


Abstract
DNA-end resection, the generation of single-stranded DNA at DNA double strand break (DSB) ends, is critical for controlling the many cellular responses to breaks. Here we show that the conserved DNA damage checkpoint sliding clamp (the 9-1-1 complex) plays two opposing roles coordinating DSB resection in budding yeast. We show that the major effect of 9-1-1 is to inhibit resection by promoting the recruitment of Rad953BP1 near DSBs. However, 9-1-1 also stimulates resection by Exo1- and Dna2-Sgs1-dependent nuclease/helicase activities, and this can be observed in the absence of Rad953BP1. Our new data resolve the controversy in the literature about the effect of the 9-1-1 complex on DSB resection. Interestingly, the inhibitory role of 9-1-1 on resection is not observed near uncapped telomeres because less Rad953BP1 is recruited near uncapped telomeres. Thus, 9-1-1 both stimulates and inhibits resection and the effects of 9-1-1 are modulated by different regions of the genome. Our experiments illustrate the central role of the 9-1-1 checkpoint sliding clamp in the DNA damage response network that coordinates the response to broken DNA ends. Our results have implications in all eukaryotic cells.
INTRODUCTION
Double strand breaks (DSBs) are one of the most toxic forms of DNA damage because both strands of DNA are damaged and therefore there is no undamaged strand of DNA available to act as a template for repair. Spontaneous or environmentally induced DSBs may cause cell death, genetic instability and cancer. On the other hand, programmed DSBs stimulate meiotic recombination and the genetic rearrangements important for immune system maturation in mammals or mating type switches in yeasts. It is therefore critical to understand how the cellular response to DSBs is coordinated in different contexts.
The eukaryotic DNA damage response (DDR) network coordinates responses to damaged DNA by regulating DNA repair, checkpoint-dependent cell cycle arrest and transcription (1,2). At DSBs, in yeast/mammalian cells the DDR initiates when the MRX/MRN (Mre11-Rad50-Xrs2/MRE11-RAD50-NBS1) complex binds to ‘blunt’ DSBs and activates the transducer checkpoint kinase Tel1/ATM. MRX also controls nuclease activities that help generate single-stranded DNA (ssDNA), another stimulus for DNA damage checkpoint activation.
The generation of ssDNA at DSBs by the process of resection is critically important for determining cellular responses to DSBs (3). In particular, resection affects DNA repair pathway choice by either non-homologous end joining (NHEJ, which requires limited or no resection) or homology-directed repair (HDR, which requires extensive resection) (4–7). Resection also controls the switch between blunt dsDNA-end-activated Tel1/ATM signalling and ssDNA-activated Mec1-Ddc2/ATR-ATRIP signalling (8–10).
Resection at DSBs is initiated by MRXMRN-Sae2CtIP, whereas more extensive DNA resection requires the nuclease Exo1 and/or the nuclease/helicase pair of Dna2 and Sgs1BLM (3,11). It is critical that Exo1 and Dna2-Sgs1 resection activities are properly regulated because excess levels of ssDNA are potentially very harmful to cell survival by inducing cell death and mutation (12–17).
The conserved, proliferating cell nuclear antigen related 9-1-1 checkpoint sliding clamp is composed of three subunits Ddc1/RAD9, Mec3/HUS1 and Rad17/RAD1. The clamp is loaded onto DNA by the Rad24-RFC (RAD17-RFC) clamp loader complex and is one of the earliest sensors recruited to sites of DNA damage (18,19). Once loaded onto the DNA, the 9-1-1 clamp performs at least three functions to activate checkpoint kinase cascades. First, 9-1-1 activates Mec1 directly via the unstructured C terminal domain of Ddc1. Second, 9-1-1 recruits Dpb11/TopBP1 near DNA lesions via phosphorylated Ddc1 (20). Third, the 9-1-1 clamp and the clamp loader Rad24-RFC promote resection (21–23).
Recently, we showed that 9-1-1 promotes resection by stimulating both Dna2-Sgs1- and Exo1-dependent resection near uncapped telomeres in yeast cells, in vivo, and using human proteins in vitro (Figure 1A) (24). We suggested that the binding of the 9-1-1 complex near sites of resection served to initiate a positive feedback loop to generate ssDNA for DNA damage checkpoint maintenance. Importantly, we showed that the human 9-1-1 complex stimulated the activities of DNA2 and EXO1 on DNA substrates in vitro (24), showing that the effect of 9-1-1 is direct and conserved. However, the effects of the 9-1-1 complex on resection in vivo, in budding yeast, have nearly all been shown using telomere-defective, cdc13-1 mutants. Whether 9-1-1 affects resection at DSBs in vivo remained unclear. One study reported that Rad17 had no role in DSB resection (25), whereas another found that Rad24 stimulated resection (26).
Figure 1.
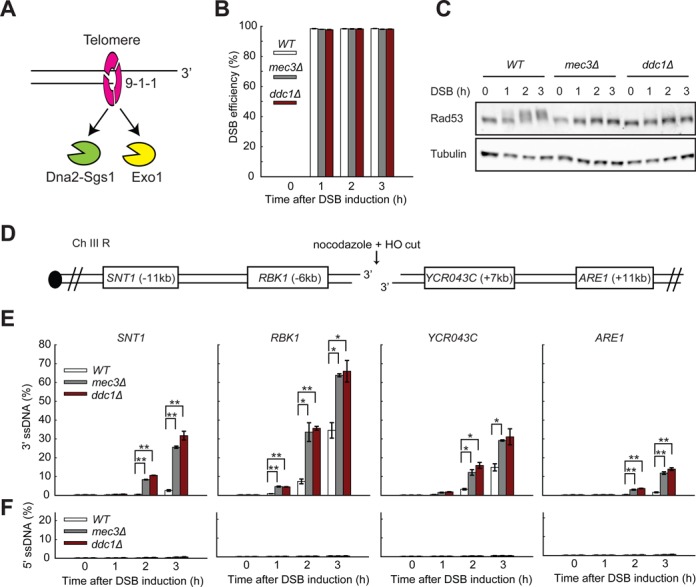
The 9-1-1 complex inhibits resection of DSBs. (A) The 9-1-1 complex promotes resection at uncapped telomeres by stimulating Exo1 and Dna2-Sgs1 recruitment to ssDNA (model based on Ngo et al. (24)). (B) The efficiencies of DSB induction at the MAT locus. (C) Yeast strains of the indicated genotypes were subjected to western blot analysis with anti-Rad53 and anti-tubulin antibodies following DSB induction. (D) Map of chromosome III showing an HO endonuclease site and loci examined in this study. (E,F) Analysis of 3′ ssDNA and 5′ ssDNA accumulation in JKM179 strains (see strain list in Supplementary Tables S1 and S2) at the indicated loci. All DSB experiments were performed in nocodazole arrested cells. The data plotted are the means and the range from two strains. P-values were calculated using two-tailed unpaired T test. *P < 0.05, **P < 0.01.
To clarify how the 9-1-1 complex affects resection at DSBs, in vivo, we have now analysed resection at DSBs in non-dividing yeast cells. Examination of non-dividing, nocodazole-arrested cells ensured that any effects of 9-1-1 were direct rather than indirectly caused by differences in cell cycle position between strains. Our experiments, in which we inactivated as many as four independent components of the DDR network, show that the 9-1-1 complex plays a complex role at DSBs. Our major finding is that 9-1-1 inhibits DSB resection by promoting the recruitment of Rad953BP1 near DSBs. In addition, in the absence of Rad9, we found that 9-1-1 stimulates DSB resection by both Dna2-Sgs1 and Exo1, as at telomeres. The role of 9-1-1 inhibiting resection is not observed near uncapped telomeres because less Rad953BP1 is recruited near uncapped telomeres in comparison to near DSBs. Our data illustrate the central role played by the conserved 9-1-1 complex in the network coordinating DNA resection and checkpoint activation at DNA ends.
MATERIALS AND METHODS
Yeast strains and plasmids
All experiments were performed on Saccharomyces cerevisiae JKM179 or W303 background strains (Supplementary Tables S1 and S2).
