


Abstract
A major challenge in the field of shotgun metagenomics is the accurate identification of organisms present within a microbial community, based on classification of short sequence reads. Though existing microbial community profiling methods have attempted to rapidly classify the millions of reads output from modern sequencers, the combination of incomplete databases, similarity among otherwise divergent genomes, errors and biases in sequencing technologies, and the large volumes of sequencing data required for metagenome sequencing has led to unacceptably high false discovery rates (FDR). Here, we present the application of a novel, gene-independent and signature-based metagenomic taxonomic profiling method with significantly and consistently smaller FDR than any other available method. Our algorithm circumvents false positives using a series of non-redundant signature databases and examines Genomic Origins Through Taxonomic CHAllenge (GOTTCHA). GOTTCHA was tested and validated on 20 synthetic and mock datasets ranging in community composition and complexity, was applied successfully to data generated from spiked environmental and clinical samples, and robustly demonstrates superior performance compared with other available tools.
INTRODUCTION
Shotgun metagenomic sequencing has become a useful tool in microbial community profiling and offers significantly less bias than amplification-dependent techniques such as 16S rRNA gene sequencing (1–3). With the tremendous sequencing capacity and cost-effectiveness of modern sequencing platforms, it is possible to sequence to sufficient depth to capture very low abundance community members in reliable proportions. Furthermore, given that shotgun metagenomic sequencing does not rely on amplification of targeted conserved regions, community profiling can be performed at a much higher level of taxonomic resolution.
Several tools for community profiling have been developed which build upon more traditional 16S or functional gene phylogenetic clustering and/or taxonomic assignment methods. A typical analysis of this type aligns next generation sequencing (NGS) read data to a reference database comprised of some subset (or all) of the known reference genes or genomes. The ability to perform alignments, or mapping, of large volumes of sequence data against large databases of thousands of genomes requires significant computational resources. To address this issue, various short-read aligners and alignment-free methods (4,5) have been developed to reduce resource requirements, some with lower memory usage, enabling such processes to run even on desktop computers. Efforts have also been made toward selectively decreasing the size of the reference databases to allow faster, but also more tailored or targeted analyses, by selecting gene subsets that are conserved within taxonomic groups (6–8) or signature fragments found among genomes, and taxonomic or phylogenetic clades (9–11).
There are limitations to current taxonomic profiling approaches, which use sequences conserved among different genomes in the classification process. First, for the creation of gene-based datasets, they are defined to coding regions, and can be annotation-dependent; in contrast, the sequencing library preparation method is not restricted to such delimited genomic regions, nor to representing data in open reading frames. Second, reliable identification of constituents of a metagenome is confounded by the conserved nature and high similarity of genomic regions such as 16S rRNA genes, housekeeping genes, orthologous/paralogous genes, and gene duplication and/or horizontal gene transfer events. Third, existing methods for calculating relative abundance rely on frequency data without taking into account the percent coverage within the targeted genes, subjecting themselves to possible high false discovery rates (FDR) [ = false positives/(true positives + false positives)] due to unaccounted for background genomic noise (e.g. host sequences). Fourth, few tools are able to consistently and reliably provide species- and strain-level classifications.
Here, we present a workflow that addresses all of these issues in shotgun metagenome data community profiling by examining Genomic Origins Through Taxonomic CHAllenge (GOTTCHA). We have implemented a semi-automated metagenomic community-profiling tool that is able to provide accurate community composition profiles at multiple taxonomic levels with reliable abundance estimates. We are able to significantly reduce the FDR by automatically eliminating genomic regions that generate the majority of false-positive signals in existing tools. We do this by analyzing the distribution and depth of coverage of only the unique fraction of each reference genome—the unique genome—to identify the true community composition and accurate relative abundance of members of the community. GOTTCHA uses empirically-derived coverage limits, supported by machine-learning approaches, to set the limits of detection. The result is a scalable, all-purpose, metagenomic community profiler with superior classification and statistical performance over all currently available tools. The GOTTCHA method is open source software available at https://github.com/LANL-Bioinformatics/GOTTCHA.
MATERIALS AND METHODS
GOTTCHA's unique reference genome databases
FASTA-encoded databases of unique signatures for prokaryotic and viral genomes were generated and used for this work (instructions provided at https://github.com/LANL-Bioinformatics/GOTTCHA). Briefly, databases of unique genome segments at multiple taxonomic levels (e.g. family, species, genus, strain-level, etc.) are used for taxonomic classification of reads. Variants of these databases, in which all human 24-mers were removed were also generated and used in this study. These 24-mers were derived from the GRCh37.p10 (Genome Reference Consortium), HuRef (J. Craig Venter Institute), and CHM1_1.0 (Washington Univesity School of Medicine) assemblies and include unplaced scaffolds.
Synthetic metagenomes
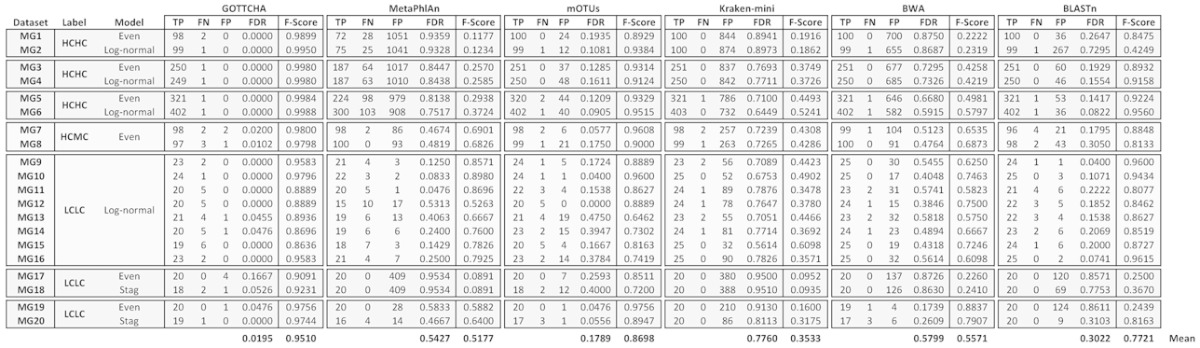
Several metagenome datasets were used to establish and validate the appropriate criteria for accurate taxonomic classification and abundance calculation using our GOTTCHA databases. Of the 16 synthetic metagenomic datasets (MG1–MG16) analyzed in this study, six were created for this study as high-complexity, high-coverage (HCHC) metagenomes with a total read amount mimicking that of a single Illumina HiSeq 2000 lane that varied in: community composition (100, >200 or >300 organisms), relative abundance (even or log-normal distribution), and per-base quality scores and error rates. Each dataset consisted of 300 million (M) 100-bp, paired-end reads. Read sets were derived from either a constant number of genomes (even: MG1, MG3, MG5) or the numbers of cells were randomly selected for each species from along a log-normal distribution curve (log-normal: MG2, MG4, MG6). Table 1 and Supplementary Table S1 summarize these synthetic metagenomic communities. Synthetic data were generated using MetaSim and a customized Illumina error model, with per-base quality assignments derived as follows. Errors were modeled by mapping real Illumina HiSeq 2000 100-bp reads against high-quality genome references whose assemblies were considered ‘finished’ (12). Sequencing errors were recorded and collated according to the read base position (1–100). An exactly-matched base was recorded in one lot while incorrectly-mapped bases were recorded in a separate, per-base lot. The per-base error probability provided to MetaSim was obtained by dividing the number of error bases at a specific position by 10M. Briefly, position-based quality scores were assigned for both error and error-free positions: error-free bases receive qualities randomly chosen from the set of exactly-matched base qualities recorded for that position in the read (of the 10M reads), whereas error-containing base qualities were randomly chosen from quality scores recorded for their position-specific lot. Ten previously published synthetic metagenomes incorporating a MAQ error model (6) were also used in the cross-tool comparison for their considerably lower sequencing coverage: two high-complexity, medium-coverage (HCMC) metagenomes, each with 100 organisms and 1M reads (MG7, MG8); and two low-complexity, low-coverage (LCLC) metagenomes, each with 25 organisms and 250k reads (MG9–MG16).
Table 1. Synthetic metagenome classification performance summary for all tools.
