


Abstract
Nucleoli are not only organelles that produce ribosomal subunits. They are also overarching sensors of different stress conditions and they control specific nucleolar stress pathways leading to stabilization of p53. During DNA replication, ATR and its activator TopBP1 initiate DNA damage response upon DNA damage and replication stress. We found that a basal level of TopBP1 protein associates with ribosomal DNA repeat. When upregulated, TopBP1 concentrates at the ribosomal chromatin and initiates segregation of nucleolar components—the hallmark of nucleolar stress response. TopBP1-induced nucleolar segregation is coupled to shut-down of ribosomal RNA transcription in an ATR-dependent manner. Nucleolar segregation induced by TopBP1 leads to a moderate elevation of p53 protein levels and to localization of activated p53 to nucleolar caps containing TopBP1, UBF and RNA polymerase I. Our findings demonstrate that TopBP1 and ATR are able to inhibit the synthesis of rRNA and to activate nucleolar stress pathway; yet the p53-mediated cell cycle arrest is thwarted in cells expressing high levels of TopBP1. We suggest that inhibition of rRNA transcription by different stress regulators is a general mechanism for cells to initiate nucleolar stress pathway.
INTRODUCTION
Preserving the integrity of DNA is vitally important to the cell. DNA needs to be constantly repaired and maintained due to breaks and structural modifications that happen especially during replication and transcription. Master regulators constantly monitor the amount of damage and are ready to initiate a checkpoint response if the threshold level of damage is exceeded.
One of the most important regulators of DNA damage response is the p53 tumour suppressor. p53 responds also to many different stress conditions that are not limited to DNA damage. Normally low p53 levels are maintained by mechanisms that are regulated by nucleoli (1). A great deal of a dividing cell's resources are put into nucleoli, where ribosomal RNA (rRNA) transcription by RNA polymerase I (RNA pol I), processing of the rRNA and ribosome assembly take place. The unique position of nucleoli within the cellular metabolism is exploited to initiate nucleolar stress response that culminates in reorganization of nucleolar structure and in stabilization and activation of p53 (2,3). Accumulating evidence indicates that an important mechanism for initiating p53 response is mediated by inhibition of ribosomal biogenesis (1,3).
Inhibition of rRNA transcription is also of clinical interest as a potential drug target against cancer (4). RNA pol I inhibitors have been used effectively to activate nucleolar stress response that results in reorganization of nucleolar components and p53-mediated cancer cell killing (5,6). Actinomycin D (ActD) is one of the oldest antineoplastic drugs still in clinical use. When used at low levels (nanomolar range), ActD binds to GC-rich sequences in the ribosomal DNA (rDNA) and specifically inhibits the RNA pol I, leading to nucleolar segregation and activation of p53 (7,8).
Problems during DNA replication result in stalled forks and initiate DNA stress response coordinated by Ataxia telangiectasia and Rad3-related (ATR) kinase (9). ATR ensures accurate replication timing in each S-phase and initiates cell cycle arrest if the threshold damage level is reached. Although ATR is activated by a broad spectrum of DNA lesions, the common activating structure seems to be single-stranded DNA (ssDNA). ATR is rapidly recruited to lesions by Replication protein A (RPA) that is bound to ssDNA. However, ATR recruitment alone is not sufficient for checkpoint activation but requires independent localization of TopBP1 to the damage site.
TopBP1 is a crucial factor in the ATR response and it appears that association of TopBP1 to chromatin is the key step in regulation of ATR activation (10–13). Even though TopBP1 can bind directly to damaged DNA, at least in vitro (13), its interaction with the Rad17 clamp loader and checkpoint clamp complex Rad9-Hus1-Rad1 (9–1–1) is critical for the checkpoint signalling at the junction between ssDNA and double-stranded DNA (14–17). By physically binding to the ATR partner protein ATRIP, TopBP1 significantly enhances ATR kinase activity (18). Ectopic expression of ATR-activation domain (AAD) of TopBP1 activates ATR in the absence of DNA damage and leads to cell cycle arrest and, if persistent, to p53-dependent senescence (19). TopBP1 is also directly involved in transcriptional regulation, and can inhibit apoptosis and p53-mediated G1 arrest by repressing E2F1 and p53 target gene expression (20–22). These findings suggest that TopBP1 can function as an activator or suppressor to balance the DNA damage response (for a review on functions of TopBP1, see (23)).
In order to better understand the function of TopBP1, we used cells designed to conditionally express eGFP-TopBP1 fusion protein. Here we report that, when present at elevated levels, TopBP1 concentrates into nucleoli, associates with transcribed region of rDNA repeat and locally activates ATR. This leads to inhibition of rRNA synthesis and concomitant nucleolar segregation, similar to that induced by ActD. However, while ActD treatment leads to transient p53-dependent cell cycle arrest lasting for several days, TopBP1-induced nucleolar segregation leads only to a modest increase in p53 levels that does not initiate cell cycle arrest. Our findings show that rRNA synthesis can be modulated by the ATR pathway and add ATR to the list of stress regulators that inhibit rRNA transcription once activated.
MATERIALS AND METHODS
Cell culture
U2OS (ATCC) cells were cultivated at 37°C and 5% CO2 in modified McCoy's 5a medium (Sigma) including 10% foetal bovine serum (FBS). Tet-On cell lines were cultivated at 37°C at 5% CO2 in modified McCoy's 5a medium (Sigma) supplemented with 10% FBS, 100 μg/ml of hygromycin and 200 μg/ml of G418 as selective antibiotics.
ATR inhibitor ETP-46464 (Millipore) and DNA intercalator ActD (Sigma) were applied directly to the plates at final concentrations of 10 μM and 30 nM, respectively. Upon release of cells from ActD they were either prepared for analysis or washed with Phosphate buffered saline (PBS) followed by addition of fresh media for further cultivation.
DNA constructs and generation of Tet-On cell lines
Previously cloned (14) human full-length TopBP1 coding sequence (CDS, Uniprot Q92547) was ligated into pEGFP-C1 mammalian expression vector (BD Biosciences, Genbank #U55763). Mutated TopBP1 in pEGFP-C1 was generated by introducing a tryptophan (W) to arginine (R) mutation at amino acid 1145 of TopBP1 using overlap extension polymerase chain reaction (PCR). For the preparation of stable Tet-On Advanced cell lines, the whole CDS of eGFP-TopBP1 WT or eGFP-TopBP1 W1145R was ligated to the pTRE-Tight vector (Clontech). TopBP1 deletion mutants were prepared and ligated into pEGFP-C1 vector using In-Fusion HD EcoDry Cloning Kit (Clontech). The correct sequences of all constructs were verified by sequencing (ABI Prism 310 Genetic Analyzer). DNA transfections were done with Effectene (Qiagen) transfection reagent.
Stable doxycycline-inducible U2OS cell lines were generated using Tet-On Advanced gene expression system (Clontech). Cells were designed to express either wild-type (WT) or W1145R mutant of human TopBP1 N-terminally fused to Enhanced Green Fluorescent Protein (eGFP).
Fluorescence microscopy
For fluorescence microscopy, cells grown on borosilicate glass coverslips were washed with PBS and immediately fixed with 3% paraformaldehyde. Cells were permeabilized with 0.2% Triton X-100-PBS and immunostained or directly stained for DNA with Hoechst 33258 and mounted on glass slides with Shandon Immu-Mount (Thermo Scientific). Primary antibodies were from Abcam (α-NCL, 4E2), Millipore (α-TopBP1, AB3245), Sigma (α-BrdU, BU-33), Life Technologies (α-NPM; FC-61991), LifeSpan Biosciences (α-Hus1; LS-C308354) and Santa Cruz Biotechnology (α-p53, DO-1; α-RPA194, C-1; α-UBF, F-9). Mouse monoclonal antibody against Rad9 was a kind gift from Dr Raimundo Freire (Universitario de Canarias, Tenerife, Spain). Secondary antibodies were Alexa Fluor 488 or 594 conjugated, highly cross-adsorbed goat α-rabbit or α-mouse IgG (Molecular Probes).
