


Abstract
SDR5C1 is an amino and fatty acid dehydrogenase/reductase, moonlighting as a component of human mitochondrial RNase P, which is the enzyme removing 5′-extensions of tRNAs, an early and crucial step in tRNA maturation. Moreover, a subcomplex of mitochondrial RNase P catalyzes the N1-methylation of purines at position 9, a modification found in most mitochondrial tRNAs and thought to stabilize their structure. Missense mutations in SDR5C1 cause a disease characterized by progressive neurodegeneration and cardiomyopathy, called HSD10 disease. We have investigated the effect of selected mutations on SDR5C1's functions. We show that pathogenic mutations impair SDR5C1-dependent dehydrogenation, tRNA processing and methylation. Some mutations disrupt the homotetramerization of SDR5C1 and/or impair its interaction with TRMT10C, the methyltransferase subunit of the mitochondrial RNase P complex. We propose that the structural and functional alterations of SDR5C1 impair mitochondrial RNA processing and modification, leading to the mitochondrial dysfunction observed in HSD10 patients.
INTRODUCTION
SDR5C1 is a member of the short-chain dehydrogenase/reductase (SDR) superfamily, a large group of nicotinamide adenine dinucleotide (phosphate)-dependent oxidoreductases (1). It was first identified as a mitochondrial short-chain l-3-hydroxy-2-methylacyl-CoA dehydrogenase, catalyzing the penultimate step in the β-oxidation of short branched-chain fatty acids and isoleucine (2). SDR5C1 was reported to be active on a wide range of substrates, including fatty acids, alcohols, and hydroxysteroids, and its gene (HSD17B10) was named according to its relationship to 17-β-hydroxysteroid dehydrogenases (3–9). In vivo, besides being essential for isoleucine degradation, SDR5C1 was suggested to be involved in the metabolism of neuroactive steroids and sex hormones (reviewed in 10,11). HSD17B10 is an essential gene (12,13) located on the X chromosome, and to date 11 pathogenic mutations have been reported: 10 missense mutations, and one affecting its splicing (12,14–26). In the latter case a decreased steady state level of SDR5C1 is associated with mild mental retardation and choreoathetosis (24). The missense mutations however cause a disease named 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency, or HSD10 disease (OMIM #300438), which is characterized by progressive neurodegeneration and cardiomyopathy (reviewed in 23). The different mutations are associated with a wide spectrum of disease manifestations, ranging from a severe neonatal form, to milder, later-onset forms. While the biochemical diagnosis of HSD10 disease is based on elevated levels of 2-methyl-3-hydroxybutyrate and tiglylglycine, two intermediates in the degradation of isoleucine, the same metabolites accumulate in patients affected by β-ketothiolase deficiency without causing neurological complications (27). Most HSD10 patients do not develop metabolic crisis, and isoleucine restriction does not prevent the progression of the disease (23). The pathogenesis and clinical presentation together with the frequent plasma and brain lactic acidosis rather remind of mitochondrial diseases (20,23), i.e. diseases due to respiratory-chain dysfunction, and, consistently, structural abnormalities of the mitochondria were observed in patient fibroblasts (12). Moreover, an SDR5C1 mutant deficient in dehydrogenase activity was found to prevent apoptosis when supplemented to SDR5C1 knock down cells (12), suggesting that other mitochondrial functions of the protein play a crucial role in mitochondrial physiology and cell survival (12).
We previously demonstrated that SDR5C1 is an essential component of a multifunctional complex catalyzing two early steps in mitochondrial tRNA ((mt)tRNA) maturation. Together with TRMT10C and the endonucleolytic subunit PRORP, it constitutes human mitochondrial RNase P (mtRNase P), the enzyme responsible for the cleavage of the polycistronic mitochondrial primary transcripts at the 5′ end of tRNA moieties (28–30). In addition, the TRMT10C-SDR5C1 subcomplex is the methyltransferase that catalyzes the N1-methylation of purines at position 9 of mitochondrial tRNAs (31), a modification crucial for proper tRNA folding (32). We have shown that the knock down of SDR5C1 in human cells is sufficient to cause the accumulation of (mt)tRNA-precursors, and to abolish purine-9 methylation of (mt)tRNAs (28,31). Consistently, it was recently reported that missense mutations that result in a decrease of SDR5C1 are associated with increased levels of unprocessed mitochondrial RNAs (33,34). SDR5C1 protein levels were assessed in a few patients (12,20,33–35) and reported to be either unchanged or decreased in carriers of the same mutation (20,34). The observed decrease of SDR5C1 alone seems not generally sufficient to explain the severity of the disease. Mechanisms underlying the possible loss of SDR5C1 protein have not been addressed either, nor have the consequences of the mutations for the function of the residual protein been clarified. Alterations of the structure and function of mutant SDR5C1 are likely to contribute to the severe impact on mitochondrial tRNA processing and modification, and the resulting mitochondrial pathology. Here, we have therefore investigated the effect of selected mutations on SDR5C1, and we provide first mechanistic insights into the impact of those mutations on SDR5C1's role in mitochondrial tRNA maturation.
MATERIALS AND METHODS
Expression and purification of recombinant proteins
The mutations were introduced into the previously described (28) plasmid for the expression of N-terminally His-tagged SDR5C1 by site-directed mutagenesis using the QuikChange protocol (Agilent Technologies). Wild type and mutant forms of SDR5C1 were expressed and purified in parallel with His SpinTrap columns (GE Healthcare) using previously described lysis, wash, and elution conditions (28). Two independent protein stock preparations of wild-type and mutant SDR5C1 gave consistent results in the different experiments. In a previous study, the purified SDR5C1R130C was reported to be unstable (12); we did not experience problems of protein stability and did not observe a loss of enzymatic activity under our experimental conditions. Native (untagged) TRMT10C, C-terminally myc-His-tagged TRMT10C, and C-terminally His-tagged PRORP were prepared as previously described (28,31).
