

Abstract
A growing body of evidence supports a role for glial-produced neuroimmune factors, including the cytokine IL-6, in CNS physiology and pathology. CNS expression of IL-6 has been documented in the normal CNS at low levels and at elevated levels in several neurodegenerative or psychiatric disease states as well as in CNS infection and injury. The altered CNS function associated with these conditions raises the possibility that IL-6 has neuronal or synaptic actions. Studies in in vitro and in vivo models confirmed this possibility and showed that IL-6 can regulate a number of important neuronal and synaptic functions including synaptic transmission and synaptic plasticity, an important cellular mechanism of memory and learning. Behavioral studies in animal models provided further evidence of an important role for IL-6 as a regulator of CNS pathways that are critical to cognitive function. This review summarizes studies that have lead to our current state of knowledge. In spite of the progress that has been made, there is a need for a greater understanding of the physiological and pathophysiological actions of IL-6 in the CNS, the mechanisms underlying these actions, conditions that induce production of IL-6 in the CNS and therapeutic strategies that could ameliorate or promote IL-6 actions.
Keywords: Hippocampus, Cerebellum, MAPK, JAK/STAT, Glia, Ca2+, NMDA, Neurologic disorders, Memory, Cognitive function
1. Introduction
The cytokine IL-6 is a small signaling glycoprotein (21–30 kD; 212 amino acids with variable glycosylation sites) first identified and characterized as an important signaling molecule in the immune system. IL-6 was initially known as B cell stimulatory factor-2 based on its action to induced proliferation and differentiation of B cells (Hirano et al., 1985; Kishimoto, 2006; Nakajima et al., 1985). In 1986, the cDNA encoding the human B cell stimulatory factor-2 was cloned and the similarity of the cDNA and protein structure to that of another immune factor with similar functional activities (i.e., interferon-β2) became apparent. This discovery led to the renaming of the two identical proteins IL-6, and the recognition that IL-6 is a pleiotropic cytokine with a diverse set of actions that vary depending on the target cell type (Yasukawa et al., 1987). Further studies resulted in the cloning of an integral plasma membrane protein, the IL-6 receptor (IL-6R), at which IL-6 acts to produce biological effects (Yamasaki et al., 1988). By the 1990’s, studies of neurological disorders and CNS injury and disease (e.g., HIV infection) established that IL-6 and other immune factors were present in the CNS at elevated levels during these conditions (Benveniste, 1992; Zhao and Schwartz, 1998). Research into the source of these immune factors eventually lead to the understanding that cells of the CNS, and in particular glial cells, produce immune factors and that glial cells serve as an innate immune system of the CNS that plays an important role in CNS physiological and pathology (Dong and Benveniste, 2001; Farina et al., 2007; Gruol and Nelson, 1997; Zhao and Schwartz, 1998). Immune factors that originate within the CNS are now referred to as neuroimmune factors to distinguish them from immune factors produced by cells of the peripheral immune system as they transverse through the CNS. Experimental research, which has primarily focused on the hippocampal and cerebellar regions of the CNS, has now established that IL-6 produced within the CNS can regulate neuronal and synaptic function and behavior. These actions may play an important role in normal CNS function and in altered CNS function associated with certain CNS disorders.
2. CNS cells produce IL-6
Like other neuroimmune factors, CNS astrocytes are a primary source of IL-6 in the CNS (Choi et al., 2014; Dong and Benveniste, 2001; Farina et al., 2007; Nakamachi et al., 2012). Microglia are also an important source of IL-6 in the CNS (Ye and Johnson, 1999). Neurons can produce IL-6 under some conditions, particularly during CNS disease and injury or during strong neuronal activity (Arruda et al., 1998; Hans et al., 1999; Juttler et al., 2002; Ringheim et al., 1995; Sallmann et al., 2000). A number of factors have been shown to induce IL-6 synthesis and release in astrocytes (Van Wagoner et al., 1999), but considerably less is known about this process in neurons. Some factors such as IL-1β have been shown to induce both astrocytes and neurons to produce IL-6 (Aloisi et al., 1995; Bergamaschi et al., 2006; Norris et al., 1994; Ringheim et al., 1995; Tsakiri et al., 2008a). Production of IL-6 typically involves synthesis and packaging in the endoplasmic reticulum with subsequent transferal to the Golgi apparatus for further processing and secretion. IL-6 processing and secretion in glial is thought to occur via a classical pathway involving sequestration of IL-6 into membrane-bound organelles that are transported to the cell membrane where fusion and exocytosis occurs (Stanley and Lacy, 2010). It is unknown if the classical pathway is used by neurons. However, studies of cultured cortical neurons revealed a different pathway. In these studies strong neuronal stimulation induced IL-6 to be transported from the Golgi to synaptic terminals through an axonal transport system and released at or near the synapse (Tsakiri et al., 2008b). IL-6 levels in the CNS are typically low under normal conditions, presumably due low levels of constitutive IL-6 expression by CNS cells. Stimulation-induced IL-6 expression by glia or neurons can significantly elevate levels of IL-6 in the CNS.
3. CNS cells express receptors for IL-6
IL-6 produces biological actions through interactions with the IL-6 receptor system, a multi-subunit complex that consists of a ligand binding subunit, IL-6Rα, and signal transducing subunit, glycoprotein 130 (gp130) (Fig 1). IL-6Rα, exist in two forms, a transmembrane bound form and a soluble form. The soluble form is generated by cleavage of the transmembrane form or by alternative splicing of the IL-6Rα mRNA (Lust et al., 1992; Mullberg et al., 1999; Rose-John et al., 2006). Signaling through the membrane bound form is referred to as classic signaling, whereas signaling through the soluble form is called trans-signaling. In classical signaling, when the membrane bound form of IL-6Rα is occupied by IL-6, the complex associates with two transmembrane-bound gp130 subunits to form a heterotrimer that then interacts with downstream signaling molecules to initiate a biological action, for example gene expression (Fig. 1A). In trans-signaling, IL-6 and the soluble receptor bind in the extracellular fluids. The IL-6/IL-6Rα complex can then bind to the membrane bound gp130 subunits and initiate downstream signaling (Fig. 1B). Because gp130 is widely expressed in the CNS (Watanabe et al.,1996), trans-signaling enables cells that do not express membrane bound IL-6Rα to respond to IL-6. Studies show that trans-signaling can play an important role in pathological actions of IL-6 (Burton and Johnson, 2012; Campbell et al., 2014; Garcia-Oscos et al., 2012).
Fig. 1.
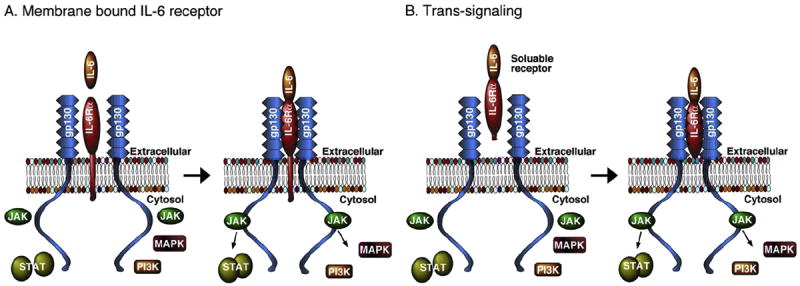
Simplified diagram showing IL-6 signal transduction components.
IL-6Rα mRNA and proteins are expressed in many CNS regions including the hippocampus and cerebellum (Dame and Juul, 2000; Gadient and Otten, 1994; Hampel et al., 2005; Morikawa et al., 2000; Schobitz et al., 1993). Both neurons and glia of the CNS express IL-6Rα and gp130 (Ha and King, 2000; Hampel et al., 2005; Nelson et al., 1999; Vollenweider et al., 2003; Watanabe et al., 1996). In the hippocampus, the distribution of IL-6Rα has been studied using immunohistochemistry. Labeling in glia was modest but neuronal labeling was prominent both in the perikaryon and outlining the apical dendrites, with dense labeling in pyramidal neurons as well as the granular neurons of the dentate gyrus (Vollenweider et al., 2003). Studies on the subcellular distribution of IL-6R subunits in rat cerebral cortex showed that both IL-6Rα and gp130 are localized in pre- and postsynaptic membranes (D’Arcangelo et al., 2000).
Several downstream signaling partners are associated with IL-6R activation (Fig. 1). Formation of the IL-6/IL-6R/gp130 complex triggers activation (i.e., phosphorylation) of tyrosine kinases of the Janus kinase (JAK) family that are associated with gp130. Activated JAKs trigger a sequence of events that results in phosphorylated gp130, which recruits and activates (i.e., phosphorylates) the signal transducer and activator of transcription-3 (STAT3). In addition to activation of the STAT pathway, the RAS/mitogen-activated protein kinase (p44/42 MAPK, also called ERK1/2; MAPK) and phosphatidylinositol-3 kinase (PI3K) pathways can be activated by the IL-6/IL-6R/gp130 complex. JAK2 and STAT3 have been shown to be located within the postsynaptic density at hippocampal synapses (Murase and McKay, 2014; Murata et al., 2000; Nicolas et al., 2012), while p44/42 MAPK is expressed in dendrites as well as the soma of hippocampal neurons (Dudek and Fields, 2001; Fiore et al., 1993). Thus, IL-6R and associated signal transduction molecules are appropriately positioned to influence synaptic function.
4. Clinical studies implicate a role for IL-6 in CNS pathology
Clinical studies have been instrumental in establishing that neuroimmune factors including IL-6 are expressed in the CNS during CNS disease, injury, neurodegenerative disorders and other adverse CNS conditions, as well as in the normal CNS. Of importance to this reviewis the growing literature fromboth clinical and animal studies that shows that IL-6 is expressed at elevated levels during CNS conditions that are associated with altered cognitive function and behavior. This association suggests the potential for neuronal or synaptic actions of IL-6, a possibility that has been supported by research using animal or in vitro models, as will be described below. Here I will briefly summarize some of the clinical literature that demonstrated that CNS levels of IL-6 are elevated in disease states that are associated with altered cognitive function. In general, the goal of these clinical studies was to find a biomarker for a particular CNS disorder rather than to gain an understanding of the role of IL-6 in the disorder. However, for some CNS disorders correlative information is available and suggests a cause–effect relationship between cognitive function and IL-6 levels. It is important to note that for most CNS diseases, IL-6 is not the only neuroimmune (or immune) factor that is elevated in the CNS. In this review, the primarily focus is on IL-6 and the involvement of other neuroimmune factors is not discussed, although results for other proinflammatory cytokines are mentioned for some studies. It is also important to note that results from clinical studies and measured values of IL-6 (e.g., Table 1) are often inconsistent, a topic that is also not discussed in this brief summary of the clinical literature. The discrepancy between different clinical studies is presumably due to difference in the characteristics of the cohorts studied (e.g., age of the subjects, stage of the disease), the number of subjects studied, the methods used to measure neuroimmune factors or other experimental variables. More information on results from clinical studies is available in several recent reviews (Atzori et al., 2012; Hiles et al., 2012; Jones and Thomsen, 2013; Sparkman and Johnson, 2008; Trapero and Cauli, 2014; Wei et al., 2013).
Table 1.
Levels of IL-6 in various CNS conditions.
| condition | Compartment | IL-6 levels | Reference |
|---|---|---|---|
| Control levels | CSF | 1-23 pg/ml | (Maier et al., 2005) |
| Controls | CSF+ | 1.4 pg/ml | (Sasayama et al., 2013) |
| Schizophrenia | CSF+ | 1.7 pg/ml | |
| Major depressive disorders | CSF+ | 1.7 pg/ml | |
| Controls (cerebellum) | Brain* | 138 ± 120 pg/μg | (Vargas et al., 2005) |
| Autism (cerebellum) | Brain* | 618 ± 222 pg/μg | |
| Control | Brain* | 9201 ± 107 pg/mg | (Li et al., 2009) |
| Autism spectrum disorders | Brain* | 2441 ± 267 pg/mg | |
| Control (median, IQR) | CSFˆ | 0.8, 0.6–1.1 pg/ml | (Lindqvist et al., 2013) |
| Non-demented Parkinson’s disease | CSFˆ | 0.7, 0.5–1.1 pg/ml | |
| Parkinson’s disease with dementia | CSFˆ | 1.0, 0.7–1.1 pg/ml | |
| Traumatic brain injury(pediatric) | CSF* | 332 ± 48 pg/ml | (Chiaretti et al., 2008) |
| Traumatic brain injury | Brain+ | 1931 pg/ml | (Hillman et al., 2005) |
| Traumatic brain injury | CSF+ | 2349 pg/ml | (Hillman et al., 2005) |
| Control | CSF* | 1.8 ± 0.5 pg/ml | (Peltola et al., 2000) |
| Tonic-clonic seizures | CSF* | 22.0 ± 31.9 pg/ml | |
| Vascular dementia | CSF* | 5.67 ± 1.7 pg/ml | (Wada-Isoe et al., 2004) |
| Alzheimer’s disease | CSF* | 2.53 ± 0.87 pg/ml | |
| Neurological disorders | CSF* | 3.15 ± 0.67 pg/ml | |
| Control | CSF* | 1.9 ± 0.09 pg/ml | (Beridze et al., 2011) |
| Ischemic stroke | CSF* | 58 ± 4.6 pg/ml |
= mean ± SD;
= median;
= median, interquartile range;
4.1. Measurement of IL-6 levels in clinical studies
While the most meaningful information about IL-6 levels in the CNS would likely come from measurements made in the interstitial space that bathes IL-6R on CNS cells or in the CNS parenchyma, these sites are not readily accessible in living patients. Therefore, in clinical studies, IL-6 levels are commonly measured in the cerebral spinal fluid (CSF) or blood of patients. IL-6 levels measured in the CSF are thought to reflect to some extent levels in the CNS parenchyma. Howclosely IL-6 levels in the CSF reflect actual levels of IL-6 in the parenchyma remains to be determined. Microdialysis studies in experimental animals during CNS injury (microdialysis probe implantation) suggest that IL-6 levels are significantly higher (~10 fold) in the CNS parenchyma (as measured in the dialysate) than in the CSF (Woodroofe et al., 1991). However, in studies of patients with severe traumatic brain injury, IL-6 levels in the CSF and CNS parenchyma (as measured in the dialysate) were similar (Roberts et al., 2013). The difference in results between these two studies may relate to the CNS region under study and/or the extent of CNS damage.