HMP mock metagenomes
Genomic mixtures of 22 organisms of fixed concentrations were created by the Human Microbiome Project (HMP) to test their sequencing protocols and analytical pipelines. Two mixture types are available: an even (EVEN) mixture, where aliquots were based on equimolar rRNA operon counts per organism; and a staggered (STAG) mixture, where the rRNA operon counts can vary by up to 4 orders of magnitude according to the following table: http://downloads.hmpdacc.org/data/HMMC/HMPRP_sT1-Mock.pdf. Each mixture was then sequenced on both the Illumina Genome Analyzer II and the 454 GS FLX Titanium. Raw sequence data was downloaded from the HMP website (http://www.hmpdacc.org/HMMC). Since it was highly unlikely that the observed sequence distribution would correlate with input concentrations at the library preparation stage (1,13–19), a round of BWA (20) mapping using only the known community members as reference was used to compute each individual's actual relative abundance. Although there were 22 organisms in the mock community, the analyses presented are limited to completed bacterial genomes, and thus exclude the eukaryote (Candida albicans) and the incomplete bacterial genome (Actinomyces odontolyticus), leaving just 20 reference organisms (MG17–MG20).
Air filter metagenome
In March 2011, genomic DNA was extracted from an air filter wash (phosphate buffered saline, 1% TritonX) and spiked with random amounts of DNA from the biothreat agent Francisella tularensis SCHU S4. Two Illumina libraries were created from the extracted 20 ng of DNA: one constructed immediately after DNA extraction and one amplified using the Qiagen Repli-G whole genome multiple displacement amplification (MDA) kit. Amplification yielded 4.3 μg of MDA DNA from which a library was prepared with the ‘Preparing Samples for Sequencing Genome DNA’ protocol without modification, and sequenced as a single read (SR) 36 bp run on the Illumina Genome Analyzer IIx, generating 91 252 832 reads. The unamplified sample was prepared with the same Illumina protocol, but required speed vacuum-concentrating to a usable volume before fragmentation on a Covaris’ E210 Focused-ultrasonicator. Because the 20 ng of starting material was much less than the 1–5 μg of DNA required by the standard protocol, the following modifications were applied to prevent large-adapter and primer dimers from forming: only 25% of the required adapter oligo mix was used and the volume made up for with water, and enriched using only 25% of the PCR primers before cleaning with Agencourt AMPure beads (Beckman Coulter), yielding 13 ng DNA/μl. The unamplified library was sequenced across six lanes of the Illumina HiSeq 2000 as a SR 36 bp run (v1 cBot kit for cluster generation; v1.5 sequencing kit), generating 631 706 030 reads; however a temporary instrumental malfunction resulted in the generation of only 30-bp reads.
Spiked human stool metagenome
Stool was collected from a single individual, divided into three samples, and spiked with various concentrations of several pathogens at the Center for Disease Control and Prevention (CDC) in Atlanta, GA. Bacterial pathogens included the A1122 vaccine strain of Yersinia pestis and the Sterne vaccine strain of Bacillus anthracis, both tested at dilutions of 108, 107 and 106 CFU/ml. Viral pathogens included Human adenovirus B (HAdV-3 strain), Mamastrovirus 1 (Human astrovirus 2) and Enterovirus C (Human poliovirus 1 strain Sabin vaccine strain). Stock concentrations for Adenovirus, Poliovirus and Astrovirus were 4.07 × 108 (diluted 1:50, 1:500, 1:5000), 4.14 × 109 (diluted 1:500, 1:5000, 1:50000), and 5.83 × 109 (diluted 1:50, 1:500, 1:5000) genome copies/ml, respectively. The organisms were radiation-inactivated and RNA was extracted using TRIzol LS (Invitrogen). The three samples were each filtered through a 0.1-μm centrifugal filter and cleaned up further using the Qiagen RNeasy kit. RNA concentrations were determined by Qubit RNA assay and confirmed using the Agilent Bioanalyzer 2100 with either the RNA Nano or RNA Pico chips. Approximately 70–80 ul of each sample averaging 371 ng RNA/μl solution was shipped in Eppendorf LoBind tubes sealed with Parafilm to Los Alamos National Laboratory for sequencing. Sequencing libraries were generated for the three fecal RNA samples using Illumina's TruSeq v2 RNA Sample Prep kit, which includes cDNA conversion and PCR enrichment. The three libraries had an average size of 330 bp with a range of 200–700 bp. Each library was sequenced in one lane each of the same HiSeq paired-end 101 bp run.
GOTTCHA-based read classification
GOTTCHA-based analyses begin with trimming input read datasets by quality, followed by fragmentation of reads into uniform sizes. Reads are first fragmented at any nucleotide < Q20, and the remaining read fragments are split into as many non-overlapping 30-mers (subreads) as possible. Currently, when using data from Pacific Biosciences (RS or RS II), lowering the quality threshold to Q10 appears to provide results comparable to less error-prone reads. Terminal fragments whose lengths are between 30 and 59 bp are retained as subreads as-is without splitting. Unlike other metagenome profiling tools that report classification accuracy on a per-read basis, GOTTCHA's classification accuracy is organism-based and, since exact read matching is currently implemented, longer read fragment lengths increase the chance of a mismatch. For this reason, sequence reads larger than 30 bp are broken down into fragments. We tested the recoverable signal-to-noise ratio of read fragments of length 24–30, 40 and 50 bp and found that the shorter fragments increase signal output, but those of 24 bp in length also increased the classification error rates, so a value of 30 bp was selected. These data suggest, however, that allowing 1–2 mismatches may be beneficial for increasing read recruitment of homologous, but slightly divergent, sequences, however this would require additional parameter optimizations. The trimmed subreads are then mapped to either the prokaryotic and/or viral GOTTCHA databases, using the maximal exact matches (mem) option of the short-read aligner BWA: bwa mem -k 24 -T 0 -B 100 -O 100 -E 100 -t 12, where k is the minimum seed length, T is the minimum alignment score, B is the mismatch penalty, O is the gap open penalty, E is the gap extension penalty, and t is the number of threads. SAM alignment results are then profiled and filtered with the GOTTCHA profiler using the following filter parameters: minLen = 100, minCov = 0.005, minHits = 10, cCov = 0.006, minMLHL = 5.
GOTTCHA terminology
Using GOTTCHA, an organism's unique genome (U) is defined as
the collection of all signatures (uj) found among its
replicons (chromosomes and plasmids) that are not present in the genomes of the other
available sequences in the taxonomic level of interest, such that:
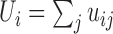 for
each of the j unique fragments of the
ith organism, where
for
each of the j unique fragments of the
ith organism, where 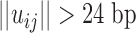 . Thus, the
unique genome of an organism is valid only for a given taxonomic level. For example,
Ui,strain for organism
i could vary greatly from
Ui,family, because
Ui,strain potentially considers many
additional organisms during the unique genome generation process. We define the
linear length (L) (of an organism's unique genome) as the sum
total non-overlapping length of each signature (lj) that
is covered by reads during the mapping process, such that each base is counted only
once (i.e. depth of coverage is not taken into account):
. Thus, the
unique genome of an organism is valid only for a given taxonomic level. For example,
Ui,strain for organism
i could vary greatly from
Ui,family, because
Ui,strain potentially considers many
additional organisms during the unique genome generation process. We define the
linear length (L) (of an organism's unique genome) as the sum
total non-overlapping length of each signature (lj) that
is covered by reads during the mapping process, such that each base is counted only
once (i.e. depth of coverage is not taken into account): 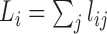 for
the j fragments mapping to organism i. The linear
coverage (LC) (of an organism's unique genome) is the percentage of the unique
genome that is covered during the mapping process:
for
the j fragments mapping to organism i. The linear
coverage (LC) (of an organism's unique genome) is the percentage of the unique
genome that is covered during the mapping process: 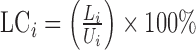 for the ith organism. This was the primary parameter used for
organism identification in a sample. The linear depth of coverage (DOC) is a measure
of the fold coverage of the linear length:
for the ith organism. This was the primary parameter used for
organism identification in a sample. The linear depth of coverage (DOC) is a measure
of the fold coverage of the linear length: 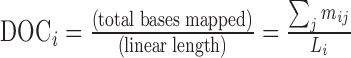 for all mij bases mapped to the unique genome of the
ith organism. This is the sole parameter used for relative
abundance calculation, whereby the relative abundance (RA) of an organism is
determined by normalizing its linear DOC to that of all organisms
detected in the sample:
for all mij bases mapped to the unique genome of the
ith organism. This is the sole parameter used for relative
abundance calculation, whereby the relative abundance (RA) of an organism is
determined by normalizing its linear DOC to that of all organisms
detected in the sample: 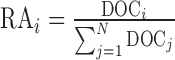 for all N organisms passing detection thresholds in the sample.
for all N organisms passing detection thresholds in the sample.