Protein concentrations were quantitated relative to bovine serum albumin standards by SDS-PAGE, Coomassie brillant blue staining, and image analysis using ImageQuant TL 7 (GE Healthcare).
Protein analysis
To determine the oligomeric state of SDR5C1, 2 μg of protein were diluted in blue native sample buffer (ε-aminocaproic acid (50 mM), imidazole (5 mM), glycerol (5%), Coomassie G250 (0.025%), pH 7) and separated by blue native, 9–19% gradient polyacrylamide gel electrophoresis (PAGE) (36) followed by Coomassie brilliant blue staining. Molecular weight reference proteins were: aldolase (158 kDa), bovine serum albumine (monomer 67 kDa, and dimer 134 kDa), ovalbumine (monomer 43 kDa, and dimer 86 kDa), and cytochrome c (13 kDa). Alternatively, SDR5C1 was subjected to size exclusion chromatography. Forty micrograms of SDR5C1 were separated on a Superdex 200 Increase PC 3.2/300 column (GE Healthcare) using an ÄKTApurifier system (GE Healthcare) with the following buffer: NaCl (150 mM), Tris·Cl pH 7.4 (50 mM), glycerol (10%). Elution fractions were collected and protein content of peaks verified by SDS-PAGE. For rapid photochemical cross-linking (37), 7.5 μg of protein were incubated on ice in 10 μl NaPO4 buffer (10 mM, pH 7.4), ammonium persulphate (5 mM), tris(2,2′-bipyridyl)dichlororuthenium(II) (250 μM). Cross-linking was induced by exposure to a halogen white light source for 5 min and quenched with Laemmli buffer; control reactions contained 1% SDS. Cross-linked samples were separated by 6–18% gradient SDS-PAGE followed by Coomassie brilliant blue staining.
To assess the interaction of SDR5C1 with TRMT10C, purified His-tagged SDR5C1 was mixed with an excess of crude bacterial lysate of native TRMT10C, and the complex captured and purified with His-affinity-coated magnetic beads (Dynal) as previously described (31). The ratio of the two proteins was assessed by SDS-PAGE and Coomassie brilliant blue staining.
The 3D-structure rendering of SDR5C1 was generated with PyMOL 1.3 (38).
Dehydrogenase assay
l-3-hydroxyacyl-CoA dehydrogenase activity was measured as acetoacetyl-CoA dependent nicotinamide adenine dinucleotide (NADH) dehydrogenation (39). SDR5C1 (10 nM) were assayed in presence of acetoacetyl-CoA (30 μM) and NADH (100 μM).
RNase P assay
In vitro transcription, 32P 5′ end labeling and purification of the (mt)tRNAIle precursor substrate, and RNase P activity assays were carried out and analyzed as previously described (28,40), with the following changes. Reactions were set at 21°C in reaction buffer: Tris·Cl pH 8 (50 mM), NaCl (20 mM), MgCl2 (4.5 mM), DTT (2 mM), BSA (20 μg/ml), Ribolock RNase inhibitor (0.5 units/μl; Fermentas). PRORP (100 nM), TRMT10C (100 nM), and SDR5C1 (200 nM; wild type or mutant) were used to reconstitute mtRNase P and were incubated with the labeled (mt)tRNAIle precursor (1–3 nM; single turnover conditions). Samples were withdrawn at defined intervals throughout substrate-to-product conversion until plateau. The percentage of cleaved tRNA-precursor was plotted against time and curves were fit by nonlinear regression (one phase exponential association) using Prism 5 (GraphPad Software).
Methyltransferase assay
In vitro transcription, internal 32P labeling at position 9 and purification of the (mt)tRNAIle and (mt)tRNALys substrates, and methyltransferase assays were carried out as described previously (31), with the following changes. Reactions were set at 30°C in the aforementioned (RNase P) reaction buffer. TRMT10C (100 nM) and SDR5C1 (200 nM; wild type or mutant) were used to reconstitute the methyltransferase subcomplex, and incubated with S-adenosyl methionine (SAM; 25 μM) and the labeled (mt)tRNA (3–10 nM; single turn-over conditions). Samples were withdrawn at defined intervals throughout substrate-to-product conversion until plateau. The percentage of methylated tRNA was plotted against time and curves were fit by nonlinear regression (one phase exponential association) using Prism 5 (GraphPad Software).
Statistical analysis
Activities and reaction rates of the different mutant forms of SDR5C1 were compared to the wild-type by one-way ANOVA followed by Dunnett's multiple comparison test, using Prism 5 (GraphPad Software). P-values < 0.05 were considered significant.
RESULTS
Among the 10 described missense mutations, we selected two found in severe neonatal cases of HSD10 disease, R226Q and N247S (21,34), and two associated with the typical infantile form, P210S and R130C, the latter being the most common mutation, accounting for about 50% of all the cases described (20,23). As control, we included SDR5C1K172A, an artificial ‘dehydrogenase-dead’ mutant that sustains cleavage and methylation activities comparable to the wild type (31); in fact, the dehydrogenase activity of SDR5C1 is dispensable for efficient cleavage and methylation of tRNA precursors by mtRNase P (31).