For nascent RNA transcription labelling, cells were given a 30-min pulse of 2 mM 5′-fluorouridine (FUrd, Sigma) before fixing them. Incorporated FUrd was detected with α-BrdU antibody, BU-33 (Sigma).
To label nascent DNA synthesis, cells were given a 30-min pulse of 5-ethynyl-2′-deoxyuridine (EdU). Incorporated EdU was detected with Click-iT EdU Alexa Fluor 594 Imaging Kit (Life Technologies).
Wide-field fluorescent images were obtained with Axiocam HR colour using a Zeiss Axioplan 2 microscope with either 40× Zeiss Plan-Neofluar or 63× Zeiss Plan-Apochromat objective. Image acquisition was done with Axiovision AxioVS40 software (v4.8.2). Confocal fluorescent images were obtained with Zeiss Axio Observer inverted microscope (40× objective) equipped with Zeiss LSM700 confocal module (Carl Zeiss Microimaging GmbH, Jena, Germany). Image acquisition was done with ZEN 2012 software (Carl Zeiss Microimaging GmbH). All images were processed with Photoshop CS5 (12.0).
Quantitative reverse-transcriptase PCR (qRT-PCR)
Cells were lysed in Trizol reagent (Invitrogen) and RNA was extracted according to instructions. Purified RNA was treated with RQ1 DNase (Promega) to remove any DNA contamination. Reverse transcription and subsequent quantitative PCR were performed with DyNAmo SYBR Green 2-Step qRT-PCR Kit (Finnzymes/Thermo scientific) according to instructions. Quantitative PCR reactions were run with the Opticon Monitor program in Chromo 4 Peltier Thermal Cycler (MJ research) and cycle program of 95°C for 15 min followed by 40 cycles with 94°C for 10 s, 57°C for 30 s and 72°C for 30 s. Pre-rRNA signals were normalized to ACTB (β-actin) signals. Primers targeting pre-rRNA 5′ external transcribed region were: FWD 5′ GGAAGGAGGTGGGTGGAC 3′ and REV 5′ GCGGTACGAGGAAACACCT 3′. Primers targeting ACTB were: FWD 5′ CTCTTCCAGCCTTCCTTCCT 3′ and REV 5′ AGCACTGTGTTGGCGTACAG 3′.
Immunoelectron microscopy (immuno-EM)
Cells were fixed with 4% paraformaldehyde in 0.1M phosphate buffer pH 7.5 for 10 min, scraped off from the plate and fixed for 1 h. Fixed cells were rinsed with PBS and mixed with 12% gelatine in PBS. After 10 min incubation at room temperature cells were centrifuged 5 min at 16 000 × g. Gelatine was solidified on ice for 30 min, after which the pellet was cut into small pieces and put into 2.3 M sucrose in PBS and rotated for 4 h at +4°C. The specimens were frozen in liquid nitrogen and thin cryosections were cut with Leica EM UC7 cryoultramicrotome (Leica Microsystems, Vienna, Austria). The sections were picked on Butvar-coated nickel grids. The grids were first incubated in 2% gelatine in PBS for 20 min and then in 0.1% glycine-PBS for 10 min. Incubation in a blocking serum containing 1% bovine serum albumin (BSA) in PBS for 5 min. One percent BSA in PBS was used in washings and dilutions of antibodies and gold conjugates. Sections were exposed to the first primary antibody for 45 min followed by incubation with protein A–gold (5 nm) for 30 min (24). After washings, 1% glutaraldehyde in 0.1 M phosphate buffer pH 7.5 was used to block free binding sites on protein A. The sections were then incubated with the second primary antibody for 45 min followed by rabbit anti-mouse IgG for 30 min (Jackson Immunoresearch Laboratories) and then incubated in the protein A–gold (10 nm) for 30 min. The controls were prepared by replacing the primary antibody with PBS. The grids were stained with neutral uranyl acetate (UA) and embedded in 2% methyl cellulose containing 0.4% UA and examined with a Tecnai Spirit transmission electron microscope (FEI, Eindhoven, The Netherlands). Images were captured by a Quemesa CCD camera (Olympus Soft Imaging Solutions GMBH, Munster, Germany). TopBP1 was detected by α-TopBP1.2 or α-TopBP1 AB3245 (raised against different regions of TopBP1), both of which gave identical staining patterns. Other antibodies were as for immunofluorescence microscopy.
Immunoblotting and subcellular fractionation
To prepare whole-cell extract for immunoblotting, cells were washed with PBS, scraped off from the plate and suspended in lysis buffer (50 mM Tris–HCl pH 7.9, 10% Glycerol, 5 mM EDTA, 100 mM NaCl, 0.5% IGEPAL CA-630, 1 mM DTT, 1 mM NaF, 1 mM Na3VO4 and 1× Complete protease inhibitor cocktail (Roche)), briefly sonicated and finally cleared by centrifugation at 16 000 × g for 10 min. Protein concentration was determined with Bradford assay.
For Immunoblotting, 10–50 μg of total protein were separated on sodium dodecyl sulphate-polyacrylamide gel electrophoresis, transferred onto a PVDF membrane and detected using chemiluminescence (SuperSignal West Pico, Thermo Scientific), and exposed on Amersham Hyperfilm ECL (GE Life Sciences).
To prepare subcellular fractions, whole cells were washed with hypotonic buffer (10 mM MOPS-NaOH pH 7.0, 10 mM NaCl, 1 mM MgCl2, 2 mM DTT) and lysed in the same buffer supplemented with 0.5% IGEPAL CA-630 for 10–30 min to give detergent-soluble fraction (S). Chromatin-bound proteins were released by incubating for 15 min in high-salt buffer (20 mM HEPES-KOH pH 7.4, 500 mM NaCl, 5 mM KCl, 0.5 mM DTT and 1× Complete protease inhibitor cocktail (Roche)) (fraction B). Remaining cell material was solubilized in SDS-lysis buffer (20 mM HEPES-KOH pH 7.4, 0.5% SDS, 100 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, 0.5 mM DTT) (fraction M).
Commercial primary antibodies were from Cell Signalling (α-ATR, #2790; α-Chk1 S345, 133D3; α-GFP, D5.1, α-p53 S15, 16G8), Millipore (α-β-Tubulin, KMX-1; α-p21, #05–345) and Santa Cruz Biotechnology (α-Chk1, G-4; α-p53, FL-393, C-1; α-UBF, F-9; α-Lamin A/C, N-18). Rabbit polyclonal antibody against TopBP1.2 for immunoblots was from our own laboratory (14). Secondary antibodies for immunoblotting were peroxidase conjugated goat α-rabbit or α-mouse IgG (Jackson Immunoresearch Laboratories). Secondary antibodies for immunofluorescence were Alexa Fluor 488 or 594 conjugated, highly cross-adsorbed goat α-rabbit or α-mouse IgG (Molecular Probes).
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) assay was performed as described (25). In brief, cells were fixed with 1% formalin, lysed in 150 mM NaCl, 50 mM Tris–HCl pH 7.5, 5 mM EDTA, 0.5% IGEPAL CA-630, 1% TX-100, 1× Roche Complete EDTA-free protease inhibitor cocktail, 1 mM NaF, 1 mM Na3VO4. Resulting nuclei were sonicated with a Hielscher UP200S at 100% amplitude for eight rounds of 15 × 0.5 s pulses to obtain 200–1000 bp sheared DNA. Antibodies used in immunoprecipitations were from Millipore (α-TopBP1 AB3245) and Santa Cruz Biotechnology (α-RNA pol I/α-RPA194, C-1). Immunocomplexes were collected with 30 μl of protein G Magnetic beads (Dynabeads, Life Technologies). DNA was extracted from the beads with 10% Chelex-resin (Bio-Rad). Input DNA sample was treated with proteinase K (Invitrogen), ethanol-precipitated and processed the same way as bead-immunoprecipitates. Real-time PCR was used to quantitate the immunoprecipitated DNA and normalized to input dilution series. Protein G bead-only precipitations were performed to check for the background level of DNA enrichment. The DNA enrichment from these controls was <0.04% of the input samples (see Supplementary Figure S5). Primers for ribosomal DNA were from (26). Quantitative PCR reactions were run with the Opticon Monitor program in Chromo 4 Peltier Thermal Cycler (MJ research) using DyNAmo SYBR Green mix (Finnzymes) and cycle program of 95°C for 2 min followed by 40 cycles with 95°C for 30 s, 58°C for 30 s and 72°C for 1 min, for all primers.