Binary classification
GOTTCHA classification occurs at the organism level rather than at the more traditional read level. For the synthetic and mock datasets, an algorithm-identified organism is labeled a true positive (TP), if the organism is known to be present in the sample, or as a false positive (FP) if it is known not to be present. A false negative (FN) is called if the algorithm does not find an organism known to be present. True negatives (TN) are dependent on the number of organisms in the reference database used, and as such, are only reported for GOTTCHA. Specifically, TN = (no. of genomes in database) – (TP + FP + FN). Binary classifications are based on the final output of each program. For the GOTTCHA profiler, this includes a filtering step. GOTTCHA results were filtered in a serial, two-stage process: the first considers only those species whose linear coverage met or exceeded 0.5%. The second rejects any species whose mean linear hit length was <5 and linear coverage <0.6%.
Statistical measures and parameter filtering
In the GOTTCHA workflow, an organism's linear coverage is GOTTCHA's primary classification parameter. In order to determine the minimum threshold above which an organism should be classified as present in a sample, we evaluated the dependence of the classification precision [ = TP/(TP + FP)] and recall [ = TP/(TP + FN)] over all valid ranges of linear coverage for metagenomes with sequencing amounts that varied over 3 orders of magnitude, from 250k to 300M reads (Supplementary Table S1, Supplementary Figure S1). Precision–recall (PR) curves (Supplementary Figure S2) were created by repeatedly profiling the SAM alignment files for different values of the linear coverage for datasets MG7–MG16. For computational efficiency, rather than computing precision and recall values throughout the entire range of linear coverage (0–100%), we computed these values up to the greatest linear coverage found in the sample. At this point and for all higher values of linear coverage, no true positives are found, and hence, precision = recall = 0. Both plots (Supplementary Figure S2A and S2B) include the random guess performance line and the precision–recall values yielding optimal F1-scores are indicated. We then plotted the best F1-scores [ = 2 × precision × recall/(precision + recall)] obtainable across the entire range of linear coverages in order to assess the best possible performance of each tool among the diverse set of metagenomes (Supplementary Figure S2C).
Profiling tool comparisons
Several tools were used to help classify metagenome reads and the results were compared (Supplementary Table S2). All tools, including GOTTCHA utilized the same quality-trimmed input dataset. MetaPhlAn v1.7.7 (6) was run with default settings, using Bowtie2 (21) against its database of unique clade-specific marker genes released on 15 October 2012. mOTUs v1 was run using default parameters (7) and reports the species directly with NCBI taxonomic IDs. Kraken v0.10.2-beta was run with default parameters with the preload option using its database available on 3 November 2013 (11), and the Kraken reporter classifies ambiguous reads with the LCA method (22). BLASTn v2.2.28+ (23) was run on 1 million randomly sampled reads for the fecal metagenomic analysis and with all reads in all other cases. BWA v0.7.4-r385 (20) used as a stand-alone tool was run locally using the aln and samse single-end reads option and the SAM file was processed with samtools (24). For both the BWA and BLASTn results, the NCBI taxonomy was assigned to each read using lowest common ancestor (LCA) method. Additionally, because BWA and BLAST are not specifically metagenome read taxonomy assignment tools, we imposed a cutoff such that only species with 10 or more read counts were reported, in order to limit the number of false positives but without compromising true positives. All tools were run with 12 threads. precision, recall, F-score, false discovery rate and accuracy, were calculated for each tool as detailed above.
Tool command lines
| BLASTn: | blastn -db [db] -num_threads 12 -query [input] |
| BWA: | (1) bwa aln [db] [input] > [input].sai |
| (2) bwa samse -t 12 -n 50 [db] [input].sai [input] > [input].sam | |
| (3) samtools view -@ 12 -uhS [input].sam | samtools sort -@ 12 - [input] | |
| (4) samtools mpileup -BQ0 -d10000000 -f [db] [input].bam > [pileup_output] | |
| MetaPhlAn: | metaphlan.py –bowtie2db bowtie2db/mpa –bowtie2out bowtie2.out |
| –nproc 12 [input] [output] | |
| mOTUs: | mOTUs.pl –processors = 12 –length-cutoff = 45 –identity-cutoff = 97 –quality-cutoff = 20 |
| Kraken-mini: | (1) kraken –db minikraken –threads 12 –preload |
| (2) kraken-report –db [db] [output].classification.csv > | |
| [output].report.csv |
Tool resource utilization
Each tool's resource utilization was tracked while processing the HMP mock datasets using 12 threads. The workstation consisted of four Intel quad-core Xeon E7440 (2.40 GHz) processors for 16 cores total, 132 GB RAM, and attached to six Seagate SAS drives (model ST9146852SS) in hardware RAID5 via a LSI Logic/Symbios Logic MegaRAID SAS 1078 controller.
Assessing the proportion of genomes that are used for classification
Using 100 randomly selected genomes, we investigated how much of their own sequence information is used (i.e. can be classified) by the various tools, and how much of this sequence information contributed deleteriously to the overall genome/taxon assignment (incorrect assignment). For this analysis the 100 genomes were decomposed into all possible 31-mers (so as to maintain minimum compatibility with all classification tools), where each consecutive 31-mer was chosen so as to overlap 30-bp with the previous 31-mer. Each tool was then used to classify these ‘reads’, according to the tool-specific protocol (above) and the read assignments recorded. A correct assignment (TP) was recorded when a 31-mer was assigned to its correct source genome, and an incorrect assignment (FP) was recorded when the 31-mer was assigned to a genome other than its true source. For each tool, the fraction of assigned (TP+FP), and the proportion of correctly assigned (TP/(TP + FP)), and incorrectly assigned reads (FP/(TP + FP)) are displayed using the boxplot function in R, and as a function of taxonomic hierarchy (such that reads placed incorrectly at the lower levels of taxonomy may still be classified correctly at higher levels of taxonomy).
Community profiling of synthetic and HMP mock metagenomes
Species identification proceeded straightforward with MetaPhlAn, mOTUs and Kraken as we applied no post-filtering to the data. Data generated from the aligners BWA and BLASTn were filtered so that species (genomes) recruiting <10 hits were discarded so as to limit the gross over-reporting of false positives. Relative abundance calculations proceed only upon finalization of species identification. Species relative abundances are provided by MetaPhlAn, mOTUs and Kraken, whereas those for BWA and BLASTn were calculated by fractions of all hit counts. GOTTCHA relative abundances were calculated by calculating the ratio of all bases mapped to the species signatures with the total linear length of all signatures mapped.
Community profiling of real, spiked metagenomes
Species identification and relative abundances proceeded as described above. The relative abundance of the organisms detected by each tool were organized into a pivot table, and a subset of these organisms—restricted only to those detected by GOTTCHA—formed the basis of a summary heat map using the matrix2png web tool (25). Extreme values were trimmed by 5% (outlier effect). The air filter metagenome values ranged from 3.1 × 10−5 (black) to 0.031 (red), whereas the human stool metagenome ranged from 2.7 × 10−8 (black) to 0.0052 (red); gray values represent organisms unidentified by the respective tool. The human stool metagenome was quite large, so the BLASTn analysis was limited to a random subset of 1M reads only. This dataset also included viral targets which mOTUs and MetaPhlAn are currently incapable of classifying. As such, no viral information is provided for these two tools. In order to correlate spike level with detectability, GOTTCHA hit counts are reported in addition to the relative abundances of the heat map.
GOTTCHA classification efficacy of novel genomes through hold-out analyses
Nearly 2000 prokaryotic draft genomes of varying degrees of novelty that were not included in the creation of the GOTTCHA databases were passed through the GOTTCHA workflow (with some modification) to attempt to identify the parent taxa. These draft genomes were obtained from NCBI in the form of draft genome assemblies. Since genome assembly collapses sequencing reads into a single, contiguous representation that is essentially devoid of the redundant input data (reads) needed to distinguish signal from noise, we used Jellyfish (26) to decompose all contigs into all possible 30-mers, where each 30-mer occurs one or more times. Since contig qualities were not readily available, no quality-based trimming was implemented. Each k-mer's multiplicity, however, was retained and these ‘read’ fragments were then mapped to the GOTTCHA database, the taxonomic level of which was determined by the taxonomic novelty of the genome analyzed. For example, a genome considered novel at the genus level was mapped to the family GOTTCHA database to attempt to place it in the proper family.