DNA damage induction and quantitative amplification of single-stranded DNA
For DSB studies, yeasts were first arrested in Yeast Extract Peptone (YEP) 3% lactate (supplemented with adenine) with 15 μg/ml nocodazole for 3 h at 30°C before addition of galactose (to a final concentration of 2%) to induce HO-endonuclease. We determined that few cells escaped nocodazole arrest by counting 4′, 6-diamidino-2-phenylindole stained cells. For telomere uncapping studies, cdc13-1 strains in Yeast Extract Peptone Dextrose (YEPD) were arrested in G1 with alpha factor at 23°C and released into 36°C. Quantitative Amplification Of Single-stranded DNA (QAOS) analyses at uncapped telomeres and DSBs were carried out as previously described (27–29), and in the DSB studies the ssDNA amounts were normalized to DSB efficiencies as previously described (30). The primers used for QAOS at DSBs are listed in Supplementary Tables S3 and S4. The efficiencies of DSB induction at the MAT and URA3 loci were determined using primer pairs which span the HO recognition sites (Supplementary Table S4), normalized to amount of DNA at PAC2, a control locus.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described (31). Briefly, yeast cultures were treated with formaldehyde (1% final) for 15 min before glycine (0.32% final) was added. The cells were washed twice in Tris-buffered saline (TBS) buffer, once in FA lysis buffer (pH 7.5, 50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 0.1% (w/v) sodium deoxycholate and 0.1% (w/v) sodium dodecylsulphate [SDS]), spun down and frozen at −80°C. Cell pellets were lysed in FA lysis buffer (with 2 mM phenylmethylsulfonyl fluoride, PMSF) by bead beating using a Precellys 24 homogenizer (Precellys). The cell lysates were incubated with anti-HA (ab9110, Abcam) or anti-Myc (ab32, Abcam) antibodies together with protein G Dynabeads (Invitrogen) on a wheel at 4°C overnight. The immunoprecipitates were washed five times in FA lysis buffer and eluted in ChIP elution buffer (50 mM Tris·Cl, pH 7.5, 10 mM EDTA and 1% (w/v) SDS). The samples were treated with proteinase K and incubated overnight at 62°C to reverse the crosslinks. Immunoprecipitated DNA was purified using PCR purification kit (QIAGEN) and quantified using the SYBR Green qPCR SuperMIX-UDG w/ROX kit (11744500, Invitrogen) by a SteponePlus qPCR machine (Life Technologies; all samples were also normalized by input DNA, quantified on the same plates).
Western Blots
Protein extracts were prepared by trichloroacetic acid (TCA) precipitation. Briefly, cells were resuspended in 10% TCA and mechanically broken using glass beads. Protein suspensions in Laemmli buffer were boiled for 3 min, spun down for 10 min and the supernatant were loaded onto 7.5% Mini-PROTEAN TGX Gels (Bio-Rad). The proteins were transferred to Hybond-ECL membranes (GE Healthcare) and probed with anti-Rad53 (sc-6749, Santa Cruz) and anti-tubulin antibodies (from Keith Gull, Oxford University).
RESULTS
The 9-1-1 complex inhibits resection at DSBs
We have shown that the 9-1-1 complex stimulates Exo1- and Dna2-Sgs1-dependent resection in vivo and in vitro (Figure 1A) (24). To extend these studies to DSBs in vivo, we deleted MEC3 and DDC1 (encoding two subunits of the 9-1-1 complex) in the widely used JKM179 genetic background (25). In the JKM179 strain background, galactose-inducible expression of the HO endonuclease leads to a DSB being induced at the MAT locus of chromosome III. Additionally, the JKM179 strain lacks HML and HMR, the silent mating type donor loci, and therefore cannot repair the DSB by gene conversion. 9-1-1 mutants are checkpoint deficient (32), and therefore we performed all the experiments in the presence of the spindle poison nocodazole to arrest cells in M phase and to ensure that any differences in ssDNA measurement were not affected by different cell cycle positions in different strains (33,34).
We arrested wild-type, mec3Δ and ddc1Δ strains for 3 h in nocodazole before adding galactose to induce HO expression. We found that DSBs were induced with high efficiencies, reaching over 95% in all strains 1 h after addition of galactose (Figure 1B). DSB induction results in the phosphorylation and activation of the downstream checkpoint effector kinase Rad53 (35). As expected, 2 h following DSB induction, we observed strong DNA damage checkpoint activation in the wild-type strain, as shown by a mobility shift of Rad53 (Figure 1C). Consistent with an essential role of 9-1-1 in activating the DSB-induced checkpoint, we observed no Rad53 phosphorylation in mec3Δ or ddc1Δ mutants (Figure 1C) (25,36). We conclude that, as expected, the 9-1-1 complex does not affect HO-induced DSB formation and is required for DSB-induced DNA damage checkpoint activation.
To examine resection, we measured ssDNA accumulation at loci on both sides of the break using QAOS (37) (Figure 1D and E, and Supplementary Figure S1A). In wild-type cells, we observed the accumulation of detectable levels of 3′ ssDNA, 6kb and 7kb from the break sites at RBK1 and YCR043C, by 2 h after DSB induction (Figure 1E). As expected, more distal to the break, 11kb away, at SNT1 and ARE1 ssDNA was detected later in the wild-type (WT) strains, by 3 h (Figure 1E). Our data show that the rate of DSB resection in the wild-type strains is about 3.5 kb/h (11 kb/3 h at SNT1/ARE1, 6–7 kb/2 h at RBK1/YCR043C and 9 kb/3 h at ELO2), consistent with previous findings (38). As expected, we did not observe 5′ ssDNA at any locus in all the strains (Figure 1F). Our results also show that even though the DSB efficiency is almost 100%, the level of 3′ ssDNA only reached 15–35% in the WT strains after 3 h at YCR043C and RBK1, consistent with previous studies (34,39,40). This finding suggests that only a fraction of breaks undergo resection or that not all breaks are resected at fast rates. Intriguingly, we also noticed an asymmetry in resection at either side of the break, with 3′ ssDNA generation being more efficient on the left than on the right side of the break (Figure 1E). This phenomenon has been observed at the MAT locus with Rad51 binding (a marker for ssDNA) more efficiently to the left side of the break in some experiments (41), but the mechanism underlying this asymmetry remains unclear.
Interestingly, and in contrast to our observations at uncapped telomeres, the 9-1-1 complex inhibited, rather than stimulated resection at DSBs (Figure 1E versus Supplementary Figure S1B) (21,23,24). For example at 3 h, at SNT1, about 11 kb from the DSB, less than 3% ssDNA was observed in WT cells but over 20% was observed in mec3Δ and ddc1Δ mutants (Figure 1E). By T test, this was statistically significant (Figure 1E). In addition, the statistically significant difference we detected between WT and 9-1-1 mutants, at the SNT1 locus at 3 h, is replicated at the SNT1 locus at 2 h, and at RBK1, YCR043C and ARE1 at all time points when ssDNA is present. In contrast to at DSBs, near uncapped telomeres, mec3Δ mutants generally showed less than half the ssDNA observed in WT cells at all loci and times (Supplementary Figure S1B). Interestingly, the rate of telomere resection near telomeres in wild-type strains is about 8 kb/h (30 kb/4 h, see DUG1) about twice as fast as at DSBs. This suggests that the telomere environment is more permissive for resection than other chromosomal locations.
In telomere uncapping experiments, checkpoint-deficient mec3Δ cells were held in late mitosis by cdc15-2-induced arrest (Supplementary Figure S1B), whereas in DSB experiments (Figure 1), mec3Δ cells were arrested earlier in mitosis by nocodazole. Therefore, one trivial reason to explain the contradictory conclusions drawn about the role of 9-1-1 on resection was that the effects of 9-1-1 were affected by cell cycle position (cdc15-2 versus nocodazole). To address this issue we chose to uncap telomeres of cdc13-1 cells in nocodazole-arrested cells. We performed these experiments at low temperatures of 32 and 34°C because nocodazole does not maintain arrest very well at higher temperatures (42) and because cdc13-1 still induces efficient uncapping at these lower temperatures (24). We observed less telomeric ssDNA generation in nocodazole-arrested cells in comparison with cdc15-2-arrested cells (Supplementary Figure S2 versus Supplementary Figure S1B). The low levels of ssDNA in cdc13-1 mutants in nocodazole has been reported before and is likely due to lower level of telomere uncapping in cells that have not gone through S phase (43). Nevertheless, in nocodazole, cdc13-1 mec3Δ cells still accumulated less ssDNA than cdc13-1 cells (Supplementary Figure S2). This contrasts to what is observed at DSBs in nocodazole where mec3Δ cells accumulate higher levels of ssDNA. We conclude that at uncapped telomeres 9-1-1 stimulates extensive resection (21,23,24) whereas at DSBs the 9-1-1 complex inhibits DNA resection.