Effect of mutations on the dehydrogenase activity of SDR5C1
The dehydrogenase activity of most mutant SDR5C1 variants was previously measured in homogenates of patient fibroblasts and found to be decreased in comparison to controls (23). However, this approach does not allow normalization of the measured activity to the actual amount of SDR5C1 protein in the tested samples. We purified recombinant, wild-type SDR5C1 and the mutant variants mentioned above, and assayed their dehydrogenase activity in parallel (Figure 1). For SDR5C1R130C and SDR5C1P210S, we observed a reduction in activity to about 20% and 25% of that of the wild type, respectively. The activity of SDR5C1R226Q and SDR5C1N247S, like that of the ‘dehydrogenase-dead’ SDR5C1K172A, was barely detectable (Figure 1). Our measurements provide a first direct comparison of the dehydrogenase activity of these SDR5C1 mutants.
Figure 1.
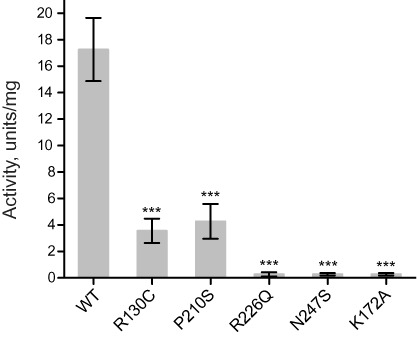
Pathogenic mutations in SDR5C1 affect its dehydrogenase activity. The l-3-hydroxyacyl-CoA dehydrogenase activity of wild-type (WT) and mutant SDR5C1 was measured with acetoacetyl-CoA as substrate and NADH as cofactor. An active-site mutant (K172A) was included as ‘dehydrogenase-dead’ control. Means and SD of three independent experiments are shown (***P < 0.001).
Pathogenic mutations impair the SDR5C1-dependent tRNA processing and modification activities of mtRNase P
Although the dehydrogenase activity and the integrity of the active site of SDR5C1 are dispensable for mitochondrial tRNA cleavage and methylation, the protein subunit is essential for efficient catalysis by mtRNase P and its methyltransferase subcomplex (28,31). Therefore, we assayed the effect of the pathogenic mutations on the function of SDR5C1 in tRNA maturation. We reconstituted the mtRNase P enzyme from purified recombinant TRMT10C, PRORP, and the different SDR5C1 variants including the wild-type protein, and compared the time course of cleavage of a (mt)tRNAIle precursor (Figure 2A and B). The enzyme reconstituted from wild-type components cleaved the tRNA precursor with a first-order rate constant (kobs) of 4.63 ± 0.61 min-1. MtRNase P reconstituted with the SDR5C1K172A showed a kobs of 5.13 ± 0.60 min-1, not significantly different from the wild type, and in agreement with our previous observation that the dehydrogenase activity of SDR5C1 is dispensable for tRNA processing (31). In the case of SDR5C1P210S, we measured a kobs of 0.59 ± 0.07 min-1, a significantly slower cleavage rate (P < 0.001) corresponding to about 13% of the wild type. In the case of the variants R130C, R226Q, and N247S, the endonucleolytic activity was too low to achieve complete substrate cleavage, and no rate could be derived.
Figure 2.
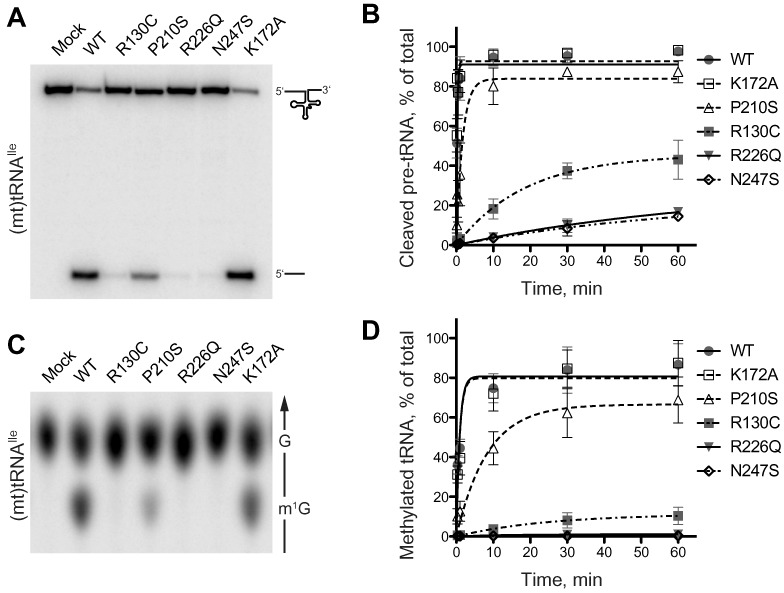
Mutations in SDR5C1 affect the tRNA-maturation activities of the mtRNase P complex. (A and B) mtRNase P was reconstituted from recombinant PRORP, TRMT10C, and wild-type or mutant SDR5C1, and its activity assayed with a 5′ labeled (mt)tRNAIle precursor. Reaction aliquots were withdrawn and stopped at different time points and resolved by denaturing PAGE. In (A), the 1-minute time point of a representative experiment is shown. (B) Cleavage data of six complete, independent experiments were plotted as means and SD, and fit by nonlinear regression. (C and D) The methyltransferase subcomplex of mtRNase P was reconstituted from recombinant TRMT10C and wild-type or mutant SDR5C1, and its activity assayed with position-9 labeled (mt)tRNAIle and SAM. Reaction aliquots were withdrawn and stopped at different time points and the tRNA hydrolysate resolved by thin-layer chromatography (TLC). In (C), the 1-minute time point of a representative experiment is shown. The direction of migration and the positions of G and m1G are indicated to the right; only the informative part of the TLC is shown. (D) Methylation data of five complete, independent experiments were plotted as means and SD, and fit by nonlinear regression.