RESULTS
The GOTTCHA workflow (Figure 1) consists of three stages: (i) trimming and fragmenting reads prior to read mapping, followed by (ii) mapping of the read fragments to the relevant GOTTCHA unique reference genome database(s) and finally (iii) profiling and filtering of the results to identify community constituents and their relative abundances. Relative abundance calculations proceed only after completion of the filtering step, and are based on the relative coverage differences between the unique genomes found in the sample. We leverage prior work applying a novel algorithm to the creation of the GOTTCHA unique genome databases (https://github.com/LANL-Bioinformatics/GOTTCHA)—a set of FASTA files representing the unique genomes of each reference organism, where all shared genomic regions (between the different genomes, or between taxonomic groupings) are removed. Here, we describe the application of the GOTTCHA trimming protocol, databases and profiler in the classification of next generation metagenomic sequencing reads from microbial communities. GOTTCHA databases are pre-computed, to allow the user to download the taxonomic rank-dependent unique genomes prior to starting the workflow. Here, we discuss the approach and its validation on a number of synthetic datasets, available mock sample data and data from spiked samples. We further compare it with existing tools to show substantial performance improvements in terms of the FDR and accuracy (F-score).
Figure 1.
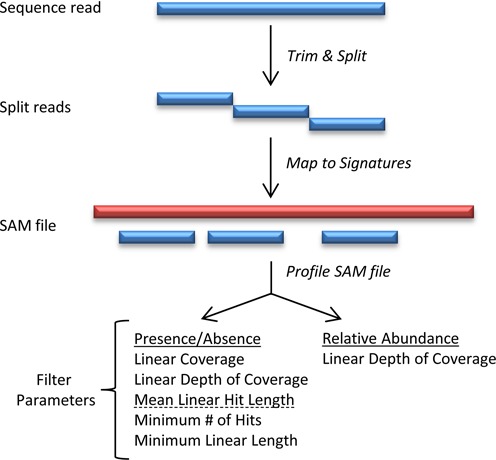
Overview of GOTTCHA workflow. Raw sequence reads are first cut on low quality bases and split into non-overlapping 30 bp fragments (see ‘Materials and Methods’ section). Read fragments are then mapped to a GOTTCHA database, after which the GOTTCHA profiler parses the alignment file and generates the community composition along with their relative abundances.
Optimal parameter selection
After analyzing various isolate genomes and metagenomes using the GOTTCHA databases, we empirically arrived at a primary classification filtering parameter that consistently provided accurate classification results—the linear coverage. This is a ratio between the length of the unique genome covered in the mapping process and the total length of the unique genome in the GOTTCHA databases. For calling a genome or taxonomic group present, values ≥0.5% tended to yield optimal results, so we tested this hypothesis against a machine learning approach across an array of community complexities of various sequencing depths and read distributions (Supplementary Figure S1). The accuracy, expressed as the F-score which incorporates both precision and recall into a single measure, overlapped maximally across all datasets in the 0.1–0.5% linear coverage range, though the high-complexity, high coverage (HCHC) metagenomes are much more tolerant than those of low coverage and span much larger ranges of linear coverage (Supplementary Figure S1A). Therefore, optimal classification performance requires only 0.1–0.5% linear coverage of the unique fraction of any genome to accurately determine its presence in samples. The low linear coverage accommodates the rare members (or intermediate abundance organisms) within samples that have not undergone substantial sequencing.
Each component of this measure of accuracy is visualized in their respective precision–recall curves (Supplementary Figure S2A and S2B) across this 0.1–0.5% range, emphasizing how performance is affected as the datasets become increasingly sparse and difficult to classify. Filtering out organisms below this coverage range (0.5% is selected by default)—coupled with a minimum hit and minimum length characteristic—is our primary approach at limiting false positives. Our secondary approach relies on tracking alignment positions within the unique genomes to discard small regions of the unique genome with disproportionately high coverage (read stacking). We have found that such stacking without sufficient linear coverage of the unique genome is a fundamental feature of a false positive. We identify these stacking events by comparing an organism's linear coverage with its mean linear hit length (MLHL), defined as the ratio of an organism's linear length to its hit count. We found empirically that most read stacking FPs were identifiable when MLHL <5 and the linear coverage did not exceed our minimal cutoff by an additional 0.1%, i.e. at least 0.6%. Additional experiments have shown that the removal of human 24-mers from the GOTTCHA signature databases removes the majority of false positives from human clinical samples, a phenomenon we refer to as background bleed-through (data not shown). As such, these database versions are preferred for profiling human microbiome samples, such as blood, sputum or feces.
GOTTCHA maximizes information reported due to its unique signatures
We examined the proportions of all possible reads derived from 100 randomly selected genomes that were correctly and incorrectly assigned by each tool. Because these 100 genomes are already integrated within each tool's database, this test is a measure of the proportion of data that can be classified, along with a measure of the proportion accurately (or incorrectly) classified. As expected, BWA was capable of assigning all reads indiscriminate of the taxonomic level (Supplementary Figure S3A), however the proportion of correctly or incorrectly assigned reads varies depending on the number and similarity of near-neighbor genomes within the database (Supplementary Figure S3B and S3C). This results in reads being more accurately assigned as the taxonomic resolution decreases (i.e. from strain to phylum), a trend expected for all tools examined. GOTTCHA had the next highest fraction of correctly assigned reads, with a median of >80% read assignment even at the strain level. Because mOTUs, Kraken-mini and MetaPhlAn focus on only a very limited component of any genome for assignment, the proportion of reads used for classification is much smaller than for BWA or GOTTCHA (Supplementary Figure S3A). Because of the nature of GOTTCHA's databases of unique signatures, the outliers in terms of percent classified reads at the genus, species, and strain levels are due to highly similar near neighbors present within the database, and are therefore the result of unmapped reads; also the proportion of correctly assigned reads was near perfect. The proportion of correctly classified data with Kraken-mini and mOTUs was generally better than for BWA at all taxonomic levels, with some mis-assignments particularly at the species or strain levels. MetaPHlAn displayed significant trouble throughout all levels of taxonomy. These data support the high information content that is retrievable from a genome using GOTTCHA, and highlights the issues with other methods of classification, imposed by high similarity among near-neighbor genomes present within the database.
Community profiling using synthetic metagenomes
We assessed GOTTCHA's profiling ability using several different metagenomes that varied in complexity (25, 100, >200 and >300 organisms), sequencing amount (250k, 1M and 300M 100-bp reads), relative abundance (even and log-normally distributed), and error content (MAQ error model, in-house Illumina error model), and compared these with existing mapping and classification tools. The HCHC metagenomes consisted of synthetic communities with 100 (MG1–MG2: Figure 2), >200 (MG3–MG4: Supplementary Figure S4A–D), and >300 members (MG5–MG6: Supplementary Figure S4E–H), each containing 300M reads—either evenly or log-normally distributed across its members—to approximate a single Illumina HiSeq lane for each of the six metagenomes. Not a single false positive was predicted by the GOTTCHA workflow in any of these six HCHC metagenomes. Other tools, however, predicted >4000 FPs, with the exception of mOTUs and BLASTn (using the LCA method and a 10-hit-minimum filter), which predicted 205 and 498 FPs respectively (for all six metagenomes). However, GOTTCHA was unable to confidently identify seven organisms (seven FNs) among the 1426 organisms used in these synthetic metagenomes. Upon closer inspection, this was due to the small number of unique signatures (<0.5 kb) within the database for the target organisms, indicating that these randomly chosen genomes are sufficiently similar to other sequenced organisms as to be essentially indistinguishable at the species level. Few organisms retain such a low amount of signatures, but these are easy to identify from the database itself. In such situations, the GOTTCHA Genus database can be used to classify these organisms (see hold-out analyses below). Relative abundance calculations were within 25% of the true value for over 91% (weighted average) of the organisms studied. For these synthetic communities covered by 300M reads, GOTTCHA's F-scores neither varied significantly across different read distributions nor across communities of varied complexity (Table 1, Supplementary Figure S2C).
Figure 2.
