


Abstract
Ribosomal ribonucleic acid (RNA), transfer RNA and other biological or synthetic RNA polymers can contain nucleotides that have been modified by the addition of chemical groups. Traditional Sanger sequencing methods cannot establish the chemical nature and sequence of these modified-nucleotide containing oligomers. Mass spectrometry (MS) has become the conventional approach for determining the nucleotide composition, modification status and sequence of modified RNAs. Modified RNAs are analyzed by MS using collision-induced dissociation tandem mass spectrometry (CID MS/MS), which produces a complex dataset of oligomeric fragments that must be interpreted to identify and place modified nucleosides within the RNA sequence. Here we report the development of RoboOligo, an interactive software program for the robust analysis of data generated by CID MS/MS of RNA oligomers. There are three main functions of RoboOligo: (i) automated de novo sequencing via the local search paradigm. (ii) Manual sequencing with real-time spectrum labeling and cumulative intensity scoring. (iii) A hybrid approach, coined ‘variable sequencing’, which combines the user intuition of manual sequencing with the high-throughput sampling of automated de novo sequencing.
INTRODUCTION
Biologically derived ribonucleic acid (RNA) polymers, such as transfer RNA (tRNA) and ribosomal RNA (rRNA), contain modified nucleotides that are not amenable to sequence determination using the standard RNA sequencing method of RT-PCR followed by Sanger sequencing (1). Instead, these complex molecules, which may be composed of any number of more than 150 naturally occurring modified nucleosides (2,3), are typically characterized using collision-induced dissociation tandem mass spectrometry (CID MS/MS) (4–9) coupled with either matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) or liquid chromatography electrospray ionization mass spectrometry (LC-ESI-MS).
CID of oligoribonucleotides typically yields c-, y-, w- and a-B-type product ions (Figure 1), although any bond within the phosphodiester backbone is susceptible to dissociation (9–12). The ultimate goal during MS/MS analysis of modified oligoribonucleotides is to utilize the detected product ions as a means of reconstructing the original sequence of the oligoribonucleotide (13). Successive product ions arising from the same phosphodiester backbone fragmentation (e.g. c1-, c2-, c3-, etc.) allow the nucleotide ordering within the oligoribonucleotide to be determined. Modified nucleosides are generally placed within the oligoribonucleotide sequence by their unique nucleoside residue mass, which will be greater than the residue mass of the four canonical ribonucleosides (cytidine (C), uridine (U), adenosine (A) and guanosine (G)) (13,14). While conceptually straightforward, with modern instrumentation the MS/MS data produced by MALDI- or LC-ESI-MS is inherently difficult to analyze due to the complexity of the resulting spectra and the minimally available software tools to aid in the analysis (15).
Figure 1.
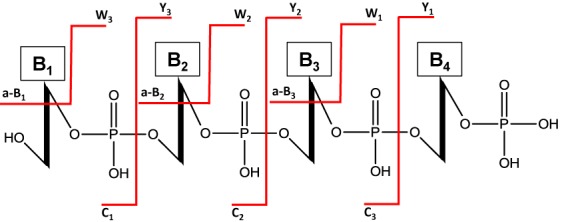
Typical RNA oligomer fragmentation products generated by collision-induced dissociation (CID). The most abundant products are typically c- and y- fragment ions, however w- and a-B (a- ions lacking the adjacent base) are often commonly observed.
One of the first attempts at computational analysis of mass spectrometric data generated from nucleic acid oligomers focused on the determination of the nucleotide composition of an ion based on the mass of the oligomer and the masses of the four canonical ribonucleosides (16). In 2002, Rozenski and McCloskey released the Simple Oligonucleotide Sequencer (SOS), which was a tool capable of assisting in the manual interpretation of oligonucleotide MS/MS data from oligomers up to 20 bases in length (17). SOS worked by displaying mass spectral peaks that corresponded to a-B– or w–type fragment ions, allowing the user to choose which nucleoside best fit the experimental data. While effective, this program is limited by the manual analysis of data, precluding its use on complex LC/MS/MS datasets and by the minimal number of modified nucleosides that can be evaluated during data analysis. More recently, Nyakas et al. (18) developed the programs OMA and OPA, which allow analysis of MS and MS/MS data with a customizable database of nucleotides, thus all known RNA modifications can be analyzed. However, this software only compares the predicted fragmentation pattern from an inputted sequence to the MS data and therefore the sequence to be analyzed must be known in advance.
Two database search strategies for RNA mass spectrometry data, RRM (19) and Ariadne (20), have been developed to approach the analysis of RNA MS and MS/MS data in a similar manner as the polypeptide analysis suite MASCOT and other similar protein-focused software (21). RRM focuses on mass spectral data only and uses the concepts originally described by Pomerantz et al. (16) to define base compositions that can be searched against genome or RNA sequence databases (19). While this strategy is effective for sourcing standard RNA, it cannot be used to characterize RNase digestion products containing modified nucleosides. Ariadne takes tandem mass spectrometry data generated from either biologically or in vitro-derived RNA and scores the comparison of the data to an inputted database of theoretical ribonuclease digested and CID-fragmented RNA sequences (20). While useful, this software can currently only be searched against sequences from Saccharomyces cerevisiae, Schizosaccharomyces pombe, Mus musculus and Homo sapiens.
Automated de novo sequencing of MS/MS data has been attempted with various strategies and degrees of success from samples composed of DNA (22–25), DNA adducts (26–28) and RNA containing 2′-O-methyl and phosphorothioate linkages (29). The global search approach, which involves generating a library of all sequence isomers of a given nucleic acid composition and then scoring these sequence isomers based on the data within the mass spectrum, has returned positive results with oligodeoxyribonucleotides of up to 12 residues (23). The limiting challenge of such an approach lies in the exponentially increasing number of potential combinations introduced by either long oligomers or large nucleotide pools. A particularly interesting solution to this problem was employed by Oberacher et al., who used a simulated annealing strategy (30) for sequence optimization of oligodeoxyribonucleotides as long as 22 residues (24). Briefly, a random sequence was generated for a nucleotide composition that matched the precursor mass. The positions of two nucleotides are computationally switched, a fitness score that evaluates theoretical fragmentation products compared to the actual spectral data is calculated and if the new score is higher than the previous score then the new sequence is kept and subjected to additional rounds of variation and selection. In this example, the use of stochastic optimization dramatically reduced the computational load for long oligomers, but the program was only designed to handle the four common DNA bases: C, T, A and G, and thus cannot be applied to the large number of modified nucleosides found in natural RNAs.
To our knowledge, a global search strategy for de novo sequence analysis has never been attempted with complex, multiple modified base-containing RNA, leaving the field of oligoribonucleotide sequencing via mass spectrometry significantly hindered at analyzing MS/MS datasets generated from RNAs containing multiple modified nucleosides. To address this need, we report on the development of RoboOligo, an interactive program equipped with abilities for the robust analysis of negative ion mode MS/MS data generated by CID. We show that an automated local search paradigm maintains the robustness of the global search paradigm and can efficiently handle modified bases. Additional data analysis flexibility is provided by manual and variable sequence capabilities that allow for user-controlled examination of MS/MS data.
MATERIALS AND METHODS
Sample preparation and LC/MS/MS conditions
Samples used for analysis include in vitro methylated tRNA transcripts, purified isoacceptor tRNA and total tRNA. In vitro Trm14 methylated Methanocaldococcus jannaschii tRNACys (31) and purified isoacceptor tRNAs Escherichia coli ΔqueC ΔqueF pGAT-queC tRNAAsp (32), E. coli tRNAGln (33) and Bacillus subtilis tRNAIle (34) served as known tRNA sequences for evaluation of the software program. Total tRNA from Lactococcus lactis (35) was purified using the tri-reagent extraction protocol followed by purification of total tRNA using a Qiagen column (36).
