK3 and a down-regulation of IIS as indicated by reduced levels of IRS1 and phospho-Akt. The feeding of HFC also resulted in a down-regulation of PSD95 and drebrin, a dendritic spine-specific protein. These findings suggest deficient trophic input and/or insulin resistance as potential mechanisms underlying tau and synaptic pathology likely contributing to cognitive impairment.
Our findings of diet-induced impairment of brain IIS are in line with increasing evidence linking insulin/IGF resistance with brain dysfunction and dementia. Insulin and IGF-I play important homeostatic roles in the brain including glucose metabolism, energy balance and trophic support as well as maintenance of normal synaptic activity and cognitive function. Therefore, a down-regulation of brain IIS can lead to deleterious neurobehavioral outcome. There are several possibilities for diet-induced altered IIS including trophic factor deficiency, IR/IGF1R down-regulation/desensitization and insulin resistance. With respect to trophic factor deficiency, it is likely that a diet-induced pre-diabetic (and atherogenic) condition induces cerebrovascular dysfunction thereby blocking insulin/IGF transport. In fact, there is evidence for an impaired transport into the brain of IGF-I due to cerebrovascular abnormalities [34, 35] as might occur in metabolic disorders. In the rabbit model of hypercholesterolemia, increased accumulation of Aβ in the hippocampus has been shown to correlate with reduced levels of IGF-1 and altered IGF-1 signaling [36]. In our studies, although insulin levels in the brain were too low to detect any changes, we did determine the levels of IGF1 by ELISA and found no evidence for its deficiency. It is of interest that an ‘insulin resistant brain state (IRBS)’ is now increasingly thought to underlie the development of sporadic AD [37–39]. There is also the suggestion that AD represents a ‘Type 3 diabetes’ [7, 24] based on post-mortem studies indicating a generalized reduction in insulin/IGF levels and associated signaling proteins [22, 24, 34]. One of the mechanisms whereby brain insulin resistance develops in AD is thought to involve Aβ oligomers interfering with neuronal insulin/IGF signaling [40, 41]. In contrast, it has been suggested that increased amyloidogenesis may follow diet-induced alterations in insulin/IGF1 receptor signaling [8]. There may be yet another mechanism at work in inducing the insulin resistant brain state in our model. This would involve previously reported neuroinflammatory changes [31]. We propose that as in systemic insulin resistance [11, 42], inflammatory mediators in the brain may negatively interact with IIS to induce brain insulin resistance. In further support of such a mechanism, there is accumulating evidence that IGF-1 and proinflammatory cytokines engage in a two-way interaction to regulate resistance/sensitivity to each other’s actions in the brain [43].
The present studies show that altered brain IIS in response to HFC diet accompanies tau hyperphosphorylation. Tau pathology is a hallmark of neurodegenerative taupathies including AD [44]. The assembly and stabilization of microtubules involved in axonal transport is a well characterized function of tau that can be compromised by abnormal phosphorylation since hyperphosphorylated tau aggregates into NFTs in neuronal perikarya as in AD. Although amyloid accumulation is thought to precede NFT formation, tau pathology more closely relates to synaptic dysfunction and neurodegeneration [45]. It is important to note that even in the absence of NFT formation, tau phosphorylation can lead to synaptic dysfunction and neurodegeneration. It is likely that the increased tau phosphorylation that we observed represents a pathological target of abnormal IIS via GSK3 activation. With regard to altered insulin signaling affecting tau phosphorylation, several studies have shown that spontaneous and experimental diabetes in animals induce tau hyperphosphorylation and that this effect is exacerbated in the Tg models of AD made diabetic [28–30, 46–49]. It has also been found that high fat diet [21, 50] and excessive sucrose intake [51] can enhance both amyloid and tau pathology in Tg mice. However, there have been only limited studies examining the effects of diet on AD-like changes in normal rodents. In one such study, Moroz et al. [52] found limited Alzheimer-type changes including tau phosphorylation with no IIS impairment in C57BL/6 mice fed a high fat diet. In contrast to these findings, we find that the dietary regimen we used (i.e., a combination of high fat and high cholesterol) is able to induce hyperphosphoryation of tau with significant induction of insulin resistance and GSK3 activation. That hypercholesterolemia itself can cause tau hyperphsophorylation in the brain has been shown using the rabbit model [53] and apoE-deficient mice fed a high cholesterol diet [54].