RNA interference
Silencer Select siRNA (Ambion) targeting ATR (Validated s536) and unspecific negative control (Negative Control #1) were transfected using HiPerfect transfection reagent (Qiagen). A total of 10 nM ATR siRNA was transfected three times at 24 h intervals.
Automated image analysis
CellProfiler version 2.2.1 (27) was used for automated counting of p53-positive and eGFP-TopBP1 positive cells. α-p53, FL-393 (Santa Cruz Biotechnology) antibody was used for immunostaining as described above. Over 600 cells were counted from each sample.
RESULTS
Elevated levels of TopBP1 induce nucleolar segregation and shut-down of nucleolar transcription
In order to study TopBP1 in cultured human cells we cloned a full-length TopBP1 coding sequence into a mammalian eGFP expression vector and prepared a stable U2OS cell line where expression of eGFP-TopBP1 can be induced by adding doxycycline in the culture medium. This cell line expressed the protein as expected, with no traces of expression observed when doxycycline was omitted (Supplementary Figure S1A). To study how ectopic expression of eGFP-TopBP1 might add to the total concentration of ToPBP1 in cells, immunoblots of extracts from cells expressing eGFP-TopBP1 were probed with an anti-TopBP1 antibody to detect both endogenous and ectopic TopBP1. Due to the appearance of ectopic TopBP1, the total concentration of TopBP1 increased 4–6 fold at 12–48 h after induction (Supplementary Figure S1B).
When cells expressing eGFP-TopBP1 were examined under fluorescent microscope, strong localization of eGFP-TopBP1 into nuclear foci was observed. More close inspection suggested that the foci were associated with the nucleoli, which were identified by weak staining of DNA with Hoechst 33258. We then looked at the localization of nucleolar marker proteins Upstream binding factor (UBF), RNA Pol I, Nucleolin (NCL) and Nucleophosmin (NPM). To our surprise, we found changes in localization of all these marker proteins upon induction of eGFP-TopBP1 (Figure 1A). UBF and RNA Pol I both co-localized extensively with the eGFP-TopBP1 foci (Figure 1A, middle panels), while in non-induced cells they localized in necklace-like structures (Figure 1A, top panels). NPM and NCL signals in the nucleoplasm were increased and both proteins formed ring-like structures around the nucleolar body in cells expressing eGFP-TopBP1, while in non-induced cells they were concentrated in the nucleoli. The relocalization of nucleolar proteins in cells expressing eGFP-TopBP1 bring to mind nucleolar segregation that is induced by inhibition of rRNA transcription after ultraviolet light irradiation or ActD treatment. Indeed, the similarity in localization of nucleolar marker proteins between eGFP-TopBP1 expressing cells and ActD-treated cells was apparent (Figure 1A, middle and bottom panels). ActD binds to the DNA duplex and inhibits specifically RNA Pol I, when used in nanomolar concentrations. Higher concentrations of ActD inhibit also RNA polymerase II. To confirm this specific inhibition of RNA pol I, we gave cells a short pulse of ribonucleotide analogue 5′-fluorouridine (FUrd) that incorporates into nascent RNA. As expected, cells treated with 30 nM ActD lost FUrd signal in nucleoli, while transcription elsewhere was visible as dispersed foci throughout the nucleoplasm (Figure 1A). Most cells displaying eGFP-TopBP1-induced segregation were devoid of nucleolar transcription (Figure 1A and Supplementary Figure S2), while transcription elsewhere in nucleoplasm looked normal, resembling also in this respect ActD-treated cells. We then further studied transcription of ribosomal RNA genes by measuring pre-ribosomal rRNA (pre-rRNA) with quantitative reverse-transcriptase PCR (qRT-PCR) assay. Upon induction of eGFP-TopBP1 expression, the pre-rRNA synthesis level dropped gradually from 100% at 1 h down to 31% at 24 h (Figure 1B). This pattern follows the microscopically observed accumulation of eGFP-TopBP1 protein levels, which appeared earliest at around 6 h and was saturated at 24 h after the induction (not shown). In cells treated with 30 nM ActD for 30 min the level of pre-rRNA was 58% compared to that of non-treated cells.
Figure 1.
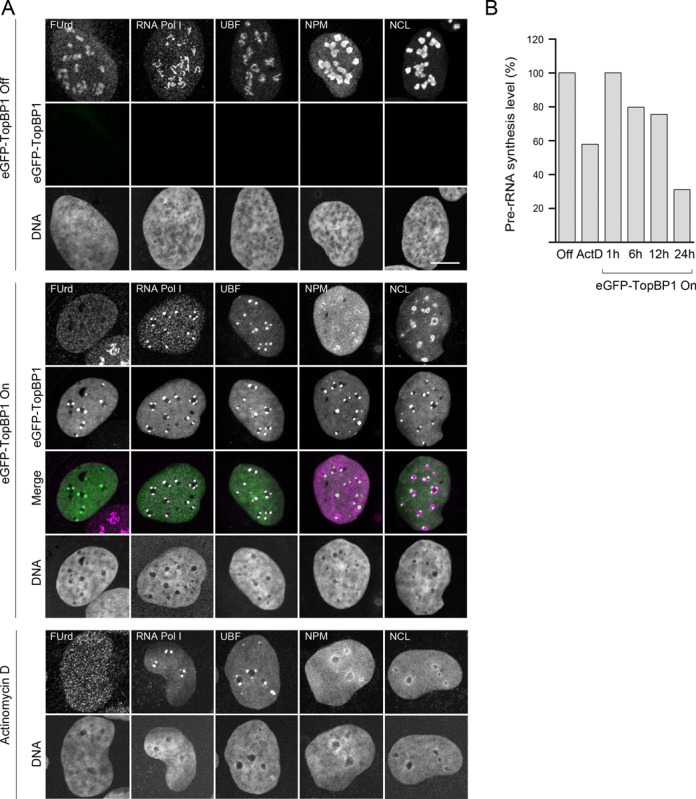
Expression of ectopic TopBP1 induces shut-down of rRNA transcription and nucleolar segregation. (A) Expression of eGFP-TopBP1 was left non-induced (eGFP-TopBP1 Off), induced for 24 h (eGFP-TopBP1 On), or cells were treated with ActD. Nascent transcripts were labelled with a short pulse of FUrd. Confocal images of FUrd, endogenous RNA Pol I, UBF, NPM (Nucleophosmin) and NCL (Nucleolin) and eGFP-TopBP1 are shown. Merge shows an overlay of panels (DNA excluded). Scale bar is 10 μm. (B) Pre-rRNA synthesis levels were determined with qRT-PCR from eGFP-TopBP1 cells left non-induced (Off), induced for the indicated times or treated with ActD. Percentages of pre-rRNA levels normalized to ACTB transcription are shown relative to non-induced cells.
Next we looked the nucleoli at the ultrastructural level using immuno-EM. In non-induced cells nucleoli were visible as strongly contrasted, round regions in the nuclei (Figure 2A). Interestingly, we found endogenous TopBP1 co-localizing with RNA Pol I, UBF and NCL inside the nucleoli in non-induced cells (Figure 2B, upper panels).The levels of endogenous TopBP1 staining by immuno-EM in the nucleolus appeared even to exceed the general staining in the nucleus. When induced to express eGFP-TopBP1, we found TopBP1 concentrating in regions that were closely associated with the nucleolus, but which appeared distinct from the more electron-dense body of nucleolus (Figure 2A and B, lower panels). These regions obviously correspond to the nucleolar TopBP1 foci visible in fluorescence microscopy.
Figure 2.

Both endogenous and ectopic TopBP1 display nucleolar localization. eGFP-TopBP1 was left non-induced (Off) or induced (On) for 24 h before fixing the cells for immunoelectron microscopy. TopBP1 was immunostained with 10 nm, and RNA pol I, UBF and NCL with 5 nm gold particles. (A) Examples of nucleoli in a non-induced and an induced cell at lower magnification. (B) Higher magnification of nucleoli in non-induced and induced cells. Endogenous TopBP1 partly co-localizes with RNA Pol I, UBF and NCL in the electron-dense regions of the nucleolus, but in induced cells it strongly concentrates in distinct regions immediately adjacent to main body of nucleolus (arrowheads). Scale bars are 100, 500 or 1000 nm, as indicated.