For sequence analysis, tRNA was digested with 50 U/μg of RNase T1 (Worthington Biochemical Corporation) or 0.01 U/μg of RNase A (Sigma-Aldrich) in 20 mM ammonium acetate (pH 6.5) for 2 h at 37°C. Digestion products from 1 μg of a single purified tRNA or 5 μg of total tRNA were separated using a Thermo Surveyor HPLC system (Thermo-Finnigan) with an XBridge BEH130 C18 3.5 μm 1.0 × 150 mm column (Waters) at room temperature with a flow rate of 40 μl/min. Before each run, the column was equilibrated for 15 min at 95% buffer A (200 mM 1,1,1,3,3,3-hexafluoroisopropanol (HFIP, Sigma-Aldrich), 8.15 mM triethylamine (TEA, Sigma-Aldrich)) and buffer B (200 mM HFIP, 8.15 mM TEA:methanol 50:50 v/v) (methanol, Burdick and Jackson). Gradient elution was used starting at 5% B held for 5 min followed by a ramp to 30% B at 7 min and a ramp to 95% B at 50 min and held at 95% B for 5 min. The eluent was directed into an LTQ-XL (Thermo Scientific) linear ion trap mass spectrometer. The instrument was operated in negative ion mode with a capillary temperature of 275°C, spray voltage of 4.5 kV, sheath gas was set to 25, auxiliary gas to 14 and sweep gas to 10 arbitrary units (a.u.). Collision-induced dissociation tandem mass spectrometry at a normalized collision energy of 42 was used to obtain sequence information of the digestion products in data-dependent mode. The four most abundant m/z values from the MS scan are selected for data-dependent tandem mass spectrometry and were excluded from MS/MS analysis after 5 scans of the precursor ion for 30 s. All MS/MS data were converted from Thermo Scientific .RAW file format to .mgf file format using the MassMatrix File Conversion Tool (www.massmatrix.net). File conversion using MSConvert (http://proteowizard.sourceforge.net/tools.shtml) was found to lead to larger .mgf files, which could lead to premature termination of automated de novo sequencing.
Data analysis
RoboOligo was developed using Visual Studio 2012 and programmed in Visual Basic .NET 4.0. The software can be downloaded at http://bearcatms.uc.edu/new/limbachgroup_publication/. All computations and results reported here were performed in the Windows 7 operating system using an Intel Core I7–3770k processor at 3.5 GHz, with 8GB DDR3–1600 RAM and a 120GB Solid-state drive. RoboOligo has been found to work on lower-speed computers such as an AMD Athlong™ 2.0 GHz dual-core processor with 3GB DDR2 RAM running Windows 7; however, computers with more cores are recommended as the automated de novo sequencing algorithm is capable of parallel processing. The ‘Nucleotides.txt’ and ‘NucleotidePools.txt’ files can be edited by a text editor, allowing users to add custom nucleotides and nucleotide pools, respectively. Only negative polarity data is supported by the program at the time of publication.
RESULTS
RoboOligo functions
The main objective in the development of this program was to establish an algorithm that was robust and accurate in the evaluation of CID MS/MS data from moderately sized RNA oligomers containing modified bases (i.e. endonuclease digestion products from tRNA and rRNA). There are three main functions of RoboOligo: (i) automated de novo sequencing via a local search paradigm with nucleotide pool, RNase digestion context and composition constraints; (ii) manual sequencing with real-time spectrum labeling and cumulative intensity scoring; (iii) a hybrid approach, coined ‘variable sequencing’, which combines the user intuition of manual sequencing with the higher-throughput of automated de novo sequencing. To handle modified nucleosides, molecular mass information for 102 uniquely massed nucleotides is available from an internal database within the program. This information can be user edited by appropriate changes to the ‘Nucleotides.txt’ file. All mass spectral data analyzed by RoboOligo can be graphically represented and appropriately labeled with sequence assignments.
Automated de novo sequencing
Algorithm design
The design approach begins with a simple calculation to generate the mass of the oligonucleotide based on the precursor ion mass-to-charge value. This target total mass is then used to calculate potential nucleotide compositions. In this process, the program finds all nucleotide combinations that, if combined as a single oligomer, would fall within the total mass range. The total mass range is defined by the calculated mass of the oligonucleotide and the user-selected total mass tolerance. The unmodified and modified nucleotides to include in the composition analysis are also chosen by the user. The de novo portion of the software then uses a local search approach to create potential sequences from this information.
The principal concept of the local search approach is that sequences are built one nucleotide at a time and after each nucleotide addition the fitness of the oligomer is evaluated by the abundance of c- and y- type product ions (17). Lack of evidence for a product ion will cause the incomplete sequence to fail, along with all of the sequences that would have been tested had it not failed. This ability to ignore entire sequence trees that lack data to support their validity provides an efficient way to logically reduce the number of oligonucleotide sequences to be tested.
The algorithm begins by attempting to match expected product ions beginning with the 5′-terminus (Figure 2). Once a potential c1- ion is identified, the program will begin building a theoretical sequence in the 5′ to 3′ direction. To do so, another nucleotide from the composition is added to the theoretical sequence and the c2- ion is calculated and compared to the data. Once a c2- ion matches the data, the nucleotide is added and the program continues until all c- ions are identified for the calculated compositions. To improve sequence reconstruction accuracy, the program then calculates all expected y- ions for this potential sequence and compares these calculated values against the experimental data to eliminate incorrect sequences. For a sequence that passes this step, the w- and (a-B)- ions are then calculated and all c-, y-, w- and (a-B)- product ions are used for theoretical sequence scoring. The entire process is then repeated for additional sequence possibilities.
Figure 2.
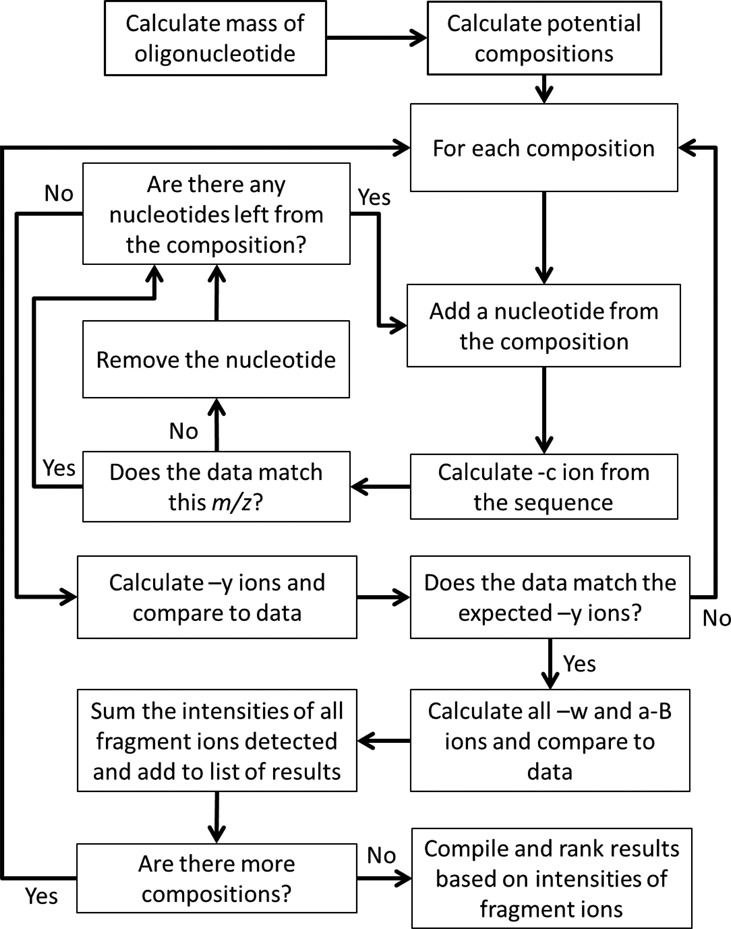
The design of the de novo sequencing algorithm. Potential compositions are produced for a calculated oligonucleotide mass and each composition is used for the de novo sequence analysis. The algorithm builds sequences one nucleotide at a time and tests the fit of the data to the expected products. This is done in the 5′ to 3′ direction using –c ions and in the 3′ to 5′ direction using –y ions. Once a potential sequence is found –w and –a-B ions are calculated and the sequences are scored based on the summed intensities of the fragment ions for that sequence.
Each composition is independently passed through the de novo sequencing algorithm, providing the identity and precise count of each nucleotide allowed within a growing oligonucleotide sequence. The benefit of confining sequence attempts through isolated compositions is that all permutations of the compositions lead to total oligonucleotide masses that fit within the target mass range and therefore reduces computational effort by never attempting sequences that would lead to an invalid total mass (25). The final list of sequences that potentially match the MS/MS data will have all c-, y-, w- and (a-B)- product ions identified including multiply charged ions if the analyzed spectrum has a charge state >1. Currently, the program supports charge states up to −3.
