

Abstract
Trimethyltin (TMT), which has a variety of applications in industry and agricultural is a neurotoxin that is known to affect the auditory system as well as central nervous system (CNS) of humans and experimental animals. However, the mechanisms underlying TMT-induced auditory dysfunction are poorly understood. To gain insights into the neurotoxic effect of TMT on the peripheral auditory system, we treated cochlear organotypic cultures with concentrations of TMT ranging from 5 to 100 μM for 24 h. Interestingly, TMT preferentially damaged auditory nerve fibers and spiral ganglion neurons in a dose-dependent manner, but had no noticeable effects on the sensory hair cells at the doses employed. TMT-induced damage to auditory neurons was associated with significant soma shrinkage, nuclear condensation and activation of caspase-3, biomarkers indicative of apoptotic cell death. Our findings show that TMT is exclusively neurotoxicity in rat cochlear organotypic culture and that TMT-induced auditory neuron death occurs through a caspase-mediated apoptotic pathway.
Keywords: Trimethyltin, neurotoxicity, cochlea, spiral ganglion neurons, apoptosis, caspase-3
Introduction
Organotin compounds (OTCs) are wieldy used in industrial and agricultural settings as plastic stabilizers, pesticides, timber preservatives, and antifouling paints. Due to their widespread application during the past 50 years a considerable amount of OTCs have been introduced into the environment (Hoch, 2001). As most of OTCs are toxic, these harmful contaminants pose great risk for human health and the ecosystem (Braman and Tompkins, 1979, Cima, 2011). Among these potentially toxic compounds, trimethyltin (TMT) has received considerable attention as numerous studies have demonstrated it can act as a potent neurotoxin.
TMT is a trialkyl-tin compound that mainly damages the central nervous system (Koczyk, 1996). TMT selectively induces neuronal death in the human and animal limbic system particularly in the hippocampus (Geloso et al., 2011a). Humans occupationally exposed to TMT develop a limbic-cerebellar syndrome characterized by hearing loss, disorientation, amnesia, aggressive behavior, complex partial and tonic-clonic seizures, ataxia and mild sensory neuropathy (Dorman, 2000). In experimental animals TMT intoxication produces a similar syndrome with spontaneous-seizures, self-mutilation, vocalizations, hyperactivity and aggressive behavior (Dyer et al., 1982). A number studies with neural cell lines and primary cultures indicate that TMT exposure produces changes indicative of programmed cell death such as nuclear condensation, DNA fragmentation, membrane blebbing, and caspase activation (Jenkins and Barone, 2004, Kuramoto et al., 2011, Mundy and Freudenrich, 2006). Similar neuronal pathologies were seen in vivo when TMT was given to rat and mice (Fiedorowicz et al., 2001, Morita et al., 2008). Although the mechanisms underlying the neurotoxicity of TMT are still obscure, the damage induced by TMT in neurons is thought to involve several pathways such as oxidative stress, intracellular calcium overload, and mitochondrial damage (Aldridge et al., 1977, Ali et al., 1992, Misiti et al., 2008, Piacentini et al., 2008). In addition, expression of stannin (Snn), a mitochondrial membrane protein mainly localized to cells with TMT sensitivity appears to play an essential role in TMT-mediated selective neuronal degeneration (Davidson et al., 2004, Toggas et al., 1992).
In addition to CNS injury, TMT can damage the peripheral auditory system (Chang and Dyer, 1983b). A single 4–6 mg/kg dose of TMT produced a frequency-dependent hearing loss that was most severe in the high frequency range (Eastman et al., 1987, Ruppert et al., 1984). TMT-induced high frequency hearing loss was closely correlated with loss of the outer hair cells (OHC) in the basal turn of the cochlea (Crofton et al., 1990, Hoeffding and Fechter, 1991). Moderate doses of TMT induced hearing loss that but partially reversible; however, the recovery period measured by startle reflex audiometry was uncharacteristically prolonged (Young and Fechter, 1986). Apart from OHC loss, both the known neurotoxic effects and the acute disruption of compound action potential (CAP) by TMT suggest a possible mechanism involving direct injury to auditory neurons in addition to the OHC (Fechter and Liu, 1995). To evaluate the relative toxicities of TMT on neurons versus sensory hair cells within the cochlea, rat postnatal cochlear organotypic cultures were exposed to varying concentrations of TMT to assess the relative damage to auditory nerve fibers (ANF) and SGN compared OHC and inner hair cells (IHC).
Materials and methods
Cochlear organotypic cultures
Postnatal day 3 (P3) SASCO Sprague-Dawley rats purchased from Charles River Laboratories were used for preparing cochlear organotypic cultures as described previously (Ding et al., 2011). In brief, after rat pups were decapitated, the cochleae were quickly removed and the whole basilar membrane containing the organ of Corti, auditory nerve fibers (ANF) and SGN were carefully dissected out and transferred on to rat tail collagen gel in a culture dish. Approximately 10 μl of collagen gel (type I collagen gel 3.76 mg/ml in 0.02 N acetic acid, 10x basal medium eagle, 2% sodium carbonate, at 9:1: 1 ratio) was applied to the surface of a 35 × 10 mm culture dish and allowed to gel for about 30 min at room temperature. Afterwards, 1.3 ml of serum-free medium (0.01 g/ml bovine serum albumin (Sigma A-4919), 1% serum-free supplement (Sigma I-1884), 1% 200 mM glutamate, 2.4% of 20% glucose, 0.2% penicillin G, 95.4% 1X basal medium eagle (Sigma B-15220) was added to the culture dish. The basilar membrane containing the sensory hair cells, SGN and supporting cells was placed on the surface of the collagen gel and then maintained in an incubator at 37 °C and 5% CO2 overnight. On the following day, fresh culture medium (2 ml) with or without various concentrations of TMT was added to each dish and cochlear explants were cultured for an additional 24 h.
Trimethyltin treatment
Cochleae were randomly divide into 5 groups (n = 10 per group). One group served as normal control; the other four groups were treated with different concentrations of TMT. TMT stock solution was freshly prepared at a concentration of 10 mM in serum-free medium and diluted to a final concentration of 0 (control) 5, 10, 50 or 100 μM for each TMT treated group. Explants were cultured in the incubator for 24 h and then harvested for histological analysis.
Immunohistochemical staining
Cochlear explants were fixed with 10% formalin for 2 h at 4 °C and rinsed three times in 0.1 M phosphate buffered saline (PBS). To stain neurofilaments, which are expressed in both type I and type II SGN and ANF as well as efferent fibers, specimens were immersed overnight at 4 °C in a monoclonal primary mouse 200 kD anti-neurofilament antibody (Sigma, No142) diluted in 1% Triton X-100 and 5% goat serum in 0.1 M PBS (1:100). Specimens were rinsed three times with PBS and incubated for an additional 2 h at room temperature in a secondary goat anti-mouse antibody conjugated with Alexa Fluor488 (Life technologies, A11001) diluted in PBS (1:200). To stain hair cell stereocilia and cuticular plate, specimens were rinsed in PBS three times and incubated for 1 h at room temperature with Alexa Fluor 568 conjugated phalloidin (Life technologies, A12380) diluted in PBS (1:200). After rinsing in PBS, the specimens were immersed for 30 minutes in fresh To-Pro-3 solution (Life technologies, T3605, 1 mM in 0.75 ml DMSO diluted in 1 ml H2O) to label the nuclei of cells in the specimen. Specimens were subsequently mounted on glass slides in glycerin, coverslipped and examined under a confocal microscope.