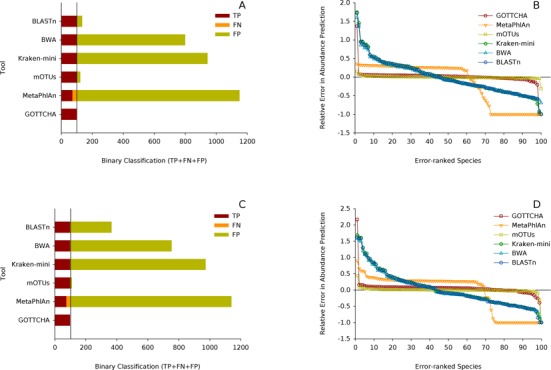
Comparison of classification and abundance profiles for two HCHC synthetic metagenomes. Species-level results for an evenly (MG1: panels A, B) and log-normally distributed (MG2: panels C, D) high complexity (n = 100) synthetic metagenome with high coverage (300M 100-bp paired-end reads) simulating one HiSeq lane. Bar charts (panels A, C) plot the sum of the binary classification results (TP + FP + FN) for each tool. A perfect classification would yield a solid maroon bar with TP = 100, FN = 0 and FP = 0. Line-and-scatter plots (panels B, D) show the relative errors in abundance calculations for each tool. Points at the zero-line predicted abundances perfectly. Those above and below are over- and under-predicted abundances, respectively, and points at −1 represent organisms the tool failed to identify.
It is expected that as the number of sequenced reads decreases, the accuracy of a predicted metagenome profile should also decrease. Therefore, we analyzed two HCMC metagenomes (MG7–MG8) with an even distribution of 1M reads each (Supplementary Figure S5). Fewer reads (lower input signal) in these lower coverage datasets resulted in a very slight decrease in GOTTCHA's performance. GOTTCHA misidentified five organisms as community members (five FPs/200) and failed to identify three organisms (three FNs/200), although GOTTCHA still outperformed all other tools (Table 1, Supplementary Figure S5), with over 91% (weighted average) of the organisms predicted within 15% of the true relative abundance values. Comparatively, for the metagenome of identical complexity and distribution in the previous HCHC set, the higher coverage resulted in 95% of the relative abundances being within 15% error (91% for the log-normally distributed metagenome), a small but important effect of sequencing coverage on accurate population profiling.
As the sequencing amount decreases further, we expect a further degradation in performance for all tools. With eight LCLC metagenomes (MG9–MG16) (Supplementary Figure S6), we observed a noticeable decrease in recall ( = TP/(TP + FN)) compared to the precision ( = TP/(TP + FP)), whereas recall remained relatively stable with the higher complexity metagenomes. Using the full-length genome sequences as references, BWA had the best mean recall at 97% (six FNs/200) whereas GOTTCHA's recall decreased to 85%, failing to identify 30 organisms (30 FNs/200) (see Table 1 for complete results). Mean precision, however, was only 50.3% for BWA, while GOTTCHA maintained a much higher precision (98.8%) than the other tools, falsely predicting only two organisms (two FPs/200). BLASTn turned out the next best precision at 85% using the LCA and 10-hit-minimum method. Incorporating both precision and recall into a single measure (F-score), the performance ranking of each tool for the LCLC metagenomes were as follows: GOTTCHA (0.914) > BLASTn (0.888) > mOTUs (0.806) > MetaPhlAn (0.762) > BWA (0.654) > Kraken (0.417). Given the relatively low number of FNs predicted by each tool, F-scores were largely impacted by FPs, and this was where our method proved superior. The other tools feature especially large FDR—a common problem with existing metagenomic profiling tools—whereas GOTTCHA's FDR was always at least one order of magnitude lower than the next best tool.
GOTTCHA displays the best performance with defined communities
To ensure that GOTTCHA's classification results were not an artifact arising from our choice of synthetic data, we tested GOTTCHA's performance on four HMP low complexity metagenomes (MG17–MG20) with controlled populations, generated from real sequencing instruments (27,28). Because both 454 and Illumina platforms were used to sequence these mock communities, these are ideal datasets to determine whether or not community profiling results are consistent between different sequencing platforms (Figure 3). The Illumina EVEN and STAG datasets contained 6.5M and 7.9M 75-bp reads, respectively, while the 454 EVEN and STAG datasets contained 1.4M, and 1.2M 533-bp reads, respectively. GOTTCHA's classification and relative abundance predictions consistently outperformed the other tools for both Illumina and 454 samples (Figure 3, Supplementary Table S1), with slightly better classification performance on the 454 datasets. This small classification improvement was likely due to the difference in scale of input number of reads—an average 5-fold larger number of reads provided more opportunity for misclassification of Illumina data. While this was of relatively little consequence for GOTTCHA—its F-score decreased only 6% from 0.975 to 0.916—the effect was much more pronounced for the other tools. In particular, the large increase in FPs reduced the F-scores for MetaPhlAn, BWA, Kraken-mini and BLASTn, decreasing the F-scores to 85%, 72%, 56% and 41% when each method was applied to the larger Illumina dataset, while the decrease in the mOTU F-score was a more moderate 16%.
Figure 3.
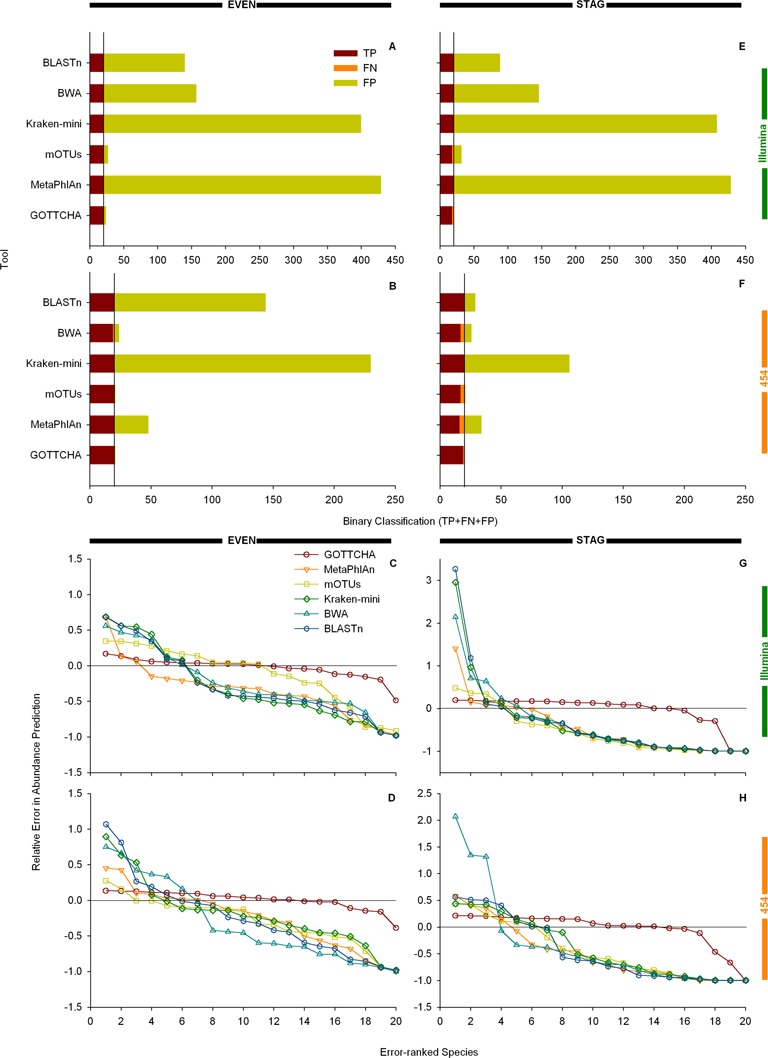
Comparison of classification and abundance profiles for the HMP mock samples. Classification and abundance performance results were generated for the Illumina even (MG17: panels A, C), Illumina staggered (MG18: panels E, G), 454 even (MG19: panels B, D) and 454 staggered (MG20: panels F, H) HMP data sets (see ‘Materials and Methods’ section). Interpretation of the bar chart and line-and-scatter plots are similar to that in Figure 2. Bar charts plot the sum of the binary classification results (TP + FP + FN) for each tool. A perfect classification would yield a solid maroon bar with TP = 20, FN = 0 and FP = 0.