To show that the localization phenotype was caused by TopBP1 and not affected by the eGFP moiety, we transiently transfected cells with a TopBP1 construct missing the eGFP tag. The localization of TopBP1 in these cells was identical to that of eGFP-TopBP1 (Supplementary Figure S3A). Furthermore, the TopBP1 phenotype was not a specific feature of U2OS cells, since we observed identical phenotype in cancer cell lines HeLa, MG63 and SaOS2, and also in human IMR90 primary fibroblasts, when they were transiently transfected with eGFP-TopBP1 (Supplementary Figure S3B). Finally, we tested if the phenotype caused by abnormally high concentration of TopBP1 might be caused also by other DNA-binding proteins. Therefore, we expressed Venus-RecQL4 in U2OS cells to obtain abnormally high cellular concentration of RecQL4, which is a DNA helicase that localizes to nucleoli (28). As expected Venus-RecQL4 was enriched in nucleoli. However, it did not induce nucleolar segregation, providing additional evidence on a specific role of TopBP1 in nucleolar segregation (Supplementary Figure S3C).
The results of the experiments presented above show that elevated levels of TopBP1 lead to inhibition of rRNA transcription and to a reorganization of nucleolar components. This TopBP1-induced nucleolar segregation is similar to that seen in ActD-treated cells.
To assess biological relevance of the results obtained with cells expressing high levels of ectopic TopBP1, we studied whether TopBP1–RNA pol I foci could be detected also in cells expressing endogenous levels of TopBP1. First, we found that ActD-induced nucleolar segregation was independent of TopBP1, since it took place normally in TopBP1-depleted U2OS cells (Supplementary Figure S4). However, endogenous TopBP1 co-localized with RNA pol I into characteristic foci even in non-treated cells, with 12% of the cells having at least one co-localization focus (Figure 3). In a typical case there were no more than one such focus per cell, and these foci were larger and more solitary than the necklace-like foci formed by RNA pol I. When cells were treated with ActD, the proportion of cells that showed co-localization of TopBP1 and RNA pol I increased to 24%. Therefore, we conclude that the foci containing TopBP1 and RNA pol I, and nucleolar segregation in cells expressing ectopic TopBP1 are not artefacts, but rather reflect a biologically relevant phenomenon. The phenotype is merely enhanced by the super-physiological concentrations of TopBP1.
Figure 3.
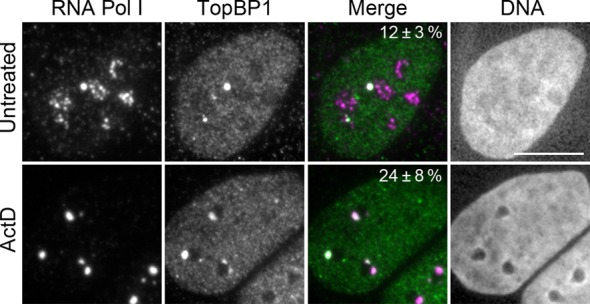
Endogenous TopBP1 co-localizes with RNA pol I. Cells were left non-treated or were treated with ActD. Wide-field images of endogenous TopBP1 and RNA pol I are shown. Percentages (± standard deviation, three independent experiments) show the proportion of cells that showed co-localization of RNA pol I and TopBP1 foci. Over 100 nuclei were counted in each experiment. Scale bar is 10 μm.
TopBP1-induced nucleolar segregation depends on ATR kinase activity
TopBP1 is an essential activator of ATR kinase (29). To study whether ATR is involved in TopBP1-induced nucleolar segregation, we first treated eGFP-TopBP1 cells with the potent ATR inhibitor ETP-46464 (30). Remarkably, treatment of cells with the ATR inhibitor resulted in complete loss of eGFP-TopBP1 foci, resumed the nucleolar RNA transcription and restored normal localization of RNA pol I and NCL (Figure 4A).
Figure 4.
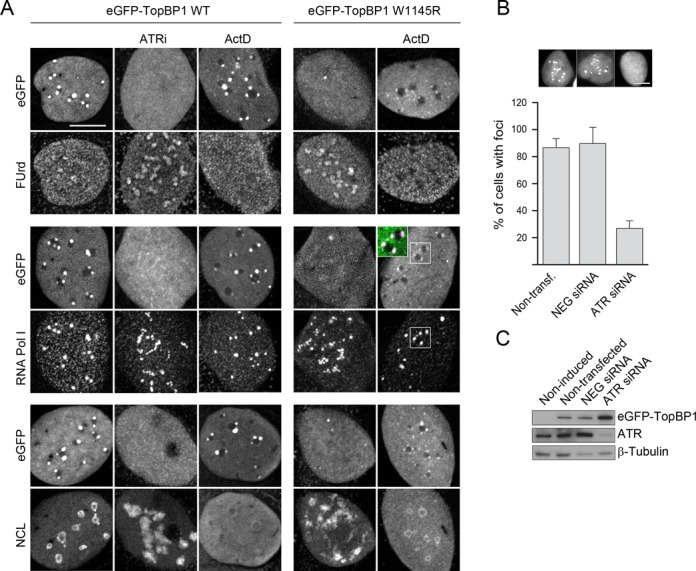
ATR is required for TopBP1-induced nucleolar segregation. (A) Chemical inhibition of ATR and expression of ATR activation mutant of TopBP1 attenuate nucleolar segregation induced by ectopic TopBP1. Expression of eGFP-TopBP1 (wild-type, WT or W1145R mutant) was induced for 15 h alone, or in the presence of ATR inhibitor ETP-46464 (ATRi), or cells expressing eGFP-TopBP1 (WT or W1145R, as indicated) were treated with ActD. Nascent transcripts were labelled with FUrd. Confocal images of FUrd, endogenous RNA Pol I and Nucleolin (NCL) and eGFP-TopBP1 are shown. (B) Downregulation of ATR suppresses nucleolar segregation induced by eGFP-TopBP1. Cells were transfected with ATR siRNA, with unspecific negative siRNA (NEG siRNA) or left non-transfected. Thereafter, expression of eGFP-TopBP1 WT was induced for 24 h prior to fixation, or left non-induced. Percentage of cells with eGFP-TopBP1 foci is shown with representative wide-field microscopy images of eGFP-TopBP1. Results are means of three independent experiments. Over 100 nuclei were counted in each experiment. Standard deviations are shown. Scale bars are 10 μm. (C) Immunoblot of the whole-cell extract shows levels of eGFP-TopBP1 and ATR. β-Tubulin is shown as control of protein loading.
Activation of ATR kinase by TopBP1 can be abrogated by a single tryptophan (W) to arginine (R) point mutation at the amino acid residue 1145 of TopBP1 (29), providing a tool to study the role of TopBP1-dependent activation of ATR kinase in nucleolar segregation. We therefore prepared a U2OS cell line inducible for the expression of this mutated form of TopBP1 (eGFP-TopBP1 W1145R). These cells lacked nucleolus-associated TopBP1 foci, showed normal distribution of RNA pol I and NCL, and nucleolar transcription was undisturbed (Figure 4A, right panels).
Given the apparent similarity between ActD- and TopBP1-induced nucleolar segregation, we wanted to see whether ActD had any additional influence on cells, beyond that of ectopically expressed TopBP1. It turned out that in cells expressing eGFP-TopBP1, ActD had no apparent influence beyond non-expressing cells on the localization of RNA pol I, and eGFP-TopBP1 and RNA pol I continued to co-localize into the characteristic foci also after the ActD treatment (Figure 4A). In cells expressing W1145R-mutated TopBP1, ActD blocked nucleolar transcription as expected, but surprisingly, caused the formation of TopBP1–RNA pol I foci (Figure 4A).