The 9-1-1 complex inhibits Dna2-Sgs1 but stimulates Exo1 at DSBs
To begin to understand how 9-1-1 inhibits DSB resection, we examined the interplay between 9-1-1 and Dna2-Sgs1 or Exo1, the two major nuclease activities involved in extensive resection. We deleted MEC3 in exo1Δ, sgs1Δ or sgs1Δ exo1Δ mutant backgrounds and compared ssDNA levels in single, double and triple mutants (Figure 2A–C). In agreement with published results (5,9,38), we found that extensive DSB resection is totally dependent on Dna2-Sgs1 and Exo1 (compare WT with sgs1Δ exo1Δ strains, Figure 2A). Importantly the mec3Δ mutation did not permit nucleases other than Dna2-Sgs1 and Exo1 to generate ssDNA since mec3Δ sgs1Δ exo1Δ and sgs1Δ exo1Δ strains each generated similarly low amounts of ssDNA (Figure 2A).
Figure 2.
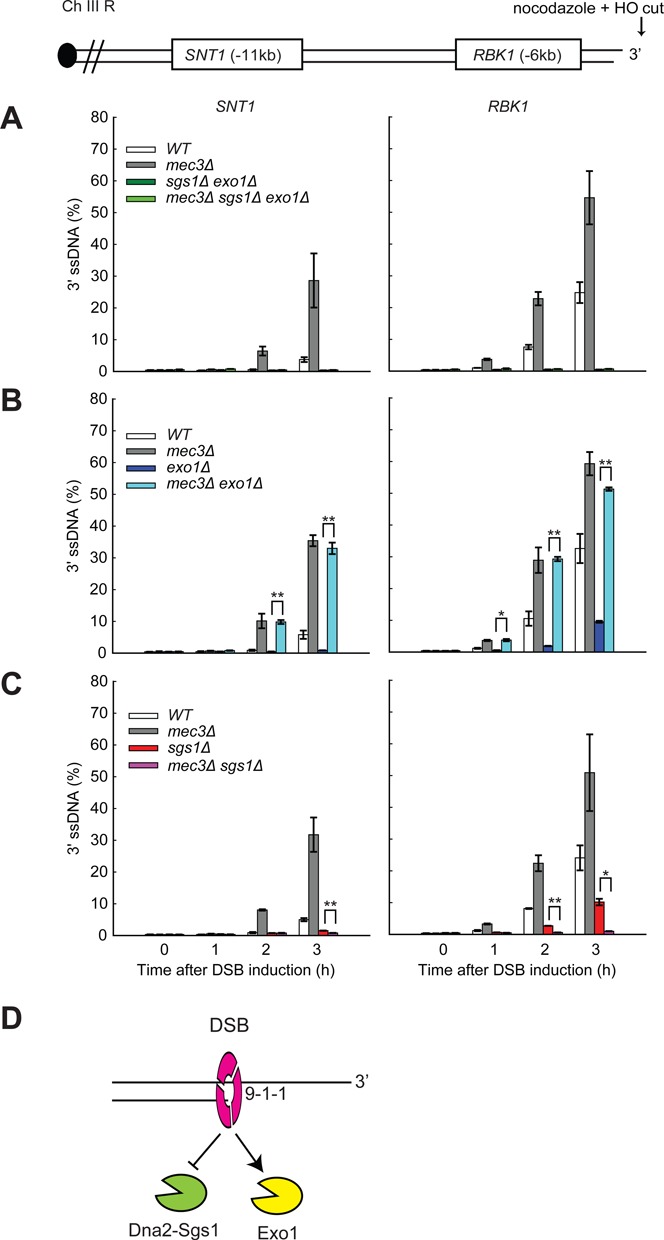
The 9-1-1 complex inhibits Dna2-Sgs1 but stimulates Exo1 at DSBs. (A-C) Analysis of 3′ ssDNA accumulation in JKM179 strains at the indicated loci. The data plotted and the P-values are as described in Figure 1. (D) The 9-1-1 complex inhibits Dna2-Sgs1 but stimulates Exo1 at DSBs.
Interestingly, we observed differing effects of Mec3 on Exo1- or Dna2-Sgs1-dependent resection. Figure 2B shows that Exo1 is critical for generating ssDNA 6 kb and 11 kb from the DSB, with much lower levels of ssDNA being detected in exo1Δ versus WT strains. We found that mec3Δ mutation strongly increased resection in exo1Δ strains (compare mec3Δ exo1Δ with exo1Δ strains). Since in exo1Δ strains resection is totally dependent on Dna2-Sgs1, we can conclude that the major inhibitory effect of Mec3 on resection is to inhibit Dna2-Sgs1-dependent nuclease activity.
We also examined how mec3Δ affects resection in sgs1Δ strains, where all the resection is due to Exo1. sgs1Δ, like exo1Δ, strongly reduced ssDNA in comparison with WT cells (compare the effects of Sgs1 and Exo1 at the RBK1 locus, in Figure 2C and B). The comparison allows us to conclude that Exo1 and Dna2-Sgs1 make similar contributions to resection in WT (MEC3) strains. Remarkably, however, 9-1-1 had opposite effects in sgs1Δ versus exo1Δ strains. mec3Δ largely eliminated ssDNA accumulation in sgs1Δ strains (compare mec3Δ sgs1Δ with sgs1Δ strains, Figure 2C), in stark contrast to the effect in exo1Δ strains where mec3Δ enhanced ssDNA accumulation (Figure 2B). To confirm the effects of Mec3 in the absence of Dna2/Sgs1, we also examined the effect of Mec3 in dna2Δ (pif1Δ background) strains (Supplementary Figure S3A), as dna2Δ and sgs1Δ mutations show similar effects on DSB resection (38). Importantly, mec3Δ largely eliminated ssDNA accumulation in dna2Δ strains (compare mec3Δ dna2Δ with dna2Δ strains, Supplementary Figure S3A). Since in sgs1Δ (and dna2Δ) strains resection is totally dependent on Exo1, we conclude that the 9-1-1 complex stimulates Exo1-dependent resection at DSBs.
Overall, examination of DSB resection in exo1Δ or sgs1Δ/dna2Δ mutants shows that 9-1-1 plays a positive role on Exo1-dependent resection but a negative role on Dna2-Sgs1-dependent resection (Figure 2D). We suggest that at DSBs the role of 9-1-1 in inhibiting Dna2-Sgs1-dependent resection predominates over its role in stimulating Exo1 and that this explains why 9-1-1 inactivation leads to increased DSB resection. In contrast at uncapped telomeres, 9-1-1 contributes more to stimulating resection than inhibiting resection (21,23,24).
The 9-1-1 complex recruits more Rad953BP1 near DSBs than uncapped telomeres
It was unclear why the effects of 9-1-1 on resection were so dramatically different at DSBs versus uncapped telomeres. One of the functions of 9-1-1 is to activate the central transducer kinase Mec1 (20). A recent study suggests that active Mec1 inhibits resection by promoting the recruitment of Rad953BP1 to DSBs (34). The clamp loader Rad24RAD17 also stimulates Rad953BP1 binding to DSBs (44). We therefore hypothesized that 9-1-1 might, like Mec1, inhibit DSB resection by promoting Rad953BP1 recruitment.
To test the effect of 9-1-1 on the recruitment of Rad953BP1, we performed ChIP experiments to examine the binding of Rad953BP1 near sites of damage. Consistent with published results (34,44), we detected Rad953BP1 recruitment to sites flanking the DSBs but not to a control locus, PDA1, 3 h following DSB induction (Figure 3A, Supplementary Figure S3B and SC). Importantly, mec3Δ and ddc1Δ strains each had much less Rad953BP1 binding at these loci, showing that 9-1-1 does indeed contribute to the recruitment of Rad953BP1 to DSBs. These ChIP data are consistent with the role of the clamp loader Rad24RAD17 in stimulating Rad953BP1 recruitment to DSBs (44). As Rad953BP1 is a strong inhibitor of DSB resection (33,34,45), the reduced Rad953BP1 binding in mec3Δ and ddc1Δ strains (6–8 fold Figure 3A) could readily account for the hyper resection phenotype observed in mec3Δ and ddc1Δ strains at DSBs (Figure 1E).