The relevance of blood levels of IL-6 to CNS function may appear to be remote, but blood levels can serve as a source for IL-6 in the CNS, especially under conditions of a leaky blood–brain barrier (Erickson et al., 2012). Moreover, a transport system for cytokines has been described in the blood–brain barrier that could account for transport of systemic IL-6 into the CNS (Threlkeld et al., 2010). Alternatively, the CNS may be a source for IL-6 in the blood (Terreni and De Simoni, 1998). Based on the ease of obtaining blood samples, in many clinical studies this compartment has been used to characterize disease associated IL-6 levels. However, the significance of blood measurements relative to disease symptoms is often elusive. Moreover, the cause and consequences of increased levels of IL-6 in the different body compartments (CNS, CSF or blood) remain to be fully elucidated. Also, in many studies mRNA levels rather than protein levels were measure. While an understanding of mRNA levels is important, mRNA levels do not always correlate with protein levels and proteins are the form that interacts with the receptor to produce a biological action. More information is needed before an understanding of the biological significance of the measured levels of IL-6 mRNA and protein in the different body compartments can be achieved. Some representative examples of measured levels of IL-6 protein in different body compartments are shown in Table 1.
4.2. CNS conditions associated with elevated levels of IL-6
A number of studies have reported elevated levels (protein or mRNA) of IL-6 and other neuroimmune factors in the CNS, CSF or blood from patients with Alzheimer’s disease, a disease characterized by dementia, memory loss and other cognitive problems (Bermejo et al., 2008; Cojocaru et al., 2011; Hampel et al., 2005; Huell et al., 1995; Luterman et al., 2000; Strauss et al., 1992; Swardfager et al., 2010; Trapero and Cauli, 2014; Wood et al., 1993). For example, in a recent meta-analysis the authors concluded that significantly higher concentrations of IL-6, TNF-α, and IL-1 β occurred in the peripheral blood of Alzheimer’s patients but not in the CSF as compared with control subjects (Swardfager et al., 2010). Attempts to correlate blood or CSF levels of IL-6 with the degree of CNS function in Alzheimer’s patient have produced inconsistent results. However, in a correlative study the severity of dementia in subjects with Alzheimer’s disease correlated with levels of IL-6 mRNA in the CNS (Luterman et al., 2000). In these studies, the cognitive and functional status of Alzheimer’s patients (78–88 years of age at death) was assessed during the last 6 months of life. IL-6 mRNA levels were measured postmortem in the entorhinal cortex, superior temporal gyrus, and occipital cortex. Patients showing severe/terminal dementia had significantly elevated IL-6 mRNA levels in the entorhinal cortex and superior temporal gyrus but not in the occipital cortex as compared with cognitively normal subjects. IL-6 mRNA levels were not elevated in these brain regions in patients showing less severe cognitive and functional status. mRNA levels for IL-1β and TGF-β1 were also assessed in these studies (Luterman et al., 2000). Results for TGF-β1 mRNA were similar to results for IL-6 mRNA, whereas IL-1β mRNA levels were very low in the three brain regions studied and showed no correlation with the severity of Alzheimer’s dementia (Luterman et al., 2000). These results suggest a complex association between CNS levels of IL-6 and Alzheimer’s disease, a situation that is likely to apply to other CNS diseases associated with elevated levels of IL-6 in the CNS.
Increased levels of IL-6 mRNA in the CNS were also observed in other diseases characterized by cognitive decline or dementia including Lewy body disease and vascular dementia. Brains of postmortem patients (65–79 yrs old) with Lewy body disease showed higher levels of IL-6 mRNA in the three brain regions studied (hippocampus, putman and cingulate cortex) compared to brains of control subjects (Imamura et al., 2005). In the same study, mRNA levels for IL-1β were decreased in the hippocampus, increased in the cingulate cortex but showed no change in the putman, whereas mRNA levels for TNF-α were increased in the putman but not in the hippocampus or cingulate cortex (Imamura et al., 2005). CSF from patients (74.5 ± 4.5 yrs old; all ages are expressed as mean ± SD unless noted) with dementia associated with vascular disease showed higher IL-6 levels (5.67 ± 1.7 pg/ml, mean ± SEM) than patients (70.0 ± 6.2 yrs old) with vascular disease without dementia (IL-6 = 2.15 ± 0.38 pg/ml, mean ± SEM) (Wada-Isoe et al., 2004). In studies of subjects with vascular dementia (mean age ~80 yrs old), plasma levels of IL-6 (range 1.95–6.98 pg/ml) correlated inversely with functional status of the subject, as measured by the ability of the subject to carry out daily activities (e.g., autonomy in dressing and bathing), whereas plasma levels of IL-1β (range 0.29–1.95 pg/ml), TNF-α (range 8.06–18.5 pg/ml), IL-8 (range 7.3–18.0 pg/ml) and TGF-β (range 15.9–48.2 ng/ml) did not correlate with functional status (Zuliani et al., 2007).
In addition to disease states, elevated levels of IL-6 in the blood have been found to correlate with poorer cognitive performance in normal subjects (Marsland et al., 2006; Wright et al., 2006) and in subjects showing age-related cognitive decline (Economos et al., 2013; Lekander et al., 2011; Yaffe et al., 2003). For example, studies of middle-aged adults (30–54 years old) revealed an inverse relationship between plasma levels of IL-6 (1.8 ± 1.6 pg/ml; unless noted all concentrations are mean ± SD) and tests scores for working memory and executive function (Marsland et al., 2006). In another study involving a racially an ethnically diverse cohort (mean age 66.8 yrs old), serum IL-6 levels (1.4 ± 1.2 pg/ml) were negatively associated with performance on a standard test of mental function (Mini–Mental Test) (Wright et al., 2006). IL-6 has also been shown to play an important role in the regulation of sleep architecture and certain sleep-related memory functions (e.g., sleep-dependent memory consolidation), which can be disrupted by abnormal levels of IL-6 in the CNS (Benedict et al., 2009; Dimitrov et al., 2006; Heffner et al., 2012; Hong et al., 2005; Irwin et al., 2004).
Studies also show a link between IL-6 and psychiatric disorders such as depression and other mood disorders. Clinical depression is a chronic, recurring illness characterized by impaired CNS function including cognitive difficulties, memory loss and a variable cluster of additional symptoms (e.g., depressed mood, sleep problems, loss of interest in activities). Sickness behavior, which is mediated by proinflammatory cytokines including IL-6, has many features in commonwith depression, although sickness behavior and depression are not identical and each has distinctive features (Dunn et al., 2005).
In a study of patients (39 ± 14 yrs old) with major depressive disorders that attempted suicide, IL-6 levels in the CSF (3.76 ± 2.65 pg/ml, mean ± SEM) were significantly higher than in the CSF (0.64 ± 0.09 pg/ml, mean ± SEM) of control subjects (37 ± 20 yrs old) with no ongoing psychiatric condition (Lindqvist et al., 2009). In the same study, no significant difference between suicide attempters and controlswas observed for CSF levels of IL-1β (0.08 ± 0.01 pg/ml vs. 0.07 ± 0.01 pg/ml, mean ± SEM, suicide attempters vs. controls) and TNF-α(0.15 ± 0.01 pg/ml vs. 0.13 ± 0.01 pg/ml, mean ± SEM, suicide attempters vs. controls) (Lindqvist et al., 2009). Increased levels of IL-6 and TNF-α were also observed in plasma of suicide attempters (39.3 ± 15.0 yrs old) and were higher than in plasma of non-suicidal depressed patients (33.8 ± 10.9 yrs old) and healthy controls (34.9 ± 11.5 yrs old) (Janelidze et al., 2011). Postmortem studies of the prefrontal cortex of young suicide victims (age range = 12–20 yrs old) revealed elevated levels of IL-6, IL-1β and TNF-α mRNA and protein in the suicide victims compared with controls (Pandey et al., 2012). In depressed and healthy elderly adults (mean age ~69 yrs old), memory performance was negatively associated with serum IL-6 levels (Elderkin-Thompson et al., 2012).
In a study of patients with major depressive disorders (42.7 ± 8.2 yrs old) or schizophrenia (40.8 ± 8.8 yrs old), IL-6 levels in the CSF were significantly higher for both conditions than in healthy controls (41.3 ± 16.4 yrs old) (Sasayama et al., 2013). In the same study, there was no significant difference in serum levels of IL-6 between patients with major depressive disorders or schizophrenia and healthy controls (Sasayama et al., 2013). Patients with systemic lupus erythematosus (SLE) with psychiatric manifestations showed higher IL-6 levels in the CSF than SLE patients without psychiatric manifestations (Fragoso-Loyo et al., 2007; Hirohata and Miyamoto, 1990; Trysberg et al., 2000). For example, in SLE patients (30.5 ± 11.5 yrs old) with neuropsychiatric manifestations, the median IL-6 levels in the CSF was 32 pg/ml compared to a median IL-6 level of 3 pg/ml with SLE patients without neuropsychiatric manifestations (Fragoso-Loyo et al., 2007). A similar ~10 fold increase in IL-6 levels in the CSF of SLE patients with neuropsychiatric manifestations was reported in another study (Trysberg et al., 2000). SLE is an autoimmune disease of unknown etiology that results in abnormal CNS function including impaired working memory, executive dysfunction, overall cognitive slowing and decreased attention (Rheumatology, 1999). Elevated levels of IL-6 were also found in the CNS, CSF and blood of autistic subjects (Masi and Quintana, 2014; Wei et al., 2012, 2011). Interestingly, autistic subjects with intellectual disability showed a higher prevalence for epilepsy than the general population (Woolfenden et al., 2012). Elevated levels of IL-6 are proposed to be a contributor to seizure predisposition, occurrence and seizure-related brain injury (Lorigados Pedre et al., 2013). Moreover, elevated levels of IL-6 in the CNS and blood are produced by seizure activity (Bauer et al., 2009; Lehtimaki et al., 2004; Mao et al., 2013; Peltola et al., 1998; Yu et al., 2012).
Taken together, these clinical studies demonstrate that elevated levels of IL-6 occur in the CNS in a variety of CNS disorders associated with altered CNS function. This association suggests a potential role for IL-6 as a regulator of CNS function. This possibility has been pursued in experimental models that have confirmed an association between elevated CNS levels of IL-6 and altered behavior and have begun to identify the synaptic, cellular and molecular mechanisms underlying these actions.
5. Animal studies provide evidence of role for IL-6 in behavior
Results from studies in animal models support the idea that CNS produced IL-6 can impact CNS function and consequently behavior. Importantly, a number of animal studies have shown that IL-6 can alter cognitive functions such as memory and learning. For example, studies in rats showed that bilateral intrahippocampal injection of IL-6 (16 and 80 ng IL-6) resulted in impaired performance in a behavioral learning task (avoidance learning) measured 8 days after IL-6 injection, while a lower dose (3.2 ng IL-6) was ineffective (Ma and Zhu, 1997). Similarly, deficits in avoidance learning were observed in homozygous and hemizygous transgenic mice (GFAP-IL-6 mice) that persistently express elevated levels of IL-6 in the CNS through increased astrocyte expression as compared to non-transgenic littermate controls (Heyser et al., 1997). In these studies, the behavioral deficits showed age- and dose-dependency in that homozygous mice exhibited deficits at a younger age than the hemizygous mice. Progressive synaptic damage and loss of inhibitory interneurons were also noted in these studies and also showed a similar age- and dose-dependency (Heyser et al., 1997). Interestingly, impaired learning in behavioral tests (Morris water maze, novel object recognition test, force swim test) was also observed in IL-6 knockout mice compared with control mice (Baier et al., 2009). Thus, both an excess and deficit of IL-6 can impact cognitive function. A role for IL-6 in cognitive function was also demonstrated by chronic intracerebroventricular administration of a neutralizing anti-IL-6 antibody. Administration of the anti-IL-6 antibody resulted in a cognitive deficit as measured by a behavioral test for reversal learning in rats (Donegan et al., 2014). In the same study, a deficit in reversal learning produced by chronic stress was reversed by elevating IL-6 in the rat orbitofrontal cortex using adeno-associated virus-mediated gene delivery (Donegan et al., 2014). Immunohistochemical studies showed that the virus was located in neurons of the orbitofrontal cortex (Donegan et al., 2014). Thus, it appears that both neuronal and glial produced IL-6 can play a regulatory role in cognitive function. Other factors besides cell source such as brain region, cognitive task, IL-6 dose and conditions of exposure are also likely to be important factors in the effects of IL-6 on cognitive function.