Detection of biothreat signatures in an air sample
To provide a more realistic scenario of deciphering the contents of true metagenomes, we utilized an older dataset that was derived from an air filter metagenome spiked with F. tularensis. This data was originally generated in 2011 as part of an exercise to examine if sequencing could be utilized to identify biothreat pathogens in complex environmental samples. While an instrument malfunction reduced the read length to 30 bp, the targeted analysis at the time was able to identify F. tularensis within a few days of sample receipt, but did not characterize any of the less abundant organisms in the sample. Reprocessing these data using GOTTCHA identified (in a matter of hours) an unsurprising set of soil/plant microbes such as Pseudomonas stutzeri, Pseudomonas fluorescens, Zymomonas mobilis and Stenotrophomonas maltophilia, as well as microbes from exfoliations and respiratory droplets such as Propionibacterium acnes, Staphylococcus epidermidis and Streptococcus pneumonia, possibly of human origin. F. tularensis stood out at relatively high abundance (Supplementary Figure S7). The only other potential biothreat agent detected by GOTTCHA was B. anthracis, although at very low abundance. Manual inspection and similar findings with the other tools suggested it was a TP, perhaps derived from local (soil) environmental aerosol trapped in the air filter. Since MetaPhlAn, BLAST, and BWA all predict >1000 species in the air filter, the organisms in the heat map of Supplementary Figure S7 are limited only to the 83 species generated by GOTTCHA and sorted in decreasing order of the GOTTCHA predicted relative abundance. In line with previous observations of classification recall, mOTUs, BWA and BLASTn identified practically all of the 83 GOTTCHA-restricted organisms, while MetaPhlAn missed 16 organisms. Since the read lengths were only 30-bp and the Kraken-mini database was constructed with k-mers of length 31, it was not used to classify any of the data in this set.
Pathogen detection in clinical samples
While detection of known organisms present at relatively high levels within real metagenomes was a useful exercise, clinical manifestations rarely require a high pathogen concentration. We tested GOTTCHA with clinical human microbiome samples that were spiked with clinically relevant levels of several pathogens. Three replicates split from a human fecal sample was spiked with the same set of pathogens in different amounts that spanned three orders of magnitude. Our goal was to assess GOTTCHA's ability to identify the spiked pathogens, and to test GOTTCHA's ability to correlate output signal with pathogen load. Out of 15 different pathogen titers (three titers/pathogen, five pathogens), GOTTCHA was able to identify all mid- and high-concentration titers. However, of the lowest titers, only Y. pestis passed GOTTCHA's default filtering thresholds (Figure 4A). Only two matches to B. anthracis hits were recoverable in the lowest titer (Figure 4B), which was below GOTTCHA's detection threshold. The other classification tools predicted from 36 to over 1000 total organisms, and given the number of FPs returned by these tools in the above described synthetic and mock community assessments, we limited the metagenomic community profiling results of the three fecal samples to that generated by the GOTTCHA classification workflow, which included just 38 bacteria and eight viruses (Figure 4A).
Figure 4.
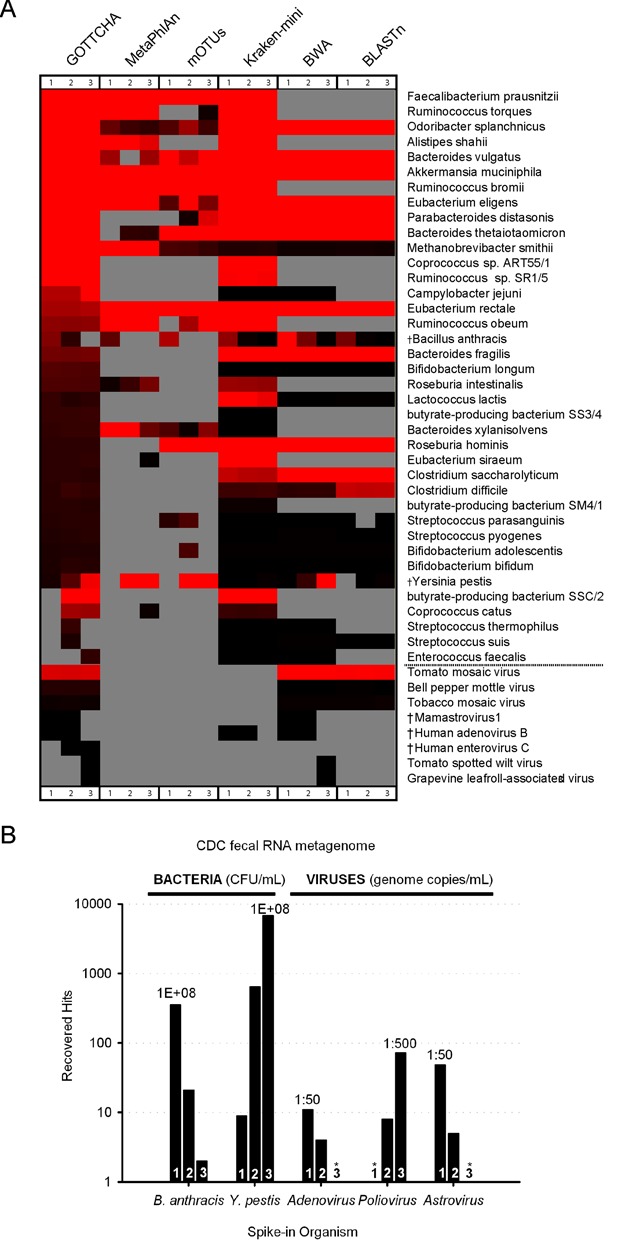
Pathogen identification in a clinical human fecal microbiome sample. Each of three aliquots from a single human fecal source was spiked with five pathogens at varying titers such that each successive titer differed 10-fold from the previous one. The range of organisms identified in the heat map (panel A) were truncated down to the 38 bacteria (upper panel) and eight viruses identified by GOTTCHA. Spike in concentrations (titers #1/2/3): B. anthracis, 108/107/106 CFU/ml; Y. pestis, 106/107/108 CFU/ml; Adenovirus (4.07 × 108 genome copies/ml), 1:50/1:500/1:5000 dilutions; Poliovirus (4.14 × 109 genome copies/ml), 1:50000/1:5000/1:500 dilutions; Astrovirus (5.83 × 109 genome copies/ml), 1:50/1:500/1:5000 dilutions. Relative abundances range from 2.7 × 10–8 (black) to 0.0052 (red), while gray cells indicate absence. Neither MetaPhlAn nor mOTUs can predict viral presence, therefore they are marked as absent (lower panel). Spiked-in pathogens are identified with a dagger (†). The total number of hits recovered for each pathogen at each titer is shown in the bar plot (panel B) and labeled where most concentrated above the bar in the triplet. Absent data points were below GOTTCHA detection thresholds and marked with asterisks (*). Pathogenic strains: Y. pestis (A1122 vaccine strain), B. anthracis (Sterne vaccine strain), Human adenovirus B (HAdV-3 strain), Mamastrovirus 1 (Human astrovirus 2), and Enterovirus C (Human poliovirus 1 strain Sabin vaccine strain).
Neither MetaPhlAn nor mOTUs can profile viral genomes, thus no viral results were reported for these tools. MetaPhlAn and mOTUs were able to identify Y. pestis in the two largest titer samples (Figure 4A, columns 2 and 3), but were only able to identify B. anthracis in the highest titer (Figure 4A, column 1). BWA and Kraken-mini were able to successfully identify both Y. pestis and B. anthracis across all three titers. For the viruses, Kraken-mini detected only Adenovirus in the two highest titer samples, while both GOTTCHA and BWA identified Astrovirus and Adenovirus in the two highest titer samples, but failed to detect them in the lowest titer due to the lack of reads. In addition, despite clear recovery of Poliovirus (Human enterovirus C) with GOTTCHA in the two highest titers, without a more sophisticated filtering mechanism, BWA was unable to detect the Poliovirus in any of the samples, due to the imposed minimum hit requirement. Because BLASTn requires an exorbitant amount of time to process these large amounts of data, we limited BLASTn analysis to a random subsampling of 1M reads when processing these samples. With this imposed limit, BLASTn failed to identify Y. pestis at its lowest titer and failed to detect all of the spiked-in viral pathogens.
Recoverable GOTTCHA hits were directly proportional to spike-in levels with R2 values of 0.9973 (B. anthracis), 0.9998 (Y. pestis), 0.8655 (Adenovirus), 0.9996 (Poliovirus) and 0.9998 (Astrovirus). Due to factors such as the unique genome size (which depends on the number and similarity of available near neighbor taxa), strain to strain variability, sample background and the fact that only 3–4 spike-in data points were available for each pathogen, it remains to be seen whether or not it is appropriate to rely on concentration curves of this sort as a general solution to predicting pathogen concentration in unknown clinical samples, as additional and more rigorous tests will be required. However, it appears clear that these mitigating factors are somewhat overcome using the GOTTCHA database, and spiked pathogens at clinically relevant concentrations are clearly detectable in NGS data using the GOTTCHA workflow.