Cochleograms
Cochlear hair cells were observed and counted under a fluorescent microscope with the appropriate filter to visualize the stereocilia and cuticular plate of hair cells labeled with Alexa Fluor 568 conjugated phalloidin. The number of hair cells were counted over 0.24 mm intervals along the enter length of the basilar membrane from apex to base. Plots showing the percent IHC and OHC loss as a function of percent distance from the apex of the cochlea were used to construct cochleograms. Cochleograms for each treatment were averaged to generate a mean cochleogram using custom software (Dong et al., 2014, Wang et al., 2014).
Caspase-3 staining
To evaluate caspase-3 activity in SGNs, cochlear explants in control and the 50 μM TMT group were labeled with a cell permeable fluorogenic caspase substrate, Red-VAD-FMK caspase, using a CaspGLOW Red Active Caspase Staining Kit (BioVision, K190) following the manufacturer instructions. Twelve hours after treatment, the unfixed specimens were incubated with Red-VAD-FMK caspase-3 diluted in culture medium (1:200) at 37 °C and 5% CO2 for 60 min, then rinsed three times with wash buffer and fixed in 10% formalin for 2 h. To optimize contrast, the red fluorescence of activated caspase-3 was converted with software to green pseudo-color in the photomicrographs. Specimens were immunolabeled with an antibody against neurofilament 200 kD (see above), rinsed in PBS, mounted in glycerin on glass slides and coverslipped.
Confocal microscopy
Cochlear specimens were examined under a confocal microscope (Zeiss LSM-510 Meta) using appropriate filters to detect the green fluorescence of Alexa Fluor 488 labeled neurofilament (excitation 488 nm, emission 519 nm), red fluorescence of Alexa Fluor 568 labeled phalloidin (excitation 578 nm, emission 600 nm), purple fluorescence of To-Pro-3 labeled nucleus (excitation 642 nm, emission 661 nm) and red fluorescence for caspase-3 (excitation 540 nm, emission 570 nm). As described previously, confocal images were processed using Confocal LSM Image Examiner and Adobe Photoshop CS 3.0 software as described previously (Ding et al., 2014, Dong et al., 2014, Wang et al., 2014).
Statistical Analysis
ANF and SGN counts were evaluated using a one-way analysis of variance (GraphPad Prism 5) and Newman-Keuls post-hoc analysis as described below.
Results
TMT damages ANF and SGN
To determine its neurotoxic effects on cochlear ANF, SGN and hair cells (HC), cochlear cultures were treated with 50 μM TMT for 24 h. Fig 1 illustrates the typical status of IHC and OHC, ANF and SGN from the upper middle turn of the cochlea in control cultures and those treated with 50 μM TMT. In control cultures, the stereocilia bundle and cuticular plate of both the IHC and OHC were heavily labeled with phalloidin (red). The three rows of OHC and one row of IHC were arranged longitudinally in a gently curving arc; there was no sign of HC loss or damage. The peripheral ANF which project out radially to the hair cells from the SGN were intensely labeled with neurofilament 200 kD (green). The radial ANF were organized into smooth and thick fascicles. The SGN in normal control cultures had large, round or oval-shaped soma with heavy neurofilament labeling of the cytoplasm and faint labeling of the nucleus (Fig. 1A). Remarkably, in cultures treated with TMT for 24 h, the IHC and OHC maintained their normal structure and position; there was no obvious loss or damage to the HC. In contrast, TMT caused massive degenerative changes in the ANF and SGN (Fig. 1B). After 24 h treatment with 50 μM TMT, most ANFs were fragmented and pixelated with nearly total loss of most peripheral fibers endings approaching the hair cells. The number of SGN was greatly reduced and the soma of the remaining SGN were shrunken and condensed and the nucleus seldom visible.
Figure 1.

Representative confocal images showing cochlear organotypic cultures from the upper middle turn of the cochlea after 24 h in culture without (A) or (B) with TMT. Three rows of outer hair cells (OHC) and one row of inner hair cells (IHC) were immunolabeled with Alexa Fluor 568 conjugated phalloidin (red). Spiral ganglion neurons (SGN) and their nerve fibers (ANF) were immunolabeled with primary antibody against neurofilament 200 kD and Alexa Fluor488 conjugated secondary antibody (green). (A) Cochlear culture in control group. (B) Cochlear culture in group treated with 50 μM TMT. Note after TMT exposure ANF and SGN were severely destroyed, whereas cochlear hair cells were intact. Mean cochleograms (n=5) showing the percentage of missing OHCs and IHCs as a function of the percent distance from the apex of cochlea in the (C) Control group and the (D) group treated with 50 μM TMT for 24 h.
Cochleograms
To evaluate the effect of TMT on cochlear HC, average cochleograms (N = 5/group) measuring the percentage of missing OHC and IHC as a function of percent distance from the apex of the cochlea were constructed after 24 h treatment with various concentrations of TMT. The mean loss of OHC and IHC for the control group (Fig. 1C) was less than 5% along the basilar membrane except for the extreme base and apex of the cochlea; losses in these regions are mainly due to mechanical damage during cochlear dissection. Mean losses of OHC and IHC in the 5, 10, 50 and 100 μM groups were similar to the control group as illustrated in Figure 1D for the 50 mM TMT dose. Mean cochleograms for all TMT doses were similar to Figure 1D. HC loss did not increase with TMT dose and even at the highest concentration.
Dose-dependent ANF damage
Since neurofilaments are heavily expressed in neurons, our neurofilament antibody would be expected to label type I and type II ANF as well as efferent fibers. Since the vast majority of fibers innervating the cochlea are type I (90–95%) or type II (5–10%) afferents, our neurofilament labeling predominantly represents the effects of TMT on ANF (Brown and Ledwith, 1990, Perkins and Morest, 1975, Spoendlin and Gacek, 1963). Figures 2A–E shows the condition of ANF in the upper middle turn of the cochlea after 24 h in culture; data are shown for controls and those treated with varying concentrations of TMT. In control cultures, the ANF fascicles were thick and smooth (Fig. 2A). However, in TMT treated cultures, the ANF fascicles degenerated in a dose-dependent manner (Fig. 2B–E). In cultures treated with 5 μM TMT, a slight decline in ANF density was evident, many blebs were present on the surviving fibers and fiber thickness had decreased. As TMT concentration increased, the numbers of surviving fibers decreased, the ANF fascicles became thinner and blebbing and fragmentation of ANF increased. At the highest TMT concentration, 100 μM, nearly all of the ANFs were absent and only the debris of ANF fascicles remained. However, a few intact fibers were still present at the highest concentration; these TMT-resistant fibers could conceivably represent type II fibers. Type II afferent fibers, which can be detected with an antibody against peripherin, tend to be resistant to trauma (Barclay et al., 2011, Lim, 1976).
