


Abstract
DNA double strand breaks (DSBs) formed during S phase are preferentially repaired by homologous recombination (HR), whereas G1 DSBs, such as those occurring during immunoglobulin class switch recombination (CSR), are repaired by non-homologous end joining (NHEJ). The DNA damage response proteins 53BP1 and BRCA1 regulate the balance between NHEJ and HR. 53BP1 promotes CSR in part by mediating synapsis of distal DNA ends, and in addition, inhibits 5’ end resection. BRCA1 antagonizes 53BP1 dependent DNA end-blocking activity during S phase, which would otherwise promote mutagenic NHEJ and genome instability. Recently, it was shown that supra-physiological levels of the E3 ubiquitin ligase RNF168 results in the hyper-accumulation of 53BP1/BRCA1 which accelerates DSB repair. Here, we ask whether increased expression of RNF168 or 53BP1 impacts physiological versus mutagenic NHEJ. We find that the anti-resection activities of 53BP1 are rate-limiting for mutagenic NHEJ but not for physiological CSR. As heterogeneity in the expression of RNF168 and 53BP1 is found in human tumors, our results suggest that deregulation of the RNF168/53BP1 pathway could alter the chemosensitivity of BRCA1 deficient tumors.
INTRODUCTION
DSBs trigger ATM/ATR/DNA-PKcs-dependent phosphorylation of histone H2AX over a large chromatin domain, which serves as a molecular platform to concentrate downstream signaling and repair factors (1). Among the multitude of proteins that home to γH2AX-decorated chromatin are two RING-domain E3 ubiquitin ligases, RNF8 and RNF168 (2–6). Together, RNF8 and RNF168 catalyze a series of ubiquitylation events on substrates such as H2A and H2AX, with the H2AK13/15 ubiquitylation being particularly important for subsequent relocation of 53BP1 into foci (7–9). Stable binding of 53BP1 to DNA ends channels DSB repair into non-homologous end joining (NHEJ) in part by suppressing end resection (10), a process that generates long stretches of single stranded DNA needed for HR. It has been proposed that BRCA1/CtIP excludes 53BP1 and/or its cofactors RIF1/PTIP from chromatin surrounding DSBs specifically in S-G2 to allow extensive end resection, while in G1, 53BP1 and its cofactor RIF1 blocks end resection (10–18). This ensures that HR is maximally activated only when sister chromatids are available to template faithful repair. In the absence of functional BRCA1, DNA lesions occurring in S-phase that are normally repaired through error-free HR are instead channeled into mutagenic NHEJ, resulting in the formation of highly aberrant end joining products such as chromosomal radials (10). Hence, BRCA1-deficiency confers exquisite sensitivity toward chemotherapeutic agents that damage DNA in S-phase cells, such as poly(ADP-ribose) polymerase inhibitors (PARPi) (10,19–20), whereas combined BRCA1/53BP1 deficiency rescues HR, resulting in resistance to PARPi.
Immunoglobulin (Ig) class switch recombination (CSR) is a physiological process in mature B cells that is critically dependent on the induction and repair of programmed DSBs in G1 (21–23). CSR is initiated by activation-induced cytidine deaminase (AID), which generates multiple DSBs at highly repetitive Ig heavy chain switch regions (23–25). Rejoining of these DSBs requires 53BP1-dependent long-range NHEJ and replaces Igμ with a downstream constant region (Igγ, Igϵ or Igα) (26,27). Unlike mutagenic NHEJ, which inappropriately joins resected DSBs, physiological NHEJ reactions in the context of 53BP1-dependent CSR are typically more accurate wherein the majority of repair junctions show only minimal nucleotide changes. In the absence of 53BP1, however, AID-induced DSBs are resected in an ATM/CtIP-dependent manner and subsequently repaired by an alternative homology-driven pathway that generates non-productive intra-switch recombination (28–31). Notably, inhibition of ATM or CtIP activity partially enhanced CSR in 53BP1-deficient B cells, suggesting that end resection is an important negative regulator of NHEJ that must be suppressed by 53BP1 to enable efficient CSR (29,30).
RNF168 is a key upstream regulator of 53BP1 and was reported to be critical for the HR defects caused by BRCA1 deficiency (32–34), suggesting that RNF168 inhibits resection by controlling 53BP1 recruitment to DNA damage sites. In addition, RNF168 promotes CSR (35,36). Recently, it was found that RNF168 stabilization led to augmented 53BP1 spreading and accelerated repair of clastogen-induced DSBs (37). This raises the possibility that DSB repair during mutagenic NHEJ in S phase and/or physiological CSR in G1 can be similarly enhanced by increasing the expression of RNF168 and/or 53BP1. Here, we demonstrate that amplification of 53BP1 spreading has deleterious consequences for genome stability during S phase yet does not influence NHEJ during CSR. Thus, the requirements for DNA damage-induced histone ubiquitylation during NHEJ-mediated CSR and misrepair of PARPi-induced DNA damage are distinct. Finally, given the heterogeneous expression of RNF168 and 53BP1 in BRCA1-deficient tumors, our data may have therapeutic implications for PARPi treatment and predicting tumor responses.
MATERIALS AND METHODS
Mice
BRCA1f(Δ11)/f(Δ11) (NCI mouse repository) and BRCA1f(Δ11)/f(Δ11)/53BP1−/− (10) mice have been described. All experiments with mice followed protocols approved by the National Institutes of Health Institutional Animal Care and Use Committee.
Cell cultures, retroviral infection and flow cytometry
Wild-type, BRCA1Δ11/Δ11, 53BP1−/− and BRCA1Δ11/Δ1153BP1−/− mouse embryonic fibroblasts (MEFs) have been described (10). Resting B cells were isolated from spleen using anti-CD43 microbeads (Miltenyi Biotec) and stimulated to proliferate with 25 μg/ml LPS, 5 ng/ml IL-4 (both Sigma-Aldrich) and 0.5 μg/ml RP105 (BD PharMingen) (10). To overexpress RNF168, constructs encoding human RNF168WT or RNF168R57D (9) were subcloned into the pMX-PIE-IRES-GFP retroviral vector and used to transfect BOSC23 cells along with the pCL-Eco helper virus. Retroviral supernatant was collected 40–48 h later for infection of MEFs and B cells as previously described (38). pMX-PIE-based retroviruses encoding 53BP1DB and 53BP1DN have been described (11,28). Class switch recombination was assayed on days 3 and 4 using biotinylated anti-IgG1 and fluorochrome-conjugated anti-B220 antibodies (BD PharMingen).