In a similar setting, we assayed the methyltransferase activity of the TRMT10C-SDR5C1 (wild type or mutant) complex (Figure 2C and D). Using as substrate (mt)tRNAIle, which contains a guanosine at position 9, we measured very similar rates for complexes containing wild-type SDR5C1 (kobs = 1.05 ± 0.16 min-1) or SDR5C1K172A (kobs = 0.90 ± 0.18 min-1). The methyltransferase complex reconstituted using SDR5C1P210S showed significantly lower methylation activity with a kobs of 0.12 ± 0.02 min-1 (P < 0.001), corresponding to about 11% of the wild type. The activity with SDR5C1R130C was too low to derive a kobs, while methylation with SDR5C1R226Q and SDR5C1N247S was undetectable. Comparable results were obtained with (mt)tRNALys, which contains an adenosine at position 9 (Supplementary Figure S1). From these results we conclude that all the pathogenic mutations analyzed cause a substantial impairment of the SDR5C1-dependent tRNA maturation activities of the mtRNase P complex.
Pathogenic mutations in SDR5C1 alter its oligomeric state and impair the interaction with TRMT10C
SDR5C1 is a protein of 27 kDa forming a homotetramer in solution (41) whose structure was solved by crystallography (42). The amino acids R130, R226, and N247 are located at the interface between the subunits of the tetramer (Figure 3), and mutations at these sites could affect the quaternary structure of SDR5C1. To test this hypothesis, we analyzed purified SDR5C1 by blue native PAGE. As shown in Figure 4A, native wild-type SDR5C1 migrated as a single band of about 120 kDa, as estimated from reference proteins, in good agreement with the 116 kDa calculated for a tetramer of the His-tagged protein. An identical band was observed for SDR5C1R130C, SDR5C1P210S, and SDR5C1K172A. In the case of SDR5C1R226Q and SDR5C1N247S, we observed a pattern of three bands, possibly corresponding to tetramers, dimers, and monomers, where the tetrameric form accounted for about 25% of the total. Consistently, SDR5C1, SDR5C1R130C, SDR5C1P210S, and SDR5C1K172A were resolved by size exclusion chromatography as a single peak at about 105 kDa, confirming the homotetrameric structure of the recombinant proteins (Supplementary Figure S2A). The elution profiles of SDR5C1R226Q and SDR5C1N247S displayed an additional broad peak at about 60 kDa accounting for about 75% of the total protein and possibly consisting of unresolved dimers and monomers. To further corroborate our results, we subjected wild-type and mutant SDR5C1 protein to rapid photochemical cross-linking (37), which induced covalent dimerization of the wild-type SDR5C1, SDR5C1P210S, and SDR5C1K172A at comparable yields (Supplementary Figure S2B); the inefficient capture of tetramers apparently reflects an insufficient cross-linking reactivity at one of the two interacting interfaces of the monomer (Figure 3). SDR5C1R226Q and SDR5C1N247S were less efficiently cross-linked, indicating a weaker interaction between the monomers. SDR5C1R130C showed an altered migration pattern, with a smeared dimer band, possibly indicating some alteration in the structure of the protein upon cross-linking. From all these experiments we conclude that the mutations R226Q and N247S severely impair the tetramerization of SDR5C1.
Figure 3.
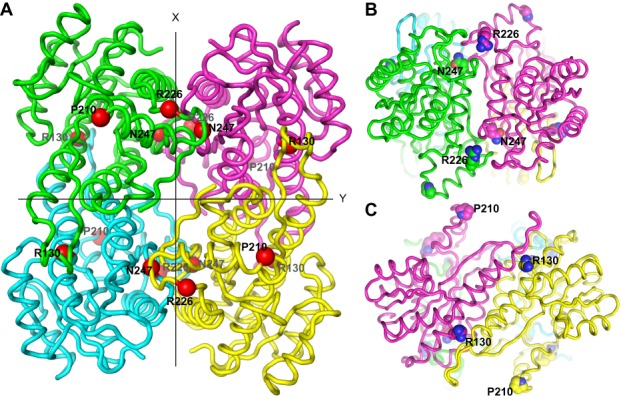
SDR5C1 structure and location of the studied pathogenic mutations. (A) Ribbon-display of the crystal structure of SDR5C1 in tetrameric form (PDB: 1U7T), as viewed along one of three mutually perpendicular dyad axes. The subunits interfaces are arranged about the two other axes, labeled X and Y. The α-carbons of the amino acids affected by the studied mutations are highlighted as red spheres and labeled. (B) SDR5C1 tetramer viewed along the X-axis indicated in (A). The side chains of the amino acids affected by the studied mutations are highlighted by ball-display and labeled. (C) SDR5C1 tetramer viewed along the Y-axis indicated in (A). The side chains of the amino acids affected by the studied mutations are highlighted by ball-display and labeled.
Figure 4.
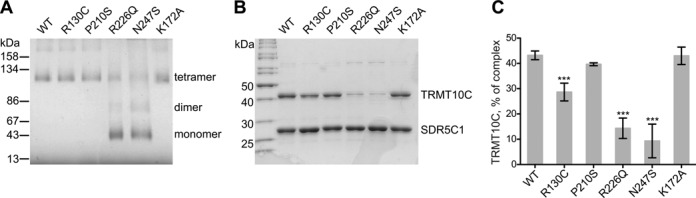
Mutations in SDR5C1 affect its tetramerization and the interaction with TRMT10C. (A) Wild-type and mutant, recombinant SDR5C1 were resolved by blue native, 9–19% gradient PAGE and stained by Coomassie brilliant blue. The migration and molecular weight of marker proteins is indicated to the left. Visible bands and oligomeric state interpretation are specified to the right. (B) Recombinant, purified His-tagged SDR5C1 (wild type or mutant) was mixed with the lysate of a bacterial strain expressing the native form of TRMT10C. The protein complex was recovered by immobilized metal-affinity chromatography, resolved by SDS-PAGE, and stained by Coomassie brilliant blue. A representative gel is shown. The molecular weight of selected marker proteins is indicated to the left. (C) Quantification of recovered TRMT10C in experiments like in (B), expressed as mass percentage of the total complex. Means and SD of six independent experiments for the wild type and three for the mutants are shown (***P < 0.001).