Mouse models of specific disease states have also provided evidence for a role for IL-6 in cognitive function. Studies in a transgenic mouse model of Alzheimer’s disease (Tg2576) with overexpression of human APP with the Swedish double mutation (APPsw) revealed age-dependent impaired cognitive function in certain behavior tests (reversal of the compound discrimination, Y-maze alternation, visible platform tasks) as compared to control mice, although no significant difference was observed in several other behavioral tests (e.g., Morris water maze, avoidance tasks) (King and Arendash, 2002; Zhuo et al., 2007). Significantly higher levels of IL-6 mRNA were observed in the cerebral cortex, hippocampus and cerebellum of the APPsw mice (4 and/or 18 months of age) as compared to wildtype mice, whereas no significant difference in the levels of IL-1β were detected (Tehranian et al., 2001). The increase levels of IL-6 were observed at ages that preceded plaque formation, suggesting that IL-6may play a role in the early stages of the disease (Tehranian et al., 2001). Increased CNS levels of IL-6 mRNA and protein were also shown to occur with normal aging in mice (Ye and Johnson, 1999, 2001). Aging is associated with a decline in general cognitive abilities in mice as well as humans (Matzel et al., 2008; Salthouse, 2010). For example, IL-6 protein levels in the brain of mice increased from ~40 pg/mg protein at 1month of age to ~80 pg/mg protein at 24 months of age (Ye and Johnson, 2001).
In a mouse model of metabolic syndrome (db/db mice), increased anxiety-like behaviors (open field and elevated maze tests) and impaired spatial working memory (special recognition test) were associated with elevated levels of mRNA for IL-6, IL-1β and TNF-α in the hippocampus (Dinel et al., 2011). Increased IL-6 protein levels in the plasma were also observed in the db/db mice, whereas there was no difference in plasma levels for IL-1β and TNF-α (Dinel et al., 2011). Metabolic syndrome is characterized in humans by a prevalence of mood symptoms and cognitive dysfunction (Dinel et al., 2011; Taylor and MacQueen, 2007). In a strain of mice that carries the main gene for catalepsy and shows spatial learning deficit in the Morris water maze, the level of IL-6 mRNA was significant higher in the hippocampus and cortex compared to a strain of mice that is catalepsy resistant (Kulikov et al., 2014). Sleep disturbance in mice result in impaired learning and memory, an effect associated with elevated IL-6 levels in the hippocampus but not the cortex (Zhu et al., 2012).
Experimental evidence also exists for a role for IL-6 in depression. In lpr/lpr mice, a mouse model of SLE, a depressive-like phenotype was observed in the forced swim and tail suspension tests and the depressive-like behaviors were shown to be associated with increased levels of IL-6 protein in the hippocampus and prefrontal cortex (Sukoff Rizzo et al., 2012). Increased levels of IL-6 and IL-1β mRNA were observed in the hippocampus and cerebellum of the lpr/lpr mice and correlated with behavioral dysfunction (Tomita et al., 2001a, 2001b). Mice with colon carcinoma cell-induced peripheral tumor showed memory impairment and depression-like behavior in behavioral tests (open field test, tail suspension test and object recognition memory test) and these behaviors were associated with increased levels of IL-6 mRNA in the hippocampus (Yang et al., 2014). Mice subjected to prolonged light deprivation, a paradigm that results in depressive-like behaviors, showed elevated levels of IL-6 in the plasma and in hippocampal tissue (Monje et al., 2011). Mice subjected to psychological stress (foot shock), another paradigm that results in depressive-like behaviors, showed elevated levels of IL-6 mRNA in the hippocampus (Chourbaji et al., 2006).
An association of IL-6 with a depressive phenotype was also demonstrated in studies where IL-6 was injected into the CNS. For example, several days’ treatment of rats with IL-6 (10–40 ng) by injection into specific brain regions (amygdala or hippocampus) resulted in decreased performance in behavioral tests (e.g., forced swim test), consistent with a depression-like behavior (Wu and Lin, 2008). Similarly, intracerebroventricular administration of IL-6 (500–1000 ng, 2 h pretreatment) produced depressive-like phenotypes in mice as determined by behavioral tests (forced swim, tail suspension) (Sukoff Rizzo et al., 2012). In agreement with these results, IL-6 knockout mice exhibited reduced depression-like behavior (e.g., forced swimming, tail suspension tests) compared with wildtype mice (Chourbaji et al., 2006). Depression is commonly comorbid with epilepsy (Pineda et al., 2010), a condition that can significantly affect CNS function and is also associated with increased CNS levels of IL-6 (Li et al., 2011; Vezzani et al., 2008). Intracerebroventricular injection of IL-6 (2500 U/kg) induces seizures in rats (Xiaoqin et al., 2005) and has a pro-convulsive effect in the CNS when applied intranasally (400 ng/40 μl) shortly before experimentally induced seizures in rats (Kalueff et al., 2004). Transgenic mice (GFAP-IL-6 mice) that persistently express elevated levels of IL-6 in the CNS show spontaneous seizures and decreased threshold for induced seizures (Campbell et al., 1993; Samland et al., 2003; Steffensen et al., 1994).
The demonstration that IL-6 is associated with specific CNS conditions or behaviors is an important step toward an understanding the roles played by IL-6 in CNS physiology or pathology. Evidence linking IL-6R activation and behavior is also important, but few studies have addressed this issue. Studies have appeared that show that signal transduction molecules used by IL-6R, including JAK2, STAT3 and p44/42 MAPK, are involved in functions that are altered in neuropsychiatric or neurologic disorders, lending support to the idea that IL-6R signaling may have a role in these disorders. For example, application of a JAK2 inhibitor (i.c.v.) reduced the levels of activated STAT3 (pSTAT3) in mouse hippocampal neurons and impaired memory in a hippocampal-dependent behavior (Chiba et al., 2009). Antagonism of JAK2 also caused memory impairment in an animal model of Alzheimer’s disease (Chiba et al., 2009). In other studies, injection of IL-6 into the hippocampus of rats increased p44/42 MAPK activation and resulted in poorer performance in a test for depression-like behavior (i.e., force swim test) (Wu and Lin, 2008). Kainic acid induced seizures resulted in activation of STAT3 in astrocytes and p42/44 MAPK in neurons and astrocytes of the hippocampus (Choi et al., 2003).
6. Studies in vivo and vitro have established that IL-6 has neuronal and synaptic actions
The presence of IL-6 in the brain, the elevated levels of IL-6 during disease states associated with altered cognitive function, and the demonstration that neurons express IL-6R are consistent with the idea that IL-6 has neuronal and synaptic actions. Recent in vivo and in vitro studies in experimental models using exogenous application of IL-6 or manipulation of endogenous levels of IL-6 support this possibility.
Studies using exogenous application of IL-6 typically employ recombinant forms of IL-6 at concentrations ranging from 0.01 ng/ml to 200 ng/ml, where the lower levels are considered to be more physiological and the higher levels are considered to be relevant to pathological conditions. Consistent with this idea, in a study thought to be reflective of a physiological process, the mean IL-6 level in the striatum of mice measured by microdialysis was approximately 30 pg/ml at 240 min after IL-6 release from astrocytes was initiated by application of an adenosine receptor agonist (Vazquez et al., 2008). In contrast, a mean value of 5.0 ng/ml was measured by microdialysis in patients with traumatic brain injury, consistent with high levels of IL-6 during pathological conditions (Roberts et al., 2013). Issues that are pertinent to interpretation of results from studies using exogenous application include the species of the IL-6 tested, IL-6 concentration, the ability of IL-6 to penetrate into the tissue, potential non-specific binding of IL-6 to non-receptor sites, and actions at sites not normally involved in pathological or physiological actions.
Studies using manipulation of endogenous levels of IL-6 involved two types of approaches: (a) the use of transgenic models or (b) viral induction of IL-6 gene expression in CNS cells. Issues that are pertinent to interpretation of results from these types of studies include the identity of the cell type expressing IL-6, the concentration of IL-6 produced, and actions at sites not normally involved in pathological or physiological actions. Studies using exogenous application and manipulation of the levels of endogenous of IL-6 have been instrumental in identifying CNS targets of IL-6 that could play an important role in regulating synaptic function under physiological or pathological conditions.
6.1. Effects of acute (min to hrs) exposure to IL-6 on neuronal and synaptic function
Studies by D’arcangelo et al. (D’Arcangelo et al., 2000) using acutely isolate nerve terminals (i.e., a synaptosomal preparation) prepared from rat neocortex showed that IL-6 exposure (15 min; 50–200 ng/ml IL-6) produced a dose-dependent reduction in the ability of pharmacological stimulation to evoke glutamate release. These results indicate that IL-6 can act presynaptically to inhibit transmitter release. In parallel studies in a more intact preparation, acutely isolated slices of somatosensory cortex, experiments using imaging and electrophysiological (field potential recordings) techniques showed that synaptic responses evoked by electrical stimulation of afferent pathways were reduced by IL-6 exposure (0.5–1 ng/ml), consistent with an inhibitory effect of IL-6 on synaptic transmission (D’Arcangelo et al., 2000). To provide evidence for the involvement of IL-6R in the inhibitory effects of IL-6, the slices used in the imaging and electrophysiological experiments were assayed by Western blot using antibodies specific for the active form (i.e., phosphorylated) of signal transduction molecules linked to IL-6R. These studies showed that the inhibitory effects of IL-6 were associated with activation of STAT3, inhibition of MAPK/ERK and no apparent effects on SAPK/JNK (D’Arcangelo et al., 2000). The cell-permeant tyrosine kinase inhibitor LavA blocked the inhibitory effects of IL-6 on synaptic transmission in imaging studies, consistent with an involvement of the JAK/STAT pathway in the inhibitory effects of IL-6 on synaptic transmission (D’Arcangelo et al., 2000). Additional studies showed that the phosphorylation state of synapsin 1 at the MAPK/ERK sites was decreased by IL-6 exposure in the nerve terminal preparation, providing evidence for the involvement of IL-6R in the presynaptic effects of IL-6 (D’Arcangelo et al., 2000). Synapsin 1 is a presynaptic protein that plays a critical role in transmitter release. Therefore, the decreased phosphorylation of synapsin 1 correlated with the IL-6 reduction of glutamate release from the nerve terminals.
An effect of IL-6 on afferent sensory transmission to the somatosensory cortex of rats measured by electrophysiological recording of evoked action potentials (i.e., single unit activity) has also been reported. In these in vivo studies, topical application of IL-6 (0.01–1 pg/10 μl saline) to the cortex produced an inhibition of the short latency component and a facilitation of the long latency component of the single unit response elicited by sensory stimulation (Shin et al., 1997). The short latency component is thought to represent sensory pathways involving a few synapses, whereas the long latency component is thought to reflect other pathways and/or processed activity (Shin et al., 1997).
In contrast to these results from somatosensory cortex, exogenous application of IL-6 (1–50 ng/ml, 10–20 min) to acutely isolated rat hippocampal slices did not alter excitatory synaptic transmission at the Schaffer collateral to CA1 pyramidal neuron synapse in electrophysiological studies (field potential recordings) (Li et al., 1997; Tancredi et al., 2000). Excitatory synaptic transmissionwas also unaltered by exogenous application of IL-6 (10 ng/mL) in acutely isolated slices from temporal cortex (Garcia-Oscos et al., 2012). However, in the study of cortical slices, IL-6 exposure produced a reduction in inhibitory synaptic transmission, an action that was identified to occur postsynaptically and to involve a decrease in GABA receptor expression in the plasma membrane due to receptor internalization (Garcia-Oscos et al., 2012).
6.2. Effects of chronic exposure to IL-6 on neuronal and synaptic function
In many CNS conditions, neuronal exposure to IL-6 occurs for a prolonged period (days to years) and results in neuroadaptive changes that persistently alter neuronal and synaptic function. Three experimental systems have been used to investigate such neuroadaptive effects of IL-6, culture preparations, transgenic mice that express elevated levels of IL-6 in the CNS, and viral induction of IL-6 gene expression in CNS cells. In culture preparations, chronic exogenous exposure typically involves addition of IL-6 to the culture medium at known concentrations. In transgenic mouse models and with viral induction of IL-6 gene expression, manipulation of neuronal or glial IL-6 gene expression is used to produce persistently elevated levels of IL-6 in specific cell types in the CNS.