To corroborate that ATR is required for focal localization of eGFP-TopBP1, we depleted cells from ATR using a cognate siRNA. Downregulation of ATR with a validated siRNA resulted in a phenotype similar to that of mutant eGFP-TopBP1 W1145R cells and to cells treated with the ATR inhibitor (Figure 4B). Immunoblotting of whole-cell extracts showed that only residual ATR remained after ATR siRNA treatment of cells, while a non-specific control siRNA did not affect the ATR protein levels (Figure 4C). Statistically, nearly 90% of the non-treated cells and control siRNA-treated eGFP-TopBP1 cells had TopBP1 foci, while ATR siRNA treatment reduced that number below 30% (Figure 4B).
Taken together, the findings above show that ATR is needed for the TopBP1-induced nucleolar segregation and for shut-down of nucleolar transcription.
TopBP1 associates with ribosomal DNA repeat
TopBP1 has been shown to associate with chromatin in vivo (31) and to bind to DNA in vitro (13). To study if eGFP-TopBP1 associates with chromatin we fractionated cells into three different fractions. First, whole cells were extracted with detergent to yield soluble fraction (S) containing cytoplasmic and soluble nuclear proteins. Remaining nuclei were extracted with high-salt to yield a chromatin-bound fraction (B), containing most of the proteins associated with DNA. Thereafter, the nuclei were lysed to yield a matrix fraction (M) containing the non-extractable nuclear proteins and more tightly attached chromatin proteins. Immunoblotting of eGFP-TopBP1 shows that both the mutant and WT proteins associate with chromatin, like the endogenous TopBP1 in normal U2OS cells (Figure 5A). In the same chromatin fraction we found, as expected, UBF. Beta-tubulin was found in the soluble fraction and the nuclear scaffold protein Lamin A/C was found solely in the remaining matrix fraction, as expected. Microscopy of detergent-treated cells revealed that both WT and W1145R TopBP1 most tightly localized into nucleoli (Figure 5B).
Figure 5.
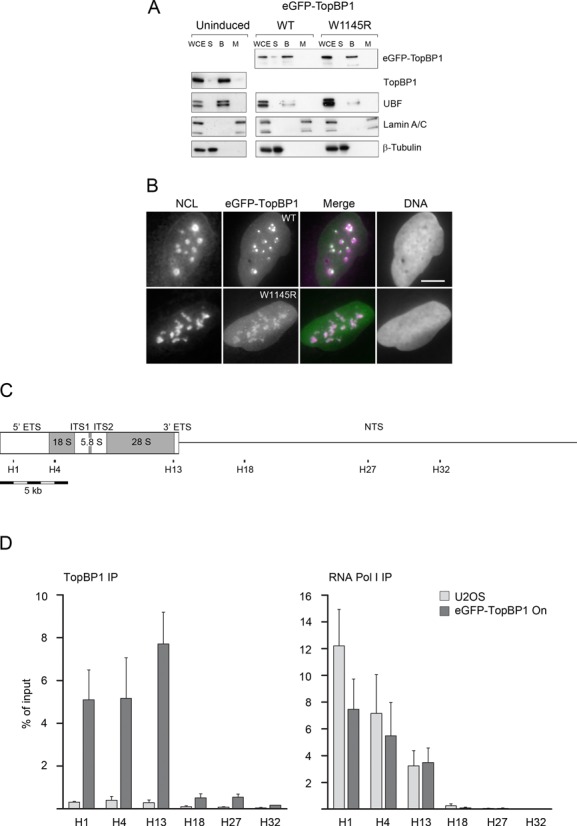
eGFP-TopBP1 associates with chromatin, localizes predominantly in nucleoli in permeabilized cells and binds to the transcribed region of the rDNA repeat. (A) Cells were left non-induced or induced to express eGFP-TopBP1, WT or W1145R, as indicated. Cells were left unfractionated (WCE, whole cell extract) or fractionated in soluble/cytoplasmic (S), chromatin bound (B) or matrix (M) fractions as described in ‘Materials and Methods’ section and subjected to immunoblotting. UBF is shown as a control for protein binding to chromatin, Lamin A/C as a nuclear matrix protein and β-Tubulin as a cytoplasmic protein. (B) Cells induced to express eGFP-TopBP1 WT or W1145R, were treated with detergent before fixing. Wide-field images show endogenous Nucleolin (NCL) or eGFP-TopBP1. Merge shows an overlay of panels (DNA excluded). Scale bar is 10 μm. (C) Schematic presentation of a complete rDNA repeat unit (U13369) that shows transcribed region (boxed area), 5′ and 3′ external transcribed spacers (ETS), internal transcribed spacers (ITS1 and ITS2), the source of 18S, 5.8S and 28S rRNAs, and the non-transcribed spacer (NTS). Positions of primer pairs H1, H4, H13, H18, H27 and H32 at the rRNA gene are shown. All positions and lengths of elements are in scale. (D) Quantification of ChIP with anti-TopBP1 (left) and anti-RNA Pol I (right) IPs in assays using nuclear material from normal U2OS cells or from cells that were induced to express eGFP-TopBP1. DNA was quantitated by qPCR using primer pairs indicated in (C). Quantification is presented as percentages of input material precipitated. Mean values of three independent experiments are shown with standard deviations.
To study if TopBP1 binds directly to ribosomal chromatin in vivo, we performed ChIP with U2OS cells containing endogenous levels of TopBP1, and with cells expressing eGFP-TopBP1. Enrichment of DNA was analysed by quantitative PCR using primer pairs spanning the transcribed and non-transcribed regions of the rDNA repeat (U13369) (Figure 5C). We found moderate enrichment of endogenous TopBP1 at the rDNA repeats indicating that a small amount binds preferentially the transcribed region of rDNA repeat (Figure 5D and Supplementary Figure S5). This is consistent with the localization of endogenous TopBP1 observed by Immuno-EM (see Figure 2). DNA enrichment in the transcribed region was dramatically increased when cells expressed eGFP-TopBP1 (Figure 5D). Mapping of RNA pol I shows, as expected, that it binds the transcribed, but not the non-transcribed portion of the rDNA repeat (Figure 5D).
These results show that eGFP-TopBP1 localizes to nucleolar chromatin and associates with transcribed region of the rDNA repeat. These and the results presented above also suggest that eGFP-TopBP1 is first recruited into the nucleoli and that nucleolar segregation is subsequently initiated by locally activated ATR.
BRCT domains 0–2 and 4–5 are required for TopBP1-induced nucleolar segregation
TopBP1 is a modular protein consisting of nine BRCT (BRCA1 C-terminal) domains (numbered from 0 to 8). The ATR activation domain (AAD) is located between BRCT domains 6 and 7. In order to gain insight into the TopBP1-induced nucleolar segregation, we prepared five mutants of eGFP-TopBP1 with BRCT domains 0–2, 3, 4–5, 6 and 7–8 deleted, respectively (Figure 6A), and examined how mutated forms of TopBP1 localized in respect to endogenous UBF. We found that TopBP1 lacking BRCT domains 0–2 or 4–5 did not induce nucleolar segregation (Figure 6B). Interestingly, BRCT 0–2 and 4–5 have been reported to mediate the interaction between the Rad9 and TopBP1 (14,15,17). Rad9 is part of the checkpoint clamp complex 9–1–1 (Rad9-Rad1-Hus1), which recruits TopBP1 into chromatin to activate ATR (15,17). This prompted us to look for endogenous Rad9 and Hus1 in eGFP-TopBP1 expressing cells. While in non-induced cells both Rad9 and Hus1 had diffusive nuclear localization, they co-localized with eGFP-TopBP1 foci in induced cells (Figure 6C). In ActD-treated cells without ectopic TopBP1, we also found co-localization of RNA pol I and Rad9 in part of the cells, similar to what was observed for TopBP1 in segregated nuclei (Figure 3, see above).
Figure 6.

Deletion of BRCT domains 0–2 or 4–5 abrogates nucleolar segregation induced by TopBP1. (A) Schematic picture of TopBP1 deletion mutant constructs. eGFP, enhanced green fluorescent protein tag; 0–8, BRCT domains 0–8; AAD, ATR activation domain; NLS, nuclear localization signal; FL, full-length. BRCT domains 0–2, 3, 4–5, 6 or 7–8 were deleted in D0–2, D3, D4–5, D6 and D7–8, respectively. All elements are in scale. (B) eGFP-TopBP1 constructs were transiently transfected into U2OS cells. Wide-field images show endogenous UBF or eGFP-TopBP1. (C) Cells were left non-induced, induced to express eGFP-TopBP1 or left non-induced and treated with ActD. Cells were immunostained for endogenous Rad9, Hus1 or RNA pol I. Non-treated and doxycycline panels are confocal images and ActD is a wide-field image. Merge shows an overlay of panels (DNA excluded). Scale bars are 10 μm.