The exact supramolecular architecture of the human mitochondrial RNase P holocomplex and its methyltransferase subcomplex have not been elucidated. However, we showed previously that SDR5C1 forms a stable complex with the methyltransferase subunit TRMT10C and seems to only loosely associate with the endonuclease subunit PRORP (28). We investigated whether the impaired tetramerization caused by the pathogenic mutations alters the interaction among the subunits of the methyltransferase subcomplex of mtRNase P. For this purpose, we made use of a strategy that we previously employed for the purification of the SDR5C1-TRMT10C complex (31). We incubated His-tagged SDR5C1 with an excess of native (untagged) TRMT10C, and recovered the formed complex again via the His-tag of SDR5C1. Analysis of the purified complex revealed a striking difference in the recovery of TRMT10C depending on whether wild-type or mutant SDR5C1 was used as a bait (Figure 4B). The complex reconstituted from wild-type components consisted of about 40% TRMT10C (43 kDa) and 60% SDR5C1 (29 kDa including His tag), compatible with a stoichiometry of 2:4 subunits of TRMT10C:SDR5C1, and in agreement with our previous findings (28). SDR5C1P210S and SDR5C1K172A showed a ratio essentially identical to the wild type; in contrast, with SDR5C1R130C, SDR5C1R226Q, and SDR5C1N247S, the recovered TRMT10C was about 30%, 15%, and 10% of the complex, respectively (Figure 4C). Therefore, the mutations disrupting the SDR5C1 tetramer, but also R130C, interfere with the interaction between subunits of the mtRNase P complex.
DISCUSSION
SDR5C1 is essential for the processing of mitochondrial primary transcripts and tRNA methylation by mtRNase P and its methyltransferase subcomplex (28,31). Alterations in the machineries involved in mitochondrial gene expression ultimately lead to a deficit in respiration, with detrimental consequences for energy metabolism, as observed in diseases caused by mutations in mitochondrial tRNA genes or in nucleus-encoded proteins involved in mitochondrial translation (reviewed in 43,44). Here, we show that pathogenic mutations of SDR5C1 severely affect all known functions of the protein, dehydrogenase and tRNA-maturation related (summarized in Table 1). The mutations R226Q and N247S, associated with the severe neonatal form of HSD10 disease, caused a dramatic impairment of SDR5C1-dependent tRNA-precursor cleavage, consistent with the recently reported accumulation of mitochondrial RNA precursors in tissues of an HSD10N247S patient (34). Moreover, we show that also the methylation at position 9 of tRNAs, a modification crucial for proper tRNA folding (32), is impaired by the mutations, likely further decreasing the pool of functional tRNAs in mitochondria. This loss of RNA maturation activity in mitochondria is expected to cause a severe impairment of protein synthesis, possibly worsened by a concomitant decrease in SDR5C1 steady-state levels.
Table 1. Molecular characterization of SDR5C1 mutant forms.
| Dehydrogenase activity (units/mg)a | RNase P activity, kobs (min-1)b | m1G9 methyltransferase activity, kobs (min-1)b | Tetramerized SDR5C1a,c | Complexed TRMT10Ca,d | |
|---|---|---|---|---|---|
| Wild type | 17.26 ± 2.39 | 4.63 ± 0.61 | 1.05 ± 0.16 | 100% | 43.2% ± 1.7 |
| R130C | 3.56 ± 0.92 | n.d. | n.d. | 100% | 28.7% ± 3.5 |
| P210S | 4.27 ± 1.32 | 0.59 ± 0.07 | 0.12 ± 0.02 | 100% | 39.7% ± 0.6 |
| R226Q | 0.26 ± 0.16 | n.d. | n.d. | 26.0% ± 9.5 | 14.3% ± 4.0 |
| N247S | 0.28 ± 0.09 | n.d. | n.d. | 25.8% ± 6.1 | 9.3% ± 6.7 |
| K172A | 0.27 ± 0.09 | 5.13 ± 0.60 | 0.90 ± 0.18 | 100% | 43.0% ± 3.5 |
Summary of SDR5C1-dependent enzymatic activities, SDR5C1 tetramerization, and interaction with TRMT10C. Data as illustrated in Figures 1, 2, and 4 (n.d., not determined).
aMean and SD.
bRate and standard error derived by non-linear regression.
cEstimated from four independent blue native PAGE experiments.
dMass percentage of the complex.
The original characterization of SDR5C1R226Q and SDR5C1N247S patients indicated a dehydrogenase activity corresponding to about 20% of wild-type controls (21,34). The discrepancy with our observations may be due to the use of different substrates, and/or the presence of other redox enzymes in cell homogenates affecting the specificity of the measurements. SDR5C1R130C and SDR5C1P210S, associated with the typical infantile form of the disease, also showed a decrease in tRNA cleavage and methylation rates, though not as drastic. In our in vitro assays, SDR5C1R130C and SDR5C1P210S showed a dehydrogenase activity of about 20% of the wild type. The reported decrease of SDR5C1R130C protein may explain the low dehydrogenase activity measured in HSD10 patient cells, apparently below 10% of controls (14–15,21,35). No solid information is available about the consequences of the mutation P210S on SDR5C1 expression levels, also because only two cases were reported to date (20).