Our findings of increased activation of GSK3 (as indicated by changes in its phosphorylations) in response to HFC are well correlated with increased tau phosphorylation and further support altered IIS as the underlying mechanism. Normally, insulin (and IGF-I) keeps GSK3 inactive through IRS-1 and PI-3K dependent mechanism via Akt-mediated phosphorylation of the enzyme isoforms. This reduces tau hyperphosphorylation mediated by GSK3β [55]. This mechanism of GSK3 control is disrupted in conditions of insulin resistance likely occurring in AD. Like the GSK3β, its α form is also thought to be involved in AD pathogenesis since activated GSK3α can trigger AβPP processing enzyme i.e., γ-secretase activity [56] thereby increasing Aβ production. There is now substantial evidence for a key role of GSK3 in AD through multiple mechanisms including tau phosphorylation, Aβ production and inflammation [56]. Its inhibition reduces Aβ while its conditional inactivation reverses AD-like phenotype in mouse models correlated with normal tau phosphorylation [57]. It has also been hypothesized that GSK3 may play a central role in late-onset AD by linking Aβ and tau [58]. Most interestingly, a recent study using induced pluripotent stem cell (iPSC) technology has suggested an intriguing relation between AβPP processing i.e., increased production of CTF (but not Aβ) and increased phospho-tau and active GSK3β in iPSC-derived neurons from both sporadic and f-AD patients [59].
In addition to tau hyperphosphorylation and GSK3 activation, we observed a down-regulation of the post-synaptic marker, PSD95 and drebrin, a dendritic spine protein in repsonse to HFC. An inadequate IIS might have led to the reduction in the levels of PSD95 since its expression is known to be regulated by insulin signaling via PI3K-Akt-mTOR pathway [60]. The actin-binding protein, drebrin is involved in shaping the dendritic spine morphology which is closely associated with spine function in learning and memory [61]. There is evidence for decreased drebrin levels in neurological diseases including AD and mild cognitive impairment (MCI) as well as in transgenic AD models showing cognitive deficits [62]. The loss of drebrin has also been shown to correlate with tau pathology in the AD brain [20] but the mechanism of its down-regulation in this case may involve direct amyloid toxicity [63]. However, since PI3K pathway seems to positively regulate the expression of drebrin [62], there is an obvious connection between IIS and drebrin expression. A recent study shows that high fat diet aggravates amyloid and tau pathologies in the 3xTg AD mouse model and that these changes are accompanied by reduced levels of drebrin [50] with unaltered levels of several presynaptic proteins (i.e., synaptophysin, SNAP-25 and syntaxin 3). Although not analyzed in that study, altered IIS was perhaps responsible for the observed postsynaptic changes. This idea is supported by another study with APP/PS1 Tg mice made diabetic by streptozotocin injection [64].
In conclusion, we have shown in this study that an atherogenic diet induces changes in the hippocampus indicative of defective IIS and insulin resistance in association with increased active GSK3 and tau phosphorylation. These changes would potentially contribute towards the cognitive impairment that we observed with this model. The mechanisms by which dietary fat/cholesterol elicits altered brain IIS cannot yet be deciphered clearly, but it is likely that neuroinflammation plays a central role in these processes, as noted above. This would be analogous to the role of systemic/metabolic inflammation caused by diet-induced prediabetic and atherogenic conditions. Perhaps, cerebrovascular dysfunction with altered BBB (unpublished observations) forms the link i.e., systemic-to-neuroinflammation transition as we have proposed [5]. A damaged cerebrovasculature would also cause brain hypoperfusion leading to neuronal energy failure and metabolic stress (Fig 6) with the outcome of exacerbated IRBS, a potential mechanism proposed for sporadic AD.
Fig 6.

Potential metabolic/signaling pathways that mediate diet-induced impairment in brain insulin signaling leading to increased GSK3 activity and tau phosphorylation.
Acknowledgments
We thank Sangeeta Mohanty and Joshua Hirschhorn for expert technical help. The studies were supported by grant # R01NS051575 from NINDS.