Taken together, BRCT domains 0–2 and 4–5 are critical in TopBP1-induced nucleolar segregation. Co-localization with Rad9 and Hus1 further suggests involvement of 9–1–1 complex in the TopBP1-induced nucleolar segregation.
TopBP1-induced nucleolar segregation does not lead to cell cycle arrest
Inhibition of RNA pol I-catalysed transcription by different chemicals, including ActD, is known to induce nucleolar segregation and stabilization of p53 (5,6,8). We studied the long-term effects of continued expression of eGFP-TopBP1 and compared them to the responses caused by ActD. We treated cells with 30 nM ActD for 2 h and monitored cycling cells over the course of 4 days. A gradual decline of DNA replication was observed, leading to a complete cell cycle arrest on the days 1–3 after the treatment (Figure 7A, top panels). However, these cells recovered DNA synthesis on the day 4.
Figure 7.
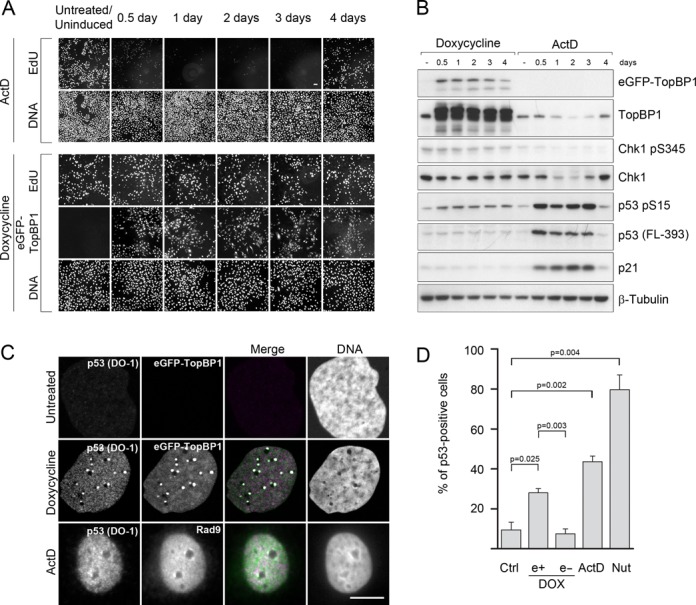
TopBP1-induced nucleolar segregation is not associated with cell cycle arrest. (A) Cells were treated with ActD for 2 h and let recover for indicated times, or induced to express eGFP-TopBP1 for the same times. Nascent DNA was labelled with a short pulse of EdU with subsequent immunolabelling of eGFP-TopBP1 before the cells were fixed and microscoped. Scale bar is 50 μm. (B) Immunoblots of TopBP1, phosphorylated Chk1, total Chk1, phosphorylated p53, total p53 and p21. β-Tubulin served as a control of protein loading. (C) Localization of activated p53 in cells expressing eGFP-TopBP1 and in cells treated with ActD. Cells were left non-induced or induced with doxycycline to express eGFP-TopBP1 for 24 h, or treated for 2 h with 30 nM ActD and fixed. Activated p53 (DO-1) and Rad9 were immunolabelled. Scale bar is 10 μm. (D) Fractions of p53-positive cells were determined in non-treated (Ctrl), ActD-treated (as in A, assayed 24 h after release) and in Nutlin 3-a-treated (5 μM, 3 h) cell populations. From induced (DOX) samples, those expressing (e+) eGFP-TopBP1 and not expressing (e-) were scored separately. p53 positive cells were identified automatically based on a threshold established in the nutlin-3a treated cells. Mean values of three independent experiments are shown with standard deviations. Statistical significance (P-value ≤0.05) was calculated with ANOVA and Dunnett's T3 post hoc tests.
Immunoblot showed that ActD-treated cells initiated p53 response, as shown by the accumulation of total p53 protein and its phosphorylation at serine 15 (S15), representing activated p53 (Figure 7B). Consistent with the replication status of the cells, p53 levels dropped to background levels on day 4. Expression of p53 target gene p21 followed accumulation of p53, suggesting that p21 mediated the cell cycle arrest. We did not find any phosphorylation of Chk1 following ActD treatment, which implies that canonical ATR signalling is not involved in the ActD-induced cell cycle arrest. Together, these results suggest that ActD-induced cell cycle arrest is mediated by the p53 pathway.
While TopBP1-induced nucleolar segregation was phenotypically similar to that induced by ActD, the continued expression of eGFP-TopBP1, surprisingly, did not cause any loss of DNA replication activity during the 4-day expression (Figure 7A, bottom panels). As detected with immunoblotting, expression of eGFP-TopBP1 did not lead to a marked accumulation of p53, but we did observe some increase in phosphorylation of S15 in p53 (Figure 7B). As after ActD, Chk1 appears not to become phosphorylated at serine 345 upon induction of eGFP-TopBP1.
ActD-treatment caused a detectable downregulation of endogenous TopBP1 and Chk1 (Figure 7B). This downregulation correlated with inhibition of DNA replication and suggests that both TopBP1 and Chk1 activity was suppressed to facilitate the p53-mediated cell cycle arrest.
Stabilization of p53 is regulated by multiple post-transcriptional modifications (32). One of the best known sites that become phosphorylated in response to different stress situations is S15. Appearance of S15-p53 in cells expressing eGFP-TopBP1 prompted us to look for activated p53 at the subcellular level. To detect activated p53, we used monoclonal DO-1 antibody which is known to bind an epitope in p53 that becomes unmasked after cells are irradiated with UV light (33). We found that activated p53 was present in the eGFP-TopBP1 foci, while in control cells activated p53 was not observed (Figure 7C). Moreover, in ActD-treated cells, we observed co-localization of activated p53 with Rad9 although the cells had not been induced to express ectopic TopBP1 (Figure 7C).
An apparent contradiction between immunoblot and immunofluorescence data prompted us to look for total p53 protein levels in individual cells expressing eGFP-TopBP1. Cells induced to express eGFP-TopBP1 showed individual cell-to-cell variation in expression levels (see e.g. Supplementary Figure S2, lower panel), which may mask small changes in protein levels. To set up a threshold level for computational analysis of p53-positive cells we exposed cells to Nutlin-3a. Nutlin-3a disrupts p53-Mdm2 interaction thus preventing p53 degradation and leading to accumulation of p53 (34). After Nutlin-3a-treatment 80% of the cells were positive for p53 at the chosen threshold, while in non-treated control cells the percentage remained at 10% (Figure 7D). When we counted those p53 positive cells in induced sample, which expressed eGFP-TopBP1, we found that 28% of the eGFP-TopBP1 positive cells were also positive for p53. On the other hand, only 7% of the cells not expressing eGFP-TopBP1, counted from the same induced sample images, were positive for p53. When cells were treated with ActD for 2 h and then let recover for 24 h, 44% of the cells were p53 positive. These results suggest that p53 is moderately stabilized in cells with high levels of TopBP1.
To study the possibility that nucleolar eGFP-TopBP1 foci represent damaged DNA, we immunostained cells expressing eGFP-TopBP1 with anti-γH2AX and anti-53BP1, two commonly used markers for DNA double strand breaks and other DNA damage types. Neither of the two DNA damage marker proteins co-localized with the nucleolar eGFP-TopBP1 foci (Supplementary Figure S6A).
Moreover, the TopBP1-induced nucleolar segregation was not restricted to S-phase of the cell cycle, since we observed this phenotype also in cells that did not incorporate EdU (Supplementary Figure S6B). Expression of eGFP-TopBP1 did not affect the proportion of replicating cells either (Supplementary Figure S6C). However, we found that in cells undergoing mitosis, eGFP-TopBP1 foci disappeared and UBF formed distinct foci not related to localization of eGFP-TopBP1 (Supplementary Figure S6D). During mitosis UBF is known to remain associated with the rDNA while the transcription of rRNA is shut down (35).