Figure 3.
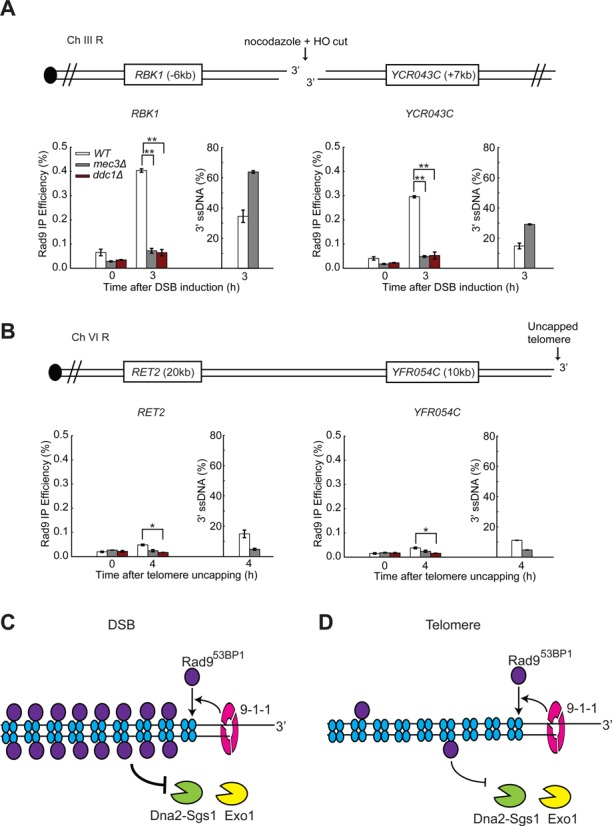
The 9-1-1 complex recruits more Rad953BP1 near DSBs than uncapped telomeres. (A, B) ChIP analyses of Rad9-HA recruitment near DSBs (A) and uncapped telomeres (B). The ssDNA data were taken from Figure 1E and Supplementary Figure S1B. The data plotted and the P-values are as described in Figure 1. (C, D) The 9-1-1 complex stimulates the recruitment of Rad953BP1 which inhibits Dna2-Sgs1 and Exo1, but Rad953BP1 binds more poorly to uncapped telomeres.
One possibility why 9-1-1 had different effects on resection at telomeres versus DSBs was that Rad953BP1 binds more poorly near telomeres compared to DSBs, perhaps due to the nature of the chromatin at telomeric and sub-telomeric regions. Rad953BP1 binds chromatin via phosphorylated H2A serine 129 and methylated H3 lysine 79 and the levels of these chromatin modifications are affected near telomeres (46–48). To test the hypothesis that Rad9 binds at different levels, we performed ChIP experiments to examine Rad953BP1 binding near uncapped telomeres and at DSBs (Figure 3B versus Figure 3A). To normalize for the amount of DNA damage, which might affect Rad953BP1 binding, we examined loci and times with comparable levels of ssDNA (Figure 3A and B). In support of the hypothesis, we observed much less Rad953BP1 recruitment to the telomeric RET2 and YFR054C loci after telomere uncapping compared to the DSB loci, RBK1 and YCR043C (compare wild type in Figure 3B and A). Even so, the binding of Rad953BP1 near uncapped telomeres was reduced in mec3Δ and ddc1Δ mutants (Figure 3B, Supplementary Figure S3D), showing that 9-1-1 also contributes to the recruitment of Rad953BP1 following telomere uncapping. Together our data suggest that 9-1-1 inhibits DSB resection by recruiting Rad953BP1 (Figure 3C). However, at uncapped telomeres less Rad953BP1 is recruited, permitting 9-1-1 to play a stimulatory role on resection (Figure 3D).
The 9-1-1 complex stimulates DSB resection in rad9Δ cells
To test the hypothesis that Rad953BP1 affects 9-1-1-dependent resection, we examined resection at DSBs in the complete absence of Rad953BP1. Consistent with results previously reported (33,34,45), Rad953BP1 inhibited resection at DSBs, since rad9Δ strains showed greatly increased levels of 3′ ssDNA at all four loci examined (Figure 4A). Interestingly, rad9Δ caused a greater increase in ssDNA than mec3Δ, at all loci and all times examined (Figure 4A), showing that the effects of Rad953BP1 were stronger. Importantly, we also observed that rad9Δ mec3Δ strains accumulated intermediate levels of ssDNA, more like mec3Δ than rad9Δ strains (Figure 4A). Thus, mec3Δ is epistatic to rad9Δ in this context. These data show that at DSBs, just as at uncapped telomeres (21,23,24), the very high levels of ssDNA that accumulate in rad9Δ strains are dependent on the 9-1-1 complex. The role of 9-1-1 seems to be particularly important in supporting extensive resection in rad9Δ strains as the effect of 9-1-1 is more obvious further from the break, at SNT1 and ARE1, 11 kb from the break (Figure 4A).
Figure 4.
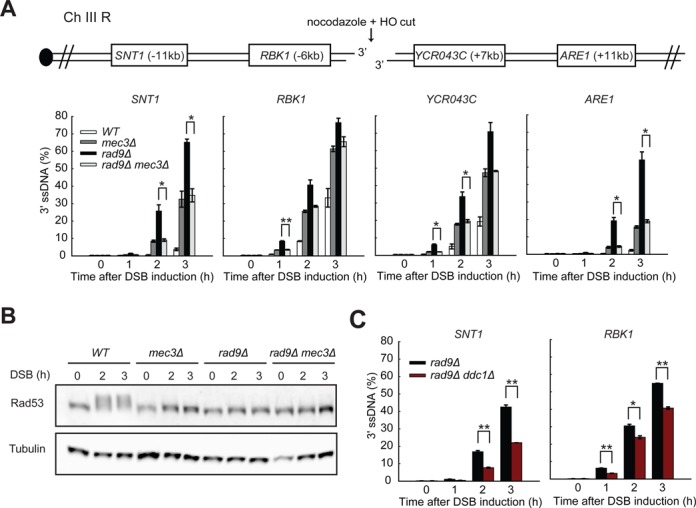
The 9-1-1 complex stimulates resection in rad9Δ cells. (A,C) Analysis of 3′ ssDNA accumulation in JKM179 strains at the indicated loci. The data plotted and the P-values are as described in Figure 1. (B) Yeast strains of the indicated genotypes were subjected to western blot analysis with anti-Rad53 and anti-tubulin antibodies following DSB induction.
We believe that the resection stimulatory role of 9-1-1 is independent of checkpoint signalling since mec3Δ, rad9Δ and rad9Δ mec3Δ mutants are all completely checkpoint defective as detected by the Rad53 phosphorylation assay (Figure 4B). We also conclude that the 9-1-1 complex per se, rather than Mec3 specifically, is important for stimulating resection in rad9Δ mutants because Ddc1, like its partner Mec3, stimulates resection (Figure 4C). Together, the results in Figures 3 and 4 suggest that 9-1-1 catalyses efficient Rad953BP1 recruitment near DSBs and this inhibits resection. However, in rad9Δ strains, 9-1-1 stimulates resection at DSBs, just as at uncapped telomeres.
The 9-1-1 complex stimulates Exo1- and Dna2-Sgs1-dependent resection in rad9Δ cells
The 9-1-1 complex promotes resection at uncapped telomeres in vivo, where Rad953BP1 binding is low (Figure 3B), and in vitro (in the absence of Rad953BP1) by stimulating both Dna2-Sgs1- and Exo1-dependent resection (24). To test whether 9-1-1 stimulates Dna2-Sgs1- and Exo1-dependent resection at DSBs in vivo, we deleted MEC3 in rad9Δ exo1Δ, rad9Δ sgs1Δ, rad9Δ dna2Δ or rad9Δ sgs1Δ exo1Δ genetic backgrounds and measured ssDNA accumulation (Figure 5A–C, Supplementary Figure S4). In agreement with the results in Figure 4A, mec3Δ decreased resection in all rad9Δ strains following the induction of DSBs (Figure 5A–C, compare rad9Δ and rad9Δ mec3Δ at SNT1). Consistent with the results in RAD9+ strains, deletion of EXO1 or SGS1/DNA2 reduced resection in rad9Δ strains and deletion of both SGS1 and EXO1 completed eliminated resection (Figure 5A–C, Supplementary Figure S4). We conclude that all resection in rad9Δ strains is dependent on Dna2-Sgs1 or Exo1. This suggests that other nucleases, such as MRX-Sae2, do not become hyperactive in the absence of Rad953BP1.