Scoring the results
The scoring algorithm used here is similar to that used originally in the SOS program (17). Those potential sequences generated by the algorithm described above are characterized by their calculated sets of c-, y-, w- and (a-B)- product ions. The experimental data from the MS/MS spectrum is evaluated by summing the ion abundances at each of the m/z values for the set of product ions calculated for each potential sequence. These ion abundances are then summed to produce the cumulative score for a sequence.
The assumption is that the cumulative product ion abundance for a correct sequence should be greater than the cumulative product ion abundance for a sequence that does not represent the oligomer being analyzed. Oberacher et al. (24) implemented a more sophisticated scoring scheme, which calculates the fitness of each sequence by applying penalties for incomplete fragmentation coverage and weighing of ion abundances that reduce the confidence in peaks that deviate more drastically from the predicted m/z value. This fitness equation performed well for their global-search automated de novo sequencing algorithm of DNA, but performed poorly with our modified base-containing RNA MS/MS data (data not shown).
Effects of constraints on the analysis
A central difficulty in de novo sequencing of oligonucleotides when considering modified nucleotides lies in the sheer number of potential sequence combinations given the length and unique entities with which these molecules may be composed. For this reason, restrictions on the possible sequence combinations to be analyzed can be beneficial. Constraints in the analysis can be applied by adjusting for the mass accuracy of the instrument in the MS or the MS/MS data. Mass constraint can have a profound impact on the number of potential compositions and on the match of experimental data to the predicted m/z as has been reported previously (16,37).
Pomerantz et al. (16) noted that oligonucleotide base compositions could be constrained by using endonucleases of high selectivity or specificity. Ribonuclease T1 (RNase T1) is known to cleave RNA at all unmodified guanosine residues (38), while RNase A is found to be more tolerant of modifications to the pyrimidine ring (39). RoboOligo incorporates such constraints within the RNase digest selection as well as the mass tolerance options. Endonuclease restrictions for RNase T1 (‘strict’—digestion at unmodified guanosine only), RNase A (‘strict’—digestion at unmodified pyrimidines only; and ‘broad’—digestion also at dihydrouridine, methylated pyrimidines and thiolated pyrimidines) and RNase U2 (digestion at unmodified purine) are supported. As the authors have also observed RNase T1 digestion of N2-methylguanosine (m2G) (40), an additional endonuclease restriction—RNase T1 ‘broad’—that enables digestion at either unmodified guanosine or methylated guanosine is also available.
For example, if the ‘RNase T1 strict’ selection was used during the composition analysis then any composition not containing only one guanosine would be considered invalid. The user is also able to bypass this constraint for scenarios in which an RNase digestion context is inappropriate; however, the quantity of valid but incorrect sequences could drastically increase, along with an increase in total computation time.
A further constraint is imbedded within the program by limiting the occurrence of particular modified nucleosides. The majority of RNA samples containing modified nucleosides should yield RNase digestion products that are not significantly large (41). Under such conditions, it is unlikely that multiple instances of the same modified nucleotide will be present within a single oligomer. This property is exploited such that only C, U, A, G, dihydrouridine (D) and singly methylated C, U, A and G are allowed to occur more than once within a composition. However, these default settings can be edited by the user in the ‘Nucleotides.txt’ file if the sample to be analyzed dictates such a change.
Missing product ions
In addition to the constraints mentioned above, a tolerance for the number of missing product ions can also be set within the program. This feature accounts for empirical aspects of typical MS/MS data, wherein occasionally a product ion may be missing in the data, especially for longer oligonucleotides or the mass range of the experimentally acquired data may not include the product ion that is expected. The user has the ability to define the number of skips that the program can use to compensate for this missing data by allowing the placement of the nucleotide being tested and then continue to iterate through and test nucleotides at the next position. In a case where all of a developing sequence's permitted skips are exhausted, the algorithm will change the direction of the local search (from 5′ → 3′ to 3′ → 5′) and will begin to examine the y- ion series for the fitness evaluation. The composition context is conserved after this sequencing direction change, ensuring that all 3′ to 5′ nt additions would eventually meet the progress made in the initial c- ion series sequencing attempt and sum to a total mass that falls within the target mass range. Skips can also compensate for modified bases that fragment in unusual ways by inferring the position of the base and then building evidence for its placement. This is accomplished by the successful incorporation of subsequent nucleotides or, if the growing oligomer reaches the precursor mass range and is evaluated as a valid complete sequence, via m/z values that correspond to predicted c-, y-, w- and (a-B)- product ions for the nucleotide at that inferred position.
User interface
The automated de novo sequencing algorithm requires the following user input (Figure 3):
Spectra to be analyzed: a single spectrum may be chosen at any time. In addition, batch processing of multiple spectra (e.g. all data from an entire LC-MS/MS run) is supported.
Nucleotide pool: these are the nucleotides to be included in the sequencing attempt. These nucleotides should be selected based on known information about the sample, such as the census of modified nucleosides (e.g. from a separate nucleoside analysis (42)) or based on the organism being studied (2,3). Due to the inability of MS/MS data to routinely identify the purine or pyrimidine ring position of modifications such as methylation, the notation for certain modified nucleosides are simplified where appropriate. For example, 1mG indicates a guanosine with a single methyl modification, which could be a methylation at any position such as 2′-O-methyl (Gm), N7-methyl (m7G), 1-methyl (m1G) or N2-methyl (m2G) guanosine. Due to an exponentially increasing computational workload (Supplementary Figure S1), it is advised to limit the nucleotides to be used in the analysis. In general, oligomers of <8 nt can utilize pools of up to 20 nt, while longer oligomers are most efficiently sequenced with pools of <12 nt.
CID product m/z tolerance: this value defines the range around a theoretical CID fragmentation product in which an m/z data point would be considered as a match. The quality of the data and accuracy of the mass spectrometer used to obtain the data should be used as a guideline when setting this tolerance. A smaller value is ideal, as it would reduce the number of sequence annotations returned as possibilities; however, larger m/z tolerances may be required to find data points that deviate substantially from their theoretical values.
Total m/z tolerance: for a ‘complete’ sequence to be evaluated as valid, its total mass must fall within the range defined by the total mass and the total mass tolerance. A lower value will reduce the number of sequence annotations returned as possibilities, but it also risks missing the correct sequence if the tolerance is too strict.
Skips: the purpose of this parameter is to provide some leeway for less than ideal data that may be missing important CID product ion m/z values. One skip is set as the default value. This skip would be used at the beginning of any sequencing attempt if the minimum m/z setting on the mass spectrometer is greater than the m/z of the potential c1- fragment ion or can be used by the program when 2′-O-methylation reduces the ion abundance of c- or y- fragment ions (10,43).
5′ and 3′ ends: the phosphate status on the 5′ and 3′ termini (linear phosphate, cyclic phosphate or no phosphate) are user-defined and employed in calculating the total mass of the oligonucleotide.
RNase digest context: if the RNA used to generate the data was first digested by an RNase, then it is suggested that this parameter be set to match the digestion condition. This selection not only limits the number of nucleotide compositions that fit a target mass, but also restricts sequences to those that are expected from the RNase used in the digestion.
Figure 3.
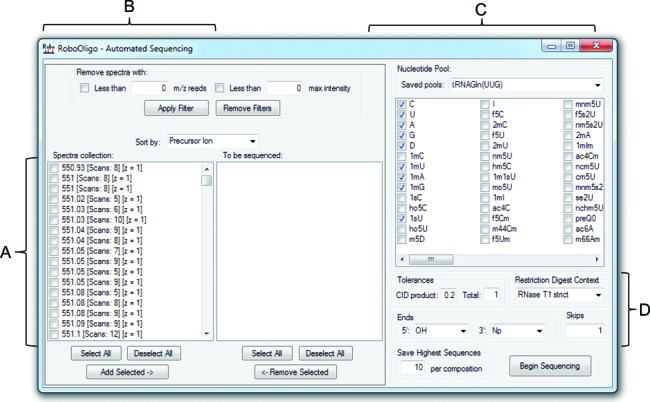
Automated de novo sequencing. (A) MS/MS data can be selected and moved to the ‘To be sequenced’ box to be included in the analysis. (B) MS/MS data with less than a user selected number of m/z reads and/or maximum ion abundance can be removed from the ‘Scan collection’ box. (C) Nucleotides to be included in the automated de novo sequencing attempt—oligomers of <8 nt can utilize pools of up to 20 nt, while longer oligomers return the best results with pools of <12 nt. New entries in the ‘Saved pools’ dropdown list can be added by editing the ‘NucleotidePools.txt’ file. (D) The CID m/z tolerance, total mass tolerance, RNase digestion context, 5′ and 3′ ends and number of allowed skips are all modifiable by the user.