FISH analysis
Stimulated B cells and MEFs were arrested at mitosis with colcemid (Invitrogen) treatment and harvested for metaphase spreads as described (38). Images were captured with an automated fluorescence microscope (Axio Imager Metasystems; Zeiss). One hundred metaphases were scored for the presence of chromosomal aberrations.
Cell cycle analysis
Cells were fixed in 70% ethanol and stained with propidium iodide for flow cytometric analysis. ModFit LT (Verity Software House) was used to assign cell cycle distribution.
Colony formation and growth competition assays
MEFs were treated with 1 μM PARPi (KU-0058948, Axon Medchem) either continuously for 10 days (wild-type, 53BP1−/−) or for 24 h followed by a 9-day post-incubation in drug-free medium (BRCA1Δ11/Δ11). Thereafter, culture dishes were stained with 0.5% crystal violet and colonies containing >50 cells were counted. Due to poor colony forming ability of BRCA1Δ11/Δ1153BP1−/− MEFs, a growth competition assay was used instead. To this end, BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with retroviral vectors encoding RNF168WT or RNF168R57D (GFP-positive) were mixed with non-transduced BRCA1Δ11/Δ1153BP1−/− MEFs (GFP-negative) at a 1:1 ratio. Alternatively BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with retroviral vectors encoding 53BP1DB (GFP-negative) were mixed with BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with the empty vector (GFP-positive). Cells were treated or not with 1 μM PARPi continuously for 9 days. Samples were collected before (day 0) as well as after PARPi treatment on days 1, 3, 5, 7 and 9 for flow cytometric analysis. Relative proliferation was calculated by normalizing the fraction of GFP-positive cells in PARPi-treated versus untreated samples collected on the same day.
Immunofluorescence
For standard immunofluorescence studies, MEFs grown on coverslips were subjected to γ-irradiation (cesium-137) or treated with PARPi. At the indicated times post-treatment, cells were first pre-extracted (20 mM HEPES, pH 7.0, 50 mM NaCl, 3 mM MgCl2, 0.3 M sucrose, 0.2% Triton X-100) for 5 min on ice to remove nucleoplasmic proteins and then sequentially fixed (4% paraformaldehyde), permeabilized (0.5% Triton X-100) and blocked (2% BSA). Samples were incubated with primary antibodies recognizing HA-tag (Santa Cruz Biotechnology), RNF168 (Millipore), conjugated ubiquitin (FK2, Sigma–Aldrich), 53BP1 (Novus Biologicals), RIF1 (14), RPA2 (Cell Signaling Technologies), RAD51 (H-92; Santa Cruz Biotechnology) followed by appropriate fluorochrome-conjugated secondary antibodies (Invitrogen). DNA was counterstained with DAPI. Foci images were captured with a fluorescence microscope at 63× magnification, unless otherwise stated. High-throughput automated imaging was used to captured chromatin-bound RPA at 20× magnification. Integrated nuclear RPA intensity was quantified using CellProfiler 2.0.
Immunoblotting
Whole cell extract (WCE) was resolved by SDS-PAGE (NuPAGE, Invitrogen) and transferred onto nitrocellulose membranes. Samples were incubated with primary antibodies recognizing RNF168 (Millipore), 53BP1 (Novus Biologicals), phospho-RPA2 (S4, S8), phospho-CHK1 (S317; all from Bethyl Laboratories), RPA2 (Cell Signaling Technologies) and α-tubulin (Sigma–Aldrich). Following incubation with the appropriate horseradish peroxidase-linked secondary antibodies (GE Healthcare), bands were visualized using enhanced chemiluminescence (GE Healthcare).
RESULTS
RNF168 and 53BP1 levels modulate PARP inhibitor-induced genome instability in BRCA1 deficient cells
53BP1 has been shown to promote mutagenic NHEJ in BRCA1-deficient cells by blocking end resection, which leads to PARPi hypersensitivity and tumorigenesis (10). For both 53BP1 and its immediate upstream regulator RNF168 we found copy number variations, genetic mutations and/or heterogeneous mRNA expression changes in a subset of BRCA1-deficient ovarian and breast tumors within the TCGA database (39–42(Figure 1). We therefore sought to determine whether modulating the chromatin loading of RNF168 and/or 53BP1 would alter PARPi sensitivity. To this end, we established mouse embryonic fibroblast (MEF) cell lines transduced with retroviral vectors encoding either wild-type RNF168 (RNF168WT) or a RNF168 mutant (RNF168R57D) that is incapable of 53BP1 recruitment (9) (Figure 2, Supplementary Figures S1A and Supplementary Figure S2). Since efficient transduction of MEFs with wild-type 53BP1 proved to be difficult due to its large size, we chose instead to overexpress 53BP1DB, a construct that lacks the extreme C-terminal BRCT domain but behaves like wild-type 53BP1 in terms of DSB end-protection (28), or a dominant negative fragment of 53BP1 (53BP1DN) (43) (Supplementary Figure S1B and C). As reported by Gudjonsson et al. (37), we found that overexpression of RNF168WT in BRCA1Δ11/Δ11 MEFs promoted the formation of conjugated ubiquitin and 53BP1 as well as RIF1 foci upon DNA damage caused by PARP inhibition or ionizing radiation (Figure 3A and Supplementary Figure S2). Interestingly, enlarged 53BP1 foci were also observed in BRCA1Δ11/Δ11 MEFs overexpressing 53BP1DB (Figure 4B). Thus, overexpression of 53BP1 itself can bypass the control of its spreading on damaged chromatin, which is normally limited by the availability of RNF168.
Figure 1.