Taken together, initiation of nucleolar stress pathway by ActD leads to stabilization of p53, accumulation of p21 and to a transient cell cycle arrest. On the contrary, while cells expressing high levels of TopBP1 show all the hallmarks of activation of nucleolar stress pathway, including a moderate activation of p53, the cell cycle is not arrested.
DISCUSSION
We describe here a novel role for TopBP1 and ATR in nucleolar dynamics, which is distinct from the canonical role of ATR pathway during DNA replication. Our study is, to our knowledge, the first one to examine the effects of high expression level of full-length TopBP1 in human cells. We demonstrated that while endogenous TopBP1 bound to nucleolar chromatin, as shown by immuno-EM and ChIP experiments, at high cellular concentrations TopBP1 activated ATR specifically at the nucleoli leading to shut-down of rRNA synthesis with concomitant nucleolar segregation.
TopBP1-induced nucleolar segregation strictly depended on ATR function. However, localization of TopBP1 into nucleoli did not depend on ATR kinase, as the mutant TopBP1, defective in ATR activation, also localized into nucleoli. This is consistent with the current model, according to which localization of TopBP1 dictates the activation of ATR (9).
Chk1 is a downstream target of ATR and becomes efficiently phosphorylated upon activation of ATR. We did not find global activation of ATR by high expression level of TopBP1, as indicated by the lack of Chk1 phosphorylation on serine 345. This differs with the reports that expression of ATR activation domain of TopBP1 alone leads to efficient activation of the ATR pathway (19,29) and to p53-dependent cell cycle arrest within few hours (19). TopBP1 is a large protein that binds multiple targets and the regulatory mechanisms of its activities are likely to be complex. One such mechanism regulates binding of TopBP1 to chromatin. In the presence of positive proliferation signalling, binding of TopBP1 to chromatin and activation of ATR is attenuated (10,11), which can switch the function of TopBP1 from checkpoint activation to transcriptional regulation (11). In our ectopic expression model it is conceivable that these regulatory mechanisms are perturbed, so that the excess of TopBP1 acts both at the checkpoint level and at the transcriptional level.
Our data show that the excess of TopBP1 is preferentially targeted to ribosomal chromatin, where it locally activates ATR. TopBP1 is known to bind to chromatin (31) and has been found to bind bulky DNA lesions to activate ATR in an in vitro system (13). It appears that bulky DNA adducts can be the driving force for nucleolar segregation (36,37). Transcription of rRNA generates RNA–DNA hybrids, G-quadruplex complexes and DNA lesions that can serve as binding sites for TopBP1. It is conceivable that TopBP1 is normally required for monitoring the integrity of DNA at the nucleoli, activating nucleolar stress response when necessary. We do not think that overexpression of TopBP1 leads to rDNA damage or stress by itself, as we did not see elevated γH2AX or 53BP1 levels. Rather, ATR seems to be activated by a positive feedback loop, caused by excessive TopBP1 binding at the ribosomal chromatin. This mode of ATR activation by TopBP1, where TopBP1 and Rad9 interact for signal amplification, has been proposed before (38). Consistently with this model, we observed co-localization of eGFP-TopBP1 with Rad9 and Hus1, and our deletion analysis revealed that the Rad9 interaction domains of TopBP1 are required for the TopBP1-induced nucleolar segregation.
An important question in understanding ATR activation is how the chromatin association of TopBP1 is regulated. The strong chromatin association of eGFP-TopBP1 observed in this study raises the possibility that TopBP1 binds chromatin by default, and in order to prevent aberrant ATR activation either TopBP1 has to be modified or its level has to be kept low. Indeed, negative regulation of TopBP1 chromatin binding by phosphorylation, and inhibition of ATR pathway has been shown before (11).
A prominent feature of nucleolar segregation is the formation of UBF foci called nucleolar caps at the periphery of nucleolar body. However, the significance of this capping is unknown. Nucleolar caps may represent rDNA that is relocated outside of the nucleolus for processing and repair. Such phenomenon has been found in yeast, where DNA double-strand breaks in the nucleolus induce transient transfer of rDNA out from the nucleoli for repair (39). Interestingly, in a similar process described for Drosophila heterochromatin, ATR plays a major role in the early steps of transient relocalization of damaged heterochromatin into repair foci (40). One possible role for TopBP1 and ATR at nucleoli could be the regulation of chromatin organization.
The nucleolus was once thought to be only responsible for ribosomal biogenesis, but this view has gradually changed after it was found that as little as 30% of the nucleolar proteins are related to the production of ribosome subunits (41,42). Today, it is accepted that the nucleolus senses stress and coordinates the stress response (2). The hallmark of nucleolar stress response is the reorganization of nucleolus, which can manifest differently in response to different stresses, but which at the end in all cases results in stabilization of p53 (1,3). We found that although activation of nucleolar stress response by ActD lead to a transient p53-mediated cell cycle arrest, this cell cycle arrest did not occur in cells expressing large quantities of TopBP1. This is consistent with the previous finding that TopBP1 physically interacts with p53 and suppresses p53 transactivation (22). p53 has also been suggested to monitor genetic stress at the nucleolus (43), and it has been shown to directly repress rRNA transcription (44). However, in our system it is unlikely that p53 mediates the rRNA transcriptional silencing as TopBP1-induced nucleolar segregation was observed also in p53 negative cell lines SaOS2 and MG63 (see Supplementary Figure S3).
Inhibition of RNA pol I appears to be an early step in the stress response. Stress-activated protein kinase JNK2 (c-Jun N-terminal kinase 2) phosphorylates and inactivates RNA pol I transcription factor TIF-1A to downregulate rRNA synthesis (45). RNA pol I is also inhibited in response to dsDNA breaks in an ATM-dependent manner (46,47). The results presented here point to a similar role for TopBP1-ATR signalling pathway in silencing of rRNA transcription at times of stress, suggesting that inhibition of rRNA synthesis by different stress-responsive kinases is a common mechanism to activate nucleolar stress pathway.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are indebted to Dr Raimundo Freire (Universitario de Canarias, Tenerife, Spain) for providing Rad9 antiserum. We are grateful for Dr Raija Sormunen for her help with the immuno-EM analyses.