We show that the formation of a tetramer of SDR5C1R226Q or SDR5C1N247S is severely impaired. Arginine 226 and asparagine 247 are located at the same subunit interface within the tetrameric structure of SDR5C1 (Figure 3). In both cases, the mutations to glutamine 226 and serine 247 introduce smaller amino acids, thus steric clashes may be excluded. However, the polar asparagine 247 forms one hydrogen bond with V193 of the same polypeptide chain, and one with F223 of the facing monomer, and the mutation to serine, which is shorter and can form only one hydrogen bond, is likely to destabilize this region of contact between monomers. The positively charged arginine 226 establishes an electrostatic interaction with E232 and a polar interaction with the backbone of L227 in the same chain. The mutation to the shorter, uncharged glutamine is likely to abolish these interactions that, although not directly linking two adjacent SDR5C1 monomers, may be important to stabilize the conformation of the dimerization surface. At the other main interaction interface, the mutation of arginine 130 to the small, uncharged cysteine abolishes three hydrogen bonds formed by the side chain of R130 with N127 and E68 of the same monomer, and H109 of the neighbouring monomer. Although apparently not affecting tetramerization, this disruption of intra- and intermolecular hydrogen bonds might possibly alter the structure of SDR5C1R130C; the different migration pattern in comparison to wild-type SDR5C1 after crosslinking may be an indication of such a subtle structural alteration. Proline 210 is located on the surface of SDR5C1, at a kink between two contiguous α-helices that form one side of the substrate-binding cavity (42). It mediates a sharp turn in the protein backbone and stabilizes the start of the following α-helical section via hydrogen bonds with V213 and R214; the substitution with serine is likely to cause a distortion of the region, possibly explaining the decreased dehydrogenase activity of the protein.
N247S and R226Q are the mutations most severely affecting the dehydrogenase activity of SDR5C1; considering their location far away from the active site and cofactor-binding pocket, our results suggest that the tetramerization is crucial for enzymatic function, and that a monomeric SDR5C1 does not retain dehydrogenase activity. In vivo, the exposed interaction surfaces of mutant SDR5C1 might induce its recognition as an unfolded protein and trigger degradation, possibly explaining the decreased steady state level of SDR5C1N247S in an HSD10 patient (34). However, a similar mechanism cannot explain the reported decrease of SDR5C1R130C in patients (33,35), since the mutation R130C does not seem to affect SDR5C1's tetramerization. While others previously did not observe an impaired binding of a Strep-tagged SDR5C1R130C to TRMT10C (33), we found a small but significant decrease in the interaction between the two proteins. The mutation R130C thus seems to cause slight alterations in the structure of SDR5C1 that decrease the binding to its partner protein, and the loss of association to TRMT10C might trigger the degradation of SDR5C1 in patient cells.
SDR5C1's dehydrogenase activity and an intact cofactor binding site are dispensable for its moonlighting function in tRNA maturation, and SDR5C1 is not directly involved in tRNA binding (31). Therefore, we suppose that SDR5C1 plays a structural role in mtRNase P and its methyltransferase subcomplex, supporting the activity of the enzyme as a sort of scaffold, rather than being directly involved in catalysis. Indeed, the active sites for endonucleolytic cleavage and methylation are located in PRORP and TRMT10C, respectively. None of the amino acid residues mutated in SDR5C1 is thus expected to be directly involved in tRNA cleavage and modification. In the cases of SDR5C1N247S and SDR5C1R226Q, the defect in tetramerization severely disrupts the interaction with TRMT10C, while SDR5C1R130C shows a milder defect in interaction with TRMT10C. SDR5C1P120S did not show an altered tetramerization or interaction with TRMT10C; however, as discussed, P210 is located at the surface of the protein and an alteration of the region might possibly hamper the scaffolding function of SDR5C1 without necessarily changing the affinity for TRMT10C. In conclusion, we propose that the detrimental effect of the investigated SDR5C1 mutations on tRNA maturation is due to structural alterations that affect the scaffolding role of SDR5C1 in the mtRNase P complex.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We acknowledge the excellent technical assistance of Ursula Toth, Aurélie Buzet, and Esther Löffler in the preparation of the recombinant proteins used in this study. We thank Roland K. Hartmann for helpful comments on the manuscript.