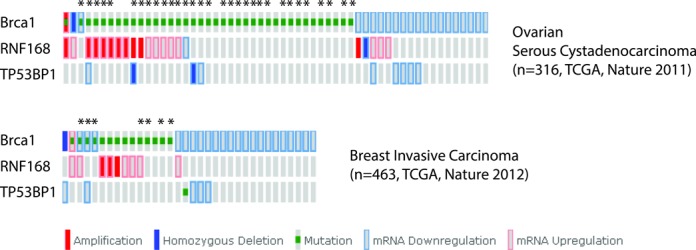
The RNF168/53BP1 pathway is altered in a subset of BRCA1-deficient tumors. Mutation data for two TCGA studies were extracted from cBioPortal (www.cbioportal.org) and used to generate oncoprints showing examples of RNF168/53BP1 pathway heterogeneity in BRCA1-deficient tumors (39–58). Asterisks denote tumors where BRCA1 is mutated in one allele and deleted in the other. All other BRCA1-mutated tumors contain heterozygous germline mutations.
Figure 2.
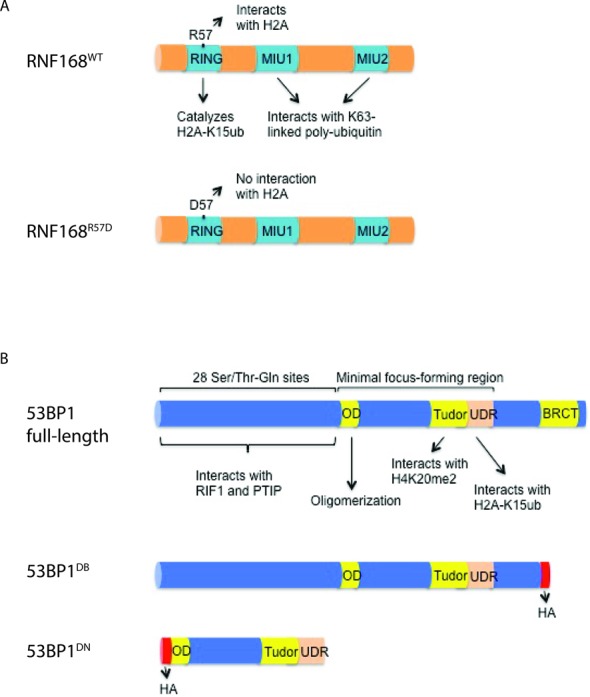
Schematic depiction of the retroviral vectors used in this study. (A) The RNF168WT and RNF168R57D differ in their ability to specifically interact with and ubiquitylate histone H2A due to a point mutation in the RING domain. MIU, motif interacting with ubiquitin. (B) 53BP1DB lacks the C-terminal BRCT domain present in full-length 53BP1 while 53BP1DN consists only of the minimal region required for foci formation. 53BP1DB is functionally wild-type with respect to end-protection and CSR (28), whereas 53BP1DN acts as a dominant negative mutant. OD, oligomerization domain; UDR, ubiquitylation-dependent recruitment.
Figure 3.
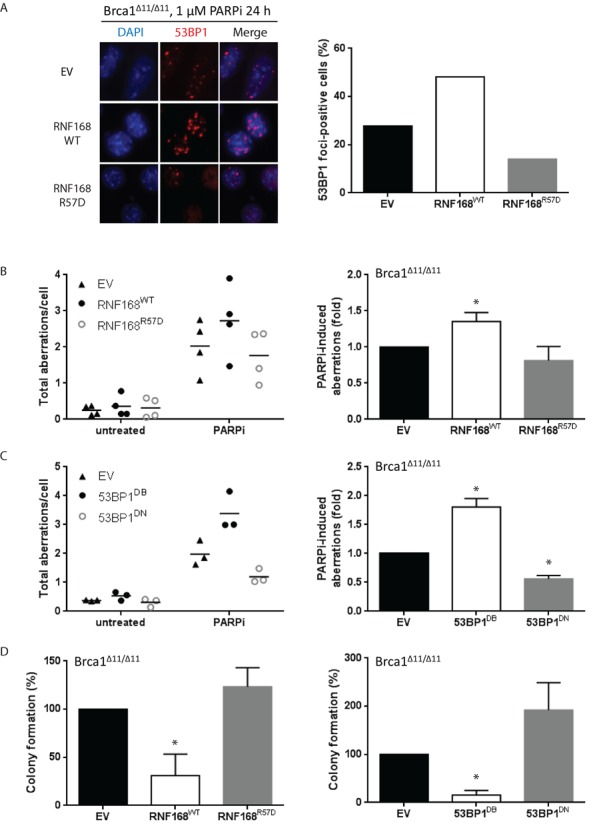
Overexpression of RNF168 or 53BP1 exacerbates PARP inhibitor-induced genomic instability and cytotoxicity. (A) BRCA1Δ11/Δ11 cells were treated with 1 μM PARPi for 24 h and processed for standard immunofluorescence. Left panel: cells were stained for 53BP1 (red) and imaged at 63× magnification. A representative experiment is shown. Right panel: the percentage of cells that contain >5 foci of 53BP1 from two independent experiments. At least 200 cells were scored for each sample and treatment condition. (B) BRCA1Δ11/Δ11 MEFs stably transduced with retroviral vectors encoding RNF168WT or RNF168R57D were treated with 1 μM PARPi (24 h) and harvested for preparation of metaphase spreads. Left panel: dot plots indicating the total amount of aberrations per cell in four independent experiments. At least 100 metaphases were analyzed for each condition. Right panel: histograms depicting PARPi-induced chromosomal aberration load relative to empty vector-transduced cells for the same experiments as shown in the corresponding left panels. (C) Similar to (B), except in BRCA1Δ11/Δ11 MEFs stably transduced with retroviral vectors encoding 53BP1DB or 53BP1DN. (D) Cells were treated with 1 μM PARPi for 24 h and then incubated in drug-free medium to allow formation of colonies. After 9 days, culture dishes were stained with crystal violet and colonies containing >50 cells were counted. Results are mean ± SD of three independent experiments. For (A)–(D), Statistical significance was determined with two-tailed unpaired Student's t-test; *, P < 0.05 compared to empty vector-transduced cells.