Figure 2.

Representative confocal images showing the condition of the peripheral auditory nerve fibers (ANF) from the upper middle turn of the cochlea. ANF in control group (A) and groups treated with indicated dose of TMT 24 h (B–E). (F) Mean (+SEM) numbers (n = 14/group) of ANF/140 μm from the upper middle turn of the cochlea in control cultures and cultures treated with various concentrations of TMT for 24 h. Individual groups significantly different from one another (* p < 0.05).
Since TMT-induced damage to ANF was similar along the length of the cochlea, we quantified the numbers of surviving ANF in the upper middle turn of the cochlea; ANF were measured per 140 μm widths in control and TMT-treated samples. Figure 2F shows the mean numbers (n = 14/group) of ANF/140 μm in control cultures and cultures treated with various concentration of TMT. In control cultures, approximately 63 fibers were present per 140 μm. The mean numbers of ANF/140 μm decreased in a dose-dependent manner and at 100 μM TMT only 4.7 fibers were present per 140 μm. A one-way analysis of variance (ANOVA) showed there was a statistically significant effect of TMT treatment on ANF survival (one-way ANOVA, P < 0.0001, F = 246.6). A Newman-Keuls post hoc test showed that the mean numbers of ANF in all comparison pairs were significantly different (Newman-Keuls post-hoc analysis, P < 0.05).
Dose-dependent SGN degeneration
Fig. 3 illustrates the status of SGN in the upper middle turn of the cochlea of control cultures and cultures treated with TMT for 24 h. The SGN in control cultures had large and round or ovoid cell bodies; strong neurofilament 200 kD immunolabeling was present throughout the cytoplasm except for the nucleus (Figure 3A). However, in TMT treated cultures, SGN soma size decreased with increasing TMT dose and at the higher doses, the size of the nucleus was substantially smaller than normal (Fig. 3B–E). After treatment with 100 μM TMT, nearly all SGNs were destroyed (Fig. 3E).
Figure 3.

Representative confocal images showing the status of the spiral ganglion neurons (SGN) from the upper middle turn of the cochlea. SGN in control cultures (A) and cultures treated with indicated dose of TMT for 24 h (B–E). (F) Mean (+SEM) numbers (n=10/group) of SGN per 105μm3 from the upper middle turn of the cochlea in control cultures and cultures treated with various concentrations of TMT for 24 h. Individual groups significantly different from one anther (*p < 0.05).
Since TMT-induced damage to SGN was similar along the length of the cochlea, we measured the density of SGN in the upper middle turn of the cochlea using procedures previously described (Wang et al., 2014). Figure 3F shows the mean numbers (n=10/group) of SGN per 105 μm3 in control cultures and those treated with varying TMT concentrations. In control cultures, there were approximately 47 SGN per 105 μm3. However, in TMT treated cultures, the mean numbers of SGN decreased in a dose-dependent manner so that at the 100 μM TMT dose there were ~3 SGN per 105 μm3. There was a significant effect of TMT treatment on SGN survival (one-way ANOVA, P < 0.0001, F = 74.68). SGN densities were also significantly different between the different TMT concentrations (Fig. 3F, Newman-Keuls post-hoc analysis, P < 0.05).
TMT-induced morphological changes
Figure 4 illustrates the characteristic morphological features of SGN before and after TMT treatment. SGN were labeled with a neurofilament primary antibody and Alexa 488 secondary antibody (green). The nuclei of all cells were labeled with To-Pro-3; the normal blue fluorescence of To-Pro-3 was converted to red pseudo-color with software to enhance contrast. In control cultures, the SGN soma had a large round or ovoid shape (Figure 4A). The cytoplasm within the SGN was heavily labeled with neurofilament 200 kD, the round or ovoid shaped nucleus in the center of the soma was heavily labeled by To-Pro-3 and ANF were labeled with neurofilament. However, in the TMT treated cultures, SGN somas were greatly reduced in size with most neurites missing, and the SGN nuclei remarkably condensed (Fig. 4B).
Figure 4.
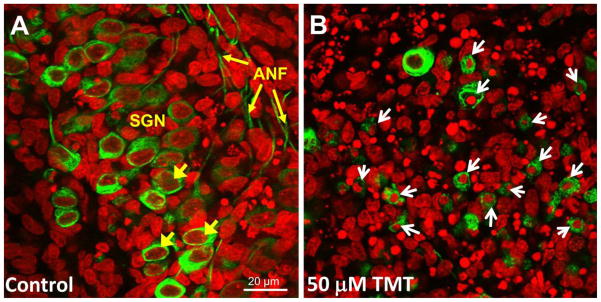
Typical confocal images showing morphological features of SGN in control culture and culture treated with 50 μM TMT for 24 h. SGN immunolabeled with Alexa Fluor488 conjugated secondary antibody (green pseudo-color). Nuclei labeled with To-Pro-3 (red pseudo-color). (A) SGN in control cultures; short yellow arrows point to SGN soma; long yellow arrow point to ANF. (B) SGN in cultures treated with 50 μM TMT. TMT exposure induced significant SGN soma shrinkage and nuclear condensation (white arrows).
To quantify the soma shrinkage, the cross sectional area of SGN soma was measured as previously reported (Wei et al., 2010). The histograms in Figure 5A–E show the numbers of SGN vs. soma size (n = 200 cells/group) in control cultures and cultures treated for 24 h with various concentrations of TMT. In controls, most SGN soma areas ranged from 150 μm2 to 250 μm2 with the major peak located at 200 μm2. In the TMT treated cultures, however, the distribution of SGN soma areas gradually shifted towards smaller values with increasing TMT dose. After treatment with 100 μM TMT, nearly all SGN had shrunken so that the major peak had decreased to 50 μm2. Figure 5F shows the mean size of SGN somas in control cultures and cultures treated with various TMT concentrations. TMT treatment caused a dose-dependent decrease in SGN soma size. Statistical analysis revealed a significant treatment effect (one-way ANOVA, P < 0.0001, F = 198.4). The differences in SGN soma sizes between all compared groups were statistically significant (Newman-Keuls post-hoc analysis, P < 0.05).
Figure 5.