6.2.1. Effects of chronic exposure to IL-6 on cerebellar neurons in culture
Chronic exposure of cultured cerebellar granule neurons (excitatory neurons that use glutamate as a transmitter) to IL-6 in the culture medium (5 ng/ml; 6–12 days) resulted in a significantly larger response to brief application of the glutamate receptor agonist NMDA than observed in granule neurons in control cultures (Qiu et al., 1995, 1998). Acute exposure to IL-6 did not produce a similar enhancement. Western blot analysis of protein expression in the granule neuron cultures revealed a significant decrease in expression of synapsin I, a presynaptic protein used as a marker for synapses, in cultures chronically exposed to IL-6 (5 ng/ml; 6–12 days) compared with control cultures, suggesting that chronic exposure to IL-6 produces a reduction in synapses (Conroy et al., 2004). In contrast, when a viral expression approach was used to induce cultured granule neurons to produce IL-6 at elevated levels, IL-6 expression resulted in increased immunostaining for two presynaptic proteins, synaptophysin, a general marker for synapses, and VGLUT1 (vesicular glutamate transporter), a presynaptic marker for excitatory synapses that use glutamate as a transmitter. There was no difference in the levels of immunoreactivity for VGAT (vesicular GABA transporter), a marker of inhibitory synapses, in IL-6 infected granule neuron cultures compared with control cultures (Wei et al., 2011). These results suggest that persistent neuronal expression of IL-6 can promote excitatory synapse formation. The apparent difference in the effects of IL-6 on synapse formation between the two studies of cultured granule neurons, one indicating reduced synapses and the second indicating increased synapses, could be due to the source of IL-6 (i.e., neuronal vs. the culture medium), the concentration of IL-6 that the cells experience (unknown for the virally induced IL-6 expression), and/or the cellular region exposed to IL-6.
Studies of Purkinje neurons in cerebellar cultures chronically exposed to IL-6 in the culturemedium(1–10 ng/ml, 8–21 days) also revealed actions of IL-6 on neuronal function. In these studies, opposing, concentration-dependent actions of IL-6 were identified. Chronic exposure to IL-6 at a low concentration (1 ng/ml) enhanced Purkinje neurons spike firing, whereas chronic exposure to a higher concentration of IL-6 (10 ng/m) resulted in an inhibition of spike firing (Nelson et al., 2002). Chronic exposure to IL-6 also elevated resting Ca2+ levels, increased the magnitude of intracellular Ca2+ signals evoked by activation of group I metabotropic glutamate receptors, enhance Ca2+ signaling induced by excitatory transmitters and membrane depolarization in the cultured Purkinje neurons (Nelson et al., 2004, 2002). All of these effects of IL-6 could significantly alter synaptic transmission involving the Purkinje neurons.
6.2.2. Effects of chronic exposure to IL-6 on hippocampal neurons in culture
Evidence for neuroadaptive effects of chronic IL-6 exposure on synaptic function was also demonstrated in cultures of hippocampal neurons chronically exposed to IL-6 in the culture medium. In theses studies, the enhancement of spontaneous synaptic network activity induced by reducing the concentration of Mg2+ in the bath saline, which removes the Mg2+ block of NMDA receptors, was significantly larger In IL-6 treated cultures (5 ng / mL, 4–16 days) compared to control cultures (Vereyken et al., 2007). In addition, Group II metabotropic receptor (mGluR2/3) ligands were less effective as modulators of synaptic network activity in IL-6-treated hippocampal cultures than in control cultures. The reduced action of mGluR2/3 ligands correlated with a reduced level of mGluR2/3 measured inWestern blots of the IL-6 treated cultures compared with control cultures. mGluR2/3s are expressed on presynaptic and postsynaptic terminals and on glial cells in the hippocampus (Petralia et al., 1996). In contrast, the differential effect of mGluR2/3 ligands on synaptic network activity between IL-6 treated vs. control cultures was not observed when spontaneous synaptic activity was enhanced by addition of GABAA receptor antagonist picrotoxin to the recording saline (normal levels of Mg2+) to block inhibitory synaptic transmission. These results imply a role for inhibitory interneurons in the actions of chronic IL-6 on hippocampal synaptic network activity (Vereyken et al., 2007).
6.2.3. Transgenic models of IL-6 exposure
Although studies using culture models have made significant contributions to an understanding of the neuronal and synaptic actions of IL-6, animal models have several important advantages over culture models for studies of synaptic function. Importantly, in animal models the cellular and synaptic organization is more reflective of the normal in vivo conditions than is possible with culture preparations. In addition, depending on the model, the source of IL-6 can be more localized and physiologically or pathologically relevant. Also, adult animals can be studied, whereas cultures are typically prepared from immature CNS tissue, and behavioral studies can be pursued with animal models. Two transgenic mouse models have been developed to investigate the effects of chronic, endogenously produced elevated levels of IL-6 on CNS function, one in which elevated levels of IL-6 are produced in the CNS by astrocytes (GFAP-IL-6 mice) (Campbell et al., 1993) and the second in which elevated levels of IL-6 are produced in the CNS by neuronal expression (Fattori et al., 1995). In the first model, elevated IL-6 production was targeted to astrocytes using a murine IL-6 expression vector derived from the murine glial fibrillary acidic protein (GFAP) gene. Several GFAP-IL-6 lines were developed that express different levels of IL-6 in the CNS. Homozygous and heterozygous GFAP-IL-6 mice also express different levels of IL-6 in the CNS. In the second model, neuronal expression of IL-6 was accomplished by use of a human IL-6 gene expression vector targeted to the neurons using the rat neuron-specific enolase promoter (NSE-IL-6 mice).
The GFAP-IL-6 mice exhibit progressive anatomical, physiological, and behavioral abnormalities (e.g., seizures, ataxia), particularly in the hippocampus and cerebellum, that become evident around 6 months of age and prominent at 12 months of age in the heterozygote and earlier in the homozygote (3–6 months of age) (Campbell, 1998; Heyser et al., 1997). In contrast, the NSE-IL-6 mice show little or no CNS pathology, although they do express elevated levels of GFAP in the CNS, as do the GFAP-IL-6 mice (Fattori et al., 1995). These differences between the GFAP-IL-6 and NSE-IL-6 mice suggest that astrocyte and neuronal expression of IL-6 serve different physiological/pathological roles. However, concentration of IL-6 in the CNS of the transgenic mice could also be a factor. Surprisingly, the mean IL-6 concentration in the supernatant of brain slice cultures prepared from the NSE-IL-6 mice was 6.3 ± 0.8 ng/ml (mean ± SD; 24 h collection period) (Fattori et al., 1995), considerably higher than 150 pg/ml and 310 pg/ml (collection period not given), measured in supernatants from astrocyte cultures prepared from brains of G167 heterozygous and G167 homozygous GFAP-IL-6 mice, respectively (Campbell et al., 1993). The reason for these differences and the potential biological significance remain to be determined.
As will be described below, several studies have examined synaptic transmission in the GFAP-IL-6 mice to identify potential consequences of astrocyte produced IL-6 on synaptic function. Astrocytes are an essential partner of neurons in synaptic transmission, are in close association with synapses and dynamically interact with neurons to regulate synaptic function (Ben Achour et al., 2010; Halassa et al., 2007; Pannasch and Rouach, 2013). Therefore, astrocyte produced IL-6 would be well situated to produce synaptic actions. Although the effects of neuronal produced IL-6 are also of interest, no comparable studies of synaptic function have appeared for the NSE-IL-6 mice.
6.2.4. Synaptic transmission in the GFAP-IL-6 cerebellum
The cerebellum shows the highest level of expression of IL-6 in the CNS of the GFAP-IL-6 mice and also the most pathology (Campbell et al., 1993). Studies of Purkinje neurons in acutely isolated slices of cerebellum from heterozygous mice from the G369 line, which expresses high levels of IL-6, exhibited a number of abnormalities in extracellular single unit recordings, indicative of IL-6-induced neuroadaptive changes. Firing rate of spontaneously active Purkinje neurons was significantly reduced in the transgenic cerebellar slices and an oscillatory pattern of spontaneous firing was more prevalent than in non-transgenic control cerebellar slices (Nelson et al., 1999). The reduced firing rate of Purkinje neurons in the transgenic cerebellum is consistent with the reduced firing rate of cultured Purkinje neurons exposed to a high concentration of IL-6 (10 ng/ml) in the culture medium noted above (Nelson et al., 2002). Excitatory synaptic responses (i.e., complex spikes) produced by stimulation of climbing fiber afferents were similar in Purkinje neurons of GFAP-IL-6 and non-transgenic cerebellar slices. However, the inhibitory period following the complex spike, referred to as the climbing fiber pause, was significantly longer in slices from the GFAP-IL-6 mice. The climbing fiber pause is thought to result from activation of Ca2+-activated potassium channels and an influence of synaptic input from cerebellar inhibitory interneurons. Studies of Purkinje neurons in culture noted above showed that chronic exposure to IL-6 enhances Ca2+ signaling induced by excitatory transmitters and membrane depolarization (Nelson et al., 2004, 2002). These actions of IL-6, if they occurred in the GFAP-IL-6 cerebellum, could play a role in the prolongation of the climbing fiber pause. The effects of IL-6 on Purkinje neuron firing and synaptic response are likely to contribute to the ataxia characteristic of the GFAP-IL-6 mice (Campbell et al., 1993).
6.2.5. Synaptic transmission in the GFAP-IL-6 hippocampus
Field potential recordings in vivo of synaptic transmission in the hippocampus of GFAP-IL-6 mice showed that the magnitude of excitatory synaptic transmission to dentate granule neurons evoked by stimulation of the perforant pathway was not altered in the homozygous GFAP-IL-6 mice compared to non-transgenic littermate controls. However, recurrent inhibition, which controls the excitability of the dentate granule neurons, was significantly increased in the GFAP-IL-6 mice compared with non-transgenic controls (Steffensen et al., 1994). Pharmacological studies indicated that the increased recurrent inhibition to the dentate granule neurons in the GFAP-IL-6 mice resulted from a reduced inhibitory influence from the medial septum onto the perforant pathway (Steffensen et al., 1994). Additional EEG recordings from the dentate gyrus showed that persistent paroxysmal discharges and reduced theta activity occurred in the GFAP-IL-6 hippocampus, but not in the non-transgenic hippocampus (Steffensen et al., 1994).
Synaptic transmission to the hippocampal dentate granule cells elicited by perforant pathway stimulation was also studied using field potential recordings in acutely isolated hippocampal slices obtained from GFAP-IL-6 and non-transgenic control mice (Bellinger et al., 1995). Consistent with the in vivo studies, excitatory synaptic transmission from the perforant pathway to dentate granule neurons was similar in the GFAP-IL-6 and non-transgenic hippocampal slices. There was also no difference in recurrent inhibition, a result that supports the proposal that the enhanced recurrent inhibition observed in the in vivo studies was due to effects of IL-6 on medial septal neurons, a part of the brain that is not present in the hippocampal slice preparation (Bellinger et al., 1995; Steffensen et al., 1994).
In the in vivo and in vitro studies of hippocampus from homozygous GFAP-IL-6 mice described above, measurements were also made of synaptic transmission at the Schaffer collaterals to CA1 pyramidal neurons and found to be of similar in magnitude in the GFAP-IL-6 and non-transgenic hippocampus (Bellinger et al., 1995; Steffensen et al., 1994). In contrast, in field potential recordings from hippocampal slices from heterozygous GFAP-IL-6 mice of the G167 line, which expresses a lower level of IL-6 in the CNS, excitatory synaptic transmission from the Schaffer collaterals to CA1 pyramidal neurons was increased in the GFAP-IL-6 hippocampus compared with the non-transgenic hippocampus, particularly at strong stimulus strengths (Nelson et al., 2012). Recurrent inhibition to the CA1 neurons by way of the GABAergic basket cells was not prominent in these studies and did not differ for the GFAPIL-6 and non-transgenic hippocampus (Nelson et al., 2012). The enhanced excitatory synaptic transmission in the CA1 pyramidal neurons may contribute to the increased susceptibility of GFAP-IL-6 mice to kainic acid- and NMDA-induced seizures (Samland et al., 2003).
In a mouse model that over-expresses IL-6 in the CNS through the use of an adenoviral gene delivery approach (Ad-GFP-IL-6 mice), field potential recordings from hippocampal slices showed that recurrent inhibition to the CA1 pyramidal neurons elicited by Schaffer collateral stimulation was decreased in the Ad-GFP-IL-6 hippocampus compared to recurrent inhibition in hippocampal slices from control mice (Wei et al., 2012). Immunostaining using antibodies to synaptic vesicle proteins in the CA1 region of hippocampus from Ad-GFP-IL-6 mice revealed increased excitatory synapses and decreased inhibitory synapses compared with control hippocampus, as evidenced by immunostaining for synaptophysin, a general marker for synapses, VGLUT1, a marker of excitatory synapses, and VGAT, a marker of inhibitory synapses (Wei et al., 2012). The reduced immunostaining for inhibitory synapses is consistent with the reduced recurrent inhibition observed in field potential recordings of the Ad-GFP-IL-6 hippocampus (Wei et al., 2012). Western blot studies of hippocampus of the GFAP-IL-6 mice (G167 line) did not show altered levels of synapsin I, VGLUT1, or VGAT compared with non-transgenic hippocampus (Nelson et al., 2012), perhaps because IL-6 levels were lower than in the Ad-GFP-IL-6 hippocampus. The exact cell types expressing the elevated levels of IL-6 in the Ad-GFP-IL-6 hippocampus were not identified in this study (Wei et al., 2012).
6.3. Summary
Taken together, the above studies involving a variety of experimental approaches and preparations demonstrate the ability of both acute and chronic IL-6 exposure to significantly affect the CNS. IL-6 actions were evident at multiple levels including protein expression, signal transduction, neuronal function, synaptic mechanisms and synaptic function. The effects of IL-6 varied for different neuronal types and CNS regions and implicate IL-6 as an important signaling molecule that could participate in a variety of physiological or pathological functions.