Figure 4.
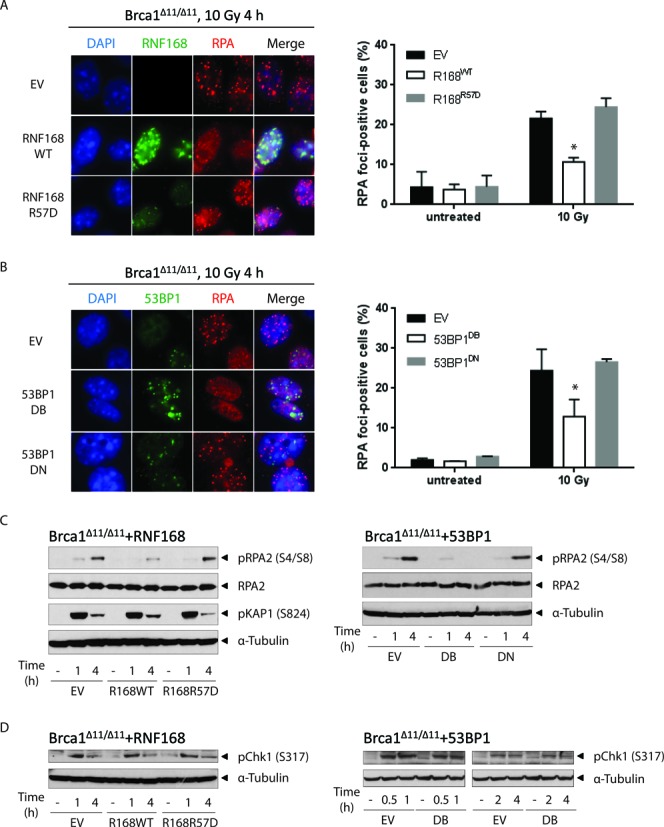
RNF168 and 53BP1 block RPA foci formation and RPA2 phosphorylation. (A) BRCA1Δ11/Δ11 MEFs stably transduced with retroviral vectors encoding RNF168WT or RNF168R57D were irradiated with 10 Gy and fixed 4 h later. Samples were processed for standard immunofluorescence. Left panel: cells were co-stained for RNF168 (green) and RPA2 (red) and imaged at 63× magnification. Note that the polyclonal anti-RNF168 antibody used in this study recognizes only the exogenously expressed human RNF168. A representative experiment is shown. Right panel: the percentage of cells that contain >10 RPA2 foci. (B) BRCA1Δ11/Δ11 MEFs stably transduced with retroviral vectors encoding 53BP1DB or 53BP1DN were irradiated and processed for standard immunofluorescence as in (A). Left panel: cells were co-stained for 53BP1 (green) and RPA2 (red) and imaged at 63x magnification. A representative experiment is shown. Right panel: the percentage of cells that contain >10 RPA2 foci. The right panels in A and B show mean ± SD of three independent experiments. At least 200 cells were scored for each sample and treatment condition. (C) Similar to (A), except cells were harvested at the indicated post-irradiation time points for western blot analysis. (D) Similar to (B), except cells were harvested at the indicated post-irradiation time points for western blot analysis. A representative blot is shown in (C) and (D). Experiments were repeated three times. For (A) and (B), statistical significance was determined with two-tailed unpaired Student's t-test; *, P < 0.05 compared to empty vector-transduced cells.
While PARPi treatment already caused substantial genome instability in BRCA1Δ11/Δ11 control cells (MEFs infected with an empty vector), PARPi-induced genome instability was further increased by 1.5–2-fold following overexpression of RNF168WT or 53BP1DB (Figure 3B and C). Conversely, BRCA1Δ11/Δ11 MEFs overexpressing RNF168R57D or 53BP1DN had less PARPi-induced chromosomal aberrations than control cells, although in the case of RNF168R57D the difference did not reach statistical significance (Figure 3B and C). Consistent with these observations, overexpression of RNF168WT or 53BP1DB conferred PARPi hypersensitivity in BRCA1Δ11/Δ11 MEFs in a colony formation assay whereas overexpression of RNF168R57D or 53BP1DN partially rescued long-term clonogenic survival (Figure 3D). Taken together, these data show that the level of chromatin-bound RNF168 and 53BP1 dictates the extent of PARPi-induced genome instability and long-term survival in BRCA1-deficient cells. Therefore, we propose that excessive spreading of either endogenous 53BP1 brought on by increased RNF168-mediated H2A ubiquitylation or its exogenously overexpressed counterpart 53BP1DB, impedes HR.
RNF168 and 53BP1 antagonize long-range end resection and RPA phosphorylation in BRCA1-deficient cells
BRCA1-mediated removal of 53BP1 is thought to be an essential prerequisite for end resection and RAD51-dependent HR (12,14–16,18). In agreement with this, 53BP1DB suppressed RAD51 foci formation in BRCA1Δ11/Δ11 MEFs (Supplementary Figure S3A). End resection enables RPA to form foci along progressively longer stretches of single-stranded DNA (ssDNA) before it is actively displaced by RAD51 (44). A recent report found that end resection can occur in the absence of functional BRCA1 but the process is slower and less efficient (45). Notably, overexpression of either RNF168WT or 53BP1DB in irradiated BRCA1Δ11/Δ11 MEFs further suppressed the chromatin loading of RPA compared to control cells (Figure 4A, B and Supplementary Figure S3B). Importantly, the cycling of BRCA1Δ11/Δ11 MEFs was not affected by overexpression of RNF168 or 53BP1, thus ruling out the possibility that differences in cell cycle progression contributed to the observed differences in RPA foci formation (Supplementary Figure S4). DNA-PK-dependent RPA2 (S4, S8) phosphorylation was also significantly attenuated in BRCA1Δ11/Δ11 MEFs overexpressing RNF168WT or 53BP1DB (Figure 4C). In contrast the phosphorylation of KAP-1, an ATM substrate that responds to DSBs, remained unchanged (Figure 4C). Interestingly, ATR-mediated phosphorylation of CHK1 was not perturbed by RNF168WT or 53BP1DB overexpression (Figure 4D). Since CHK1 activation by ATR requires only minimal resection (46,47), these results suggest that RNF168 regulates 53BP1 chromatin binding to antagonize long-range DSB end resection in BRCA1-deficient cells. Consistent with this notion, RNF168WT failed to block RPA loading (Figure 5A) or to enhance PARPi sensitivity in BRCA1Δ11/Δ11 MEFs when 53BP1 was co-deleted (Figure 5B and C). By contrast, 53BP1DB overexpression re-imparted susceptibility to PARPi-induced genome instability and decreased the survival of BRCA1Δ11/Δ1153BP1−/− MEFs (Figure 5B and C).