Figure 5.
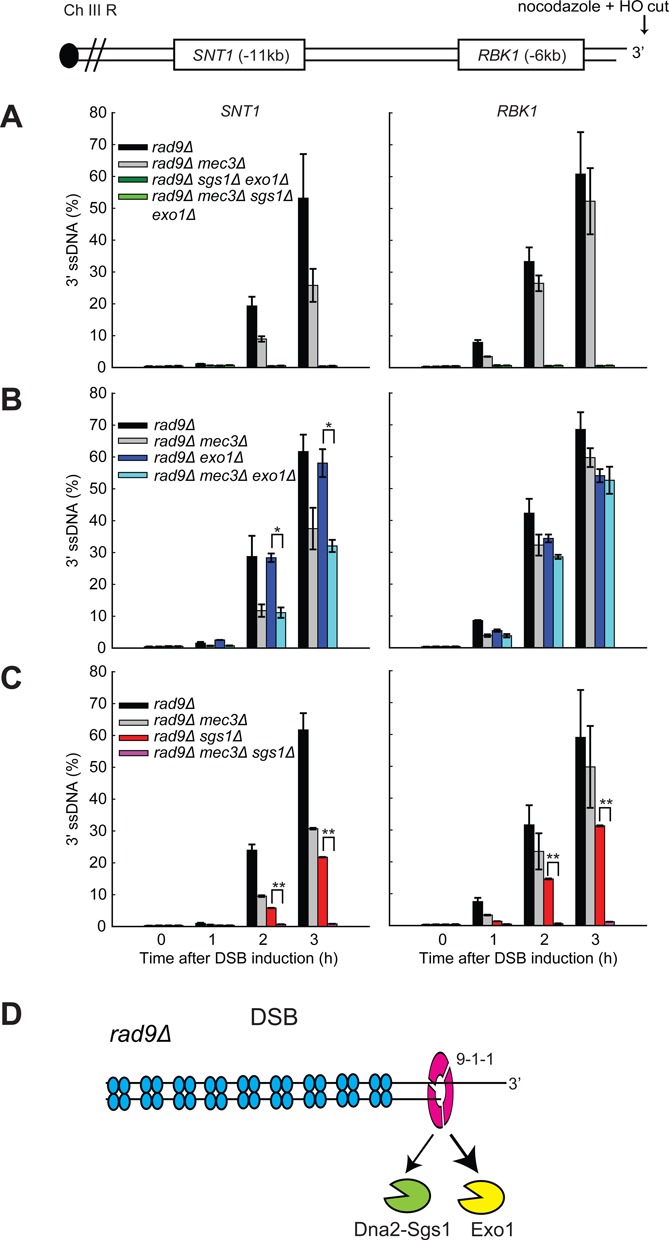
The 9-1-1 complex stimulates both Exo1- and Dna2-Sgs1-dependent resection in rad9Δ cells. (A-C) Analysis of 3′ ssDNA accumulation in rad9Δ background strains at the indicated loci. The data plotted and the P-values are as described in Figure 1. (D) The 9-1-1 complex stimulates both Exo1 and Dna2-Sgs1 in rad9Δ cells.
Interestingly, resection in rad9Δ strains (as in mec3Δ strains, Figure 2) appears to be more dependent on Dna2-Sgs1 than Exo1 (compare rad9Δ exo1Δ strains in Figure 5B with rad9Δ dna2Δ strains in Figure 5C, and in Supplementary Figures S5 and S6). This contrasts to what is observed in WT strains where resection is more or less equally dependent on Exo1 or Dna2-Sgs1 (Figure 2). These comparisons suggest that Rad953BP1, like 9-1-1 (Figure 2), inhibits Dna2-Sgs1 more than Exo1.
To determine whether 9-1-1 promotes Dna2-Sgs1-dependent resection in the absence of Rad953BP1, we examined how mec3Δ affects ssDNA accumulation in rad9Δ exo1Δ strains, where all resection is due to Dna2-Sgs1. It was clear that the mec3Δ mutation reduced resection in rad9Δ exo1Δ strains (Figure 5B, compare rad9Δ mec3Δ exo1Δ with rad9Δ exo1Δ strains at SNT1). The effect of 9-1-1 on Dna2-Sgs1 is more clearly seen at loci further away from the breaks likely because in the absence of 9-1-1, Sgs1-Dna2 can still resect DNA, but not as efficiently. We conclude that 9-1-1 facilitates Dna2-Sgs1-dependent resection at DSBs in rad9Δ strains.
To determine whether 9-1-1 promotes Exo1-dependent resection in the absence of Rad953BP1, we examined how mec3Δ affects ssDNA accumulation in rad9Δ sgs1Δ or rad9Δ dna2Δ strains, where all resection is due to Exo1. Similar to the results obtained in RAD9+ sgs1Δ or RAD9+ dna2Δ strains (Figure 2C, Supplementary Figure S3A), the mec3Δ mutation completely eliminated resection in rad9Δ sgs1Δ or rad9Δ dna2Δ strains (Figure 5C, compare rad9Δ mec3Δ sgs1Δ with rad9Δ sgs1Δ strains, Supplementary Figure S4, compare rad9Δ mec3Δ dna2Δ with rad9Δ dna2Δ strains), confirming that 9-1-1 is critical for Exo1-dependent resection in rad9Δ strains. The observation that rad9Δ mec3Δ sgs1Δ or rad9Δ mec3Δ dna2Δ strains had much less ssDNA than rad9Δ mec3Δ exo1Δ strains (Figure 5B and C, Supplementary Figure S5 and S6) shows that Exo1 is more dependent on 9-1-1 activity than Dna2-Sgs1. We conclude that the 9-1-1 complex stimulates both Dna2-Sgs1- and Exo1-dependent resection, but that Exo1 is more dependent on 9-1-1 (Figure 5D).
The 9-1-1 complex affects the binding of both Exo1 and Dna2-Sgs1 to DNA
To better understand how the 9-1-1 complex and Rad953BP1 affect resection by Dna2-Sgs1 and Exo1, we examined the effects of Mec3 and Rad953BP1 on the binding of Dna2 and Exo1 near DSB sites (Figure 6A and B). We found that rad9Δ strongly increased the binding of Dna2 and Exo1 to ARE1 and SNT1 near a DSB, but not to a control locus PDA1 (compare WT and rad9Δ, Figure 6A and B). This observation suggests that Rad953BP1 inhibits the binding of both Dna2 and Exo1 to DSBs. mec3Δ also increased binding of Dna2 and Exo1 to ARE1 and SNT1 (compare mec3Δ and WT, Figure 6A and B), likely due to decreased Rad953BP1 recruitment to DSBs (Figure 3A). Importantly, rad9Δ mec3Δ strains had reduced binding of Dna2 and Exo1 to these DSB loci compared to rad9Δ strains (Figure 6A and B, Supplementary Figure S7A), suggesting that 9-1-1 stimulates Dna2-Sgs1 and Exo1 recruitment (and resection) in rad9Δ cells.
Figure 6.
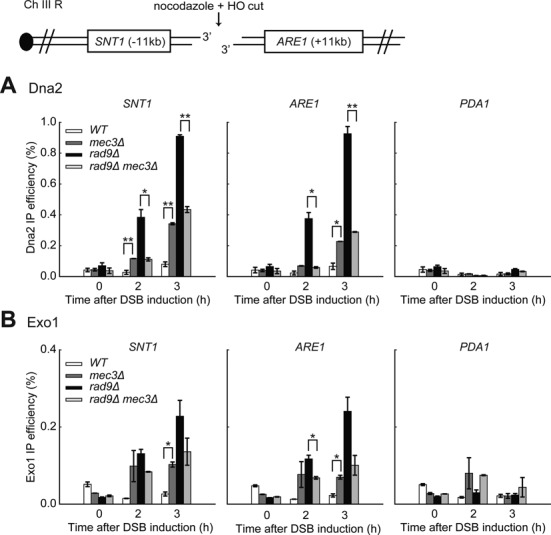
The 9-1-1 complex affects the binding of both Exo1 and Dna2-Sgs1 to DNA. (A,B) ChIP analysis of Dna2-Myc and Exo1-Myc recruitment to DSBs in JKM179 background strains. The data plotted and the P-values are as described in Figure 1.