7. IL-6 and synaptic plasticity in the hippocampus
The hippocampal region of the brain plays a central role in memory and learning. It is generally accepted that memories formed in the hippocampus are encoded by changes in the strength of synaptic transmission, a process called synaptic plasticity (Lynch, 2004). Information inherent in these synaptic changes is later reorganized and stored in other brain regions that are responsible for higher cognitive functions. For example, synaptic plasticity at the Schaffer collateral to CA1 pyramidal neuron synapse is thought to be critical for information processing and memory formation. CA1 neurons monosynaptically innervate neurons of the prefrontal cortex, a brain region responsible for higher cognitive functions (i.e., executive functions) that are often impaired in CNS disorders. Synaptic plasticity that involves a long-term increase in synaptic strength is referred to as long-term potentiation (LTP), whereas synaptic plasticity that involves a long-term decrease in synaptic strength is referred to as long-term depression (LTD). Both LTP and LTD are important memory mechanisms and have been associated with hippocampal-dependent behaviors (Eichenbaum, 2004; Ge et al., 2010; Shapiro, 2001; Smith and Squire, 2005). In addition, a short-term (min) form of synaptic plasticity, post-tetanic potentiation (PTP), contributes to short-term memory mechanisms. PTP is a short-term increase in synaptic strength resulting from changes in transmitter release at presynaptic terminals. LTP and LTD primarily involve postsynaptic mechanisms, although presynaptic mechanisms can also contribute (Blitzer et al., 2005; Lauri et al., 2007; Massey and Bashir, 2007).
7.1. Effects of acute exposure to IL-6 on synaptic plasticity in the hippocampus
Effects of IL-6 on synaptic plasticity were studied at the Schaffer collateral to CA1 pyramidal neuron synapse using field potential recordings in acutely isolated hippocampal slices and exogenous application of IL-6. Acute exposure to IL-6 (5–50 ng/ml) reduced both LTP and PTP at the Schaffer collateral to CA1 pyramidal neuron synapse in a dose-dependent manner (Li et al., 1997; Tancredi et al., 2000). Studies using pharmacological agents showed that the depression of LTP and PTP by IL-6was associated with alterations in the levels of the active form of signal transduction molecules used by IL-6R, indicating the involvement of IL-6R with the altered synaptic plasticity (Tancredi et al., 2000). Thus, the depression of LTP and PTPwas associated with a prolonged increase in the level of the activated form of STAT3 (pSTAT3) and a reduction in the level of the active form of p44/42 MAPK (pp44/42 MAPK). In addition, the effects of IL-6 on LTP and PTP were blocked by pharmacological inhibition of JAK2 (Tancredi et al., 2000). Increase activation of p44/42 MAPK is known to be essential for the induction of LTP (Sweatt, 2001). Therefore, the reduced levels of pp44/42 MAPK is consistent with the reduced LTP produced by IL-6. However, the consequence of the increased levels of pSTAT3 was not investigated and remains to be elucidated.
7.2. Synaptic plasticity in the hippocampus of GFAP-IL-6 transgenic mice
Studies in acutely isolated hippocampal slices from GFAP-IL-6 (167 line) and non-transgenic littermates were carried out using field potential recordings to determine the neuroadaptive effects of prolonged exposure to IL-6 on LTP (Nelson et al., 2012). No significant difference was observed in the magnitude of LTP at the Schaffer collateral to CA1 pyramidal neuron synapse between the GFAP-IL-6 and non-transgenic hippocampus in young (1–2 months of age) and adult mice (3–6 months of age). However, in hippocampus of young GFAP-IL-6 mice, PTP was significantly reduced compared to PTP in the hippocampus of non-transgenic young mice, an effect not observed in the adult mice (Nelson et al., 2012). This reduction of PTP suggests neuroadaptive effects of IL-6 at presynaptic sites.
LTP in dentate granule cells elicited by perforant pathway stimulation was also studied in acutely isolated hippocampal slices obtained from GFAP-IL-6 mice. However, in these studies, homozygous animals from the 16 line were used, which express higher levels of IL-6. Field potential recordings showed that LTP was significantly reduced at this synapse in the GFAP-IL-6 hippocampus compared with the non-transgenic hippocampus (Bellinger et al., 1995). The difference in sensitivity of LTP to elevated levels of IL-6 between the Schaffer collateral to CA1 pyramidal neuron synapse vs. the perforant pathway to granule neuron synapse is likely due to differences in the levels of IL-6 expressed in the two lines of GFAP-IL-6 mice. However, differences in the type of LTP studied could also be a contributing factor.
7.3. IL-6 expression during hippocampal LTP
Although studies using exogenous application of IL-6 provided support for a role of IL-6 in LTP, studies that investigated IL-6 expression during LTP were instrumental in defining a regulatory role for IL-6 in LTP. Studies of LTP at the Schaffer collateral to CA1 pyramidal neuron synapse in hippocampus from normal animals in vivo and in vitro revealed that neuronal activity that induces hippocampal LTP results in increased levels of IL-6 in the hippocampus. Thus, when anesthetized animals implanted with electrodes in the hippocampus were subjected to Schaffer collateral stimulation using a protocol known to induce LTP, the level of IL-6 mRNA was significantly increased (approximately 20 fold) in the CA1 region by the Schaffer collateral stimulation (Jankowsky et al., 2000). The expression of IL-6 mRNA was localized to astrocytes in the parenchyma near the stimulation site but not in neurons (Jankowsky et al., 2000). Similar results were obtained in LTP experiments carried out in in vitro hippocampal slices. In these studies, LTP at the Schaffer collateral to CA1 pyramidal neuron synapse resulted in significantly increased levels of IL-6 mRNA by 1 h post stimulation (Balschun et al., 2004). LTP induction at the perforant path to granule neuron synapse in freelymoving rats also resulted in Increased IL-6 mRNA expression in the hippocampus 8 h after the LTP induction protocol (Balschun et al., 2004). The increase in IL-6 mRNA was observed only when successful LTP had been induced and was blocked by infusion of an NMDA receptor antagonist, which blocks LTP, prior to the LTP induction protocol (Balschun et al., 2004). Additional experiments involving infusion of an IL-6 neutralizing antibody prior to the LTP induction protocol showed that blocking IL-6 enhanced the late phase of LTP (i.e., long-lasting LTP) (Balschun et al., 2004). Infusion of an IL-6 neutralizing antibody also enhanced behavior in a hippocampus-dependent task (spatial alternation) shown to be associated with increased levels of IL-6 in the hippocampus (del Rey et al., 2013). The increase of IL-6 in the behavioral task was about 2 fold compared with a 5–7 fold increase in LTP experiments. From these studies, the authors concluded that endogenous IL-6 induced in the hippocampus by neuronal activity plays a role in the maintenance phase of LTP (i.e., long-lasting LTP), where it acts as a negative regulator of LTP maintenance by limiting long-term storage of certain types of memory information. Negative regulation of memory mechanisms is essential for fine-tuning memory consolidation and resetting memory mechanisms so that new memories can be encoded. A negative role for IL-6 in memory formation was also supported by studies of IL-6 deficient mice (IL-6 KO), where a facilitatory effect of the IL-6 deficiency on learning and memory was observed (Baier et al., 2009).
7.4. IL-6 and LTD
While studies have shown that IL-6 can regulate LTP in the adult hippocampus, no studies have addressed potential actions of IL-6 on LTD. Effects of IL-6 on LTD is of interest because recent studies show that the JAK2/STAT3 signal transduction pathway, the signal transduction pathway used by IL-6 (Fig. 1), plays an essential role in the induction of LTD at the Schaffer collateral to CA1 synapse in hippocampal slices from juvenile mice (13–18 days postnatal) (Nicolas et al., 2012). These studies used field potential recordings and a combination of pharmacological, biochemical and genetic tools. They identified that: (a) the induction of LTD depended on Ca2+ influx through NMDA receptors and activation of the JAK2/STAT3 pathway through phosphorylation of JAK2 and STAT3, (b) that the interaction took place at the synapses and did not involve gene expression, (c) that new protein synthesis was not required, and (d) that the JAK2/STAT3 pathway was not involved in baseline synaptic transmission. If IL-6 can engage this pathway, it would provide a novel mechanism for IL-6 regulation of CNS function.
7.5. Summary
Taken together, the above studies provide convincing evidence that IL-6 plays a role in homeostatic control of memory mechanisms, at least in the hippocampus. The studies also show that signal transduction molecules such as JAK2, STAT3, and p42/44 MAPK are important players in IL-6 regulation of memory mechanisms, but many details are still missing and need to be revealed before the role of IL-6 in memory and other cognitive functions will be fully understood.
8. Conclusions
Research has provided clear evidence that IL-6 is produced by cells of the CNS, that IL-6 is an important signaling molecule in the CNS, and that IL-6 is involved in a variety of activities, ranging from neuronal physiology, an issue addressed in this review, to neurodevelopment, neuroprotection and neurotoxicity (Gadient and Otten, 1997; Gruol and Nelson, 1997). However, our understanding of roles of IL-6 in CNS physiology and pathology and the molecular mechanisms involved are still limited. Confounding this situation is the fact that typically multiple neuroimmune factors are produced under similar conditions and that other neuroimmune factors have neuronal actions, a complexity that challenges efforts to identify the functional role of individual factors, such as IL-6. Moreover, interaction between neuroimmune factors at the signal transduction level could render the downstream effects dependent on the complement of neuroimmune factors present, their concentrations and the cell types responding. Fortunately, recognition of the importance of neuroimmune aspects of CNS function is rapidly growing and the research base is expanding, changes that will likely lead to significant advances in the field and to the development of new therapeutic strategies for treating CNS disorders.
Acknowledgments
The author acknowledges the support of her research over the past years from the following NIH grants: AA019261, AA07456, MH083723, MH63712, MH62261, MH47680, HD38231, DA10187.