FUNDING
Academy of Finland [Grant number 251576 to J.E.S.]; Federal Government of Germany and the State of Thuringia (to The Fritz Lipmann Institute). Funding for open access charge: Academy of Finland [Grant number 251576 to J.E.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Vlatkovic N., Boyd M.T., Rubbi C.P. Nucleolar control of p53: a cellular achilles’ heel and a target for cancer therapy. Cell Mol. Life Sci. 2014;71:771–791. doi: 10.1007/s00018-013-1361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boulon S., Westman B.J., Hutten S., Boisvert F.M., Lamond A.I. The nucleolus under stress. Mol. Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubbi C.P., Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003;22:6068–6077. doi: 10.1093/emboj/cdg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hein N., Hannan K.M., George A.J., Sanij E., Hannan R.D. The nucleolus: an emerging target for cancer therapy. Trends Mol. Med. 2013;19:643–654. doi: 10.1016/j.molmed.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Bywater M.J., Poortinga G., Sanij E., Hein N., Peck A., Cullinane C., Wall M., Cluse L., Drygin D., Anderes K., et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peltonen K., Colis L., Liu H., Trivedi R., Moubarek M.S., Moore H.M., Bai B., Rudek M.A., Bieberich C.J., Laiho M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014;25:77–90. doi: 10.1016/j.ccr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraschini A., Bottone M.G., Scovassi A.I., Denegri M., Risueno M.C., Testillano P.S., Martin T.E., Biggiogera M., Pellicciari C. Changes in extranucleolar transcription during actinomycin D-induced apoptosis. Histol. Histopathol. 2005;20:107–117. doi: 10.14670/HH-20.107. [DOI] [PubMed] [Google Scholar]
- 8.Choong M.L., Yang H., Lee M.A., Lane D.P. Specific activation of the p53 pathway by low dose actinomycin D: A new route to p53 based cyclotherapy. Cell Cycle. 2009;8:2810–2818. doi: 10.4161/cc.8.17.9503. [DOI] [PubMed] [Google Scholar]
- 9.Cimprich K.A., Cortez D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herold S., Hock A., Herkert B., Berns K., Mullenders J., Beijersbergen R., Bernards R., Eilers M. Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J. 2008;27:2851–2861. doi: 10.1038/emboj.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K., Graves J.D., Scott J.D., Li R., Lin W.C. Akt switches TopBP1 function from checkpoint activation to transcriptional regulation through phosphoserine binding-mediated oligomerization. Mol. Cell. Biol. 2013;33:4685–4700. doi: 10.1128/MCB.00373-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindsey-Boltz L.A., Sancar A. Tethering DNA damage checkpoint mediator proteins topoisomerase IIbeta-binding protein 1 (TopBP1) and claspin to DNA activates ataxia-telangiectasia mutated and RAD3-related (ATR) phosphorylation of checkpoint kinase 1 (Chk1) J. Biol. Chem. 2011;286:19229–19236. doi: 10.1074/jbc.M111.237958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi J.H., Lindsey-Boltz L.A., Sancar A. Cooperative activation of the ATR checkpoint kinase by TopBP1 and damaged DNA. Nucleic Acids Res. 2009;37:1501–1509. doi: 10.1093/nar/gkn1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mäkiniemi M., Hillukkala T., Tuusa J., Reini K., Vaara M., Huang D., Pospiech H., Majuri I., Westerling T., Mäkelä T.P., et al. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J. Biol. Chem. 2001;276:30399–30406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- 15.Delacroix S., Wagner J.M., Kobayashi M., Yamamoto K., Karnitz L.M. The Rad9-Hus1-Rad1 (9–1–1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J., Dunphy W.G. Rad17 plays a central role in establishment of the interaction between TopBP1 and the Rad9-Hus1-Rad1 complex at stalled replication forks. Mol. Biol. Cell. 2010;21:926–935. doi: 10.1091/mbc.E09-11-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J., Kumagai A., Dunphy W.G. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 2007;282:28036–28044. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- 18.Mordes D.A., Glick G.G., Zhao R., Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008;22:1478–1489. doi: 10.1101/gad.1666208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toledo L.I., Murga M., Gutierrez-Martinez P., Soria R., Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22:297–302. doi: 10.1101/gad.452308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu K., Lin F.T., Ruppert J.M., Lin W.C. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell. Biol. 2003;23:3287–3304. doi: 10.1128/MCB.23.9.3287-3304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu K., Luo Y., Lin F.T., Lin W.C. TopBP1 recruits Brg1/brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 2004;18:673–686. doi: 10.1101/gad.1180204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K., Bellam N., Lin H.Y., Wang B., Stockard C.R., Grizzle W.E., Lin W.C. Regulation of p53 by TopBP1: a potential mechanism for p53 inactivation in cancer. Mol. Cell. Biol. 2009;29:2673–2693. doi: 10.1128/MCB.01140-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sokka M., Parkkinen S., Pospiech H., Syväoja J.E. Function of TopBP1 in genome stability. Subcell. Biochem. 2010;50:119–141. doi: 10.1007/978-90-481-3471-7_7. [DOI] [PubMed] [Google Scholar]
- 24.Slot J.W., Geuze H.J. A new method of preparing gold probes for multiple-labeling cytochemistry. Eur. J. Cell Biol. 1985;38:87–93. [PubMed] [Google Scholar]
- 25.Nelson J.D., Denisenko O., Sova P., Bomsztyk K. Fast chromatin immunoprecipitation assay. Nucleic Acids Res. 2006;34:e2. doi: 10.1093/nar/gnj004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grandori C., Gomez-Roman N., Felton-Edkins Z.A., Ngouenet C., Galloway D.A., Eisenman R.N., White R.J. C-myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 27.Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo L.L., Futami K., Shimamoto A., Furuichi Y., Frank K.M. The rothmund-thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp. Cell Res. 2006;312:3443–3457. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 29.Kumagai A., Lee J., Yoo H.Y., Dunphy W.G. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 30.Toledo L.I., Murga M., Zur R., Soria R., Rodriguez A., Martinez S., Oyarzabal J., Pastor J., Bischoff J.R., Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J.E., McAvoy S.A., Smith D.I., Chen J. Human TopBP1 ensures genome integrity during normal S phase. Mol. Cell. Biol. 2005;25:10907–10915. doi: 10.1128/MCB.25.24.10907-10915.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavin M.F., Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 33.Craig A.L., Blaydes J.P., Burch L.R., Thompson A.M., Hupp T.R. Dephosphorylation of p53 at Ser20 after cellular exposure to low levels of non-ionizing radiation. Oncogene. 1999;18:6305–6312. doi: 10.1038/sj.onc.1203085. [DOI] [PubMed] [Google Scholar]
- 34.Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 35.Gebrane-Younes J., Fomproix N., Hernandez-Verdun D. When rDNA transcription is arrested during mitosis, UBF is still associated with non-condensed rDNA. J. Cell. Sci. 1997;110:2429–2440. doi: 10.1242/jcs.110.19.2429. [DOI] [PubMed] [Google Scholar]
- 36.Al-Baker E.A., Boyle J., Harry R., Kill I.R. A p53-independent pathway regulates nucleolar segregation and antigen translocation in response to DNA damage induced by UV irradiation. Exp. Cell Res. 2004;292:179–186. doi: 10.1016/j.yexcr.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Moore H.M., Bai B., Boisvert F.M., Latonen L., Rantanen V., Simpson J.C., Pepperkok R., Lamond A.I., Laiho M. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.009241. doi:10.1074/mcp.M111.009241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohashi E., Takeishi Y., Ueda S., Tsurimoto T. Interaction between Rad9-Hus1-Rad1 and TopBP1 activates ATR-ATRIP and promotes TopBP1 recruitment to sites of UV-damage. DNA Repair. 2014;21:1–11. doi: 10.1016/j.dnarep.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Torres-Rosell J., Machin F., Farmer S., Jarmuz A., Eydmann T., Dalgaard J.Z., Aragon L. SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat. Cell Biol. 2005;7:412–419. doi: 10.1038/ncb1239. [DOI] [PubMed] [Google Scholar]
- 40.Chiolo I., Minoda A., Colmenares S.U., Polyzos A., Costes S.V., Karpen G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. 2011;144:732–744. doi: 10.1016/j.cell.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andersen J.S., Lyon C.E., Fox A.H., Leung A.K., Lam Y.W., Steen H., Mann M., Lamond A.I. Directed proteomic analysis of the human nucleolus. Curr. Biol. 2002;12:1–11. doi: 10.1016/s0960-9822(01)00650-9. [DOI] [PubMed] [Google Scholar]
- 42.Scherl A., Coute Y., Deon C., Calle A., Kindbeiter K., Sanchez J.C., Greco A., Hochstrasser D., Diaz J.J. Functional proteomic analysis of human nucleolus. Mol. Biol. Cell. 2002;13:4100–4109. doi: 10.1091/mbc.E02-05-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubbi C.P., Milner J. Non-activated p53 co-localizes with sites of transcription within both the nucleoplasm and the nucleolus. Oncogene. 2000;19:85–96. doi: 10.1038/sj.onc.1203378. [DOI] [PubMed] [Google Scholar]
- 44.Zhai W., Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000;20:5930–5938. doi: 10.1128/mcb.20.16.5930-5938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayer C., Bierhoff H., Grummt I. The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis. Genes Dev. 2005;19:933–941. doi: 10.1101/gad.333205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruhlak M., Crouch E.E., Orlov M., Montano C., Gorski S.A., Nussenzweig A., Misteli T., Phair R.D., Casellas R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007;447:730–734. doi: 10.1038/nature05842. [DOI] [PubMed] [Google Scholar]
- 47.Larsen D.H., Hari F., Clapperton J.A., Gwerder M., Gutsche K., Altmeyer M., Jungmichel S., Toledo L.I., Fink D., Rask M.B., et al. The NBS1-treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell Biol. 2014;16:792–803. doi: 10.1038/ncb3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.