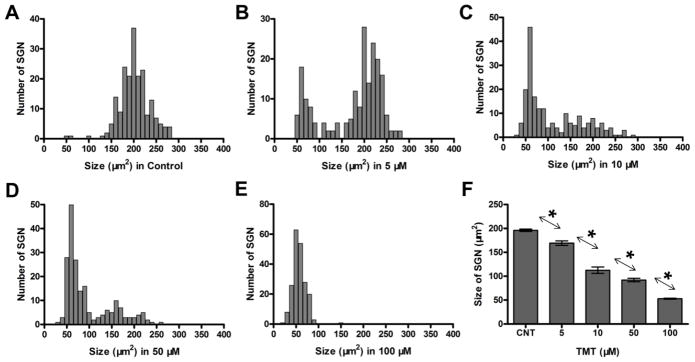
Effect of TMT on SGN soma size. Distributions (n = 200 cells/group) of SGN soma size in control group (A) and cultures treated with TMT for 24 h; treatment/dose indicated below abscissa (B–E). (F) Mean (+/−SEM, n=200 cells) size of SGN soma in control cultures and cultures treated with various concentrations of TMT. Arrows/asterisks indicate significant between group differences (*p < 0.05).
Caspase-3 activation induced by TMT
A fluorogenic probe was used to detect activated executioner caspase-3 in cochlear cultures. When the fluorogenic probe was left out of the culture, there was a complete absence of green caspase-3 labeling in the negative control (Figure 3A). When the fluorogenic probe was added to positive control cultures (Figure 6B), green caspase-3 labeling was present on some non-neuronal cells (negative for neurofilament 200 kD, dashed squares). However, a few shrunken SGN which stained positive for neurofilament and caspase-3 (circles, green/yellow) were detected. After 12 h treatment with 50 μM TMT, there were many neurofilament-positive, shrunken SGN with apoptotic features; these cells exhibited green/yellow cytoplasmic labeling (open circles) indicative of caspase-mediated programmed cell death. Green caspase-3 labeling was also present in non-neuronal cells (dashed square, neurofilament negative)
Figure 6.
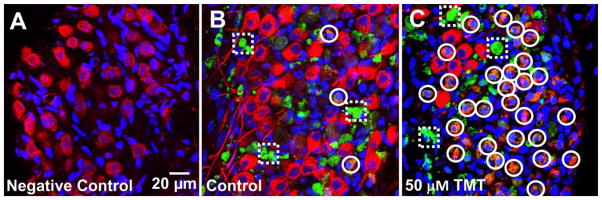
Representative confocal images of SGN showing caspase-3 labeling (pseudo-green) after 12 h in culture. (A) Note absence of green label in negative control without caspase-3 probe. (B) Positive control with caspase-3 probe, but without TMT. (C) Caspase-3 label during 50 μM TMT treatment. SGN labeled with antibody against neurofilament (pseudo-red); nuclei labeled with To-Pro-3 (blue), green caspase-3 labeling (pseudo-green). Dashed squares identify non-neuronal caspase-3 (pseudo-green) positive cells. Circles label neurofilament-positive (red/reddish pink) SGN with shrunken soma and nuclei and green or yellow cytoplasmic labeling of caspase-3.
Discussion
TMT is one of a several alkyltin derivatives that have a variety of industrial applications which include heat stabilizers in polyvinylchloride tubing, curing agents for rubber production, disinfectants in hospitals, and biocides. Human exposure to these compounds can result in a variety of clinical disorders resulting from damage or destruction of cells in the central nervous system, immune system, spleen, lung, and kidney (Brown et al., 1979, Geloso et al., 2011b, Philbert et al., 2000, Snoeij et al., 1985). The distinct range of cellular targets of these compounds make these toxins somewhat unique as they are structurally related though mechanistically function quite differently. Some of these differences can be attributed to divergences in their hydrophobicity which has the potential to control their toxic behavior (Komulainen and Bondy, 1987, Mushak et al., 1982). Differences in the affected targeted organs identified for each derivative implies there are distinct biochemical pathways disrupted within these cells resulting in the toxic specificity. For example, TMT preferentially induces neuronal damage whereas dimethyltin (DMT) induces myelin edema and astrocytes degeneration (O’Callaghan et al., 1989). The reason for this is unclear as TMT exposure as well as other alkyltin derivatives can all provoke mitochondrial dysfunction and increase ROS, yet the final disrupted tissues are different (LeBel et al., 1990, Stine et al., 1988). It has been suggested that the intrinsic discrepancy between susceptibility of neuronal and other cells to TMT may be dependent on differential regulation of the mitochondrial permeability transition pore (Qu et al., 2011). The lack of TMT damage to postnatal hair cells may be related to the immature development of proteins, cellular organelles such mitochondria and aerobic respiration (Bruce et al., 2000, Ito et al., 1995, Weaver and Schweitzer, 1994). This hypothesis could be tested by applying TMT to more mature cochlear cultures. Another possibility is that Snn, a transmembrane protein that sensitizes cells to TMT damage, is not expressed in postnatal hair cells (Billingsley et al., 2006, Buck-Koehntop et al., 2005, Davidson et al., 2004, Toggas et al., 1992). This hypothesis could be evaluated by evaluating developmental changes in Snn expression in hair cells. A third possibility is that postnatal hair cells can be damaged by TMT, but only at higher doses or longer treatment durations than those employed in this study. This could be tested by using higher doses of TMT or applying TMT for longer durations.
Although TMT toxicity has been extensively studied characterization of the actual molecular processes and its intracellular targets have not been adequately described. In general, TMT toxicity results in apoptotic cell death, which includes chromatin condensation, nuclear fragmentation, mitochondrial dysfunction, ROS production, membrane blebbing, and caspase activation (Geloso et al., 2002, Jenkins and Barone, 2004). TMT selectively destroyed neurons located in several brain areas including the neocortex, amygdala, and olfactory tubercle, although the most prominent effects were seen in the hippocampus (Balaban et al., 1988, Brown et al., 1979, Thompson et al., 1996). Recent studies suggest that the highly conserved, 88-amino acid protein, Snn may mediate the selective toxicity of organotins (Buck-Koehntop et al., 2005, Davidson et al., 2004, Toggas et al., 1992). This is based on the observation that Snn predominates in tissues with high toxicity to TMT, which include the CNS, immune system, spleen, kidney and lung but is essentially lacking in cells that exhibit considerable resistance to TMT. Snn peptide has been shown to bind TMT in a 1:1 ratio and can dealkylate TMT to DMT (Davidson et al., 2004). Transfection of TMT-resistant NIH-3T3 mouse fibroblasts with Snn increased cytotoxicity to both TMT and DMT. The selective localization of Snn, along with its mitochondrial localization, coupled with its ability to bind and dealkylate TMT, suggests a possible mechanism by which alkyltin toxicity may be mediated.