FUNDING
Austrian Science Fund (FWF) [P25983]; Vienna Science and Technology Fund (WWTF) [LS09–032]. Funding for open access charge: FWF [P25983].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kallberg Y., Oppermann U., Jörnvall H., Persson B. Short-chain dehydrogenases/reductases (SDRs) Eur. J. Biochem. 2002;269:4409–4417. doi: 10.1046/j.1432-1033.2002.03130.x. [DOI] [PubMed] [Google Scholar]
- 2.Luo M.J., Mao L.-F., Schulz H. Short-chain 3-hydroxy-2-methylacyl-CoA dehydrogenase from rat liver: purification and characterization of a novel enzyme of isoleucine metabolism. Arch. Biochem. Biophys. 1995;321:214–220. doi: 10.1006/abbi.1995.1388. [DOI] [PubMed] [Google Scholar]
- 3.He X.-Y., Merz G., Mehta P., Schulz H., Yang S.-Y. Human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase is a single-domain multifunctional enzyme. Characterization of a novel 17β-hydroxysteroid dehydrogenase. J. Biol. Chem. 1999;274:15014–15019. doi: 10.1074/jbc.274.21.15014. [DOI] [PubMed] [Google Scholar]
- 4.He X.-Y., Merz G., Yang Y.-Z., Pullakart R., Mehta P., Schulz H., Yang S.-Y. Function of human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase in androgen metabolism. Biochim. Biophys. Acta. 2000;1484:267–277. doi: 10.1016/s1388-1981(00)00014-7. [DOI] [PubMed] [Google Scholar]
- 5.He X.-Y., Yang Y.-Z., Schulz H., Yang S.-Y. Intrinsic alcohol dehydrogenase and hydroxysteroid dehydrogenase activities of human mitochondrial short-chain L-3-hydroxyacyl-CoA dehydrogenase. Biochem. J. 2000;345:139–143. [PMC free article] [PubMed] [Google Scholar]
- 6.Shafqat N., Marschall H.-U., Filling C., Nordling E., Wu X.-Q., Björk L., Thyberg J., Mårtensson E., Salim S., Jörnvall H., et al. Expanded substrate screenings of human and Drosophila type 10 17β-hydroxysteroid dehydrogenases (HSDs) reveal multiple specificities in bile acid and steroid hormone metabolism: characterization of multifunctional 3α/7α/7β/17β/20β/21-HSD. Biochem. J. 2003;376:49–60. doi: 10.1042/BJ20030877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He X.-Y., Wegiel J., Yang Y.-Z., Pullarkat R., Schulz H., Yang S.-Y. Type 10 17beta-hydroxysteroid dehydrogenase catalyzing the oxidation of steroid modulators of γ-aminobutyric acid type A receptors. Mol. Cell. Endocrinol. 2005;229:111–117. doi: 10.1016/j.mce.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 8.He X.-Y., Wegiel J., Yang S.-Y. Intracellular oxidation of allopregnanolone by human brain type 10 17beta-hydroxysteroid dehydrogenase. Brain Res. 2005;1040:29–35. doi: 10.1016/j.brainres.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Adamski J., Jakob F.J. A guide to 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2001;171:1–4. doi: 10.1016/s0303-7207(00)00383-x. [DOI] [PubMed] [Google Scholar]
- 10.Yang S.-Y., He X.-Y., Schulz H. Multiple functions of type 10 17β-hydroxysteroid dehydrogenase. Trends Endocrinol. Metab. 2005;16:167–175. doi: 10.1016/j.tem.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Yang S.-Y., He X.-Y., Miller D. HSD17B10: a gene involved in cognitive function through metabolism of isoleucine and neuroactive steroids. Mol. Genet. Metab. 2007;92:36–42. doi: 10.1016/j.ymgme.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Rauschenberger K., Schöler K., Sass J.O., Sauer S., Djuric Z., Rumig C., Wolf N.I., Okun J.G., Kölker S., Schwarz H., et al. A non-enzymatic function of 17β-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol. Med. 2010;2:51–62. doi: 10.1002/emmm.200900055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torroja L., Ortuño-Sahagún D., Ferrús A., Hämmerle B., Barbas J.A. Scully, an essential gene of Drosophila, is homologous to mammalian mitochondrial type II L-3-hydroxyacyl-CoA dehydrogenase/amyloid-β peptide-binding protein. J. Cell Biol. 1998;141:1009–1017. doi: 10.1083/jcb.141.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zschocke J., Ruiter J.P.N., Brand J., Lindner M., Hoffmann G.F., Wanders R.J.A., Mayatepek E. Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: a novel inborn error of branched-chain fatty acid and isoleucine metabolism. Pediatr. Res. 2000;48:852–855. doi: 10.1203/00006450-200012000-00025. [DOI] [PubMed] [Google Scholar]
- 15.Ensenauer R., Niederhoff H., Ruiter J.P.N., Wanders R.J.A., Schwab K.O., Brandis M., Lehnert W. Clinical variability in 3-hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. Ann. Neurol. 2002;51:656–659. doi: 10.1002/ana.10169. [DOI] [PubMed] [Google Scholar]
- 16.Olpin S.E., Pollitt R.J., McMenamin J., Manning N.J., Besley G., Ruiter J.P.N., Wanders R.J.A. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency in a 23-year-old man. J. Inherit. Metab. Dis. 2002;25:477–482. doi: 10.1023/a:1021251202287. [DOI] [PubMed] [Google Scholar]
- 17.Sutton V.R., O'Brien W.E., Clark G.D., Kim J., Wanders R.J.A. 3-Hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2003;26:69–71. doi: 10.1023/a:1024083715568. [DOI] [PubMed] [Google Scholar]
- 18.Sass J.O., Forstner R., Sperl W. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: impaired catabolism of isoleucine presenting as neurodegenerative disease. Brain Dev. 2004;26:12–14. doi: 10.1016/s0387-7604(03)00071-8. [DOI] [PubMed] [Google Scholar]
- 19.Poll-The B.T., Wanders R.J.A., Ruiter J.P.N., Ofman R., Majoie C.B.L.M., Barth P.G., Duran M. Spastic diplegia and periventricular white matter abnormalities in 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency, a defect of isoleucine metabolism: differential diagnosis with hypoxic-ischemic brain diseases. Mol. Genet. Metab. 2004;81:295–299. doi: 10.1016/j.ymgme.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Villoria J., Navarro-Sastre A., Fons C., Pérez-Cerdá C., Baldellou A., Fuentes-Castelló M.Á., González I., Hernández-Gonzalez A., Fernández C., Campistol J., et al. Study of patients and carriers with 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency: difficulties in the diagnosis. Clin. Biochem. 2009;42:27–33. doi: 10.1016/j.clinbiochem.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Perez-Cerdá C., Garcia-Villoria J., Ofman R., Sala P.R., Merinero B., Ramos J., Garcia-Silva M.T., Beseler B., Dalmau J., Wanders R.J.A., et al. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency: an X-linked inborn error of isoleucine metabolism that may mimic a mitochondrial disease. Pediatr. Res. 2005;58:488–491. doi: 10.1203/01.pdr.0000176916.94328.cd. [DOI] [PubMed] [Google Scholar]