Our previous biochemical studies showed that the 9-1-1 complex directly stimulates the activities of DNA2 and EXO1 (24). Consistent with this hypothesis, we observed increased binding of Mec3 and Ddc1 (the two subunits of the 9-1-1 complex) near DSBs in rad9Δ strains (Supplementary Figure S7B and SC), which explains the increased recruitment of Dna2 and Exo1 in this mutant background. Collectively, the data in Figure 6 suggest that the 9-1-1 complex affects Dna2-Sgs1 and Exo1-dependent resection by regulating the recruitment of these nuclease and helicase activities near DSBs.
The 9-1-1 complex coordinates DSB resection at another internal locus
We find that the 9-1-1 complex stimulates resection at an HO-induced DSB but other groups have reported that 9-1-1 showed no effect or inhibited resection (25,26). Therefore to clarify the role of 9-1-1 in DSB resection, we examined resection after DSB induction at another locus in a different chromosome and in a different genetic background. We used a strain where an HO DSB can be induced at the URA3 locus on chromosome V (26). As before, we performed all the experiments in the presence of nocodazole to ensure that any effects we observed were not affected by cell cycle position. We found that at URA3, in W303 strains, DSBs were induced with lower efficiencies compared to the MAT locus in JKM179 strains, reaching just over 60% in all strains 1 h after addition of galactose, and 90% after 3 h (compare Supplementary Figure S7D with Figure 1). However, mec3Δ did not affect DSB formation (Supplementary Figure S7D).
To examine resection, we measured ssDNA accumulation at loci on both sides of the break using QAOS (Figure 7A). Interestingly, we found that resection at URA3 generated more ssDNA (60% at 5 kb after 3 h in the wild-type strains) than at the MAT locus (35% at 6 kb after 3 h), possibly suggesting that a higher fraction of breaks were resected or resected at fast rates at the URA3 locus in W303 strains. Importantly, in our hands, mec3Δ strains consistently showed higher levels of ssDNA compared to the wild type (Figure 7B). This result is consistent with our finding at the MAT locus (Figure 1), but contradicts Aylon and Kupiec (26), who showed that rad24Δ mutants, which cannot load the 9-1-1 complex on DNA, had less resection. We believe this difference arises likely because Aylon and Kupiec performed their resection experiments in asynchronous cultures, and rad24Δ mutants, which are checkpoint deficient, escape G2/M arrest and enter G1, where resection is less active due to lower CDK1 activity. We conclude that the 9-1-1 complex inhibits DSB resection at the URA3 locus.
Figure 7.

The 9-1-1 complex coordinates DSB resection at the URA3 locus. (A) Map of chromosome V showing an HO endonuclease site and loci examined in this study. (B,D) Analysis of 3′ ssDNA accumulation in the indicated strains. (C) ChIP analyses of Rad9-HA recruitment near DSBs. All the experiments were performed in nocodazole arrested cells. The data plotted and the P-values are as described in Figure 1.
To determine whether 9-1-1 inhibits DSB resection at the URA3 locus via recruitment of Rad953BP1, we examined Rad953BP1 binding to GEA2 and EAF5 by ChIP. Consistent with our results at the MAT locus, DSB induction induced enrichment of Rad953BP1 near DSBs, and recruitment is dependent on Mec3 (Figure 7C, Supplementary Figure S7E). We conclude that 9-1-1 inhibits resection at URA3 by affecting Rad953BP1 recruitment near DSBs.
Finally, we tested whether 9-1-1 has the ability to stimulate DSB resection in rad9Δ mutants (Figure 7D). Importantly, rad9Δ mec3Δ accumulated much less ssDNA than rad9Δ strains (Figure 7D), showing that 9-1-1 strongly stimulates DSB resection at URA3. We conclude that 9-1-1 both inhibits and stimulates DSB resection at the URA3 locus, as well as at the MAT locus, and presumably does at most non-telomeric DSB loci.
DISCUSSION
ssDNA is a critical intermediate in the DNA damage response because it is required for homologous recombination and DNA damage checkpoint maintenance. In all eukaryotic cells two major nucleases, Exo1 and Dna2-Sgs1, contribute to long-range resection in vivo. Why two nucleases, rather than one, are required and how they are coordinated remains unclear. One hypothesis to explain the use of two nucleases is that each nuclease has preference for different types of chromatin substrate and the nucleases interchange to ensure the most efficient control over resection. Since ssDNA is potentially very harmful to cells, there is clear need to regulate Exo1 and Dna2-Sgs1 to ensure that sufficient ssDNA is generated to help the DNA damage response, but not too much to harm cell survival.
A number of DDR proteins regulate DNA resection. Tel1 and its mammalian homologue ATM stimulate resection initiation by activating MRX and CtIP (8,49,50). Mec1ATR also stimulates resection but this mechanism is poorly understood (22,34). Other DDR proteins inhibit resection, presumably reducing the harmful effects of ssDNA on genome stability. Rad953BP1 and its mammalian homologue 53BP1 inhibit resection (21,51). Mec1ATR inhibits resection in yeast by at least two mechanisms: (i) stimulating Rad53-dependent phosphorylation of Exo1 which leads to downregulation of Exo1 activity (15,16) and (ii) promoting the binding of the resection inhibitor Rad953BP1 close to DNA lesions (34).
Here we establish the central role of the 9-1-1 complex in coordinating Exo1 and Dna2-Sgs1 activities. Figure 8A and B illustrate the roles of the 9-1-1 complex, Rad953BP1, Exo1 and Dna2-Sgs1 at DSBs and uncapped telomeres. We propose that at DSBs and uncapped telomeres, 9-1-1 helps recruit Exo1 and Dna2-Sgs1 to DNA to facilitate resection (Pathway 1, P1). 9-1-1 also helps recruit Rad953BP1, a resection inhibitor to inhibit resection (Pathway 2, P2) (33,45). We suggest that the essential difference between DSBs and uncapped telomeres is due to the balance between Pathways 1 and 2 at telomeres versus internal chromosomal loci, with Pathway 2, the recruitment of Rad953BP1, working much better at internal DSBs versus telomeres. This difference likely explains why resection at uncapped telomeres (8 kb/h) is faster than at DSBs (3.5 kb/h), as well as explains the different effects of 9-1-1 on resection at telomeres versus DSBs.
Figure 8.