Abbreviations
- Aβ
Beta amyloid
- AD
Alzheimer’s disease
- AβPP
Amyloid-β precursor protein
- GSK3
Glycogen synthase kinase3
- HFC
High fat/high cholesterol
- IIS
Insulin/IGF-I signaling
- IRS-1
Insulin receptor substrate-1
- T1DM
Type 1 diabetes
- T2DM
Type 2 diabetes
References
- 1.Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet. 2004;363:1139–46. doi: 10.1016/S0140-6736(04)15900-X. [DOI] [PubMed] [Google Scholar]
- 2.Sambamurti K, Granholm AC, Kindy MS, Bhat NR, Greig NH, Lahiri DK, et al. Cholesterol and Alzheimer’s disease: clinical and experimental models suggest interactions of different genetic, dietary and environmental risk factors. Curr Drug Targets. 2004;5:517–28. doi: 10.2174/1389450043345335. [DOI] [PubMed] [Google Scholar]
- 3.Cechetto DF, Hachinski V, Whitehead SN. Vascular risk factors and Alzheimer’s disease. Expert Rev Neurother. 2008;8:743–50. doi: 10.1586/14737175.8.5.743. [DOI] [PubMed] [Google Scholar]
- 4.de la Torre JC. Cerebrovascular and cardiovascular pathology in Alzheimer’s disease. Int Rev Neurobiol. 2009;84:35–48. doi: 10.1016/S0074-7742(09)00403-6. [DOI] [PubMed] [Google Scholar]
- 5.Bhat NR. Linking cardiometabolic disorders to sporadic Alzheimer’s disease: a perspective on potential mechanisms and mediators. J Neurochem. 2010;115:551–62. doi: 10.1111/j.1471-4159.2010.06978.x. Research Support, N.I.H Extramural Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milionis HJ, Florentin M, Giannopoulos S. Metabolic syndrome and Alzheimer’s disease: a link to a vascular hypothesis? CNS Spectr. 2008;13:606–13. doi: 10.1017/s1092852900016886. [DOI] [PubMed] [Google Scholar]
- 7.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–5. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasinetti GM, Eberstein JA. Metabolic syndrome and the role of dietary lifestyles in Alzheimer’s disease. J Neurochem. 2008;106:1503–14. doi: 10.1111/j.1471-4159.2008.05454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2:231–7. doi: 10.1242/dmm.001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond) 2008;32(Suppl 7):S52–4. doi: 10.1038/ijo.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. 2009;6:399–409. doi: 10.1038/nrcardio.2009.55. [DOI] [PubMed] [Google Scholar]
- 12.Martins IJ, Hone E, Foster JK, Sunram-Lea SI, Gnjec A, Fuller SJ, et al. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–36. doi: 10.1038/sj.mp.4001854. [DOI] [PubMed] [Google Scholar]
- 13.Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging. 2005;26(Suppl 1):46–9. doi: 10.1016/j.neurobiolaging.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Wu A, Ying Z, Gomez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur J Neurosci. 2004;19:1699–707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- 15.Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–8. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sparks DL, Scheff SW, Hunsaker JC, 3rd, Liu H, Landers T, Gross DR. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol. 1994;126:88–94. doi: 10.1006/exnr.1994.1044. [DOI] [PubMed] [Google Scholar]
- 17.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, et al. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–31. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 18.Levin-Allerhand JA, Lominska CE, Smith JD. Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J Nutr Health Aging. 2002;6:315–9. [PubMed] [Google Scholar]
- 19.Hooijmans CR, Van der Zee CE, Dederen PJ, Brouwer KM, Reijmer YD, van Groen T, et al. DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiol Dis. 2009;33:482–98. doi: 10.1016/j.nbd.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Julien C, Tremblay C, Bendjelloul F, Phivilay A, Coulombe MA, Emond V, et al. Decreased drebrin mRNA expression in Alzheimer disease: correlation with tau pathology. J Neurosci Res. 2008;86:2292–302. doi: 10.1002/jnr.21667. [DOI] [PubMed] [Google Scholar]
- 21.Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–89. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 31:224–43. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Sun MK, Alkon DL. Links between Alzheimer’s disease and diabetes. Drugs Today (Barc) 2006;42:481–9. doi: 10.1358/dot.2006.42.7.973588. [DOI] [PubMed] [Google Scholar]
- 24.de la Monte SM. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009;42:475–81. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from model organisms to human disease. Curr Alzheimer Res. 2009;6:213–23. doi: 10.2174/156720509788486527. [DOI] [PubMed] [Google Scholar]
- 26.Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer’s disease: an epidemiological perspective. Eur J Pharmacol. 2008;585:119–29. doi: 10.1016/j.ejphar.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim Biophys Acta. 2009;1792:482–96. doi: 10.1016/j.bbadis.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 28.Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–24. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 29.Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, et al. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 2008;86:3265–74. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. 2009;150:5294–301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thirumangalakudi L, Prakasam A, Zhang R, Bimonte-Nelson H, Sambamurti K, Kindy MS, et al. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem. 2008;106:475–85. doi: 10.1111/j.1471-4159.2008.05415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun XJ, Liu F. Phosphorylation of IRS proteins Yin-Yang regulation of insulin signaling. Vitam Horm. 2009;80:351–87. doi: 10.1016/S0083-6729(08)00613-4. [DOI] [PubMed] [Google Scholar]
- 33.Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296:E581–91. doi: 10.1152/ajpendo.90437.2008. [DOI] [PubMed] [Google Scholar]