Performance of automated de novo sequencing
Previously acquired LC-MS/MS data from two tRNA isoacceptors, E. coli ΔqueC ΔqueF pGAT-queC Asp-tRNAGUC (32) and E. coli Gln-tRNAUUG (33) (Supplementary Figure S2), were used to assess the performance of the automated de novo sequencing algorithm. The nucleotide pools used for each sequencing attempt were determined by independent LC-MS/MS nucleoside analysis and found to be: C, U, A, G, 1mA, 1mG, 1sU, preQ0, D and G+ for Asp-tRNAGUC; and C, U, A, G, 1sU, 1mG, 1mA, 1mU, D and cmnm5s2U for Gln-tRNAUUG. All attempts were allowed one skip, a total mass tolerance of 1.0, a CID product m/z tolerance of 0.3 and the RNase digestion context of ‘RNase T1 strict’ unless otherwise stated.
RNase T1 digestion of the RNA sequence of Asp-tRNAGUC is predicted to yield 27 digestion products ranging from monomers (Gp) to an 8-mer. When this tRNA was previously analyzed by LC-MS/MS, manual interpretation of the MS/MS spectra identified 16 unique oligomer sequences matching the tRNA sequence at the dimer level and higher (32). Subjecting the same LC-MS/MS dataset to automated sequencing by RoboOligo revealed that 15 out of 16 of these oligomers were evaluated by the algorithm and annotated with the correct modified nucleosides. For 14 of these oligomers, the RoboOligo annotated sequence was found to yield the highest cumulative ion abundance score (Table 1). The most complex sequence evaluated, based on length (9 nt) and the number of modified nucleotides, was CCU[preQ0]UC[m2A]CGp. This sequence had a cumulative ion abundance score that was 5% higher than the next highest sequence, which was the isomer CUC[preQ0]UC[m2A]CGp that differs only by a C and U base switch between positions two and three. This result highlights a general challenge with RNA sequencing in that C and U isomers are particularly troublesome as the mass difference between the two is 1.0 if singly charged, 0.5 if doubly charged and 0.33 if triply charged. The 3′ end of tRNAAsp, CCA-OH, was not found to be the top-ranked oligomer, yet it differed from the highest ranked sequence, CAC, by only 1.5% in cumulative ion abundance. The one failed sequencing attempt occurred with the digestion product [m7G]UCGp, which is likely due to the unusual fragmentation pattern of m7G (44,45). When m7G is present in an oligonucleotide the weak glycosidic bond fragments producing a large peak that is 165 Da smaller than the precursor and often very few c- and y- product ions are observed.
Table 1. Summary of results generated by the automated sequencing of E. coli ΔqueC ΔqueF pGAT-queC tRNAAsp and E. coli tRNAGln(UUG).
| Sequence | Precursor m/z | Charge state | Rank | % Diff | Complete sequences | Compositions | Time (ms) |
|---|---|---|---|---|---|---|---|
| CGp | 667.37 | −1 | 1 | 17 | 2 | 2 | 41 |
| AGp | 691.27 | −1 | 1 | NA | 1 | 1 | 40 |
| [s4U]AGpa | 1013.4 | −1 | 1 | 37 | 4 | 2 | 58 |
| [D]CGp | 975.52 | −1 | 1 | 40 | 4 | 2 | 150 |
| CAGp | 996.39 | −1 | 1 | 20 | 4 | 2 | 52 |
| CCAb | 876.26 | −1 | 2 | 1.5 | 8 | 2 | 44 |
| [m5U][Ψ]CGpc,d | 1293.4 | −1 | 1 | 12 | 12 | 6 | 122 |
| [D][D]AGp | 653.53 | −1 | 1 | 33 | 3 | 1 | 47 |
| [Ψ]CCGpd | 1278.5 | −1 | 1 | 36 | 5 | 2 | 73 |
| [m7G]UCGpe | None found | ||||||
| UCCCGpf | 791.83 | −2 | 1 | 3 | 30 | 6 | 71 |
| UCCCGp | 791.83 | −2 | 1 | 8 | 14 | 3 | 60 |
| UUCAGp | 803.91 | −2 | 1 | 8 | 25 | 3 | 100 |
| AAUACCUGpf | 1286.0 | −2 | 1 | 1 | 367 | 15 | 891 |
| CCU[preQ0]UC[m2A]CGpf,g | 1453.5 | −2 | 1 | 5 | 720 | 34 | 491 |
| CCU[G+]UC[m2A]CGpf,g | 1461.9 | −2 | 1 | 6 | 195 | 44 | 1019 |
The T1 restriction digest confinement was used to match the sample preparation. Unless otherwise noted, all automated sequencing attempts were allowed one skip, a target mass tolerance of 1 Da and a mass-to-charge tolerance of 0.3. The ‘% Diff.’ is the cumulative intensity difference between the correct oligomer sequence and second highest scoring oligomer if the correct sequence is the highest scoring result. Otherwise, it measures the difference between the highest scoring sequence and the independently verified correct sequence. ‘Complete sequences’ is the number of sequences which the m/z data supports, given user-defined sequencing parameters. ‘Compositions’ is the number of nucleotide compositions that fit the target mass range, given the user-defined nucleotide pool. ‘Time’ measures the time in milliseconds that the program takes to return the automated sequencing results.
a[s4U] = 1sU.
bCAC scored 1.5% higher than CCA and was analyzed with the 3′-OH setting and no digestion constraint.
c[m5U] = 1mU.
d[Ψ] = U.
e[m7G] = 1mG.
fTarget mass tolerance = 2.
g[m2A] = 1mA.
Similarly, Gln-tRNAUUG was analyzed. The prior manual analysis of LC-MS/MS data from the RNase T1 digest of this tRNA yielded 13 digestion products including those containing the six modified nucleosides (33). Again, the prior LC-MS/MS dataset was analyzed using the automated de novo sequencing function of RoboOligo. RoboOligo was able to annotate those 13 digestion products, with all of the correct sequence assignments being the top ranked possibility (Table 2). The sequence with the most modified nucleotides, U[Um]U[cmnm5s2U]UGp, had a cumulative intensity score 12% higher than that of the second ranked sequence. The longest sequence in this dataset was the 9-mer CAUUCCCUGp, which was correctly identified.
Table 2. Summary of results generated by the automated sequencing of E. coli tRNAGln(UUG).
| Sequence | Precursor m/z | Charge state | Rank | % Diff | Complete sequences | Compositions | Time (ms) |
|---|---|---|---|---|---|---|---|
| CGp | 667.14 | −1 | 1 | 20 | 2 | 2 | 61 |
| C[Gm]Gpa | 1026.1 | −1 | 1 | 14 | 6 | 4 | 55 |
| CCAg | 876.14 | −1 | 1 | 14 | 8 | 2 | 57 |
| [D]AAGp | 663.73 | −2 | 1 | 18 | 4 | 3 | 65 |
| [m5U][Ψ]CGpb,c | 646.64 | −2 | 1 | 23 | 14 | 6 | 79 |
| UA[s4U]CGpd | 811.68 | −2 | 1 | 8 | 68 | 6 | 94 |
| CCAAGp | 814.89 | −2 | 1 | 10 | 14 | 4 | 207 |
| CACCGp | 802.76 | −2 | 1 | 17 | 11 | 3 | 113 |
| U[Um]U[cmnm5s2U]UGpb | 1004.5 | −2 | 1 | 12 | 41 | 24 | 186 |
| [m2A]UACCGpe | 974.84 | −2 | 1 | 1 | 91 | 12 | 231 |
| AAUCCAGpf | 1132.4 | −2 | 1 | 12 | 12 | 40 | 588 |
| UACCCCAGp | 1273.0 | −2 | 1 | 8 | 56 | 19 | 137 |
| CAUUCCCUGpf | 942.91 | −3 | 1 | 11 | 428 | 12 | 380 |
The T1 restriction digest confinement was used to match the sample preparation. Unless otherwise noted, all automated sequencing attempts were allowed one skip, a target mass tolerance of 1 Da and a mass-to-charge tolerance of 0.3.
a[Gm] = 1mG
b[m5U], [Um] = 1mU
c[Ψ] = U
d[s4U] = 1sU
e[m2A] = 1mA
fTarget mass tolerance = 2
gCCA was analyzed with the 3′-OH setting and no digestion constraint.