Figure 5.
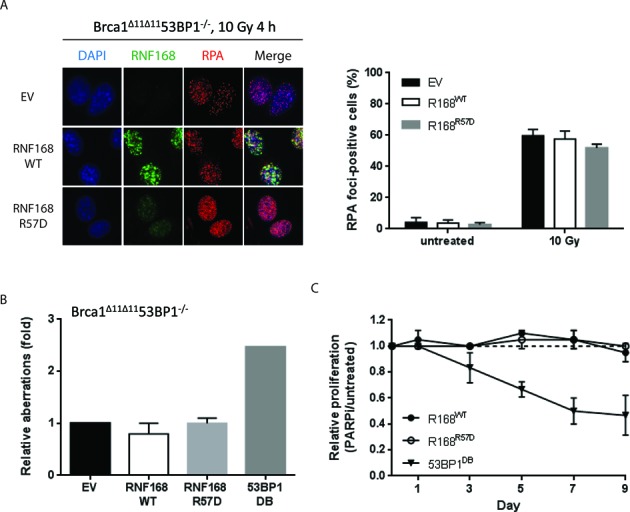
RNF168 uses 53BP1 to block RPA loading in BRCA1-deficient cells. (A) BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with retroviral vectors encoding RNF168WT or RNF168R57D were irradiated with 10 Gy and fixed 4 h later. Samples were processed for standard immunofluorescence. Left panel: cells were co-stained for RNF168 (green) and RPA2 (red) and imaged at 20x magnification (optovar: 1.25×). A representative experiment is shown. Right panel: the percentage of cells that contain >10 RPA2 foci. At least 100 cells were scored for each sample and treatment condition. (B) BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with retroviral vectors encoding RNF168WT, RNF168R57D or 53BP1DB were treated with 1 μM PARPi (24 h) and harvested for preparation of metaphase spreads. Histograms depict PARPi-induced chromosomal aberration load relative to empty vector-transduced cells for two-three independent experiments. At least 100 metaphases were analyzed for each condition. (C) BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with retroviral vectors encoding RNF168WT or RNF168R57D (GFP-positive) were plated with non-transduced BRCA1Δ11/Δ1153BP1−/− MEFs (GFP-negative) at a 1:1 ratio. Alternatively BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with retroviral vectors encoding 53BP1DB (GFP-negative) were mixed 1:1 with BRCA1Δ11/Δ1153BP1−/− MEFs stably transduced with the empty vector (GFP-positive). Cells were treated or not with 1 μM PARPi continuously for 9 days. Samples were collected before (day 0) as well as after PARPi treatment on days 1, 3, 5, 7 and 9 for flow cytometric analysis. Relative proliferation is calculated by normalizing the fraction of GFP-positive cells in PARPi-treated versus untreated samples collected on the same day. Data shown represents mean ± SD of three independent experiments.
In wild-type and 53BP1−/− MEFs, where BRCA1 is fully operational, ectopic expression of 53BP1DB also led to increased genome instability in response to PARPi treatment (Figure 6A and Supplementary Figure S1D). Moreover, 53BP1DB suppressed RAD51 loading in wild-type MEFs, as evidenced by the reduced numbers and smaller sizes of RAD51 foci (Supplementary Figure S3C), although this effect was milder than in BRCA1-deficient MEFs. As a result, 53BP1DB overexpression partially enhanced the toxicity of PARPi in wild-type MEFs (Figure 6A and C). Similarly, overexpression of RNF168WT increased PARPi-induced genome instability and cytotoxicity in wild-type MEFs (Figure 6B and D). Taken altogether, our data suggest that while a functional BRCA1 pathway can fully counteract endogenous RNF168/53BP1 during a normal S-phase DNA damage response, increased levels of RNF168 and 53BP1 can still suppress HR, increase toxicity and limit long-range end resection in BRCA1-proficient cells.
Figure 6.
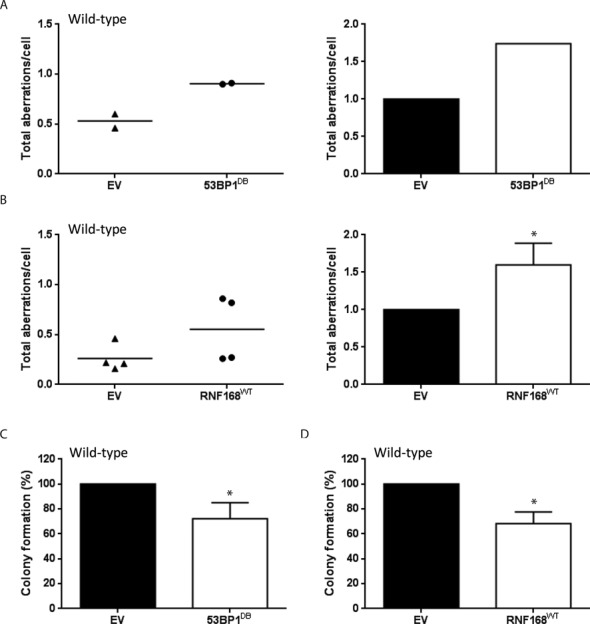
53BP1 suppresses HR in wild-type cells. Wild-type cells were stably transduced with retroviral vectors encoding 53BP1DB or RNF168WT. (A, B) Cells were treated with 1 μM PARPi (24 h) and harvested for preparation of metaphase spreads. Left panels: dot plots indicating the total amount of aberrations per cell. Right panels: histograms depict PARPi-induced chromosomal aberration load relative to empty vector-transduced cells. At least 100 metaphases were scored for chromosomal aberrations for each condition. Results are mean ± SD of two (A) and four (B) independent experiments, respectively. (C, D) Cells were treated with 1 μM PARPi continuously for 10 days, after which culture dishes were stained with crystal violet and colonies containing >50 cells were counted. Results are mean ± SD of three independent experiments. For (B), (C) and (D), statistical significance was determined with two-tailed unpaired and paired Student's t-test, respectively; *P < 0.05 compared to empty vector-transduced cells.