In the context of hearing and the inner ear, TMT induces a permanent hearing loss and cochlear damage which is most pronounced at the high frequencies (Chang and Dyer, 1983a, Eastman et al., 1987, Ruppert et al., 1984). Consistent with this is the observation that OHC in the base of the cochlea appear to be sensitive to TMT toxicity (Crofton et al., 1990, Fechter and Liu, 1995, Hoeffding and Fechter, 1991). Taken together, these results suggest that TMT is mainly toxic to hair cells in adult animals, although there is evidence that is also damages SGN (Chang and Dyer, 1983b). On the other hand, the acute effects of TMT in vivo indicate that it has limited effects on both the cochlear microphonic generated by the OHC, the endolymphatic potential generated by the stria vascularis and the summating potential largely generated by IHC (Clerici et al., 1991, Durrant et al., 1998, Fechter and Liu, 1994). In contrast, TMT rapidly reduced the CAP. Since the CAP reflects the summed neural response from the auditory nerve, these results suggest that the acute effects of TMT predominantly damage the SGN consistent with our in vitro results. The large TMT-induced effects on the CAP occurred at a dose of 0.2 mg/kg, equivalent to 1.5 μM assuming a uniform distribution throughout the body (Fechter and Liu, 1994). This predicted dose is only slightly lower than the 5 μM TMT dose that caused a 30% loss of SGN (Fig. 3F). Given the obvious experimental differences (e.g., postnatal vs. adult, function vs. structure), the TMT anatomical damage seen in our cochlear cultures is in reasonable accord with the CAP data.
The TMT toxicity profile in the developing inner ear is largely unknown. Because of the known neurotoxic profile of TMT in the CNS, it is also reasonable to speculate that auditory neurons within the developing cochlea may, in fact, be a more sensitive target to this alkyltin derivative. To test this hypothesis, we used rat postnatal cochlear organotypic culture as a model system to determine the selective neurotoxic actions of TMT on the hair cells, supporting cells and neurons in the cochlea. Our results clearly demonstrate that TMT preferentially destroys ANF and SGN in rat cochlear cultures in a dose-dependent manner. Qualitatively, damage to SGN and ANF appeared relatively uniform along the length of the cochlea; therefore we quantified SGN and ANF losses in the middle of the cochlea. SGN and ANF degeneration occurred at concentrations as low as 5 μM after only 24 h treatment and nearly all the ANF and SGN were destroyed after 24 h treatment with 100 μM TMT. In contrast to prior studies in adult rats, there was little or no damage to either OHC or IHC even at the highest TMT concentration of 100 μM that destroyed virtually all SGN. This is not totally surprising as hair cells share some similarities with astrocytes, which contain the astrocytic marker, glial fibrillary acidic protein (GFAP) (Moriya et al., 1993) and are less sensitive to TMT toxicity (Gunasekar et al., 2001).
The mechanisms underlying the differences in TMT sensitivity between postnatal vs. adult SGN and hair cells are currently unknown. As noted above, similarities shared with astrocyte may promote the survival of hair cells relative to neurons after TMT exposure. Additionally, it has been demonstrated that hippocampal neurons containing the calcium-binding protein calretinin (CR) and parvalbumin (PV) are resistant to TMT-induced neuronal degeneration whereas those containing calbindin (CB) are selectively damaged by TMT (Geloso et al., 1996, 1997). In contrast, both PV and CB-positive neurons are sensitive to TMT neurotoxicity and only CR-positive neurons are resistant (Geloso et al., 1998, Reuhl et al., 1983). This, however, seems unlikely to account for this difference in the cochlea because CR and PV are also highly expressed by SGN (Dechesne et al., 1991, Soto-Prior et al., 1995). Unfortunately, expression of Snn, identified as a critical factor in TMT-induced selective cytotoxicity in neurons in the CNS (Davidson et al., 2004, Reese et al., 2005) has not been measured in the inner ear. Thus, further studies are needed to determine which cells in the cochlea express Snn and whether Snn expression changes during development.
Our data further demonstrate, for the first time, that TMT induces SGN cell death by apoptosis in cochlear cultures. TMT exposure caused significant morphological alterations in SGN characterized by soma shrinkage and nuclear condensation, morphological hallmarks of cells undergoing apoptosis. In addition, activation of caspase-3, a major executioner in the caspase-dependent apoptotic pathway, was also observed in the SGN during the early stage of TMT treatment, consistent with prior findings in CNS (Fiedorowicz et al., 2001, Jenkins et al., 2004). TMT exposure has also been shown to enhance ROS formation leading to apoptotic cell death in a number of different cell lines and primary cultures (Wang et al., 2008, Zhang et al., 2006). ROS production could damage mitochondria and activate an apoptotic pathway mediated by the activation of mitochondrial permeability transition pore, release of cytochrome c and activation of effector caspase-3 (Qu et al., 2011). Consistent with ROS involvement, TMT-induced neuronal apoptosis can be reduced by antioxidants that reduce ROS expression (Cookson et al., 1998, Qu et al., 2011, Shin et al., 2005).
Model of SGN Degeneration
Our results clearly demonstrated that TMT selectively destroys ANF and SGN in P3 cochlear organotypic cultures in a dose-dependent manner. Since there was no evidence of hair cell damage even at the highest dose employed, 100 μM, TMT could conceivably be used to develop several experimental models of selective SGN death. If TMT were applied to the round window of the postnatal cochlea, it could conceivably result in partial destruction of SGN with retention of OHC and IHC. In cases such as this, the cochlear amplification and sharp tuning provided by the OHC would remain intact, but the neural input to the central nervous system would be greatly reduced creating a model that could be used to examine the perceptual and electrophysiological consequences of sensory deprivation (Lobarinas et al., 2013). Efforts are underway to use stem cells and trophic factors to regenerate SGN and their peripheral ANF (Diensthuber et al., 2014). The TMT model of selective SGN degeneration could conceivably be used as experimental platform to determine if and how stem cells form synaptic connections to hair cells in the organ of Corti and neurons in the cochlear nucleus. Efforts aimed at replenishing missing SGN are critical for the transmission of acoustic or electrical stimulation from the cochlea to the auditory brainstem (Bas et al., 2014, Berkingali et al., 2008, Schmiedt et al., 2002, Zhang et al., 2013).
Summary
Our results show that TMT selectively damages SGN and ANF in postnatal rat cochlear cultures without damaging the hair cells. Whether local application of TMT produces a similar pattern of damage in vivo in postnatal or adult cochlea remains an important unanswered question. TMT-induced auditory neuronal death occurs via a caspase-mediated apoptotic cell death pathway consistent with soma shrinkage and nuclear condensation, morphological hallmarks of programmed cell death. Further studies are required to determine the molecular mechanisms underlying the selective destruction of postnatal SGN and ANF in vitro and the reasons why postnatal hair cells are resistant to TMT. An important unanswered question is whether a development shift occurs in TMT toxicity such that hair cells become more vulnerable with advancing age due to neural myelination or developmental changes in the antioxidant enzymes expressed in hair cells and neurons.
Acknowledgments
Funding: This research was supported in part by grant from the National Institute for Occupational Safety and Health, R01 OH010235, the National Nature Science Foundation of China (No. 81170912), the Major State Basic Research Development Program of China (No. 2014CB943003) and Hunan Provincial Innovation Foundation for Postgraduate 71380100016.
Footnotes
Conflict of interest: The authors declare that there are no conflicts of interest.
Animal welfare: This research was approved by the University at Buffalo Institutional Animal Care and Use Committee. All applicable international, national and institutional guidelines for the care and use of animals were followed.