Predicting parent taxa with GOTTCHA using novel genomes
Real metagenomes will undoubtedly contain previously unobserved genomes (that are not in reference databases). For these ‘novel’ genomes, we must rely on their assumed higher similarity with their nearest taxonomic neighbors, whose genomes can be found in the databases. Generally, taxonomic classification of novel organisms within a metagenomic sample relies on extrapolating observed read matches to known organisms that share the same parent taxa (e.g. a novel Y. pestis strain will undoubtedly match many known Y. pestis strains). Because the GOTTCHA databases are built specifically for each taxonomic level, we explored GOTTCHA's ability to properly classify novel genomes into the appropriate parent taxa (e.g. can a novel Y. pestis be placed into the Yersinia genus). We examined nearly 2000 draft genomes with varying degrees of taxonomic novelty (Supplementary Figure S8): 1027 novel strains, 658 novel species, 150 novel genera, 10 novel families and four novel classes.
It is important to note that draft genome assemblies present a unique challenge in that their genomes are incomplete, can be riddled with errors (SNPs, indels, rearrangements, chimeras), may be contaminated with exogenous DNA (or have vector, primer, adapters, etc.), and have not necessarily been validated for appropriate taxonomic placement (i.e. the taxonomy identifier may not match the true identity of the sequenced genome). After bioinformatically shredding each draft into all possible overlapping 30-mers, GOTTCHA was able to identify the proper parent taxa as the top result for 75% of the novel classes, 80% of the families, 61% of the genera, 85% of the species and 92% of the strains, despite the aforementioned drawbacks and the focus of many of these projects to investigate genomic novelty (29). In real metagenomes, the genome coverage is unlikely to be so uniform or complete, thus more detailed investigations are required to ascertain how best to utilize this information to highlight putative novel organisms found within complex mixtures.
In terms of misclassification, some causes arose from the anthropogenic nature of our microbial taxonomic tree (which only approximates a true phylogenetic representation). For example, there were issues with Shigella drafts being classified as Escherichia, which is partially expected as Shigella is more properly classified as a member of the same lineage, under Escherichia (30,31). There were other situations of possible horizontal gene transfer between closely related organisms within the same Genus (e.g. among the closely related Pseudomonads, Rhodococci, or within the B. cereus-group species), and even outside the same genus (Xanthomonas campestris and Methylobacterium radiotolerans, where previous work has already discovered evidence of horizontal gene transfer between these distantly related organisms (32)), that resulted in incorrect primary classifications.
In some instances, however, the distinction between organisms suggested possible contamination of the genome project(s), otherwise large portions of unique genomes are shared amongst them. For example, the novel Serratia symbiotica Tucson draft genome shared very little signatures with the only S. symbiotica reference genome available (∼1 kb), but shared 20-fold more linear coverage with Sodalis glossinidius, an endosymbiont of the tsetse fly Glossina palpalis gambiensis, an organism which also happens to harbor a novel S. glossinae organism in its midgut (33). Though contamination of some draft genome projects are likely, this and other examples must be more closely examined before being definitively labeled as such. Other genome projects appear to be entirely mislabeled, such as the Staphylococcus epidermidis VCU006 genome project that contained only 38 bp of identifiable signatures from S. epidermidis, whereas ∼2.2 Mb of S. aureus signatures were identified. This project has recently been relabeled as a S. aureus isolate in NCBI (Taxonomy and GenBank).
Computational performance
Both the RAM usage (gigabytes) and processing rate (reads per second per core) were tracked consistently among the various community profiling tools used in this study. We focus on the analysis of each of the HMP mock datasets in Supplementary Figure S9 using an isolated workstation employing 12 out of 16 available cores (four quad-core CPUs) writing to local disk. As expected, processing rates are primarily dependent on the size of the input data. GOTTCHA's memory requirements are somewhat larger than several of the other classifiers tested but can still be comfortably run on a modest laptop. GOTTCHA was a mid-performer in terms of speed, performing 2–5× slower than Kraken and MetaPhlAn, but 2–4× faster than mOTUs and BWA, and between 75–100× faster than BLASTn. However, GOTTCHA's superior performance in terms of both FDR and abundance measurements justify this small additional computational expense.
DISCUSSION
The crux of metagenomic community profiling is to determine the presence and abundance of individual community members, as well as the functional capabilities of the community as a whole. Here we focus on a solution to the first two challenges, incorporating the GOTTCHA method with tailored databases of the unique portions of genomes to provide unambiguous reference sequence to which shotgun metagenomic reads can be aligned. Currently, GOTTCHA expects a sequence alignment file (SAM) output file to parse, although the method is robust to future changes in sequencing and bioinformatics methods as it is easily amenable to almost any aligner output format and has been shown to work equally well with disparate sequence data types (e.g. Illumina and 454). Parsing of the SAM file provides coordinates indicating where each read fragment maps, and allows the GOTTCHA profiler to detect stacked read fragments—a typical feature of false positive signal which can arise from background genomes that were unaccounted for (i.e. not in the original database, and thus not subtracted from the unique reference genomes). The background depends on the community studied, but for the two real metagenomic communities that included human cells (Figure 4, Supplementary Figure S7), variants of the bacterial and viral GOTTCHA databases were created with all human 24-mers removed. Tracking of the individual coordinates also allows expansion of GOTTCHA to identify unique genes and associated functions in the metagenome.
Metagenomic community profiling is a fast-moving field, so only the most recent tools were evaluated. At the time this paper was written, mOTUs had recently been published, and a newer tool, Kraken was available but still under review. MetaPhlAn had been used widely since its publication (27,28) and has been shown to outperform tools such as the Naïve Bayes classifier (NBC), PhyloPythiaS, Phymm, PhymmBL and the Rapid Identification of Taxonomic Assignments (RITA) pipeline (see (6) for this comparison). Tools such as Sequedex, MetaPhyler and MetaCV were tested but were incapable of producing species-level results and were thus excluded from these analyses. When comparing the output, GOTTCHA consistently produced superior classification and relative abundance predictions compared to the three other classifiers considered—MetaPhlAn, mOTUs and Kraken—as well as those results obtained by BWA and BLASTn using a combined LCA and 10-hit-minimum filter. In addition, even without further improvements, GOTTCHA can be run on a laptop, allowing deployable analytical capabilities rather than requiring a link to a large server or cluster.
Rarely are metagenomes sequenced to exhaustion, given that many true communities contain thousands to millions of members (34–37) with a wide range in relative abundance. We therefore tested GOTTCHA using a wide range of sequencing amounts: from a full Illumina HiSeq lane (300M reads) (Figure 2, Supplementary Figure S4) to only 2–3% (6–9M reads) (Figure 3), 0.3% (∼1M reads) (Supplementary Figure S5) and ∼0.01% (250k reads) of a lane (Supplementary Figure S6). Community profiling accuracy – both in organism identification and relative abundance prediction—consistently exceeded that of the other tools among all datasets tested, regardless of read distribution type (evenly distributed among community members or following a log-normal distribution). GOTTCHA's classification performance is the result of a low number of FPs leading to a high precision and correspondingly high F-score. Not surprisingly, community-profiling accuracy of rare members tended to degrade as sequencing amount decreased. For example, maximum F-scores derived from the LCLC metagenomes show a decrease due to lower recall (more FNs) (Supplementary Figure S2B and S2C). These are, however, realistic and typical scenarios, giving us a first measure of providing a relative error estimate that correlates with the estimated abundance.
Accurate and rapid metagenomic community profiling has many important applications in both applied and basic science. Clinical diagnosis, exploring fundamental physiogenomic relationships, environmental biosurveillance, agriculture and water quality monitoring, and monitoring bioreactor yields for biofuel production are examples that will all benefit from algorithmic profiling improvements. In particular, critical applications that can be severely undermined by high FP rates, such as clinical diagnosis and biosurveillance, will strongly benefit from a very accurate profiling method like GOTTCHA.
As applications of metagenomics become more widespread, avoiding FPs when assessing real microbial communities taken from diverse environments becomes an increasingly important feature of reference-based shotgun metagenome classification tools. Using only the unique portions of reference genomes, our method is designed to avoid FPs, and will continue to improve as genome databases expand. In addition, the GOTTCHA process classifies the greatest amount of genomic information among the tested classifiers, and incorporates several detection metrics in addition to simple frequency data (number of reads) that other tools base their abundance calculations on. Further, because we use unique reference genome databases at each level of taxonomy, we can classify never before seen genomes within their respective parent taxonomic groups. The improved accuracy of predicted organism presence within samples will immediately aid in ascertaining important players within sampled environments, and will make significant contributions to robust identification of pathogens in environmental and clinical samples.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Jason Gans for critical discussions on classification and machine learning techniques, and Shihai Feng for the generation of synthetic datasets.