References
- Alexander GM, Perreault MJ, Reichenberger ER, Schwartzman RJ. Changes in immune and glial markers in the CSF of patients with complex regional pain syndrome. Brain Behav Immun. 2007;21:668–676. doi: 10.1016/j.bbi.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Aloisi F, Borsellino G, Care A, Testa U, Gallo P, Russo G, Peschle C, Levi G. Cytokine regulation of astrocyte function: in-vitro studies using cells from the human brain. Int J Dev Neurosci. 1995;13:265–274. doi: 10.1016/0736-5748(94)00071-a. [DOI] [PubMed] [Google Scholar]
- Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- Arruda JL, Colburn RW, Rickman AJ, Rutkowski MD, DeLeo JA. Increase of interleukin-6 mRNA in the spinal cord following peripheral nerve injury in the rat: potential role of IL-6 in neuropathic pain. Brain Res Mol Brain Res. 1998;62:228–235. doi: 10.1016/s0169-328x(98)00257-5. [DOI] [PubMed] [Google Scholar]
- Atzori M, Garcia-Oscos F, Mendez JA. Role of IL-6 in the etiology of hyperexcitable neuropsychiatric conditions: experimental evidence and therapeutic implications. Future Med Chem. 2012;4:2177–2192. doi: 10.4155/fmc.12.156. [DOI] [PubMed] [Google Scholar]
- Baier PC, May U, Scheller J, Rose-John S, Schiffelholz T. Impaired hippocampus-dependent and -independent learning in IL-6 deficient mice. Behav Brain Res. 2009;200:192–196. doi: 10.1016/j.bbr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Balschun D, Wetzel W, Del Rey A, Pitossi F, Schneider H, Zuschratter W, Besedovsky HO. Interleukin-6: a cytokine to forget. FASEB J. 2004;18:1788–1790. doi: 10.1096/fj.04-1625fje. [DOI] [PubMed] [Google Scholar]
- Bauer S, Cepok S, Todorova-Rudolph A, Nowak M, Koller M, Lorenz R, Oertel WH, Rosenow F, Hemmer B, Hamer HM. Etiology and site of temporal lobe epilepsy influence postictal cytokine release. Epilepsy Res. 2009;86:82–88. doi: 10.1016/j.eplepsyres.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Bellinger FP, Madamba SG, Campbell IL, Siggins GR. Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci Lett. 1995;198:95–98. doi: 10.1016/0304-3940(95)11976-4. [DOI] [PubMed] [Google Scholar]
- Ben Achour S, Pont-Lezica L, Bechade C, Pascual O. Is astrocyte calcium signaling relevant for synaptic plasticity? Neuron Glia Biol. 2010;6:147–155. doi: 10.1017/S1740925X10000207. [DOI] [PubMed] [Google Scholar]
- Benedict C, Scheller J, Rose-John S, Born J, Marshall L. Enhancing influence of intranasal interleukin-6 on slow-wave activity and memory consolidation during sleep. FASEB J. 2009;23:3629–3636. doi: 10.1096/fj.08-122853. [DOI] [PubMed] [Google Scholar]
- Benveniste EN. Inflammatory cytokines within the central nervous system: sources, function, and mechanism of action. Am J Physiol. 1992;263:C1–C16. doi: 10.1152/ajpcell.1992.263.1.C1. [DOI] [PubMed] [Google Scholar]
- Bergamaschi A, Corsi M, Garnier MJ. Synergistic effects of cAMPdependent signalling pathways and IL-1 on IL-6 production by H19-7/IGF-IR neuronal cells. Cell Signal. 2006;18:1679–1684. doi: 10.1016/j.cellsig.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Beridze M, Sanikidze T, Shakarishvili R, Intskirveli N, Bornstein NM. Selected acute phase CSF factors in ischemic stroke: findings and prognostic value. BMC Neurol. 2011;11:41. doi: 10.1186/1471-2377-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo P, Martin-Aragon S, Benedi J, Susin C, Felici E, Gil P, Ribera JM, Villar AM. Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer’s disease. Immunol Lett. 2008;117:198–202. doi: 10.1016/j.imlet.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Iyengar R, Landau EM. Postsynaptic signaling networks: cellular cogwheels underlying long-term plasticity. Biol Psychiatry. 2005;57:113–119. doi: 10.1016/j.biopsych.2004.02.031. [DOI] [PubMed] [Google Scholar]
- Burton MD, Johnson RW. Interleukin-6 trans-signaling in the senescent mouse brain is involved in infection-related deficits in contextual fear conditioning. Brain Behav Immun. 2012;26:732–738. doi: 10.1016/j.bbi.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IL. Transgenic mice and cytokine actions in the brain: bridging the gap between structural and functional neuropathology. Brain Res Brain Res Rev. 1998;26:327–336. doi: 10.1016/s0165-0173(97)00038-6. [DOI] [PubMed] [Google Scholar]
- Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci U S A. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IL, Erta M, Lim SL, Frausto R, May U, Rose-John S, Scheller J, Hidalgo J. Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J Neurosci. 2014;34:2503–2513. doi: 10.1523/JNEUROSCI.2830-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaretti A, Antonelli A, Mastrangelo A, Pezzotti P, Tortorolo L, Tosi F, Genovese O. Interleukin-6 and nerve growth factor upregulation correlates with improved outcome in children with severe traumatic brain injury. J Neurotrauma. 2008;25:225–234. doi: 10.1089/neu.2007.0405. [DOI] [PubMed] [Google Scholar]
- Chiba T, Yamada M, Aiso S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin Ther Targets. 2009;13:1155–1167. doi: 10.1517/14728220903213426. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim SY, Park HJ, Cha JH, Choi YS, Kang JE, Chung JW, Chun MH, Lee MY. Upregulation of gp130 and differential activation of STAT and p42/44 MAPK in the rat hippocampus following kainic acid-induced seizures. Brain Res Mol Brain Res. 2003;119:10–18. doi: 10.1016/j.molbrainres.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Choi SS, Lee HJ, Lim I, Satoh J, Kim SU. Human astrocytes: secretome profiles of cytokines and chemokines. PloS One. 2014;9:e92325. doi: 10.1371/journal.pone.0092325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourbaji S, Urani A, Inta I, Sanchis-Segura C, Brandwein C, Zink M, Schwaninger M, Gass P. IL-6 knockout mice exhibit resistance to stressinduced development of depression-like behaviors. Neurobiol Dis. 2006;23:587–594. doi: 10.1016/j.nbd.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Cojocaru IM, Cojocaru M, Miu G, Sapira V. Study of interleukin-6 production in Alzheimer’s disease. Romanian J Intern Med. 2011;49:55–58. [PubMed] [Google Scholar]
- Conroy SM, Nguyen V, Quina LA, Blakely-Gonzales P, Ur C, Netzeband JG, Prieto AL, Gruol DL. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J Neuroimmunol. 2004;155:43–54. doi: 10.1016/j.jneuroim.2004.06.014. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Tancredi V, Onofri F, D’Antuono M, Giovedi S, Benfenati F. Interleukin-6 inhibits neurotransmitter release and the spread of excitation in the rat cerebral cortex. Eur J Neurosci. 2000;12:1241–1252. doi: 10.1046/j.1460-9568.2000.00011.x. [DOI] [PubMed] [Google Scholar]
- Dame JB, Juul SE. The distribution of receptors for the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum Dev. 2000;58:25–39. doi: 10.1016/s0378-3782(00)00064-5. [DOI] [PubMed] [Google Scholar]
- del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO. A cytokine network involving brain-borne IL-1beta, IL-1ra, IL-18, IL-6, and TNFalpha operates during long-term potentiation and learning. Brain Behav Immun. 2013;33:15–23. doi: 10.1016/j.bbi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Dimitrov S, Lange T, Benedict C, Nowell MA, Jones SA, Scheller J, Rose-John S, Born J. Sleep enhances IL-6 trans-signaling in humans. FASEB J. 2006;20:2174–2176. doi: 10.1096/fj.06-5754fje. [DOI] [PubMed] [Google Scholar]
- Dinel AL, Andre C, Aubert A, Ferreira G, Laye S, Castanon N. Cognitive and emotional alterations are related to hippocampal inflammation in a mouse model of metabolic syndrome. PloS One. 2011;6:e24325. doi: 10.1371/journal.pone.0024325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donegan JJ, Girotti M, Weinberg MS, Morilak DA. A novel role for brain interleukin-6: facilitation of cognitive flexibility in rat orbitofrontal cortex. J Neurosci. 2014;34:953–962. doi: 10.1523/JNEUROSCI.3968-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Fields RD. Mitogen-activated protein kinase/extracellular signal-regulated kinase activation in somatodendritic compartments: roles of action potentials, frequency, and mode of calcium entry. J Neurosci. 2001;21:Rc122. doi: 10.1523/JNEUROSCI.21-02-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, de Beaurepaire R. Cytokines as mediators of depression: what can we learn from animal studies? Neurosci Biobehav Rev. 2005;29:891–909. doi: 10.1016/j.neubiorev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Economos A, Wright CB, Moon YP, Rundek T, Rabbani L, Paik MC, Sacco RL, Elkind MS. Interleukin 6 plasma concentration associates with cognitive decline: the northern Manhattan study. Neuroepidemiology. 2013;40:253–259. doi: 10.1159/000343276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H. Hippocampus: cognitive processes and neural representations that underlie declarative memory. Neuron. 2004;44:109–120. doi: 10.1016/j.neuron.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Elderkin-Thompson V, Irwin MR, Hellemann G, Kumar A. Interleukin-6 and memory functions of encoding and recall in healthy and depressed elderly adults. Am J Geriatr Psychiatry. 2012;20:753–763. doi: 10.1097/JGP.0b013e31825d08d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MA, Dohi K, Banks WA. Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation. 2012;19:121–130. doi: 10.1159/000330247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Fattori E, Lazzaro D, Musiani P, Modesti A, Alonzi T, Ciliberto G. IL-6 expression in neurons of transgenic mice causes reactive astrocytosis and increase in ramified microglial cells but no neuronal damage. Eur J Neurosci. 1995;7:2441–2449. doi: 10.1111/j.1460-9568.1995.tb01042.x. [DOI] [PubMed] [Google Scholar]
- Fiore RS, Bayer VE, Pelech SL, Posada J, Cooper JA, Baraban JM. p42 mitogen-activated protein kinase in brain: prominent localization in neuronal cell bodies and dendrites. Neuroscience. 1993;55:463–472. doi: 10.1016/0306-4522(93)90516-i. [DOI] [PubMed] [Google Scholar]
- Fragoso-Loyo H, Richaud-Patin Y, Orozco-Narvaez A, Davila-Maldonado L, Atisha-Fregoso Y, Llorente L, Sanchez-Guerrero J. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 2007;56:1242–1250. doi: 10.1002/art.22451. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten U. Expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat brain during postnatal development. Brain Res. 1994;637:10–14. doi: 10.1016/0006-8993(94)91211-4. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten UH. Interleukin-6 (IL-6)–a molecule with both beneficial and destructive potentials. Prog Neurobiol. 1997;52:379–390. doi: 10.1016/s0301-0082(97)00021-x. [DOI] [PubMed] [Google Scholar]
- Garcia-Esparcia P, Llorens F, Carmona M, Ferrer I. Complex deregulation and expression of cytokines and mediators of the immune response in Parkinson’s disease brain is region dependent. Brain Pathol. 2014 doi: 10.1111/bpa.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Oscos F, Salgado H, Hall S, Thomas F, Farmer GE, Bermeo J, Galindo LC, Ramirez RD, D’Mello S, Rose-John S, Atzori M. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol Psychiatry. 2012;71:574–582. doi: 10.1016/j.biopsych.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, Wang YT. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci U S A. 2010;107:16697–16702. doi: 10.1073/pnas.1008200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruol DL, Nelson TE. Physiological and pathological roles of interleukin-6 in the central nervous system. Mol Neurobiol. 1997;15:307–339. doi: 10.1007/BF02740665. [DOI] [PubMed] [Google Scholar]
- Ha BK, King JS. Localization of gp130 in the developing and adult mouse cerebellum. J Chem Neuroanat. 2000;19:129–141. doi: 10.1016/s0891-0618(00)00056-9. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Hampel H, Haslinger A, Scheloske M, Padberg F, Fischer P, Unger J, Teipel SJ, Neumann M, Rosenberg C, Oshida R, Hulette C, Pongratz D, Ewers M, Kretzschmar HA, Moller HJ. Pattern of interleukin-6 receptor complex immunoreactivity between cortical regions of rapid autopsy normal and Alzheimer’s disease brain. Eur Arch Psychiatry Clin Neurosci. 2005;255:269–278. doi: 10.1007/s00406-004-0558-2. [DOI] [PubMed] [Google Scholar]
- Hans VH, Kossmann T, Lenzlinger PM, Probstmeier R, Imhof HG, Trentz O, Morganti-Kossmann MC. Experimental axonal injury triggers interleukin-6 mRNA, protein synthesis and release into cerebrospinal fluid. J Cereb Blood Flow Metab. 1999;19:184–194. doi: 10.1097/00004647-199902000-00010. [DOI] [PubMed] [Google Scholar]
- Heffner KL, Ng HM, Suhr JA, France CR, Marshall GD, Pigeon WR, Moynihan JA. Sleep disturbance and older adults’ inflammatory responses to acute stress. Am J Geriatr Psychiatry. 2012;20:744–752. doi: 10.1097/JGP.0b013e31824361de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyser CJ, Masliah E, Samimi A, Campbell IL, Gold LH. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc Natl Acad Sci U S A. 1997;94:1500–1505. doi: 10.1073/pnas.94.4.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiles SA, Baker AL, de Malmanche T, Attia J. A meta-analysis of differences in IL-6 and IL-10 between people with and without depression: exploring the causes of heterogeneity. Brain Behav Immun. 2012;26:1180–1188. doi: 10.1016/j.bbi.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Hillman J, Aneman O, Anderson C, Sjogren F, Saberg C, Mellergard P. A microdialysis technique for routine measurement of macromolecules in the injured human brain. Neurosurgery. 2005;56:1264–1268. doi: 10.1227/01.neu.0000159711.93592.8d. discussion 1268e1270. [DOI] [PubMed] [Google Scholar]