References
- Aldridge WN, Street BW, Skilleter DN. Oxidative phosphorylation. Halide-dependent and halide-independent effects of triorganotin and trioganolead compounds on mitochondrial functions. Biochem J. 1977;168:353–64. doi: 10.1042/bj1680353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SF, LeBel CP, Bondy SC. Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxicology. 1992;13:637–48. [PubMed] [Google Scholar]
- Balaban CD, O’Callaghan JP, Billingsley ML. Trimethyltin-induced neuronal damage in the rat brain: comparative studies using silver degeneration stains, immunocytochemistry and immunoassay for neuronotypic and gliotypic proteins. Neuroscience. 1988;26:337–61. doi: 10.1016/0306-4522(88)90150-9. [DOI] [PubMed] [Google Scholar]
- Barclay M, Ryan AF, Housley GD. Type I vs type II spiral ganglion neurons exhibit differential survival and neuritogenesis during cochlear development. Neural Dev. 2011;6:33. doi: 10.1186/1749-8104-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas E, Van De Water TR, Lumbreras V, Rajguru S, Goss G, Hare JM, et al. Adult human nasal mesenchymal-like stem cells restore cochlear spiral ganglion neurons after experimental lesion. Stem Cells Dev. 2014;23:502–14. doi: 10.1089/scd.2013.0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkingali N, Warnecke A, Gomes P, Paasche G, Tack J, Lenarz T, et al. Neurite outgrowth on cultured spiral ganglion neurons induced by erythropoietin. Hear Res. 2008;243:121–6. doi: 10.1016/j.heares.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Billingsley ML, Yun J, Reese BE, Davidson CE, Buck-Koehntop BA, Veglia G. Functional and structural properties of stannin: roles in cellular growth, selective toxicity, and mitochondrial responses to injury. J Cell Biochem. 2006;98:243–50. doi: 10.1002/jcb.20809. [DOI] [PubMed] [Google Scholar]
- Braman RS, Tompkins MA. Separation and determination of nanogram amounts of inorganic tin and methyltin compounds in the environment. Anal Chem. 1979;51:12–9. doi: 10.1021/ac50037a011. [DOI] [PubMed] [Google Scholar]
- Brown AW, Aldridge WN, Street BW, Verschoyle RD. The behavioral and neuropathologic sequelae of intoxication by trimethyltin compounds in the rat. Am J Pathol. 1979;97:59–82. [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Ledwith JVIII. Projections of thin (type-II) and thick (type-I) auditory-nerve fibers into the cochlear nucleus of the mouse. Hear Res. 1990;49:105–18. doi: 10.1016/0378-5955(90)90098-a. [DOI] [PubMed] [Google Scholar]
- Bruce LL, Christensen MA, Warr WB. Postnatal development of efferent synapses in the rat cochlea. J Comp Neurol. 2000;423:532–48. doi: 10.1002/1096-9861(20000731)423:3<532::aid-cne14>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Buck-Koehntop BA, Mascioni A, Buffy JJ, Veglia G. Structure, dynamics, and membrane topology of stannin: a mediator of neuronal cell apoptosis induced by trimethyltin chloride. J Mol Biol. 2005;354:652–65. doi: 10.1016/j.jmb.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Chang LW, Dyer RS. A time-course study of trimethyltin induced neuropathology in rats. Neurobehav Toxicol Teratol. 1983a;5:443–59. [PubMed] [Google Scholar]
- Chang LW, Dyer RS. Trimethyltin induced pathology in sensory neurons. Neurobehav Toxicol Teratol. 1983b;5:673–96. [PubMed] [Google Scholar]
- Cima F. Tin: Environmental Pollution and Health Effects. In: Jerome ON, editor. Encyclopedia of Environmental Health. Burlington: Elsevier; 2011. pp. 351–9. [Google Scholar]
- Clerici WJ, Ross B, JrFechter LD. Acute ototoxicity of trialkyltins in the guinea pig. Toxicol Appl Pharmacol. 1991;109:547–56. doi: 10.1016/0041-008x(91)90017-9. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Slamon ND, Pentreath VW. Glutathione modifies the toxicity of triethyltin and trimethyltin in C6 glioma cells. Arch Toxicol. 1998;72:197–202. doi: 10.1007/s002040050488. [DOI] [PubMed] [Google Scholar]
- Crofton KM, Dean KF, Menache MG, Janssen R. Trimethyltin effects on auditory function and cochlear morphology. Toxicol Appl Pharmacol. 1990;105:123–32. doi: 10.1016/0041-008x(90)90364-z. [DOI] [PubMed] [Google Scholar]
- Davidson CE, Reese BE, Billingsley ML, Yun JK. Stannin, a protein that localizes to the mitochondria and sensitizes NIH-3T3 cells to trimethyltin and dimethyltin toxicity. Mol Pharmacol. 2004;66:855–63. doi: 10.1124/mol.104.001719. [DOI] [PubMed] [Google Scholar]
- Dechesne CJ, Winsky L, Kim HN, Goping G, Vu TD, Wenthold RJ, et al. Identification and ultrastructural localization of a calretinin-like calcium-binding protein (protein 10) in the guinea pig and rat inner ear. Brain Res. 1991;560:139–48. doi: 10.1016/0006-8993(91)91224-o. [DOI] [PubMed] [Google Scholar]
- Diensthuber M, Zecha V, Wagenblast J, Arnhold S, Edge AS, Stover T. Spiral ganglion stem cells can be propagated and differentiated into neurons and glia. Biores Open Access. 2014;3:88–97. doi: 10.1089/biores.2014.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D, Roth J, Salvi R. Manganese is toxic to spiral ganglion neurons and hair cells in vitro. Neurotoxicology. 2011;32:233–41. doi: 10.1016/j.neuro.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D, Salvi R, Roth JA. Cellular localization and developmental changes of Zip8, Zip14 and transferrin receptor 1 in the inner ear of rats. Biometals. 2014;27:731–44. doi: 10.1007/s10534-014-9765-0. [DOI] [PubMed] [Google Scholar]
- Dong Y, Ding D, Jiang H, Shi JR, Salvi R, Roth JA. Ototoxicity of paclitaxel in rat cochlear organotypic cultures. Toxicol Appl Pharmacol. 2014;280:526–33. doi: 10.1016/j.taap.2014.08.022. [DOI] [PubMed] [Google Scholar]
- Dorman DC. An integrative approach to neurotoxicology. Toxicol Pathol. 2000;28:37–42. doi: 10.1177/019262330002800106. [DOI] [PubMed] [Google Scholar]
- Durrant JD, Wang J, Ding DL, Salvi RJ. Are inner or outer hair cells the source of summating potentials recorded from the round window? J Acoust Soc Am. 1998;104:370–7. doi: 10.1121/1.423293. [DOI] [PubMed] [Google Scholar]
- Dyer RS, Walsh TJ, Wonderlin WF, Bercegeay M. The trimethyltin syndrome in rats. Neurobehav Toxicol Teratol. 1982;4:127–33. [PubMed] [Google Scholar]