To examine the limits of oligomers that could be sequenced by RoboOligo, select MS/MS spectra from the RNase T1 digest of L. lactis total tRNA were chosen. Within the LC-MS/MS data, two 14-mers were identified manually, ACUCUU[t6A]AUCUAUGp and ACU[cmnm5s2U]UU[t6A]AUCAAAGp (35). Automated sequencing of the spectra associated with these 14-mers found that the former sequence was correctly identified as the top scorer while the latter sequence scored as the second highest and had a cumulative abundance that differed from the top result by 1%. Another 48 MS/MS spectra from various RNA samples (Supplementary Table S1) were analyzed by RoboOligo in the same manner as the two tRNA isoacceptors described above. Regarded as a whole, the RoboOligo automated de novo sequencing algorithm correctly annotated the sequences of 73 out of 77 (94%) independently verified oligomers. Of the four incorrect sequence annotations, two of these sequences were the second highest scoring choices, one sequence was the fifth highest scoring result and one sequence could not be determined (Figure 4).
Figure 4.
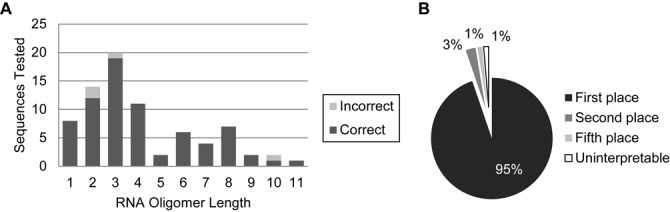
Sequencing accuracy in relation to oligomer length. (A) Accuracy as a function of RNA oligomer length. (B) Relative rank of all tested oligomers. First 95% (73/77) second 3% (2/77), fifth 1% (1/77) and uninterpreted 1% (1/77).
MS/MS spectral annotation
Figure 5 provides a representative illustration of de novo MS/MS annotation using selections from the Gln-tRNAUUG dataset. Figure 5A is a typical MS/MS annotation showing matching c- and y-type fragment ions, along with a few a-B and w-type ion assignments. The complete c- and y-type ion series’ are annotated in this spectrum and the major unannotated peak corresponds to loss of the terminal phosphate group (−98 Da), which is a fragment ion that is not yet implemented in the current version of RoboOligo. Figure 5B provides a summary of the fragment ions detected for all scans that yielded 1mUUCG as the top-ranked annotation. For this tetramer, there are three informative fragment ions (n1–n3) for each ion series (Figure 1), although the 3′-dehydration product could lead to anticipated a4-B and c4 fragment ions. Of the 15 MS/MS scans annotated with m1UUCG as the top choice, 13/15 scans annotated all expected c1–c3 fragment ions and 7/15 scans annotated the complementary y1-y3 fragment ion series. All individual MS/MS scan annotation information is directly accessible to the user, either through visualization in the program main window or by review of the exported results in a comma-separated values (CSV) file.
Figure 5.
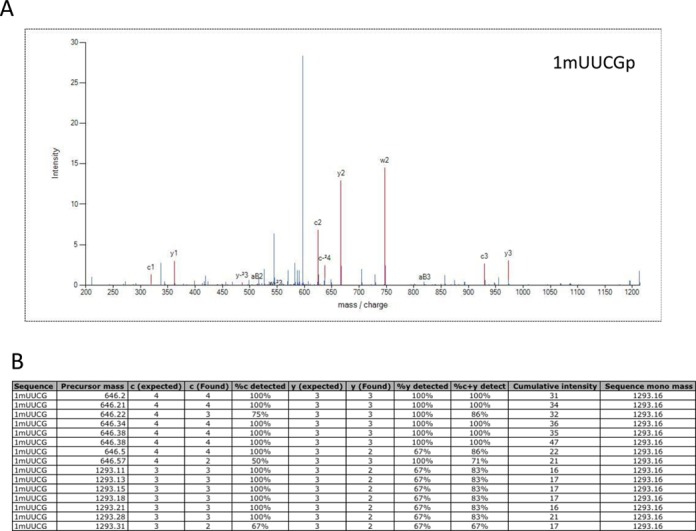
(A) Annotated MS/MS spectrum from the program output of a sequence identified as 1mUUCG, which would correspond to the expected [m5U][Ψ]CGp digestion product from Escherichia coli Gln-tRNAUUG. (B) The 15 MS/MS scans annotated as 1mUUCG from the E. coli Gln-tRNAUUG dataset. Nearly every MS/MS scan yielded the complete, expected c-ion series, while almost half of the MS/MS scans could be annotated with the complementary y-ion series.
Manual sequencing
The manual sequencing portion of RoboOligo is the conceptual successor of SOS (17) in that it provides a platform for a researcher to build RNA sequences ab initio in either the 5′ or 3′ direction using the c-, y-, w- and (a-B)- product ions series’ alone or in any combination. Upon selection of the spectrum to be analyzed, the program generates a graphical representation of the data and automatically interprets the charge and mass of the precursor to calculate a theoretical oligomer target mass. This number, when coupled with the target mass tolerance, determines the mass range in which the oligomers built by the user will be evaluated as a valid potential sequence.
The m/z tolerance is the range used to determine if a spectral data point fits a theoretical CID product ion. The required size of this value depends on the mass spectrometer technology and the data quality. Testing of this software was performed with data generated by a Thermo LTQ-XL in normal scan mode during LC-MS/MS analysis of tRNAs digested with RNase T1 or RNase A. The minimum MS/MS m/z tolerance tested with this data was 0.3, which was sufficient for finding product ions without significant false positives. Choosing too small of an MS/MS tolerance could miss relevant data points that differ slightly from theoretical fragmentation values. It is recommended that various precursor mass tolerances and MS/MS m/z tolerances be tested using known RNA or oligoribonucleotide standards to identify which combination of values produces the most accurate results while also limiting the number of valid but incorrect sequences.
A useful starting point when analyzing an oligomer is to determine the possible nucleotide compositions by using the ‘Composition Analysis’ form. The composition analysis requires a nucleotide pool, target mass, target mass tolerance, the nature of the 5′ and 3′ ends of the oligomer and the RNase digestion context if applicable. All of these factors can be edited by the user to fit the profile of the sample and the instrumentation used. The nucleotide pool can contain any number of standard and modified nucleotides, but the interpretation of the results is simplified if the pool is limited to only the expected nucleotides. In a case where the ‘RNase T1strict’ digestion context is selected, only compositions containing one unmodified guanosine will be evaluated as valid and compositions that do not fit the context will be filtered away. The resulting compositions are transferred back to the manual sequencing form where they can be individually selected, limiting the usable nucleotide list to only those within the composition. Alternatively, if a composition is not chosen, then all of the nucleotides within the program's database will be accessible for manual sequencing operations.
Manual sequencing enables the user to build sequences one position at a time by clicking on the symbol representation of the nucleotide with the ‘Nucleotide’ list (Supplementary Figure S3). The program will calculate the theoretical CID product ion m/z values and then attempt to find and label the matching m/z values within the mass spectrum data displayed in the graph. Sequences can be built in either the 5′ to 3′ direction (generating c- and a-B- fragments) or the 3′ to 5′ direction (generating y- and w- fragments). Sequences may be saved in any state of progress to the ‘5′ series’ or the ‘3′ series’ tabs in the workbench (depending on sequencing direction chosen by the user). Built sequences with total masses that fit within the range of the target mass can be saved to the ‘Complete’ tab of the workbench. Once there, all theoretical c-, y-, w- and (a-B)- product ions are calculated and labeled in the spectrum. Each workbench list is sorted in order of high to low cumulative intensities, with the idea that sequences with higher summed intensities are more likely to be correct. Manual sequencing in RoboOligo not only allows for quick prototyping of putative sequences, but also serves as a capable tool for the detailed analysis of long oligomers that may prove troublesome for the automated de novo sequencing algorithm.