- Hirano T, Taga T, Nakano N, Yasukawa K, Kashiwamura S, Shimizu K, Nakajima K, Pyun KH, Kishimoto T. Purification to homogeneity and characterization of human B-cell differentiation factor (BCDF or BSFp-2) Proc Natl Acad Sci U S A. 1985;82:5490–5494. doi: 10.1073/pnas.82.16.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirohata S, Miyamoto T. Elevated levels of interleukin-6 in cerebrospinal fluid from patients with systemic lupus erythematosus and central nervous system involvement. Arthritis Rheum. 1990;33:644–649. doi: 10.1002/art.1780330506. [DOI] [PubMed] [Google Scholar]
- Hong S, Mills PJ, Loredo JS, Adler KA, Dimsdale JE. The association between interleukin-6, sleep, and demographic characteristics. Brain Behav Immun. 2005;19:165–172. doi: 10.1016/j.bbi.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hu Z, Liu G, Zhou W, Zhang Y. Cytokines induced by long-term potentiation (LTP) recording: a potential explanation for the lack of correspondence between learning/memory performance and LTP. Neuroscience. 2013;231:432–443. doi: 10.1016/j.neuroscience.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Huell M, Strauss S, Volk B, Berger M, Bauer J. Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol. 1995;89:544–551. doi: 10.1007/BF00571510. [DOI] [PubMed] [Google Scholar]
- Ichiyama T, Ito Y, Kubota M, Yamazaki T, Nakamura K, Furukawa S. Serum and cerebrospinal fluid levels of cytokines in acute encephalopathy associated with human herpesvirus-6 infection. Brain Dev. 2009;31:731–738. doi: 10.1016/j.braindev.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Imamura K, Hishikawa N, Ono K, Suzuki H, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. 2005;109:141–150. doi: 10.1007/s00401-004-0919-y. [DOI] [PubMed] [Google Scholar]
- Irwin M, Rinetti G, Redwine L, Motivala S, Dang J, Ehlers C. Nocturnal proinflammatory cytokine-associated sleep disturbances in abstinent African American alcoholics. Brain Behav Immun. 2004;18:349–360. doi: 10.1016/j.bbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Janelidze S, Mattei D, Westrin A, Traskman-Bendz L, Brundin L. Cytokine levels in the blood may distinguish suicide attempters from depressed patients. Brain Behav Immun. 2011;25:335–339. doi: 10.1016/j.bbi.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Derrick BE, Patterson PH. Cytokine responses to LTP induction in the rat hippocampus: a comparison of in vitro and in vivo techniques. Learn Mem. 2000;7:400–412. doi: 10.1101/lm.32600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Thomsen C. The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci. 2013;53:52–62. doi: 10.1016/j.mcn.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Juttler E, Tarabin V, Schwaninger M. Interleukin-6 (IL-6): a possible neuromodulator induced by neuronal activity. Neuroscientist. 2002;8:268–275. doi: 10.1177/1073858402008003012. [DOI] [PubMed] [Google Scholar]
- King DL, Arendash GW. Behavioral characterization of the Tg2576 transgenic model of Alzheimer’s disease through 19 months. Physiol Behav. 2002;75:627–642. doi: 10.1016/s0031-9384(02)00639-x. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther. 2006;8(Suppl 2):S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov AV, Fursenko DV, Khotskin NV, Bazovkina DV, Kulikov VA, Naumenko VS, Bazhenova EY, Popova NK. Spatial learning in the Morris water maze in mice genetically different in the predisposition to catalepsy: the effect of intraventricular treatment with brain-derived neurotrophic factor. Pharmacol Biochem Behav. 2014;122:266–272. doi: 10.1016/j.pbb.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Palmer M, Segerstrale M, Vesikansa A, Taira T, Collingridge GL. Presynaptic mechanisms involved in the expression of STP and LTP at CA1 synapses in the hippocampus. Neuropharmacology. 2007;52:1–11. doi: 10.1016/j.neuropharm.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lehtimaki KA, Keranen T, Huhtala H, Hurme M, Ollikainen J, Honkaniemi J, Palmio J, Peltola J. Regulation of IL-6 system in cerebrospinal fluid and serum compartments by seizures: the effect of seizure type and duration. J Neuroimmunol. 2004;152:121–125. doi: 10.1016/j.jneuroim.2004.01.024. [DOI] [PubMed] [Google Scholar]
- Lekander M, von Essen J, Schultzberg M, Andreasson AN, Garlind A, Hansson LO, Nilsson LG. Cytokines and memory across the mature life span of women. Scand J Psychol. 2011;52:229–235. doi: 10.1111/j.1467-9450.2010.00865.x. [DOI] [PubMed] [Google Scholar]
- Li AJ, Katafuchi T, Oda S, Hori T, Oomura Y. Interleukin-6 inhibits longterm potentiation in rat hippocampal slices. Brain Res. 1997;748:30–38. doi: 10.1016/s0006-8993(96)01283-8. [DOI] [PubMed] [Google Scholar]
- Li G, Bauer S, Nowak M, Norwood B, Tackenberg B, Rosenow F, Knake S, Oertel WH, Hamer HM. Cytokines and epilepsy. Seizure. 2011;20:249–256. doi: 10.1016/j.seizure.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Lindqvist D, Hall S, Surova Y, Nielsen HM, Janelidze S, Brundin L, Hansson O. Cerebrospinal fluid inflammatory markers in Parkinson’s disease – associations with depression, fatigue, and cognitive impairment. Brain Behav Immun. 2013;33:183–189. doi: 10.1016/j.bbi.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Lindqvist D, Janelidze S, Hagell P, Erhardt S, Samuelsson M, Minthon L, Hansson O, Bjorkqvist M, Traskman-Bendz L, Brundin L. Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol Psychiatry. 2009;66:287–292. doi: 10.1016/j.biopsych.2009.01.030. [DOI] [PubMed] [Google Scholar]
- Lorigados Pedre L, Morales Chacon LM, Orozco Suarez S, Pavon Fuentes N, Estupinan Diaz B, Serrano Sanchez T, Garcia Maeso I, Rocha Arrieta L. Inflammatory mediators in epilepsy. Curr Pharm Des. 2013;19:6766–6772. doi: 10.2174/1381612811319380009. [DOI] [PubMed] [Google Scholar]
- Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. 1992;4:96–100. doi: 10.1016/1043-4666(92)90043-q. [DOI] [PubMed] [Google Scholar]
- Luterman JD, Haroutunian V, Yemul S, Ho L, Purohit D, Aisen PS, Mohs R, Pasinetti GM. Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch Neurol. 2000;57:1153–1160. doi: 10.1001/archneur.57.8.1153. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Ma TC, Zhu XZ. Intrahippocampal infusion of interleukin-6 impairs avoidance learning in rats. Zhongguo Yao Li Xue Bao Acta Pharmacol Sin. 1997;18:121–123. [PubMed] [Google Scholar]
- Maier B, Laurer HL, Rose S, Buurman WA, Marzi I. Physiological levels of pro- and anti-inflammatory mediators in cerebrospinal fluid and plasma: a normative study. J Neurotrauma. 2005;22:822–835. doi: 10.1089/neu.2005.22.822. [DOI] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao LY, Ding J, Peng WF, Ma Y, Zhang YH, Fan W, Wang X. Interictal interleukin-17A levels are elevated and correlate with seizure severity of epilepsy patients. Epilepsia. 2013;54:e142–145. doi: 10.1111/epi.12337. [DOI] [PubMed] [Google Scholar]
- Marsland AL, Petersen KL, Sathanoori R, Muldoon MF, Neumann SA, Ryan C, Flory JD, Manuck SB. Interleukin-6 covaries inversely with cognitive performance among middle-aged community volunteers. Psychosom Med. 2006;68:895–903. doi: 10.1097/01.psy.0000238451.22174.92. [DOI] [PubMed] [Google Scholar]
- Masi A, Quintana DS. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. 2014 doi: 10.1038/mp.2014.59. [DOI] [PubMed] [Google Scholar]
- Massey PV, Bashir ZI. Long-term depression: multiple forms and implications for brain function. Trends Neurosci. 2007;30:176–184. doi: 10.1016/j.tins.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Matzel LD, Grossman H, Light K, Townsend D, Kolata S. Age-related declines in general cognitive abilities of Balb/C mice are associated with disparities in working memory, body weight, and general activity. Learn Mem. 2008;15:733–746. doi: 10.1101/lm.954808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje FJ, Cabatic M, Divisch I, Kim EJ, Herkner KR, Binder BR, Pollak DD. Constant darkness induces IL-6-dependent depression-like behavior through the NF-kappaB signaling pathway. J Neurosci. 2011;31:9075–9083. doi: 10.1523/JNEUROSCI.1537-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa Y, Tohya K, Tamura S, Ichihara M, Miyajima A, Senba E. Expression of interleukin-6 receptor, leukemia inhibitory factor receptor and glycoprotein 130 in the murine cerebellum and neuropathological effect of leukemia inhibitory factor on cerebellar Purkinje cells. Neuroscience. 2000;100:841–848. doi: 10.1016/s0306-4522(00)00302-x. [DOI] [PubMed] [Google Scholar]
- Mullberg J, Vollmer P, Althoff K, Marz P, Rose-John S. Generation and function of the soluble interleukin-6 receptor. Biochem Soc Trans. 1999;27:211–219. doi: 10.1042/bst0270211. [DOI] [PubMed] [Google Scholar]
- Murase S, McKay RD. Neuronal activity-dependent STAT3 localization to nucleus is dependent on Tyr-705 and Ser-727 phosphorylation in rat hippocampal neurons. Eur J Neurosci. 2014;39:557–565. doi: 10.1111/ejn.12412. [DOI] [PubMed] [Google Scholar]
- Murata S, Usuda N, Okano A, Kobayashi S, Suzuki T. Occurrence of a transcription factor, signal transducer and activators of transcription 3 (Stat3), in the postsynaptic density of the rat brain. Brain Res Mol Brain Res. 2000;78:80–90. doi: 10.1016/s0169-328x(00)00077-2. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Hirano T, Takatsuki F, Sakaguchi N, Yoshida N, Kishimoto T. Physicochemical and functional properties of murine B cell-derived B cell growth factor II (WEHI-231-BCGF-II) J Immunol. 1985;135:1207–1212. [PubMed] [Google Scholar]
- Nakamachi T, Tsuchida M, Kagami N, Yofu S, Wada Y, Hori M, Tsuchikawa D, Yoshikawa A, Imai N, Nakamura K, Arata S, Shioda S. IL-6 and PACAP receptor expression and localization after global brain ischemia in mice. J Mol Neurosci. 2012;48:518–525. doi: 10.1007/s12031-012-9819-0. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Campbell IL, Gruol DL. Altered physiology of Purkinje neurons in cerebellar slices from transgenic mice with chronic central nervous system expression of interleukin-6. Neuroscience. 1999;89:127–136. doi: 10.1016/s0306-4522(98)00316-9. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Netzeband JG, Gruol DL. Chronic interleukin-6 exposure alters metabotropic glutamate receptor-activated calcium signalling in cerebellar Purkinje neurons. Eur J Neurosci. 2004;20:2387–2400. doi: 10.1111/j.1460-9568.2004.03706.x. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Olde Engberink A, Hernandez R, Puro A, Huitron Resendiz S, Hao C, DeGraan PN, Gruol DL. Altered synaptic transmission in the hippocampus of transgenic mice with enhanced central nervous systems expression of interleukin-6. Brain Behav Immun. 2012;26:959–971. doi: 10.1016/j.bbi.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson TE, Ur CL, Gruol DL. Chronic interleukin-6 exposure alters electrophysiological properties and calcium signaling in developing cerebellar Purkinje neurons in culture. J Neurophysiol. 2002;88:475–486. doi: 10.1152/jn.2002.88.1.475. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, Collingridge GL. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JG, Tang LP, Sparacio SM, Benveniste EN. Signal transduction pathways mediating astrocyte IL-6 induction by IL-1 beta and tumor necrosis factor-alpha. J Immunol. 1994;152:841–850. [PubMed] [Google Scholar]
- Pandey GN, Rizavi HS, Ren X, Fareed J, Hoppensteadt DA, Roberts RC, Conley RR, Dwivedi Y. Proinflammatory cytokines in the prefrontal cortex of teenage suicide victims. J Psychiatr Res. 2012;46:57–63. doi: 10.1016/j.jpsychires.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannasch U, Rouach N. Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci. 2013;36:405–417. doi: 10.1016/j.tins.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Peltola J, Hurme M, Miettinen A, Keranen T. Elevated levels of interleukin- 6 may occur in cerebrospinal fluid from patients with recent epileptic seizures. Epilepsy Res. 1998;31:129–133. doi: 10.1016/s0920-1211(98)00024-2. [DOI] [PubMed] [Google Scholar]
- Peltola J, Palmio J, Korhonen L, Suhonen J, Miettinen A, Hurme M, Lindholm D, Keranen T. Interleukin-6 and interleukin-1 receptor antagonist in cerebrospinal fluid from patients with recent tonic-clonic seizures. Epilepsy Res. 2000;41:205–211. doi: 10.1016/s0920-1211(00)00140-6. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- Pineda E, Shin D, Sankar R, Mazarati AM. Comorbidity between epilepsy and depression: experimental evidence for the involvement of serotonergic, glucocorticoid, and neuroinflammatory mechanisms. Epilepsia. 2010;51(Suppl 3):110–114. doi: 10.1111/j.1528-1167.2010.02623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Parsons KL, Gruol DL. Interleukin-6 selectively enhances the intracellular calcium response to NMDA in developing CNS neurons. J Neurosci. 1995;15:6688–6699. doi: 10.1523/JNEUROSCI.15-10-06688.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Sweeney DD, Netzeband JG, Gruol DL. Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J Neurosci. 1998;18:10445–10456. doi: 10.1523/JNEUROSCI.18-24-10445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheumatology, T. A. C. o. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42:599–608. doi: 10.1002/1529-0131(199904)42:4<599::AID-ANR2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Ringheim GE, Burgher KL, Heroux JA. Interleukin-6 mRNA expression by cortical neurons in culture: evidence for neuronal sources of interleukin-6 production in the brain. J Neuroimmunol. 1995;63:113–123. doi: 10.1016/0165-5728(95)00134-4. [DOI] [PubMed] [Google Scholar]