- Eastman CL, Young JS, Fechter LD. Trimethyltin ototoxicity in albino rats. Neurotoxicol Teratol. 1987;9:329–32. doi: 10.1016/0892-0362(87)90026-2. [DOI] [PubMed] [Google Scholar]
- Fechter LD, Liu Y. Trimethyltin disrupts N1 sensitivity, but has limited effects on the summating potential and cochlear microphonic. Hear Res. 1994;78:189–96. doi: 10.1016/0378-5955(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Fechter LD, Liu Y. Elevation of intracellular calcium levels in spiral ganglion cells by trimethyltin. Hear Res. 1995;91:101–9. doi: 10.1016/0378-5955(95)00176-x. [DOI] [PubMed] [Google Scholar]
- Fiedorowicz A, Figiel I, Kaminska B, Zaremba M, Wilk S, Oderfeld-Nowak B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001;912:116–27. doi: 10.1016/s0006-8993(01)02675-0. [DOI] [PubMed] [Google Scholar]
- Geloso MC, Corvino V, Michetti F. Trimethyltin-induced hippocampal degeneration as a tool to investigate neurodegenerative processes. Neurochem Int. 2011a;58:729–38. doi: 10.1016/j.neuint.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Geloso MC, Corvino V, Michetti F. Trimethyltin-induced hippocampal degeneration as a tool to investigate neurodegenerative processes. Neurochem Int. 2011b;58:729–38. doi: 10.1016/j.neuint.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Geloso MC, Vercelli A, Corvino V, Repici M, Boca M, Haglid K, et al. Cyclooxygenase-2 and Caspase 3 Expression in Trimethyltin-Induced Apoptosis in the Mouse Hippocampus. Experimental Neurology. 2002;175:152–60. doi: 10.1006/exnr.2002.7866. [DOI] [PubMed] [Google Scholar]
- Geloso MC, Vinesi P, Michetti F. Parvalbumin-immunoreactive neurons are not affected by trimethyltin-induced neurodegeneration in the rat hippocampus. Exp Neurol. 1996;139:269–77. doi: 10.1006/exnr.1996.0100. [DOI] [PubMed] [Google Scholar]
- Geloso MC, Vinesi P, Michetti F. Calretinin-containing neurons in trimethyltin-induced neurodegeneration in the rat hippocampus: an immunocytochemical study. Exp Neurol. 1997;146:67–73. doi: 10.1006/exnr.1997.6491. [DOI] [PubMed] [Google Scholar]
- Geloso MC, Vinesi P, Michetti F. Neuronal subpopulations of developing rat hippocampus containing different calcium-binding proteins behave distinctively in trimethyltin-induced neurodegeneration. Exp Neurol. 1998;154:645–53. doi: 10.1006/exnr.1998.6949. [DOI] [PubMed] [Google Scholar]
- Gunasekar PG, Mickova V, Kotyzova D, Li L, Borowitz JL, Eybl V, et al. Role of astrocytes in trimethyltin neurotoxicity. J Biochem Mol Toxicol. 2001;15:256–62. doi: 10.1002/jbt.10004. [DOI] [PubMed] [Google Scholar]
- Hoch M. Organotin compounds in the environment — an overview. Applied Geochemistry. 2001;16:719–43. [Google Scholar]
- Hoeffding V, Fechter LD. Trimethyltin disrupts auditory function and cochlear morphology in pigmented rats. Neurotoxicol Teratol. 1991;13:135–45. doi: 10.1016/0892-0362(91)90003-f. [DOI] [PubMed] [Google Scholar]
- Ito M, Spicer SS, Schulte BA. Cytological changes related to maturation of the organ of Corti and opening of Corti’s tunnel. Hear Res. 1995;88:107–23. doi: 10.1016/0378-5955(95)00106-e. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Barone S. The neurotoxicant trimethyltin induces apoptosis via caspase activation, p38 protein kinase, and oxidative stress in PC12 cells. Toxicol Lett. 2004;147:63–72. doi: 10.1016/j.toxlet.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Ehman K, Barone S., Jr Structure-activity comparison of organotin species: dibutyltin is a developmental neurotoxicant in vitro and in vivo. Brain Res Dev Brain Res. 2004;151:1–12. doi: 10.1016/j.devbrainres.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Koczyk D. How does trimethyltin affect the brain: facts and hypotheses. Acta Neurobiol Exp (Wars) 1996;56:587–96. doi: 10.55782/ane-1996-1164. [DOI] [PubMed] [Google Scholar]
- Komulainen H, Bondy SC. Transient elevation of intrasynaptosomal free calcium by putrescine. Brain Res. 1987;401:50–4. doi: 10.1016/0006-8993(87)91162-0. [DOI] [PubMed] [Google Scholar]
- Kuramoto N, Seko K, Sugiyama C, Shuto M, Ogita K. Trimethyltin initially activates the caspase 8/caspase 3 pathway for damaging the primary cultured cortical neurons derived from embryonic mice. J Neurosci Res. 2011;89:552–61. doi: 10.1002/jnr.22588. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ali SF, McKee M, Bondy SC. Organometal-induced increases in oxygen reactive species: the potential of 2′,7′-dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicol Appl Pharmacol. 1990;104:17–24. doi: 10.1016/0041-008x(90)90278-3. [DOI] [PubMed] [Google Scholar]
- Lim DJ. Ultrastructural cochlear changes following acoustic hyperstimulation and ototoxicity. Ann Otol Rhinol Laryngol. 1976;85:740–51. doi: 10.1177/000348947608500604. [DOI] [PubMed] [Google Scholar]
- Lobarinas E, Salvi R, Ding D. Insensitivity of the audiogram to carboplatin induced inner hair cell loss in chinchillas. Hear Res. 2013;302:113–20. doi: 10.1016/j.heares.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misiti F, Orsini F, Clementi ME, Lattanzi W, Giardina B, Michetti F. Mitochondrial oxygen consumption inhibition importance for TMT-dependent cell death in undifferentiated PC12 cells. Neurochem Int. 2008;52:1092–9. doi: 10.1016/j.neuint.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Morita M, Imai H, Liu Y, Xu X, Sadamatsu M, Nakagami R, et al. FK506-protective effects against trimethyltin neurotoxicity in rats: hippocampal expression analyses reveal the involvement of periarterial osteopontin. Neuroscience. 2008;153:1135–45. doi: 10.1016/j.neuroscience.2008.01.078. [DOI] [PubMed] [Google Scholar]
- Moriya K, Sekitani T, Yamashita H. Glial fibrillary acidic protein (GFAP)-like immunoreactivity in the vestibular endorgan of the rat. Acta Otolaryngol Suppl. 1993;503:119–20. doi: 10.3109/00016489309128091. [DOI] [PubMed] [Google Scholar]