Variable sequencing
The variable sequencing technique is a hybrid of the manual sequencing approach and the automated de novo sequencing algorithm, and has the benefit of automated sequence investigation while providing more direct control of the sequences being tested. This technique introduces the variable nucleotide pools that the user is able to define. In addition to the variable nucleotides, the defined nucleotides, which behave in the same manner as those in the manual sequencing form, can be thought of as constants. Using variable and defined nucleotides, the user can generate a sequence of any combination of the two. For instance if the 5′ and 3′ of the oligonucleotide sequence is known, then the sequence CUXXXXAG can be entered where CU at the 5′ end and AG at the 3′ end are defined and the XXXX in the middle are unknown nucleotides. The position and identity of the defined nucleotides will be conserved, while each variable nucleotide will iterate through its user-defined nucleotide pool and attempt to find m/z values within the data that match the theoretical CID fragmentation of each sequence generated. The variable sequencing approach is especially useful when parts of the oligonucleotide are known, or at least suspected, and the user wants to quickly test unknown residues. It also works well for generating the first or last few nucleotides of sequences, which can be used as a foundation for the user to manually sequence the rest of the oligonucleotide.
The variable sequencing algorithm, like the automated de novo sequencing algorithm, utilizes the local-search (17) paradigm for checking whether the data in the spectrum supports the growing polynucleotide. As such, if the variable nucleotide iterates to a nucleotide whose primary fragment m/z (5′ to 3′: c- ions and 3′ to 5′: y- ions) is not supported then the algorithm will attempt to skip to the next nucleotide. This process involves incorporating the unsupported nucleotide and moving on to the next residue. If the user-allotted skips for a sequence are exhausted then that sequence will fail, along with all of the potential sequences that would have arisen if the data had supported its placement.
As an example, MS/MS data with a precursor m/z of 1004.49 and a charge of 2− generated from RNase T1 digested E. coli Gln-tRNAUUG was generated for analysis using the variable sequencing algorithm (Supplementary Figure S4). The sequence X-X-X-cmnm5s2U-X-X, where each unidentified sequence position (X) could be C,U,A, G, 1mU, 1mA, 1mG, 1sU or cmnm5s2U (all of the unique masses in the published sequence of E. coli Gln-tRNAUUG), was attempted with a CID product m/z tolerance of 0.3 and a precursor mass tolerance of 1.0. The results of variable sequencing are returned after summation of m/z ion abundances and are sorted from high to low cumulative ion abundances. The highest ranked sequence corresponded to the known sequence of U-Um-U-cmnm5s2U-U-Gp, which is found in the anticodon loop of Gln-tRNAUUG (33).
RNA modification mapping with RoboOligo
A particularly useful application of RoboOligo is to generate annotated MS/MS data for mapping modified nucleosides onto one or more primary RNA sequences. This RNA Modification Mapping approach using mass spectrometry data was first described by McCloskey et al. (14) and members of his laboratory later described using SOS to assist in MS/MS spectra interpretation (44,46). With automated and batch processing capabilities, RoboOligo should enhance RNA Modification Mapping experiments and can complement database strategies (19,20). RoboOligo also allows the export of sequencing results into a CSV file, which can be analyzed in a variety of external programs.
To illustrate how RoboOligo can enhance RNA modification mapping, the LC-MS/MS data files from the RNase T1 digests of Asp-tRNAGUC and Gln-tRNAUUG were subjected to an automated de novo sequencing batch analysis. For each data file, the top ranking annotated MS/MS spectral results were exported and then compared against the primary (unmodified) tRNA sequence. With the exception of pseudouridine, which cannot be identified in RoboOligo and m7G in Asp-tRNAGUC (discussed above), all other previously identified modification sites (32,33) were represented in the RoboOligo-annotated sequences, with many spectra containing modified nucleosides characterized multiple times (Supplementary Figure S5).
Modification mapping using RoboOligo annotated MS/MS spectra also revealed several annotated sequences that did not map onto the expected primary RNA sequences. In certain cases, these sequences can be excluded given other known information about the sample. For example, the annotated oligomer [preQ0][1mG][G+]G from Asp-tRNAGUC can be excluded, as such hypermodified nucleosides are not found in the variable loop of tRNAs (41). In other instances, these top-ranked annotation results are actually found to have relatively few abundant fragment ions—a property that is easily determined by investigating the annotated data within the main window of the program. In certain cases, high quality (i.e. high sequence coverage and ion abundance) annotated MS/MS spectra that cannot be mapped onto a given RNA sequence may reveal the existence of additional RNAs within the sample. One advantage of an unbiased, de novo interpretation of MS/MS data is the potential for identifying sample components that are not necessarily expected or immediately obvious within the LC-MS/MS results.
DISCUSSION
RoboOligo was created for the analysis of tandem mass spectrometry data of modified RNAs, where it may serve as a productivity-increasing aid to those with expertise and also as an entry point for researchers with rudimentary knowledge of the data produced by CID-MS/MS of RNase-digested modified RNAs. The sequencing algorithm utilizes c- and y- fragment ions as the primary means of reconstructing the oligomer sequence from the experimental data. These fragment ions were chosen as they are the most common and abundant for tRNA and rRNA samples that are typically analyzed by MALDI- or LC-MS (8,10,44,47,48). In cases where specific c- or y- fragment ions may not be present in the dataset, the algorithm allows for a nucleotide placement skip and then looks at possible dinucleotide compositions that match the experimental data. The final scoring and ranking of annotated sequences then includes (a-B)- and w- fragment ions. Although any covalent bond in the phosphodiester backbone may be generated and thus used for scoring, the current iteration of the program is limited to these commonly detected fragment ions. Future iterations of the program may incorporate additional fragment ions such as phosphate loss. Through this selection of commonly detected backbone fragment ions, we anticipate that RoboOligo will be capable of handling the vast majority of MS/MS data generated by modern MS instruments. Moreover, to minimize vendor-specific file formats, RoboOligo utilizes *.mgf file input.
The automated de novo sequencing algorithm could correctly annotate MS/MS spectra from 72 of 77 previously characterized RNA oligomers of lengths from 2 to 12 bases. We show that the local-search paradigm is effective for sequencing RNase digestion products and with the support of 102 unique masses for canonical and modified bases, RoboOligo is a suitable tool for analyzing biologically derived tRNA, rRNA and other modified RNAs. Oligomers >9 nt will often require smaller nucleotide pools or higher mass accuracy as the number of valid compositions and sequences increases exponentially (Supplementary Figure S1), potentially creating computational workloads that push the capabilities of the modern desktop computer. Nucleotide pool sizes should be tempered to the oligomer length and reasonable expectations of the modified nucleosides within the sample. In batch sequencing mode, the number of precursors that must be examined will also influence program performance. Appropriate precursor selection filters are available to enable the user to match data analysis needs with the user's computational resources.
Although most of the RNase digestion products examined here were corroborated by external evaluation of the data, it should be noted that not all MS/MS scans with similar precursor masses and charge states will produce the same results. Incomplete MS/MS data, improper charge designation and the presence of different oligomers with similar precursor mass and charge all contribute to explain this discrepancy. Thus, while RoboOligo can be a useful tool to simplify the tedious process of MS/MS spectral annotation, as with any de novo mass spectrometry interpretation software, ultimately the user will have to make the final judgment in sequence determination. In this manner, RoboOligo is similar in characteristics to common peptide de novo sequencing software, which is incapable of universal identification of the correct sequence (49). Despite that limitation, de novo annotation software has proven useful to the proteomics community in cases where a database approach is limited (by modification, splice variants, SNPs, etc.). RoboOligo should fulfill a similar need for the mass spectrometry community interested in nucleic acids and complements the database approaches that are already available (19,20).
We also introduce a method coined as ‘variable sequencing’ that combines the hands-on approach of manual sequencing with the high-throughput sequencing analysis of the automated de novo sequencing algorithm. This technique allows for quick sequence prototyping and specific nucleotide position evaluation given fixed sequence constraint. Additionally, RoboOligo dramatically simplifies and expedites manual sequencing by providing the user with a simple interface that searches for and automatically labels m/z values that correspond with the inputted oligonucleotide data.