- Roberts DJ, Jenne CN, Leger C, Kramer AH, Gallagher CN, Todd S, Parney IF, Doig CJ, Yong VW, Kubes P, Zygun DA. Association between the cerebral inflammatory and matrix metalloproteinase responses after severe traumatic brain injury in humans. J Neurotrauma. 2013;30:1727–1736. doi: 10.1089/neu.2012.2842. [DOI] [PubMed] [Google Scholar]
- Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–236. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- Sallmann S, Juttler E, Prinz S, Petersen N, Knopf U, Weiser T, Schwaninger M. Induction of interleukin-6 by depolarization of neurons. J Neurosci. 2000;20:8637–8642. doi: 10.1523/JNEUROSCI.20-23-08637.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754–760. doi: 10.1017/S1355617710000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samland H, Huitron-Resendiz S, Masliah E, Criado J, Henriksen SJ, Campbell IL. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J Neurosci Res. 2003;73:176–187. doi: 10.1002/jnr.10635. [DOI] [PubMed] [Google Scholar]
- Sasayama D, Hattori K, Wakabayashi C, Teraishi T, Hori H, Ota M, Yoshida S, Arima K, Higuchi T, Amano N, Kunugi H. Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. J Psychiatr Res. 2013;47:401–406. doi: 10.1016/j.jpsychires.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Schobitz B, de Kloet ER, Sutanto W, Holsboer F. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur J Neurosci. 1993;5:1426–1435. doi: 10.1111/j.1460-9568.1993.tb00210.x. [DOI] [PubMed] [Google Scholar]
- Shapiro M. Plasticity, hippocampal place cells, and cognitive maps. Arch Neurol. 2001;58:874–881. doi: 10.1001/archneur.58.6.874. [DOI] [PubMed] [Google Scholar]
- Shin HC, Oh S, Jung SC, Park J, Won CK. Differential modulation of short and long latency sensory responses in the SI cortex by IL-6. Neuroreport. 1997;8:2841–2844. doi: 10.1097/00001756-199709080-00007. [DOI] [PubMed] [Google Scholar]
- Silvestroni A, Faull RL, Strand AD, Moller T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport. 2009;20:1098–1103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- Smith C, Squire LR. Declarative memory, awareness, and transitive inference. J Neurosci. 2005;25:10138–10146. doi: 10.1523/JNEUROSCI.2731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation. 2008;15:323–330. doi: 10.1159/000156474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley AC, Lacy P. Pathways for cytokine secretion. Physiol Bethesda. 2010;25:218–229. doi: 10.1152/physiol.00017.2010. [DOI] [PubMed] [Google Scholar]
- Steffensen SC, Campbell IL, Henriksen SJ. Site-specific hippocampal pathophysiology due to cerebral overexpression of interleukin-6 in transgenic mice. Brain Res. 1994;652:149–153. doi: 10.1016/0006-8993(94)90329-8. [DOI] [PubMed] [Google Scholar]
- Strauss S, Bauer J, Ganter U, Jonas U, Berger M, Volk B. Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer’s disease patients. Lab Investig. 1992;66:223–230. [PubMed] [Google Scholar]
- Sukoff Rizzo SJ, Neal SJ, Hughes ZA, Beyna M, Rosenzweig-Lipson S, Moss SJ, Brandon NJ. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Transl Psychiatry. 2012;2:e199. doi: 10.1038/tp.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swardfager W, Lanctot K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biol Psychiatry. 2010;68:930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Takahashi W, Nakada TA, Abe R, Tanaka K, Matsumura Y, Oda S. Use-fulness of interleukin 6 levels in the cerebrospinal fluid for the diagnosis of bacterial meningitis. J Crit Care. 2014;29:693.e691–693.e696. doi: 10.1016/j.jcrc.2014.02.020. [DOI] [PubMed] [Google Scholar]
- Tan H, Cao J, Zhang J, Zuo Z. Critical role of inflammatory cytokines in impairing biochemical processes for learning and memory after surgery in rats. J Neuroinflammation. 2014;11:93. doi: 10.1186/1742-2094-11-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tancredi V, D’Antuono M, Cafe C, Giovedi S, Bue MC, D’Arcangelo G, Onofri F, Benfenati F. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem. 2000;75:634–643. doi: 10.1046/j.1471-4159.2000.0750634.x. [DOI] [PubMed] [Google Scholar]
- Taylor VH, MacQueen GM. Cognitive dysfunction associated with metabolic syndrome. Obes Rev. 2007;8:409–418. doi: 10.1111/j.1467-789X.2007.00401.x. [DOI] [PubMed] [Google Scholar]
- Tehranian R, Hasanvan H, Iverfeldt K, Post C, Schultzberg M. Early induction of interleukin-6 mRNA in the hippocampus and cortex of APPsw transgenic mice Tg2576. Neurosci Lett. 2001;301:54–58. doi: 10.1016/s0304-3940(01)01592-0. [DOI] [PubMed] [Google Scholar]
- Terreni L, De Simoni MG. Role of the brain in interleukin-6 modulation. Neuroimmunomodulation. 1998;5:214–219. doi: 10.1159/000026339. [DOI] [PubMed] [Google Scholar]
- Threlkeld SW, Lynch JL, Lynch KM, Sadowska GB, Banks WA, Stonestreet BS. Ovine proinflammatory cytokines cross the murine blood-brain barrier by a common saturable transport mechanism. Neuroimmunomodulation. 2010;17:405–410. doi: 10.1159/000288265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita M, Holman BJ, Santoro TJ. Aberrant cytokine gene expression in the hippocampus in murine systemic lupus erythematosus. Neurosci Lett. 2001a;302:129–132. doi: 10.1016/s0304-3940(01)01679-2. [DOI] [PubMed] [Google Scholar]
- Tomita M, Holman BJ, Williams LS, Pang KC, Santoro TJ. Cerebellar dysfunction is associated with overexpression of proinflammatory cytokine genes in lupus. J Neurosci Res. 2001b;64:26–33. doi: 10.1002/jnr.1050. [DOI] [PubMed] [Google Scholar]
- Trapero I, Cauli O. Interleukin 6 and cognitive dysfunction. Metab Brain Dis. 2014 doi: 10.1007/s11011-014-9551-2. [DOI] [PubMed] [Google Scholar]
- Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus. 2000;9:498–503. doi: 10.1177/096120330000900704. [DOI] [PubMed] [Google Scholar]
- Tsakiri N, Kimber I, Rothwell NJ, Pinteaux E. Differential effects of interleukin-1 alpha and beta on interleukin-6 and chemokine synthesis in neurones. Mol Cell Neurosci. 2008a;38:259–265. doi: 10.1016/j.mcn.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Tsakiri N, Kimber I, Rothwell NJ, Pinteaux E. Mechanisms of interleukin- 6 synthesis and release induced by interleukin-1 and cell depolarisation in neurones. Mol Cell Neurosci. 2008b;37:110–118. doi: 10.1016/j.mcn.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Van Wagoner NJ, Oh JW, Repovic P, Benveniste EN. Interleukin-6 (IL-6) production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J Neurosci. 1999;19:5236–5244. doi: 10.1523/JNEUROSCI.19-13-05236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Vazquez JF, Clement HW, Sommer O, Schulz E, van Calker D. Local stimulation of the adenosine A2B receptors induces an increased release of IL-6 in mouse striatum: an in vivo microdialysis study. J Neurochem. 2008;105:904–909. doi: 10.1111/j.1471-4159.2007.05191.x. [DOI] [PubMed] [Google Scholar]
- Vereyken EJ, Bajova H, Chow S, de Graan PN, Gruol DL. Chronic interleukin-6 alters the level of synaptic proteins in hippocampus in culture and in vivo. Eur J Neurosci. 2007;25:3605–3616. doi: 10.1111/j.1460-9568.2007.05615.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Balosso S, Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun. 2008;22:797–803. doi: 10.1016/j.bbi.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Vollenweider F, Herrmann M, Otten U, Nitsch C. Interleukin-6 receptor expression and localization after transient global ischemia in gerbil hippocampus. Neurosci Lett. 2003;341:49–52. doi: 10.1016/s0304-3940(03)00136-8. [DOI] [PubMed] [Google Scholar]
- Wada-Isoe K, Wakutani Y, Urakami K, Nakashima K. Elevated interleukin- 6 levels in cerebrospinal fluid of vascular dementia patients. Acta Neurol Scand. 2004;110:124–127. doi: 10.1111/j.1600-0404.2004.00286.x. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Yoshimura R, Khalil M, Yoshida K, Kishimoto T, Taga T, Kiyama H. Characteristic localization of gp130 (the signal-transducing receptor component used in common for IL-6/IL-11/CNTF/LIF/OSM) in the rat brain. Eur J Neurosci. 1996;8:1630–1640. doi: 10.1111/j.1460-9568.1996.tb01307.x. [DOI] [PubMed] [Google Scholar]
- Wei H, Alberts I, Li X. Brain IL-6 and autism. Neuroscience. 2013;252:320–325. doi: 10.1016/j.neuroscience.2013.08.025. [DOI] [PubMed] [Google Scholar]
- Wei H, Chadman KK, McCloskey DP, Sheikh AM, Malik M, Brown WT, Li X. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim Biophys Acta. 2012;1822:831–842. doi: 10.1016/j.bbadis.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, Li X. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation. 2011;8:52. doi: 10.1186/1742-2094-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JA, Wood PL, Ryan R, Graff-Radford NR, Pilapil C, Robitaille Y, Quirion R. Cytokine indices in Alzheimer’s temporal cortex: no changes in mature IL-1 beta or IL-1RA but increases in the associated acute phase proteins IL-6, alpha 2-macroglobulin and C-reactive protein. Brain Res. 1993;629:245–252. doi: 10.1016/0006-8993(93)91327-o. [DOI] [PubMed] [Google Scholar]
- Woodroofe MN, Sarna GS, Wadhwa M, Hayes GM, Loughlin AJ, Tinker A, Cuzner ML. Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol. 1991;33:227–236. doi: 10.1016/0165-5728(91)90110-s. [DOI] [PubMed] [Google Scholar]
- Woolfenden S, Sarkozy V, Ridley G, Coory M, Williams K. A systematic review of two outcomes in autism spectrum disorder – epilepsy and mortality. Dev Med Child Neurol. 2012;54:306–312. doi: 10.1111/j.1469-8749.2012.04223.x. [DOI] [PubMed] [Google Scholar]
- Wright CB, Sacco RL, Rundek T, Delman J, Rabbani L, Elkind M. Interleukin-6 is associated with cognitive function: the Northern Manhattan Study. J Stroke Cerebrovasc Dis. 2006;15:34–38. doi: 10.1016/j.jstrokecerebrovasdis.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TH, Lin CH. IL-6 mediated alterations on immobile behavior of rats in the forced swim test via ERK1/2 activation in specific brain regions. Behav Brain Res. 2008;193:183–191. doi: 10.1016/j.bbr.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Wullschleger A, Kapina V, Molnarfi N, Courvoisier DS, Seebach JD, Santiago-Raber ML, Hochstrasser DF, Lalive PH. Cerebrospinal fluid interleukin- 6 in central nervous system inflammatory diseases. PloS One. 2013;8:e72399. doi: 10.1371/journal.pone.0072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiaoqin Z, Zhengli L, Changgeng Z, Xiaojing W, Li L. Changes in behavior and amino acid neurotransmitters in the brain of rats with seizure induced by IL-1beta or IL-6. J Huazhong Univ Sci Technol Med Sci. 2005;25:236–239. doi: 10.1007/BF02828129. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Lindquist K, Penninx BW, Simonsick EM, Pahor M, Kritchevsky S, Launer L, Kuller L, Rubin S, Harris T. Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology. 2003;61:76–80. doi: 10.1212/01.wnl.0000073620.42047.d7. [DOI] [PubMed] [Google Scholar]
- Yamasaki K, Taga T, Hirata Y, Yawata H, Kawanishi Y, Seed B, Taniguchi T, Hirano T, Kishimoto T. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science. 1988;241:825–828. doi: 10.1126/science.3136546. [DOI] [PubMed] [Google Scholar]
- Yang M, Kim J, Kim JS, Kim SH, Kim JC, Kang MJ, Jung U, Shin T, Wang H, Moon C. Hippocampal dysfunctions in tumor-bearing mice. Brain Behav Immun. 2014;36:147–155. doi: 10.1016/j.bbi.2013.10.022. [DOI] [PubMed] [Google Scholar]
- Yasukawa K, Hirano T, Watanabe Y, Muratani K, Matsuda T, Nakai S, Kishimoto T. Structure and expression of human B cell stimulatory factor-2 (BSF-2/IL-6) gene. EMBO J. 1987;6:2939–2945. doi: 10.1002/j.1460-2075.1987.tb02598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from brain of aged mice. J Neuroimmunol. 1999;93:139–148. doi: 10.1016/s0165-5728(98)00217-3. [DOI] [PubMed] [Google Scholar]
- Ye SM, Johnson RW. Regulation of interleukin-6 gene expression in brain of aged mice by nuclear factor kappaB. J Neuroimmunol. 2001;117:87–96. doi: 10.1016/s0165-5728(01)00316-2. [DOI] [PubMed] [Google Scholar]
- Yu N, Di Q, Hu Y, Zhang YF, Su LY, Liu XH, Li LC. A meta-analysis of pro-inflammatory cytokines in the plasma of epileptic patients with recent seizure. Neurosci Lett. 2012;514:110–115. doi: 10.1016/j.neulet.2012.02.070. [DOI] [PubMed] [Google Scholar]
- Zhao B, Schwartz JP. Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res. 1998;52:7–16. doi: 10.1002/(SICI)1097-4547(19980401)52:1<7::AID-JNR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Zhu B, Dong Y, Xu Z, Gompf HS, Ward SA, Xue Z, Miao C, Zhang Y, Chamberlin NL, Xie Z. Sleep disturbance induces neuroinflammation and impairment of learning and memory. Neurobiol Dis. 2012;48:348–355. doi: 10.1016/j.nbd.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo JM, Prescott SL, Murray ME, Zhang HY, Baxter MG, Nicolle MM. Early discrimination reversal learning impairment and preserved spatial learning in a longitudinal study of Tg2576 APPsw mice. Neurobiol Aging. 2007;28:1248–1257. doi: 10.1016/j.neurobiolaging.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Zuliani G, Guerra G, Ranzini M, Rossi L, Munari MR, Zurlo A, Ble A, Volpato S, Atti AR, Fellin R. High interleukin-6 plasma levels are associated with functional impairment in older patients with vascular dementia. Int J Geriatr Psychiatry. 2007;22:305–311. doi: 10.1002/gps.1674. [DOI] [PubMed] [Google Scholar]