RNF168 and 53BP1 are not limiting factors during immunoglobulin class switch recombination
53BP1 promotes productive long-range NHEJ during CSR and VDJ recombination at least in part by blocking end resection (11,26–28,31,48). A recent study found that increased 53BP1 spreading caused by stabilization of RNF168 accelerates NHEJ (37); we therefore asked whether overexpression of exogenous RNF168 and/or 53BP1 might augment CSR. However, overexpression of RNF168 or 53BP1 did not affect CSR in wild-type B cells (Supplementary Figure S5). Next, we tested whether exogenous RNF168 and/or 53BP1 might augment CSR in the context of a resection defect imparted by BRCA1 deficiency. As expected (37), retroviral-mediated overexpression of RNF168WT led to enhanced chromatin loading of 53BP1 (Figure 7A). Despite this, however, overexpression of RNF168WT or 53BP1DB did not significantly enhance CSR in BRCA1-deficient B cells (Figure 7B and C). Conversely, CSR was not reduced by enforced expression of RNF168R57D (Figure 7B and C). Together, these data demonstrate that levels of chromatin-bound 53BP1 are not rate-limiting for physiological NHEJ during CSR (Figure 7D).
Figure 7.
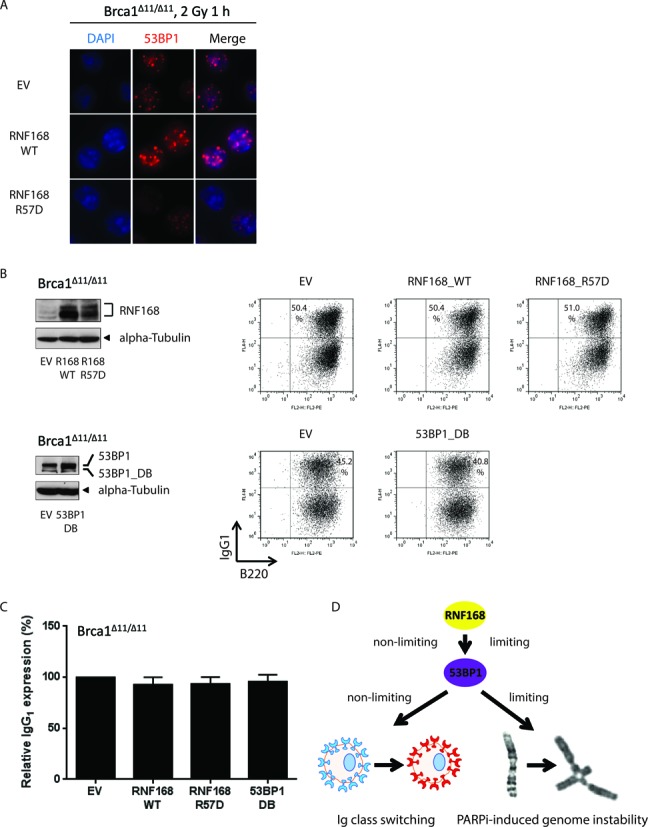
RNF168 and 53BP1 are not limiting factors during immunoglobulin class switch recombination (CSR). (A) BRCA1Δ11/Δ11 splenic B cells transduced with retroviral vectors encoding RNF168WT or RNF168R57D were cultured in the presence of LPS/IL-4/RP105 to stimulate CSR. On day 3, cells were irradiated with 2 Gy and fixed 1 h later. Samples were stained for 53BP1 (red) and imaged at 63× magnification. (B) BRCA1Δ11/Δ11 splenic B cells were transduced with retroviral vectors encoding RNF168WT, RNF168R57D or 53BP1DB and cultured in the presence of LPS/IL-4/RP105 to stimulate CSR. Left panels: overexpression of RNF168 and 53BP1 were confirmed by western blotting. The upper band in the 53BP1 blot corresponds to the endogenous protein. Right panels: two-color flow cytometric analysis of IgG1 expression in B220-positive cells on day 4; scatter plots for B cells overexpressing RNF168 and its empty vector counterpart were gated on GFP. A representative experiment is shown. (C) Frequency of IgG1 expression in B220-positive BRCA1Δ11/Δ11 B cells from three-four independent experiments. (D) Model depicting the differential roles of 53BP1 and RNF168 in physiological and mutagenic NHEJ.
DISCUSSION
End resection is a key determinant of DSB repair choice in mammalian cells (44). It peaks in the S-G2 phase of the cell cycle, when sister chromatids are available to template error-free repair by HR, but is actively suppressed by 53BP1 in G1, where HR would be harmful (49–51). A critical function of BRCA1 during S phase is to exclude 53BP1 and its cofactors from chromatin surrounding DSBs allowing long-range resection by nucleases (12–13,15). Loss of BRCA1, which occurs in a significant proportion of human ovarian and breast cancers, allows 53BP1 to aberrantly accumulate at DSB sites in S-G2 and block resection, thereby impairing HR and shifting the repair of replication-associated DSBs into error-prone NHEJ, leading to the formation of toxic repair intermediates and tumorigenesis (10,52). As such, BRCA1-deficient tumors are exquisitely sensitive to treatment with drugs that induce DSBs in S-phase, such as PARPi (19). Conversely, loss of 53BP1 expression has been implicated as a potential mechanism of acquired resistance to PARPi in BRCA1-deficient tumors (53–55). We found that a subset of BRCA1-deficient tumors within the TCGA database show copy number variation, mutation and/or mRNA expression changes in RNF168 or 53BP1 (Figure 1). Moreover, we have shown that overexpression of RNF168 or 53BP1DB confers PARPi hypersensitivity in BRCA1Δ11/Δ11 cells by promoting 53BP1 spreading and further suppressing the already sub-optimal resection machinery (Figures 3 and 4). These data indicate that heterogeneity in the RNF168/53BP1 pathway can be an important and therapeutically relevant modifier of chemosensitivity in BRCA1-deficient tumors.