Limitations of RoboOligo for RNA modification mapping RNA modification mapping, as pioneered by McCloskey et al. (14,44), has some inherent limitations that cannot be addressed solely by a program that aids in the analysis of LC-MS/MS data of oligonucleotides. One primary limitation is the inability to distinguish the structural arrangement of modifications known to occur in one or more ring (or sugar) locations; for example, methylation. In general, MS/MS data will not reveal whether the methylation is on the ring or sugar as all such methylations will increase the nucleoside mass by 14 Da. The only common exception is that of N7-methylguanosine (m7G), which can be differentiated from other methylated guanosines (e.g. m1G, m2G and Gm) by the facile base loss (44,45). While RoboOligo can identify a sequence location as being methylated, additional information will be required to denote the specific methylation present.
Another limitation arises with the common modified nucleoside pseudouridine (50). Pseudouridine has the same elemental composition as uridine, precluding its identification within RoboOligo. Although McCloskey et al. reported that the CID mass spectra of pseudouridine result in changes to specific fragment ion abundances as compared to uridine (51), the current algorithm is not able to compare w- and a- fragment ion abundances for pseudouridine identification. Another common strategy to identify pseudouridine using mass spectrometry is by selectively derivatizing pseudouridine (52,53). The derivatization reaction results in a specific mass increase attributable to pseudouridine, which can be easily detected during mass spectral analysis. While the program does not contain these derivatized masses in the default .txt files, they can be added to the potential nucleotide list as required.
Similar to different modifications with identical masses, another limitation in this method is determining the sequence of two or more precursor ions with the same or nearly identical mass to charge ratios if the precursor ions are not separated chromatographically. This issue can arise when complex mixtures of RNase digestion products are analyzed by LC-MS/MS (54). In these cases, usually one of the sequences (the most abundant ion) can be identified but the other sequences can be difficult to determine with great confidence. In spite of these limitations, RoboOligo should significantly reduce the person-hours required to interpret CID MS/MS data, thereby facilitating higher-throughput RNA modification mapping by mass spectrometry.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank the labs of Valerie de Crecy-Lagard, John J. Perona and Uttam L. RajBhandary for their generous contributions of isolated tRNAs used to test the program and numerous colleagues, in particular Yang Jiao and Ningxi Yu, for beta testing the software.
FUNDING
National Institutes of Health [GM058843 to P.A.L., GM084065-07 to J.D.A.]. Funding for open access charge: National Institutes of Health [GM058843].
Conflict of interest statement. None declared.
REFERENCES
- 1.Sanger F., Coulson A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975;94:441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 2.Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M., et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantara W.A., Crain P.F., Rozenski J., McCloskey J.A., Harris K.A., Zhang X., Vendeix F.A., Fabris D., Agris P.F. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huber C.G., Oberacher H. Analysis of nucleic acids by on-line liquid chromatography–Mass spectrometry. Mass Spectrom. Rev. 2001;20:310–343. doi: 10.1002/mas.10011. [DOI] [PubMed] [Google Scholar]
- 5.McCloskey J., Whitehill A., Rozenski J., Qiu F., Crain P. New techniques for the rapid characterization of oligonucleotides by mass spectrometry. Nucleosides Nucleotides. 1999;18:1549–1553. doi: 10.1080/07328319908044782. [DOI] [PubMed] [Google Scholar]
- 6.McLuckey S.A., Habibi-Goudarzi S. Decompositions of multiply charged oligonucleotide anions. J. Am. Chem. Soc. 1993;115:12085–12095. [Google Scholar]
- 7.McLuckey S.A., Van Berker G.J., Glish G.L. Tandem mass spectrometry of small, multiply charged oligonucleotides. J. Am. Soc. Mass Spectrom. 1992;3:60–70. doi: 10.1016/1044-0305(92)85019-G. [DOI] [PubMed] [Google Scholar]
- 8.Meng Z., Limbach P.A. Mass spectrometry of RNA: linking the genome to the proteome. Brief Funct. Genomic Proteomic. 2006;5:87–95. doi: 10.1093/bfgp/ell012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu J., McLuckey S.A. Gas-phase fragmentation of oligonucleotide ions. Int. J. Mass Spectrom. 2004;237:197–241. [Google Scholar]
- 10.Tromp J.M., Schürch S. Gas-phase dissociation of oligoribonucleotides and their analogs studied by electrospray ionization tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2005;16:1262–1268. doi: 10.1016/j.jasms.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Monn S.T.M., Schürch S. New aspects of the fragmentation mechanisms of unmodified and methylphosphonate-modified oligonucleotides. J. Am. Soc. Mass Spectrom. 2007;18:984–990. doi: 10.1016/j.jasms.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Nyakas A., Stucki S.R., Schürch S. Tandem mass spectrometry of modified and platinated oligoribonucleotides. J. Am. Soc. Mass Spectrom. 2011;22:875–887. doi: 10.1007/s13361-011-0106-z. [DOI] [PubMed] [Google Scholar]
- 13.Nordhoff E., Kirpekar F., Roepstorff P. Mass spectrometry of nucleic acids. Mass Spectrom. Rev. 1996;15:67–138. doi: 10.1002/(SICI)1098-2787(1996)15:2<67::AID-MAS1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 14.Kowalak J.A., Pomerantz S.C., Crain P.F., McCloskey J.A. A novel method for the determination of post-transcriptional modification in RNA by mass spectrometry. Nucleic Acids Res. 1993;21:4577–4585. doi: 10.1093/nar/21.19.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakayama H., Takahashi N., Isobe T. Informatics for mass spectrometry-based RNA analysis. Mass Spectrom. Rev. 2011;30:1000–1012. doi: 10.1002/mas.20325. [DOI] [PubMed] [Google Scholar]
- 16.Pomerantz S.C., Kowalak J.A., McCloskey J.A. Determination of oligonucleotide composition from mass spectrometrically measured molecular weight. J. Am. Soc. Mass Spectrom. 1993;4:204–209. doi: 10.1016/1044-0305(93)85082-9. [DOI] [PubMed] [Google Scholar]
- 17.Rozenski J., McCloskey J.A. SOS: a simple interactive program for ab initio oligonucleotide sequencing by mass spectrometry. J. Am. Soc. Mass Spectrom. 2002;13:200–203. doi: 10.1016/S1044-0305(01)00354-3. [DOI] [PubMed] [Google Scholar]
- 18.Nyakas A., Blum L.C., Stucki S.R., Reymond J.-L., Schu¨rch S. OMA and OPA—software-supported mass spectra analysis of native and modified nucleic acids. J. Am. Soc. Mass Spectrom. 2013;24:249–256. doi: 10.1007/s13361-012-0529-1. [DOI] [PubMed] [Google Scholar]
- 19.Matthiesen R., Kirpekar F. Identification of RNA molecules by specific enzyme digestion and mass spectrometry: software for and implementation of RNA mass mapping. Nucleic Acids Res. 2009;37:e48. doi: 10.1093/nar/gkp139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayama H., Akiyama M., Taoka M., Yamauchi Y., Nobe Y., Ishikawa H., Takahashi N., Isobe T. Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data. Nucleic Acids Res. 2009;37:e47. doi: 10.1093/nar/gkp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapp E.A., Schütz F., Connolly L.M., Chakel J.A., Meza J.E., Miller C.A., Fenyo D., Eng J.K., Adkins J.N., Omenn G.S., et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5:3475–3490. doi: 10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- 22.Muddiman D.C., Anderson G.A., Hofstadler S.A., Smith R.D. Length and base composition of PCR-amplified nucleic acids using mass measurements from electrospray ionization mass spectrometry. Anal. Chem. 1997;69:1543–1549. doi: 10.1021/ac961134r. [DOI] [PubMed] [Google Scholar]