- 34.Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links & clinical implications. Curr Alzheimer Res. 2008;5:438–47. doi: 10.2174/156720508785908919. [DOI] [PubMed] [Google Scholar]
- 35.Aleman A, Torres-Aleman I. Circulating insulin-like growth factor I and cognitive function: neuromodulation throughout the lifespan. Prog Neurobiol. 2009;89:256–65. doi: 10.1016/j.pneurobio.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 36.Sharma S, Prasanthi RPJ, Schommer E, Feist G, Ghribi O. Hypercholesterolemia-induced Abeta accumulation in rabbit brain is associated with alteration in IGF-1 signaling. Neurobiol Dis. 2008;32:426–32. doi: 10.1016/j.nbd.2008.08.002. Research Support, N.I.H Extramural. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Salkovic-Petrisic M, Osmanovic J, Grunblatt E, Riederer P, Hoyer S. Modeling sporadic Alzheimer’s disease: the insulin resistant brain state generates multiple long-term morphobiological abnormalities including hyperphosphorylated tau protein and amyloid-beta. J Alzheimers Dis. 2009;18:729–50. doi: 10.3233/JAD-2009-1184. Research Support, Non-U.S Gov’t Review. [DOI] [PubMed] [Google Scholar]
- 38.Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Insulin-resistant brain state: the culprit in sporadic Alzheimer’s disease? Ageing research reviews. 2011;10:264–73. doi: 10.1016/j.arr.2011.01.001. Research Support, N.I.H Extramural Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–38. doi: 10.1172/JCI59903. Research Support, N.I.H., Extramural Research Support, Non-U.S Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao FF, Xu H. Insulin signaling in sporadic Alzheimer’s disease. Sci Signal. 2009;2:pe36. doi: 10.1126/scisignal.274pe36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J Clin Invest. 2012;122:1339–53. doi: 10.1172/JCI57256. Research Support, N.I.H., Extramural Research Support, Non-U.S Gov’t Video-Audio Media. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 43.O’Connor JC, McCusker RH, Strle K, Johnson RW, Dantzer R, Kelley KW. Regulation of IGF-I function by proinflammatory cytokines: at the interface of immunology and endocrinology. Cellular immunology. 2008;252:91–110. doi: 10.1016/j.cellimm.2007.09.010. Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 45.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–9. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Clodfelder-Miller BJ, Zmijewska AA, Johnson GV, Jope RS. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes. 2006;55:3320–5. doi: 10.2337/db06-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp Neurol. 2009 doi: 10.1016/j.expneurol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007;27:13635–48. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ke YD, Delerue F, Gladbach A, Gotz J, Ittner LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2009;4:e7917. doi: 10.1371/journal.pone.0007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–31. doi: 10.1016/j.neurobiolaging.2008.08.022. Research Support, Non-U.S Gov’t. [DOI] [PubMed] [Google Scholar]
- 51.Cao D, Lu H, Lewis TL, Li L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2007;282:36275–82. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- 52.Moroz N, Tong M, Longato L, Xu H, de la Monte SM. Limited Alzheimer-type neurodegeneration in experimental obesity and type 2 diabetes mellitus. J Alzheimers Dis. 2008;15:29–44. doi: 10.3233/jad-2008-15103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen X, Wagener JF, Morgan DH, Hui L, Ghribi O, Geiger JD. Endolysosome mechanisms associated with Alzheimer’s disease-like pathology in rabbits ingesting cholesterol-enriched diet. J Alzheimers Dis. 2010;22:1289–303. doi: 10.3233/JAD-2010-101323. Research Support, N.I.H Extramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rahman A, Akterin S, Flores-Morales A, Crisby M, Kivipelto M, Schultzberg M, et al. High cholesterol diet induces tau hyperphosphorylation in apolipoprotein E deficient mice. FEBS Lett. 2005;579:6411–6. doi: 10.1016/j.febslet.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 55.Balaraman Y, Limaye AR, Levey AI, Srinivasan S. Glycogen synthase kinase 3beta and Alzheimer’s disease: pathophysiological and therapeutic significance. Cell Mol Life Sci. 2006;63:1226–35. doi: 10.1007/s00018-005-5597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104:1433–9. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Engel T, Hernandez F, Avila J, Lucas JJ. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci. 2006;26:5083–90. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60:534–42. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–20. doi: 10.1038/nature10821. Research Support, N.I.H., Extramural Research Support, Non-U.S Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem. 2005;280:18543–50. doi: 10.1074/jbc.M414112200. Comparative Study In Vitro Research Support, Non-U.S Gov’t. [DOI] [PubMed] [Google Scholar]
- 61.Sekino Y, Kojima N, Shirao T. Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem Int. 2007;51:92–104. doi: 10.1016/j.neuint.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 62.Kojima N, Shirao T. Synaptic dysfunction and disruption of postsynaptic drebrin-actin complex: a study of neurological disorders accompanied by cognitive deficits. Neurosci Res. 2007;58:1–5. doi: 10.1016/j.neures.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 63.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burdo JR, Chen Q, Calcutt NA, Schubert D. The pathological interaction between diabetes and presymptomatic Alzheimer’s disease. Neurobiol Aging. 2009;30:1910–7. doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]