Deregulation of the RNF168/53BP1 pathway is not limited to BRCA1-deficient tumors. For ovarian and breast cancers that are wild-type for BRCA1, elevated expression of 53BP1 is associated with decreased survival and lymph node metastasis, respectively (56,57). A possible scenario is that increased expression of 53BP1 per se promotes genome instability by compromising the ability of HR to overcome replication stress in rapidly cycling tumor cells. Consistent with this, we found that overexpression of 53BP1 enhanced the accumulation of chromosomal aberrations in wild-type MEFs and led to a mild decrease in colony formation after PARPi treatment (Figure 6). However, PARPi-induced cytotoxicity was more severe in BRCA1-deficient cells relative to wild-type when 53BP1 was overexpressed (compare Figures 3D and 6C). This is presumably because the extent of damage in absolute terms is still relatively low and can thereby be tolerated in wild-type cells. Our data therefore suggest that tumors with functional BRCA1 and elevated RNF168/53BP1 levels may derive only limited benefit from PARPi-based therapy. Nevertheless, 53BP1 overexpression can increase mutagenic NHEJ by suppressing HR in both WT and BRCA1-deficient cells.
We found that the requirements of 53BP1 in mutagenic repair of PARPi-induced DNA damage and physiological NHEJ during CSR are dissimilar (Figure 7D). As it has been proposed that the anti-resection function of 53BP1 is critical for CSR (28–29,31), we reasoned that further inhibiting end resection with exogenously overexpressed 53BP1 might lead to enhanced CSR. To our surprise, however, neither RNF168 nor 53BP1 had any significant impact on CSR efficiency when overexpressed. Therefore, the expression levels of RNF168 or 53BP1 proteins are clearly more than sufficient to handle all AID-generated breaks under physiological conditions. Since the levels of RNF168 render ubiquitin signaling rate-limiting when the number of DSBs exceeds 20–40 (37), one possible explanation for these findings is that PARPi-induced DSBs in BRCA1-deficient cells may far exceed the number of DSBs generated by AID during CSR. The increased DSBs in PARPi-treated cells, in contrast to the fewer number of CSR-associated breaks could saturate histone ubiquitylation. To date, the only known limiting factor for CSR is AID itself (58), which when overexpressed causes supra-physiological accumulation of cleavage events at Ig switch regions, additional substrates for 53BP1-dependent NHEJ and increased CSR. Taken together, these data showed that the anti-resection activity of 53BP1 is limiting during mutagenic but not physiological NHEJ.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank members of Nussenzweig laboratory for helpful discussions.
FUNDING
Intramural Research Program of the National Institutes of Health, the National Cancer Institute, the Center for Cancer Research; Department of Defense grant [BCRP DOD Idea Expansion Award, 11557134 to A.N.]; Novo Nordisk Foundation [NNF14CC0001 to J.L. and C.L.]. Funding for open access charge: NIH Intramural Research Program.
Conflict of interest statement. None declared.
REFERENCES
- 1.Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R. 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y., et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 2.Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D.H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J., et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 3.Huen M.S., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolas N.K., Chapman J.R., Nakada S., Ylanko J., Chahwan R., Sweeney F.D., Panier S., Mendez M., Wildenhain J., Thomson T.M., et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mailand N., Bekker-Jensen S., Faustrup H., Melander F., Bartek J., Lukas C., Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 6.Stewart G.S., Panier S., Townsend K., Al-Hakim A.K., Kolas N.K., Miller E.S., Nakada S., Ylanko J., Olivarius S., Mendez M., et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 7.Fradet-Turcotte A., Canny M.D., Escribano-Diaz C., Orthwein A., Leung C.C., Huang H., Landry M.C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F., et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatti M., Pinato S., Maspero E., Soffientini P., Polo S., Penengo L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle. 2012;11:2538–2544. doi: 10.4161/cc.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mattiroli F., Vissers J.H., van Dijk W.J., Ikpa P., Citterio E., Vermeulen W., Marteijn J.A., Sixma T.K. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150:1182–1195. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L., et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callen E., Di Virgilio M., Kruhlak M.J., Nieto-Soler M., Wong N., Chen H.T., Faryabi R.B., Polato F., Santos M., Starnes L.M., et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–1280. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman J.R., Barral P., Vannier J.B., Borel V., Steger M., Tomas-Loba A., Sartori A.A., Adams I.R., Batista F.D., Boulton S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman J.R., Sossick A.J., Boulton S.J., Jackson S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012;125:3529–3534. doi: 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A.D., Feldhahn N., Resch W., Oliveira T.Y., Chait B.T., et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Escribano-Diaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkac J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Feng L., Fong K.W., Wang J., Wang W., Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013;288:11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakarougkas A., Ismail A., Katsuki Y., Freire R., Shibata A., Jeggo P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013;41:10298–10311. doi: 10.1093/nar/gkt802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C., et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 20.Shen S.X., Weaver Z., Xu X., Li C., Weinstein M., Chen L., Guan X.Y., Ried T., Deng C.X. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17:3115–3124. doi: 10.1038/sj.onc.1202243. [DOI] [PubMed] [Google Scholar]