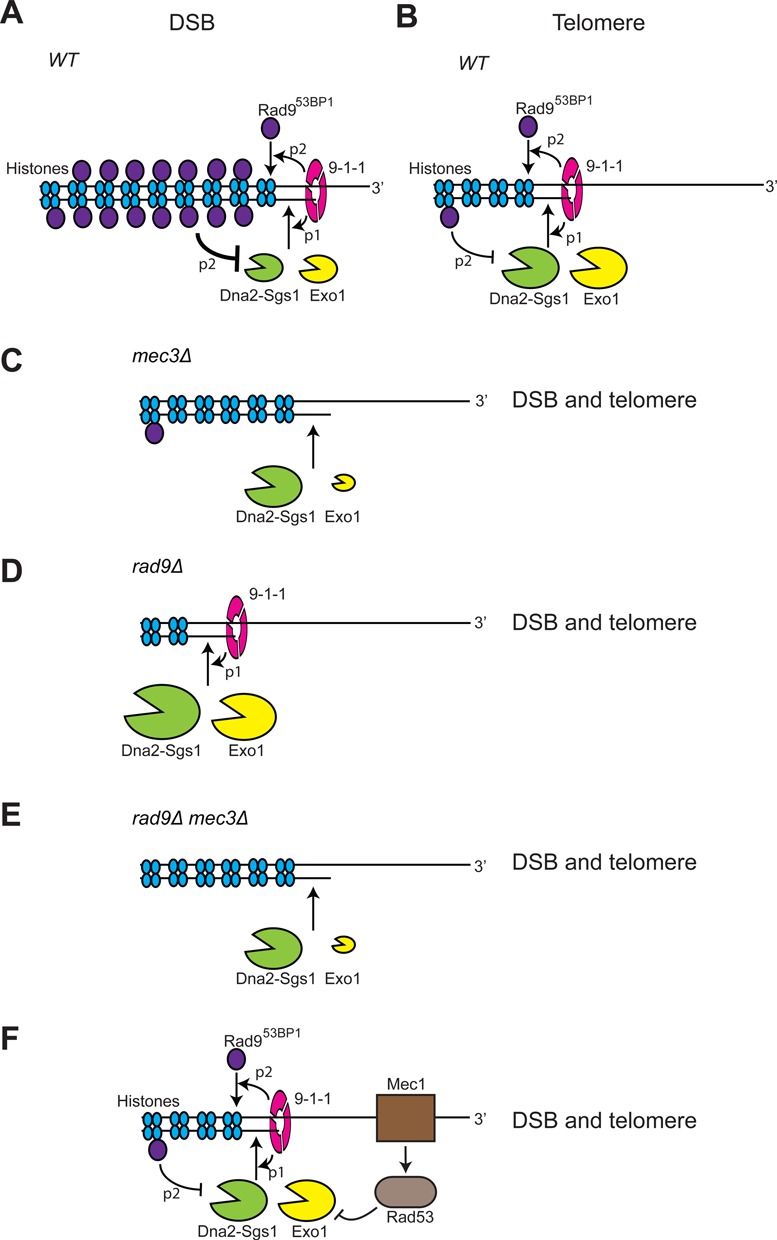
Control of resection at DSBs and telomeres. Models for the roles of 9-1-1, Rad953BP1, Exo1 and Dna2-Sgs1 on resection near DSBs and at uncapped telomeres. The size of the nucleases in each schematic indicates relative resection activities and is deduced by ssDNA measurements in the different genetic settings. Data supporting these figures are taken from Figures 1–7 and Ngo et al. (24). (A, B) 9-1-1 stimulates recruitment of Exo1 and Dna2-Sgs1 to facilitate resection (pathway 1, p1). 9-1-1 stimulates recruitment of Rad953BP1 to inhibit resection (pathway 2, p2). Rad953BP1 binds more near DSBs than uncapped telomeres. (C) In mec3Δ cells, there is less Rad953BP1 recruitment (lack of p2), but there is no 9-1-1 to stimulate activity of Exo1 and Dna2-Sgs1 (lack of p1). At DSBs Exo1 is less active (lack of p1) but Dna2-Sgs1 is more active (lack of p2) (C compared to A). The overall effect is increased resection in mec3Δ mutants (C compared to A). At telomeres Exo1 is less active (lack of p1) but Dna2-Sgs1 activity remains little changed because little Rad953BP1 binds (p2 less active at telomeres), and so the overall effect of mec3Δ is reduced resection (C compared to B). (D) In rad9Δ cells, there is no Rad953BP1 recruitment. Therefore, Dna2-Sgs1 and Exo1 are more active than in (A, B). Dna2-Sgs1 is more active than Exo1 in the absence of Rad953BP1. (E) In rad9Δ mec3Δ cells, there is no 9-1-1 to stimulate Exo1 or Dna2-Sgs1. Therefore, Dna2-Sgs1 and Exo1 are less active than in D. Exo1 is more dependent on 9-1-1 than Dna2-Sgs1. (F) Mec1ATR also initiates a checkpoint cascade to inhibit Exo1.
Interestingly, in the absence of 9-1-1 (mec3Δ cells), Rad953BP1, or both, resection at DSBs and uncapped telomeres is similar (Figure 8C–E). In mec3Δ cells, at both types of loci, there is less Rad953BP1 recruitment (lack of Pathway 2) and there is no 9-1-1 to stimulate activity of Exo1 and Dna2-Sgs1 (lack of Pathway 1; Figure 8C). The overall effect of 9-1-1 inactivation is increased resection at DSBs but decreased resection at uncapped telomeres (compare Figure 8C with Figure 8A and B). At DSBs, in mec3Δ strains, Exo1 is less active due to lack of Pathway 1 but Dna2-Sgs1 more than compensates for this, and is more active, due to lack of Pathway 2 (compare Figure 8C with Figure 8A). At telomeres, in mec3Δ strains, Exo1 is less active (lack of Pathway 1), but in contrast to at DSBs, Dna2-Sgs1 activity remains little changed, because Pathway 2 is less active near uncapped telomeres (compare Figure 8C with Figure 8B). Thus, the overall effect of loss of 9-1-1 at uncapped telomeres is reduced resection. Importantly, we find that the 9-1-1 complex strongly stimulates resection at both DSBs and uncapped telomeres and this is most clearly seen in the absence of Rad953BP1 (compare Figure 8D and Figure 8E).
We are unclear why Rad953BP1 binds less well near uncapped telomeres but this could be due to the nature of the chromatin at telomeric and sub-telomeric regions with reduced methylation of H3 lysine 79, which is needed for Rad953BP1 binding (47). Alternatively, there might be insufficient Rad953BP1 available to bind all the uncapped telomeres in comparison with the smaller number of ends induced by a single DSB. Irrespective of the reason, the weak binding of Rad953BP1 to telomeres appears to be conserved, since Crb2, the Rad953BP1 orthologue in fission yeast, also binds less efficiently near uncapped telomeres than DSBs (52). Even though Rad953BP1 binds weakly to telomeres, it still inhibits resection since rad9Δ strains clearly show increased resection at uncapped telomeres (and DSBs; Figure 8D) (21,23,24).
Interestingly, all our experiments suggest that Exo1 is more reliant on 9-1-1 than Dna2-Sgs1 for its activity in vivo. Furthermore, Rad953BP1 inhibits Dna2-Sgs1 at DSBs more than Exo1, consistent with a recent study (53). Thus, it seems that the combined effects of the checkpoint sliding clamp and the Rad953BP1 checkpoint mediator protein are to stimulate Exo1 and inhibit Dna2-Sgs1-dependent nuclease activities. Why DNA damage checkpoint proteins should favour Exo1 over Dna2-Sgs1 is unclear. However, we note that Exo1 is also targeted and inhibited by DNA damage checkpoint kinase cascades (Figure 8F) (15,16).
We propose that the ring-shaped 9-1-1 clamp recruits and tethers Exo1 to DNA, and that tethered Exo1 is better able to resect through Rad953BP1 containing chromatin. We suggest that 9-1-1 also recruits Dna2-Sgs1 to damaged sites but that Dna2-Sgs1 activity (presumably tethered on DNA by the hexameric Sgs1 helicase ring) is more active on Rad953BP1 free chromatin. Thus, 9-1-1 may play a critical role in promoting the interchange of nucleases to facilitate resection through different types of chromatin. It has also been reported that Dna2-Sgs1 and Exo1 are recruited to DSBs by the MRXMRN complex and replication protein A (RPA) (54,55). However, MRX dissociates from the ends following extensive resection (56). Thus, the recruitment role of 9-1-1 would appear to be more important at sites distal to the initial lesion.
Overall, our experiments reinforce the idea that 9-1-1 checkpoint sliding clamp binding to DNA lesions modulates DNA damage metabolism as well as stimulating checkpoint-dependent cell cycle arrest. We suggest that whether 9-1-1 promotes or inhibits resection is regulated by type of chromatin found near the DNA damage and other factors such as the activity of the central checkpoint kinase Mec1ATR. Mec1ATR-dependent phosphorylation of Ddc1 subunit of 9-1-1 stimulates the interaction of 9-1-1 with Dpb11TopBP1, which in turn interacts with Rad953BP1 and Mec1ATR to help activate the DNA damage checkpoint (57,58). We suggest that Mec1ATR phosphorylation of Ddc1 may switch the role of 9-1-1 from stimulating resection (Pathway 1) to facilitating Rad953BP1 recruitment and checkpoint activation (Pathway 2) (57), and therefore reduce the potential harm caused by excess ssDNA accumulation.