- 23.Oberacher H., Mayr B.M., Huber C.G. Automated de novo sequencing of nucleic acids by liquid chromatography-tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2004;15:32–42. doi: 10.1016/j.jasms.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Oberacher H., Pitterl F. Tandem mass spectrometric de novo sequencing of oligonucleotides using simulated annealing for stochastic optimization. Int. J. Mass Spectrom. 2011;304:124–129. [Google Scholar]
- 25.Oberacher H., Wellenzohn B., Huber C.G. Comparative sequencing of nucleic acids by liquid chromatography-tandem mass spectrometry. Anal. Chem. 2002;74:211–218. doi: 10.1021/ac015595a. [DOI] [PubMed] [Google Scholar]
- 26.Liao Q., Chiu N.H., Shen C., Chen Y., Vouros P. Investigation of enzymatic behavior of benzonase/alkaline phosphatase in the digestion of oligonucleotides and DNA by ESI-LC/MS. Anal. Chem. 2007;79:1907–1917. doi: 10.1021/ac062249q. [DOI] [PubMed] [Google Scholar]
- 27.Liao Q., Shen C., Vouros P. GenoMass-a computer software for automated identification of oligonucleotide DNA adducts from LC-MS analysis of DNA digests. J. Mass Spectrom. 2009;44:549–560. doi: 10.1002/jms.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma V.K., Glick J., Liao Q., Shen C., Vouros P. GenoMass software: a tool based on electrospray ionization tandem mass spectrometry for characterization and sequencing of oligonucleotide adducts. J. Mass Spectrom. 2012;47:490–501. doi: 10.1002/jms.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kretschmer M., Lavine G., McArdle J., Kuchimanchi S., Murugaiah V., Manoharan M. An automated algorithm for sequence confirmation of chemically modified oligonucleotides by tandem mass spectrometry. Anal. Biochem. 2010;405:213–223. doi: 10.1016/j.ab.2010.06.023. [DOI] [PubMed] [Google Scholar]
- 30.Kirkpatrick S., Gelatt D. Jr, Vecchi M.P. Optimization by simulated annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 31.Menezes S., Gaston K.W., Krivos K.L., Apolinario E.E., Reich N.O., Sowers K.R., Limbach P.A., Perona J.J. Formation of m2G6 in Methanocaldococcus jannaschii tRNA catalyzed by the novel methyltransferase Trm14. Nucleic Acids Res. 2011;39:7641–7655. doi: 10.1093/nar/gkr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips G., Swairjo M.A., Gaston K.W., Bailly M., Limbach P.A., Iwata-Reuyl D., de Crécy-Lagard V. Diversity of archaeosine synthesis in crenarchaeota. ACS Chem. Biol. 2012;7:300–305. doi: 10.1021/cb200361w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez-Hernandez A., Spears J.L., Gaston K.W., Limbach P.A., Gamper H., Hou Y.-M., Kaiser R., Agris P.F., Perona J.J. Structural and mechanistic basis for enhanced translational efficiency by 2-thiouridine at the tRNA anticodon wobble position. J. Mol. Biol. 2013;425:3888–3906. doi: 10.1016/j.jmb.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Köhrer C., Mandal D., Gaston K.W., Grosjean H., Limbach P.A., RajBhandary U.L. Life without tRNAIle-lysidine synthetase: translation of the isoleucine codon AUA in Bacillus subtilis lacking the canonical tRNA2Ile. Nucleic Acids Res. 2014;42:1904–1915. doi: 10.1093/nar/gkt1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puri P., Wetzel C., Saffert P., Gaston K.W., Russell S.P., Varela J.A.C., van der Vlies P., Zhang G., Limbach P.A., Ignatova Z., et al. Systematic identification of tRNAome and its dynamics in Lactococcus lactis. Mol. Microbiol. 2014;93:944–956. doi: 10.1111/mmi.12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spears J., Gaston K., Alfonzo J. In: RNA and DNA Editing. Aphasizhev R, editor. Vol. 718. Humana Press; 2011. pp. 209–226. [Google Scholar]
- 37.Meng Z., Limbach P.A. RNase mapping of intact nucleic acids by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (ESI-FTICRMS) and 18O labeling. Int. J. Mass Spectrom. 2004;234:37–44. [Google Scholar]
- 38.Steyaert J. A decade of protein engineering on ribonuclease T1. Atomic dissection of the enzyme-substrate interactions. Eur. J. Biochem. 1997;247:1–11. doi: 10.1111/j.1432-1033.1997.t01-1-00001.x. [DOI] [PubMed] [Google Scholar]
- 39.Krog J.S., Español Y., Giessing A.M.B., Dziergowska A., Malkiewicz A., Pouplana L.R., Kirpekar F. 3-(3-amino-3-carboxypropyl)-5,6-Dihydrouridine is one of two novel post-transcriptional modifications in tRNA(Lys) (UUU) from Trypanosoma brucei. FEBS J. 2011;278:4782–4796. doi: 10.1111/j.1742-4658.2011.08379.x. [DOI] [PubMed] [Google Scholar]
- 40.Zallot R., Brochier-Armanet C., Gaston K.W., Forouhar F., Limbach P.A., Hunt J.F., de Crecy-Lagard V. Plant, animal, and fungal micronutrient queuosine is salvaged by members of the DUF2419 protein family. ACS Chem. Biol. 2014;9:1812–1825. doi: 10.1021/cb500278k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grosjean H. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Austin, TX: Landes Bioscience; 2009. [Google Scholar]
- 42.Pomerantz S.C., McCloskey J.A. Analysis of RNA hydrolyzates by liquid chromatography-mass spectrometry. Methods Enzymol. 1990;193:796–824. doi: 10.1016/0076-6879(90)93452-q. [DOI] [PubMed] [Google Scholar]
- 43.Gao Y., McLuckey S.A. Collision-induced dissociation of oligonucleotide anions fully modified at the 2′-position of the ribose: 2′-F/-H and 2′-F/-H/-OMe mix-mers. J. Mass Spectrom. 2012;47:364–369. doi: 10.1002/jms.2044. [DOI] [PubMed] [Google Scholar]
- 44.Guymon R., Pomerantz S.C., Crain P.F., McCloskey J.A. Influence of phylogeny on posttranscriptional modification of rRNA in thermophilic prokaryotes: the complete modification map of 16S rRNA of Thermus thermophilus. Biochemistry. 2006;45:4888–4899. doi: 10.1021/bi052579p. [DOI] [PubMed] [Google Scholar]
- 45.Wong S.Y., Javid B., Addepalli B., Piszczek G., Strader M.B., Limbach P.A., Barry C.E. Functional role of methylation of G518 of the 16S rRNA 530 loop by GidB in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013;57:6311–6318. doi: 10.1128/AAC.00905-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emmerechts G., Barbé S., Herdewijn P., Anné J., Rozenski J. Post-transcriptional modification mapping in the Clostridium acetobutylicum 16S rRNA by mass spectrometry and reverse transcriptase assays. Nucleic Acids Res. 2007;35:3494–3503. doi: 10.1093/nar/gkm248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Douthwaite S., Kirpekar F. Identifying modifications in RNA by MALDI mass spectrometry. Methods Enzymol. 2007;425:3–20. doi: 10.1016/S0076-6879(07)25001-3. [DOI] [PubMed] [Google Scholar]
- 48.Schürch S., Tromp J., Monn S. Mass spectrometry of oligonucleotides. Nucleosides Nucleotides Nucleic Acids. 2007;26:1629–1633. doi: 10.1080/15257770701549053. [DOI] [PubMed] [Google Scholar]
- 49.Medzihradszky K.F., Chalkley R.J. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom. Rev. 2015;34:43–63. doi: 10.1002/mas.21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durairaj A., Limbach P.A. Mass spectrometry of the fifth nucleoside: a review of the identification of pseudouridine in nucleic acids. Anal. Chim. Acta. 2008;623:117–125. doi: 10.1016/j.aca.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pomerantz S.C., McCloskey J.A. Detection of the common RNA nucleoside pseudouridine in mixtures of oligonucleotides by mass spectrometry. Anal. Chem. 2005;77:4687–4697. doi: 10.1021/ac058023p. [DOI] [PubMed] [Google Scholar]
- 52.Durairaj A., Limbach P.A. Improving CMC-derivatization of pseudouridine in RNA for mass spectrometric detection. Anal. Chim. Acta. 2008;612:173–181. doi: 10.1016/j.aca.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mengel-Jørgensen J., Kirpekar F. Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry. Nucleic Acids Res. 2002;30:e135. doi: 10.1093/nar/gnf135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wetzel C., Limbach P.A. The global identification of tRNA isoacceptors by targeted tandem mass spectrometry. Analyst. 2013;138:6063–6072. doi: 10.1039/c3an01224g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.