- 21.Honjo T., Kinoshita K., Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu. Rev. Immunol. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 22.Matthews A.J., Zheng S., DiMenna L.J., Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv. Immunol. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen S., Casellas R., Reina-San-Martin B., Chen H.T., Difilippantonio M.J., Wilson P.C., Hanitsch L., Celeste A., Muramatsu M., Pilch D.R., et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniel J.A., Nussenzweig A. The AID-induced DNA damage response in chromatin. Mol. Cell. 2013;50:309–321. doi: 10.1016/j.molcel.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rada C., Williams G.T., Nilsen H., Barnes D.E., Lindahl T., Neuberger M.S. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 26.Manis J.P., Morales J.C., Xia Z., Kutok J.L., Alt F.W., Carpenter P.B. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 27.Ward I.M., Reina-San-Martin B., Olaru A., Minn K., Tamada K., Lau J.S., Cascalho M., Chen L., Nussenzweig A., Livak F., et al. 53BP1 is required for class switch recombination. J. Cell Biol. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bothmer A., Robbiani D.F., Di Virgilio M., Bunting S.F., Klein I.A., Feldhahn N., Barlow J., Chen H.T., Bosque D., Callen E., et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bothmer A., Robbiani D.F., Feldhahn N., Gazumyan A., Nussenzweig A., Nussenzweig M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bothmer A., Rommel P.C., Gazumyan A., Polato F., Reczek C.R., Muellenbeck M.F., Schaetzlein S., Edelmann W., Chen P.L., Brosh R.M. Jr, et al. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J. Exp. Med. 2013;210:115–123. doi: 10.1084/jem.20121975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reina-San-Martin B., Chen J., Nussenzweig A., Nussenzweig M.C. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1-/- B cells. Eur. J. Immunol. 2007;37:235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- 32.Munoz M.C., Laulier C., Gunn A., Cheng A., Robbiani D.F., Nussenzweig A., Stark J.M. RING finger nuclear factor RNF168 is important for defects in homologous recombination caused by loss of the breast cancer susceptibility factor BRCA1. J. Biol. Chem. 2012;287:40618–40628. doi: 10.1074/jbc.M112.410951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munoz M.C., Yanez D.A., Stark J.M. An RNF168 fragment defective for focal accumulation at DNA damage is proficient for inhibition of homologous recombination in BRCA1 deficient cells. Nucleic Acids Res. 2014;12:7720–7733. doi: 10.1093/nar/gku421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakada S., Yonamine R.M., Matsuo K. RNF8 regulates assembly of RAD51 at DNA double-strand breaks in the absence of BRCA1 and 53BP1. Cancer Res. 2012;72:4974–4983. doi: 10.1158/0008-5472.CAN-12-1057. [DOI] [PubMed] [Google Scholar]
- 35.Bohgaki T., Bohgaki M., Cardoso R., Panier S., Zeegers D., Li L., Stewart G.S., Sanchez O., Hande M.P., Durocher D., et al. Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the RIDDLE syndrome. PLoS Genet. 2011;7:e1001381. doi: 10.1371/journal.pgen.1001381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramachandran S., Chahwan R., Nepal R.M., Frieder D., Panier S., Roa S., Zaheen A., Durocher D., Scharff M.D., Martin A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2010;107:809–814. doi: 10.1073/pnas.0913790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gudjonsson T., Altmeyer M., Savic V., Toledo L., Dinant C., Grofte M., Bartkova J., Poulsen M., Oka Y., Bekker-Jensen S., et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 2012;150:697–709. doi: 10.1016/j.cell.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 38.Santos M.A., Huen M.S., Jankovic M., Chen H.T., Lopez-Contreras A.J., Klein I.A., Wong N., Barbancho J.L., Fernandez-Capetillo O., Nussenzweig M.C., et al. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J. Exp. Med. 2010;207:973–981. doi: 10.1084/jem.20092308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie A., Hartlerode A., Stucki M., Odate S., Puget N., Kwok A., Nagaraju G., Yan C., Alt F.W., Chen J., et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell. 2007;28:1045–1057. doi: 10.1016/j.molcel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cruz-Garcia A., Lopez-Saavedra A., Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014;9:451–459. doi: 10.1016/j.celrep.2014.08.076. [DOI] [PubMed] [Google Scholar]
- 46.Kousholt A.N., Fugger K., Hoffmann S., Larsen B.D., Menzel T., Sartori A.A., Sorensen C.S. CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J. Cell Biol. 2012;197:869–876. doi: 10.1083/jcb.201111065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shiotani B., Nguyen H.D., Hakansson P., Marechal A., Tse A., Tahara H., Zou L. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 2013;3:1651–1662. doi: 10.1016/j.celrep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Difilippantonio S., Gapud E., Wong N., Huang C.Y., Mahowald G., Chen H.T., Kruhlak M.J., Callen E., Livak F., Nussenzweig M.C., et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daley J.M., Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 2014;34:1380–1388. doi: 10.1128/MCB.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell. Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 51.Zimmermann M., de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014;24:108–117. doi: 10.1016/j.tcb.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao L., Xu X., Bunting S.F., Liu J., Wang R.H., Cao L.L., Wu J.J., Peng T.N., Chen J., Nussenzweig A., et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouwman P., Aly A., Escandell J.M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q., et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grotsky D.A., Gonzalez-Suarez I., Novell A., Neumann M.A., Yaddanapudi S.C., Croke M., Martinez-Alonso M., Redwood A.B., Ortega-Martinez S., Feng Z., et al. BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. J. Cell Biol. 2013;200:187–202. doi: 10.1083/jcb.201204053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaspers J.E., Kersbergen A., Boon U., Sol W., van Deemter L., Zander S.A., Drost R., Wientjens E., Ji J., Aly A., et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacot W., Thezenas S., Senal R., Viglianti C., Laberenne A.C., Lopez-Crapez E., Bibeau F., Bleuse J.P., Romieu G., Lamy P.J. BRCA1 promoter hypermethylation, 53BP1 protein expression and PARP-1 activity as biomarkers of DNA repair deficit in breast cancer. BMC Cancer. 2013;13:523. doi: 10.1186/1471-2407-13-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pennington K.P., Wickramanayake A., Norquist B.M., Pennil C.C., Garcia R.L., Agnew K.J., Taniguchi T., Welcsh P., Swisher E.M. 53BP1 expression in sporadic and inherited ovarian carcinoma: Relationship to genetic status and clinical outcomes. Gynecol. Oncol. 2013;128:493–499. doi: 10.1016/j.ygyno.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robbiani D.F., Bunting S., Feldhahn N., Bothmer A., Camps J., Deroubaix S., McBride K.M., Klein I.A., Stone G., Eisenreich T.R., et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol. Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.