FUNDING
U.S. Defense Threat Reduction Agency [R-00059-12-0 and R-00332-13-0 to P.S.G.C.]. Funding for open access charge: PI at Los Alamos National Laboratory, provided by the US Defense Threat Reduction Agency [R-00332-13-0 to P.S.G.C.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Degnan P.H., Ochman H. Illumina-based analysis of microbial community diversity. ISME J. 2012;6:183–194. doi: 10.1038/ismej.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrosino J.F., Highlander S., Luna R.A., Gibbs R.A., Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin. Chem. 2009;55:856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scholz M.B., Lo C.C., Chain P.S.G. Next generation sequencing and bioinformatic bottlenecks: the current state of metagenomic data analysis. Curr. Opin. Biotech. 2012;23:9–15. doi: 10.1016/j.copbio.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 4.Hatem A., Bozdag D., Toland A.E., Catalyurek U.V. Benchmarking short sequence mapping tools. BMC Bioinform. 2013;14:184. doi: 10.1186/1471-2105-14-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schbath S., Martin V., Zytnicki M., Fayolle J., Loux V., Gibrat J.F. Mapping reads on a genomic sequence: An algorithmic overview and a practical comparative analysis. J. Comput. Biol. 2012;19:796–813. doi: 10.1089/cmb.2012.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segata N., Waldron L., Ballarini A., Narasimhan V., Jousson O., Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sunagawa S., Mende D.R., Zeller G., Izquierdo-Carrasco F., Berger S.A., Kultima J.R., Coelho L.P., Arumugam M., Tap J., Nielsen H.B., et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods. 2013;10:1196–1199. doi: 10.1038/nmeth.2693. [DOI] [PubMed] [Google Scholar]
- 8.Liu B., Gibbons T., Ghodsi M., Treangen T., Pop M. Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences. BMC Genomics. 2011;12(Suppl. 2):S4. doi: 10.1186/1471-2164-12-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berendzen J., Bruno W.J., Cohn J.D., Hengartner N.W., Kuske C.R., McMahon B.H., Wolinsky M.A., Xie G. Rapid phylogenetic and functional classification of short genomic fragments with signature peptides. BMC Res. Notes. 2012;5:460. doi: 10.1186/1756-0500-5-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J.M., Wang H.F., Yang H.X., Zhang Y.Z., Wang J.F., Zhao F.Q., Qi J. Composition-based classification of short metagenomic sequences elucidates the landscapes of taxonomic and functional enrichment of microorganisms. Nucleic Acids Res. 2013:41. doi: 10.1093/nar/gks828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis M.P.A., van Dongen S., Abreu-Goodger C., Bartonicek N., Enright A.J. Kraken: a set of tools for quality control and analysis Of high-throughput sequence data. Methods. 2013;63:41–49. doi: 10.1016/j.ymeth.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chain P.S.G., Grafham D.V., Fulton R.S., FitzGerald M.G., Hostetler J., Muzny D., Ali J., Birren B., Bruce D.C., Buhay C., et al. Genome project standards in a new era of sequencing. Science. 2009;326:236–237. doi: 10.1126/science.1180614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beszteri B., Temperton B., Frickenhaus S., Giovannoni S.J. Average genome size: a potential source of bias in comparative metagenomics. ISME J. 2010;4:1075–1077. doi: 10.1038/ismej.2010.29. [DOI] [PubMed] [Google Scholar]
- 14.Lazarevic V., Gaia N., Girard M., Francois P., Schrenzel J. Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLoS One. 2013;8:e67699. doi: 10.1371/journal.pone.0067699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lombard N., Prestat E., van Elsas J.D., Simonet P. Soil-specific limitations for access and analysis of soil microbial communities by metagenomics. Fems Microbiol. Ecol. 2011;78:31–49. doi: 10.1111/j.1574-6941.2011.01140.x. [DOI] [PubMed] [Google Scholar]
- 16.Pan Y., Bodrossy L., Frenzel P., Hestnes A.G., Krause S., Luke C., Meima-Franke M., Siljanen H., Svenning M.M., Bodelier P.L.E. Impacts of inter- and intralaboratory variations on the reproducibility of microbial community analyses. Appl. Environ. Microb. 2010;76:7451–7458. doi: 10.1128/AEM.01595-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosseel T., Van Borm S., Vandenbussche F., Hoffmann B., van den Berg T., Beer M., Hoper D. The origin of biased sequence depth in sequence-independent nucleic acid amplification and optimization for efficient massive parallel sequencing. PLoS One. 2013;8:e76144. doi: 10.1371/journal.pone.0076144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solonenko S.A., Ignacio-Espinoza J.C., Alberti A., Cruaud C., Hallam S., Konstantinidis K., Tyson G., Wincker P., Sullivan M.B. Sequencing platform and library preparation choices impact viral metagenomes. BMC Genomics. 2013;14:320. doi: 10.1186/1471-2164-14-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J.Z., Jiang Y.H., Deng Y., Shi Z., Zhou B.Y., Xue K., Wu L.Y., He Z.L., Yang Y.F. Random sampling process leads to overestimation of beta-diversity of microbial communities. Mbio. 2013;4 doi: 10.1128/mBio.00324-13. doi:10.1128/mBio.00324-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Federhen S. The NCBI Handbook. NCBI; 2002. [Google Scholar]
- 23.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J.H., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Proc G.P.D. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavlidis P., Noble W.S. Matrix2png: a utility for visualizing matrix data. Bioinformatics. 2003;19:295–296. doi: 10.1093/bioinformatics/19.2.295. [DOI] [PubMed] [Google Scholar]
- 26.Marcais G., Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huttenhower C., Gevers D., Knight R., Abubucker S., Badger J.H., Chinwalla A.T., Creasy H.H., Earl A.M., FitzGerald M.G., Fulton R.S., et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Methe B.A., Nelson K.E., Pop M., Creasy H.H., Giglio M.G., Huttenhower C., Gevers D., Petrosino J.F., Abubucker S., Badger J.H., et al. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu D., Hugenholtz P., Mavromatis K., Pukall R., Dalin E., Ivanova N.N., Kunin V., Goodwin L., Wu M., Tindall B.J., et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lan R., Reeves P.R. Escherichia coli in disguise: molecular origins of Shigella. Microb. Infect./Inst. Pasteur. 2002;4:1125–1132. doi: 10.1016/s1286-4579(02)01637-4. [DOI] [PubMed] [Google Scholar]
- 31.Chaudhuri R.R., Henderson I.R. The evolution of the Escherichia coli phylogeny. Infect. Genet. Evol. 2012;12:214–226. doi: 10.1016/j.meegid.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Studholme D.J., Kemen E., MacLean D., Schornack S., Aritua V., Thwaites R., Grant M., Smith J., Jones J.D. Genome-wide sequencing data reveals virulence factors implicated in banana Xanthomonas wilt. FEMS Microbiol. Lett. 2010;310:182–192. doi: 10.1111/j.1574-6968.2010.02065.x. [DOI] [PubMed] [Google Scholar]
- 33.Geiger A., Fardeau M.L., Falsen E., Ollivier B., Cuny G. Serratia glossinae sp. nov., isolated from the midgut of the tsetse fly Glossina palpalis gambiensis. Int. J. Syst. Evol. Microbiol. 2010;60:1261–1265. doi: 10.1099/ijs.0.013441-0. [DOI] [PubMed] [Google Scholar]
- 34.Breitbart M., Salamon P., Andresen B., Mahaffy J.M., Segall A.M., Mead D., Azam F., Rohwer F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edwards R.A., Rohwer F. Viral metagenomics. Nat. Rev. Microbiol. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- 36.Breitbart M., Felts B., Kelley S., Mahaffy J.M., Nulton J., Salamon P., Rohwer F. Diversity and population structure of a near-shore marine-sediment viral community. Proc. Biol. Sci./R. Soc. 2004;271:565–574. doi: 10.1098/rspb.2003.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wylie K.M., Truty R.M., Sharpton T.J., Mihindukulasuriya K.A., Zhou Y., Gao H., Sodergren E., Weinstock G.M., Pollard K.S. Novel bacterial taxa in the human microbiome. PLoS One. 2012;7:e35294. doi: 10.1371/journal.pone.0035294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.