- 22.Cazorla M.R., Verdu A., Perez-Cerda C., Ribes A. Neuroimage findings in 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency. Pediatr. Neurol. 2007;36:264–267. doi: 10.1016/j.pediatrneurol.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Zschocke J. HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J. Inherit. Metab. Dis. 2012;35:81–89. doi: 10.1007/s10545-011-9415-4. [DOI] [PubMed] [Google Scholar]
- 24.Lenski C., Kooy R.F., Reyniers E., Loessner D., Wanders R.J.A., Winnepenninckx B., Hellebrand H., Engert S., Schwartz C.E., Meindl A., et al. The reduced expression of the HADH2 protein causes X-linked mental retardation, choreoathetosis, and abnormal behavior. Am. J. Hum. Genet. 2007;80:372–377. doi: 10.1086/511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seaver L.H., He X.-Y., Abe K., Cowan T., Enns G.M., Sweetman L., Philipp M., Lee S., Malik M., Yang S.-Y. A novel mutation in the HSD17B10 gene of a 10-year-old boy with refractory epilepsy, choreoathetosis and learning disability. PLoS ONE. 2011;6:e27348. doi: 10.1371/journal.pone.0027348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukao T., Akiba K., Goto M., Kuwayama N., Morita M., Hori T., Aoyama Y., Venkatesan R., Wierenga R., Moriyama Y., et al. The first case in Asia of 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) with atypical presentation. J Hum Genet. 2014;59:609–614. doi: 10.1038/jhg.2014.79. [DOI] [PubMed] [Google Scholar]
- 27.Korman S.H. Inborn errors of isoleucine degradation: a review. Mol. Genet. Metab. 2006;89:289–299. doi: 10.1016/j.ymgme.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Holzmann J., Frank P., Löffler E., Bennett K.L., Gerner C., Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–474. doi: 10.1016/j.cell.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Rossmanith W. Of P and Z: mitochondrial tRNA processing enzymes. Biochim. Biophys. Acta. 2012;1819:1017–1026. doi: 10.1016/j.bbagrm.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossmanith W., Holzmann J. Processing mitochondrial (t)RNAs: new enzyme, old job. Cell Cycle. 2009;8:1650–1653. doi: 10.4161/cc.8.11.8502. [DOI] [PubMed] [Google Scholar]
- 31.Vilardo E., Nachbagauer C., Buzet A., Taschner A., Holzmann J., Rossmanith W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012;40:11583–11593. doi: 10.1093/nar/gks910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motorin Y., Helm M. RNA nucleotide methylation. WIREs RNA. 2011;2:611–631. doi: 10.1002/wrna.79. [DOI] [PubMed] [Google Scholar]
- 33.Deutschmann A.J., Amberger A., Zavadil C., Steinbeisser H., Mayr J.A., Feichtinger R.G., Oerum S., Yue W.W., Zschocke J. Mutation or knock-down of 17β-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum. Mol. Genet. 2014;23:3618–3628. doi: 10.1093/hmg/ddu072. [DOI] [PubMed] [Google Scholar]
- 34.Chatfield K.C., Coughlin C.R.2nd, Friederich M.W., Gallagher R.C., Hesselberth J.R., Lovell M.A., Ofman R., Swanson M.A., Thomas J.A., Wanders R.J.A., et al. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion. 2015;21:1–10. doi: 10.1016/j.mito.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ofman R., Ruiter J.P.N., Feenstra M., Duran M., Poll-The B.T., Zschocke J., Ensenauer R., Lehnert W., Sass J.O., Sperl W., et al. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency is caused by mutations in the HADH2 gene. Am. J. Hum. Genet. 2003;72:1300–1307. doi: 10.1086/375116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wittig I., Braun H.P., Schägger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 37.Vollers S.S., Teplow D.B., Bitan G. Determination of Peptide oligomerization state using rapid photochemical crosslinking. Methods Mol. Biol. 2005;299:11–18. doi: 10.1385/1-59259-874-9:011. [DOI] [PubMed] [Google Scholar]
- 38.Schrödinger L.L.C. The PyMOL Molecular Graphics System. 2010. Version 1.3r1. [Google Scholar]
- 39.Binstock J.F., Schulz H. Fatty acid oxidation complex from Escherichia coli. Methods Enzymol. 1981;71:403–411. doi: 10.1016/0076-6879(81)71051-6. [DOI] [PubMed] [Google Scholar]
- 40.Rossmanith W., Tullo A., Potuschak T., Karwan R., Sbisà E. Human mitochondrial tRNA processing. J. Biol. Chem. 1995;270:12885–12891. doi: 10.1074/jbc.270.21.12885. [DOI] [PubMed] [Google Scholar]
- 41.He X.-Y., Schulz H., Yang S.-Y. A human brain L-3-hydroxyacyl-coenzyme A dehydrogenase is identical to an amyloid β-peptide-binding protein involved in Alzheimer's disease. J. Biol. Chem. 1998;273:10741–10746. doi: 10.1074/jbc.273.17.10741. [DOI] [PubMed] [Google Scholar]
- 42.Kissinger C.R., Rejto P.A., Pelletier L.A., Thomson J.A., Showalter R.E., Abreo M.A., Agree C.S., Margosiak S., Meng J.J., Aust R.M., et al. Crystal structure of human ABAD/HSD10 with a bound inhibitor: implications for design of Alzheimer's disease therapeutics. J. Mol. Biol. 2004;342:943–952. doi: 10.1016/j.jmb.2004.07.071. [DOI] [PubMed] [Google Scholar]
- 43.Rötig A. Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta. 2011;1807:1198–1205. doi: 10.1016/j.bbabio.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 44.Pearce S., Nezich C.L., Spinazzola A. Mitochondrial diseases: translation matters. Mol. Cell. Neurosci. 2013;55:1–12. doi: 10.1016/j.mcn.2012.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.