- Mundy WR, Freudenrich TM. Apoptosis of cerebellar granule cells induced by organotin compounds found in drinking water: involvement of MAP kinases. Neurotoxicology. 2006;27:71–81. doi: 10.1016/j.neuro.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mushak P, Krigman MR, Mailman RB. Comparative organotin toxicity in the developing rat: somatic and morphological changes and relationship to accumulation of total tin. Neurobehav Toxicol Teratol. 1982;4:209–15. [PubMed] [Google Scholar]
- O’Callaghan JP, Niedzwiecki DM, Means JC. Variations in the neurotoxic potency of trimethyltin. Brain Res Bull. 1989;22:637–42. doi: 10.1016/0361-9230(89)90083-x. [DOI] [PubMed] [Google Scholar]
- Perkins RE, Morest DK. A study of cochlear innervation patterns in cats and rats with the Golgi method and Nomarkski Optics. J Comp Neurol. 1975;163:129–58. doi: 10.1002/cne.901630202. [DOI] [PubMed] [Google Scholar]
- Philbert MA, Billingsley ML, Reuhl KR. Mechanisms of injury in the central nervous system. Toxicol Pathol. 2000;28:43–53. doi: 10.1177/019262330002800107. [DOI] [PubMed] [Google Scholar]
- Piacentini R, Gangitano C, Ceccariglia S, Del Fa A, Azzena GB, Michetti F, et al. Dysregulation of intracellular calcium homeostasis is responsible for neuronal death in an experimental model of selective hippocampal degeneration induced by trimethyltin. J Neurochem. 2008;105:2109–21. doi: 10.1111/j.1471-4159.2008.05297.x. [DOI] [PubMed] [Google Scholar]
- Qu M, Zhou Z, Chen C, Li M, Pei L, Chu F, et al. Lycopene protects against trimethyltin-induced neurotoxicity in primary cultured rat hippocampal neurons by inhibiting the mitochondrial apoptotic pathway. Neurochem Int. 2011;59:1095–103. doi: 10.1016/j.neuint.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Reese BE, Davidson C, Billingsley ML, Yun J. Protein kinase C epsilon regulates tumor necrosis factor-alpha-induced stannin gene expression. J Pharmacol Exp Ther. 2005;314:61–9. doi: 10.1124/jpet.105.084236. [DOI] [PubMed] [Google Scholar]
- Reuhl KR, Smallridge EA, Chang LW, Mackenzie BA. Developmental effects of trimethyltin intoxication in the neonatal mouse. I. Light microscopic studies. Neurotoxicology. 1983;4:19–28. [PubMed] [Google Scholar]
- Ruppert PH, Dean KF, Reiter LW. Trimethyltin disrupts acoustic startle responding in adult rats. Toxicol Lett. 1984;22:33–8. doi: 10.1016/0378-4274(84)90042-0. [DOI] [PubMed] [Google Scholar]
- Schmiedt RA, Okamura HO, Lang H, Schulte BA. Ouabain application to the round window of the gerbil cochlea: a model of auditory neuropathy and apoptosis. J Assoc Res Otolaryngol. 2002;3:223–33. doi: 10.1007/s1016200220017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin EJ, Suh SK, Lim YK, Jhoo WK, Hjelle OP, Ottersen OP, et al. Ascorbate attenuates trimethyltin-induced oxidative burden and neuronal degeneration in the rat hippocampus by maintaining glutathione homeostasis. Neuroscience. 2005;133:715–27. doi: 10.1016/j.neuroscience.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Snoeij NJ, van Iersel AA, Penninks AH, Seinen W. Toxicity of triorganotin compounds: comparative in vivo studies with a series of trialkyltin compounds and triphenyltin chloride in male rats. Toxicol Appl Pharmacol. 1985;81:274–86. doi: 10.1016/0041-008x(85)90164-4. [DOI] [PubMed] [Google Scholar]
- Soto-Prior A, Cluzel M, Renard N, Ripoll C, Lavigne-Rebillard M, Eybalin M, et al. Molecular cloning and expression of α parvalbumin in the guinea pig cochlea. Molecular Brain Research. 1995;34:337–42. doi: 10.1016/0169-328x(95)00205-7. [DOI] [PubMed] [Google Scholar]
- Spoendlin HH, Gacek RR. Electron Microscopic Study of the Efferent and Afferent Innervation of the Organ of Corti in the Cat. Ann Otol Rhinol Laryngol. 1963;72:660–86. doi: 10.1177/000348946307200307. [DOI] [PubMed] [Google Scholar]
- Stine KE, Reiter LW, Lemasters JJ. Alkyltin inhibition of ATPase activities in tissue homogenates and subcellular fractions from adult and neonatal rats. Toxicol Appl Pharmacol. 1988;94:394–406. doi: 10.1016/0041-008x(88)90280-3. [DOI] [PubMed] [Google Scholar]
- Thompson TA, Lewis JM, Dejneka NS, Severs WB, Polavarapu R, Billingsley ML. Induction of apoptosis by organotin compounds in vitro: neuronal protection with antisense oligonucleotides directed against stannin. J Pharmacol Exp Ther. 1996;276:1201–16. [PubMed] [Google Scholar]
- Toggas SM, Krady JK, Billingsley ML. Molecular neurotoxicology of trimethyltin: identification of stannin, a novel protein expressed in trimethyltin-sensitive cells. Mol Pharmacol. 1992;42:44–56. [PubMed] [Google Scholar]
- Wang L, Ding D, Salvi R, Roth JA. Nicotinamide adenine dinucleotide prevents neuroaxonal degeneration induced by manganese in cochlear organotypic cultures. Neurotoxicology. 2014;40:65–74. doi: 10.1016/j.neuro.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Wang X, Cai J, Zhang J, Wang C, Yu A, Chen Y, et al. Acute trimethyltin exposure induces oxidative stress response and neuronal apoptosis in Sebastiscus marmoratus. Aquat Toxicol. 2008;90:58–64. doi: 10.1016/j.aquatox.2008.07.017. [DOI] [PubMed] [Google Scholar]
- Weaver SP, Schweitzer L. Development of gerbil outer hair cells after the onset of cochlear function: an ultrastructural study. Hear Res. 1994;72:44–52. doi: 10.1016/0378-5955(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Wei L, Ding D, Salvi R. Salicylate-induced degeneration of cochlea spiral ganglion neurons-apoptosis signaling. Neuroscience. 2010;168:288–99. doi: 10.1016/j.neuroscience.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JS, Fechter LD. Trimethyltin exposure produces an unusual form of toxic auditory damage in rats. Toxicol Appl Pharmacol. 1986;82:87–93. doi: 10.1016/0041-008x(86)90441-2. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li L, Prabhakaran K, Borowitz JL, Isom GE. Trimethyltin-induced apoptosis is associated with upregulation of inducible nitric oxide synthase and Bax in a hippocampal cell line. Toxicol Appl Pharmacol. 2006;216:34–43. doi: 10.1016/j.taap.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Zhang PZ, He Y, Jiang XW, Chen FQ, Chen Y, Shi L, et al. Stem cell transplantation via the cochlear lateral wall for replacement of degenerated spiral ganglion neurons. Hear Res. 2013;298:1–9. doi: 10.1016/j.heares.2013.01.022. [DOI] [PubMed] [Google Scholar]