Although we have found different roles for 9-1-1, Rad953BP1, Exo1 and Dna2-Sgs1 at DSBs in comparison to uncapped telomeres, we do not consider the two loci to be distinct classes. In reality, we imagine that there is a spectrum of locus types between, and perhaps beyond, the DSB-like and telomere-like loci we have examined. The loci will differ, for example, in their affinity for Rad953BP1. It is interesting that Rad953BP1 has recently been reported to constitutively bind chromatin in combination with Aft1 transcription factor, and to bind with preference to fragile genomic regions (59). This pattern of binding is consistent with a role for Rad953BP1, limiting resection at those genomic locations that are more likely to suffer damage. In contrast, at telomeres, it is perhaps useful if cells favour resection because homologous recombination between the repetitive, non-coding telomeric DNAs is essentially harmless, whereas the alternative NHEJ pathway of repair is potentially much more dangerous because it could induce dicentric chromosomes and genetic instability.
In conclusion, our findings reveal the central role of the 9-1-1 checkpoint sliding clamp coordinating DNA resection in addition to its better known role of stimulating cell cycle arrest. 9-1-1 recruits both an inhibitor (Rad953BP1) and two activators of resection (Exo1 and Sgs1/Dna2) to DSBs. It is clear that mammalian 53BP1, like its yeast orthologue Rad953BP1, inhibits resection (51). It will be interesting to see if the 9-1-1 complex regulates DSB resection in vivo in mammalian cells and whether this role contributes to the involvement of 9-1-1 in processes like recombination and cancer progression (60–62). It seems reasonable to assume that control over resection in mammalian cells will be at least as sophisticated as in yeast and that the 9-1-1 complex is a central player in the DNA damage response network across all eukaryotic cells.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Marco Muzi-Falconi, Keith Gull and Martin Kupiec for the generous gifts of strains and antibodies, and Anton Gartner and members of the Lydall lab for comments on the manuscript.
FUNDING
Wellcome Trust [075294 and 093088 to D.L.]. Funding for open access charge: Wellcome Trust [093088].
Conflict of interest statement. None declared.
REFERENCES
- 1.Finn K., Lowndes N.F., Grenon M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 2012;69:1447–1473. doi: 10.1007/s00018-011-0875-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 4.Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mimitou E.P., Symington L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huertas P., Cortes-Ledesma F., Sartori A.A., Aguilera A., Jackson S.P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ira G., Pellicioli A., Balijja A., Wang X., Fiorani S., Carotenuto W., Liberi G., Bressan D., Wan L., Hollingsworth N.M., et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jazayeri A., Falck J., Lukas C., Bartek J., Smith G.C., Lukas J., Jackson S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 9.Gravel S., Chapman J.R., Magill C., Jackson S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiotani B., Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannavo E., Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- 12.Maringele L., Lydall D. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Delta mutants. Genes Dev. 2002;16:1919–1933. doi: 10.1101/gad.225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ngo H.P., Lydall D. Survival and growth of yeast without telomere capping by Cdc13 in the absence of Sgs1, Exo1, and Rad9. PLoS Genet. 2010;6:e1001072. doi: 10.1371/journal.pgen.1001072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts S.A., Sterling J., Thompson C., Harris S., Mav D., Shah R., Klimczak L.J., Kryukov G.V., Malc E., Mieczkowski P.A., et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morin I., Ngo H.P., Greenall A., Zubko M.K., Morrice N., Lydall D. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008;27:2400–2410. doi: 10.1038/emboj.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segurado M., Diffley J.F. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008;22:1816–1827. doi: 10.1101/gad.477208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y., Sterling J., Storici F., Resnick M.A., Gordenin D.A. Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLoS Genet. 2008;4:e1000264. doi: 10.1371/journal.pgen.1000264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo T., Wakayama T., Naiki T., Matsumoto K., Sugimoto K. Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science. 2001;294:867–870. doi: 10.1126/science.1063827. [DOI] [PubMed] [Google Scholar]
- 19.Melo J.A., Cohen J., Toczyski D.P. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 2001;15:2809–2821. doi: 10.1101/gad.903501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navadgi-Patil V.M., Burgers P.M. Cell-cycle-specific activators of the Mec1/ATR checkpoint kinase. Biochem. Soc. Trans. 2011;39:600–605. doi: 10.1042/BST0390600. [DOI] [PubMed] [Google Scholar]
- 21.Lydall D., Weinert T. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- 22.Jia X., Weinert T., Lydall D. Mec1 and Rad53 inhibit formation of single-stranded DNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 2004;166:753–764. doi: 10.1534/genetics.166.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zubko M.K., Guillard S., Lydall D. Exo1 and Rad24 differentially regulate generation of ssDNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 2004;168:103–115. doi: 10.1534/genetics.104.027904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ngo G.H., Balakrishnan L., Dubarry M., Campbell J.L., Lydall D. The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 2014;42:10516–10528. doi: 10.1093/nar/gku746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S.E., Moore J.K., Holmes A., Umezu K., Kolodner R.D., Haber J.E. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 26.Aylon Y., Kupiec M. The checkpoint protein Rad24 of Saccharomyces cerevisiae is involved in processing double-strand break ends and in recombination partner choice. Mol. Cell. Biol. 2003;23:6585–6596. doi: 10.1128/MCB.23.18.6585-6596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dubrana K., van Attikum H., Hediger F., Gasser S.M. The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of ATR-kinase foci in yeast. J. Cell Sci. 2007;120:4209–4220. doi: 10.1242/jcs.018366. [DOI] [PubMed] [Google Scholar]
- 28.Dewar J.M., Lydall D. Simple, non-radioactive measurement of single-stranded DNA at telomeric, sub-telomeric, and genomic loci in budding yeast. Methods Mol. Biol. 2012;920:341–348. doi: 10.1007/978-1-61779-998-3_24. [DOI] [PubMed] [Google Scholar]
- 29.Holstein E.M., Lydall D. Quantitative amplification of single-stranded DNA. Methods Mol. Biol. 2012;920:323–339. doi: 10.1007/978-1-61779-998-3_23. [DOI] [PubMed] [Google Scholar]
- 30.van Attikum H., Fritsch O., Hohn B., Gasser S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 31.Aparicio O., Geisberg J.V., Struhl K. Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Curr. Protoc. Cell. Biol. 2004 doi: 10.1002/0471143030.cb1707s23. Chapter 17, Unit 17.7. [DOI] [PubMed] [Google Scholar]
- 32.Weinert T.A., Kiser G.L., Hartwell L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 33.Lazzaro F., Sapountzi V., Granata M., Pellicioli A., Vaze M., Haber J.E., Plevani P., Lydall D., Muzi-Falconi M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008;27:1502–1512. doi: 10.1038/emboj.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clerici M., Trovesi C., Galbiati A., Lucchini G., Longhese M.P. Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J. 2014;33:198–216. doi: 10.1002/embj.201386041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pellicioli A., Lee S.E., Lucca C., Foiani M., Haber J.E. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 36.Paciotti V., Lucchini G., Plevani P., Longhese M.P. Mec1p is essential for phosphorylation of the yeast DNA damage checkpoint protein Ddc1p, which physically interacts with Mec3p. EMBO J. 1998;17:4199–4209. doi: 10.1093/emboj/17.14.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Booth C., Griffith E., Brady G., Lydall D. Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res. 2001;29:4414–4422. doi: 10.1093/nar/29.21.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zierhut C., Diffley J.F. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008;27:1875–1885. doi: 10.1038/emboj.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrari M., Dibitetto D., De Gregorio G., Eapen V.V., Rawal C.C., Lazzaro F., Tsabar M., Marini F., Haber J.E., Pellicioli A. Functional Interplay between the 53BP1-Ortholog Rad9 and the Mre11 Complex Regulates Resection, End-Tethering and Repair of a Double-Strand Break. PLoS Genet. 2015;11:e1004928. doi: 10.1371/journal.pgen.1004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugawara N., Wang X., Haber J.E. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 42.Tavormina P.A., Wang Y., Burke D.J. Differential requirements for DNA replication in the activation of mitotic checkpoints in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997;17:3315–3322. doi: 10.1128/mcb.17.6.3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vodenicharov M.D., Wellinger R.J. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol. Cell. 2006;24:127–137. doi: 10.1016/j.molcel.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 44.Naiki T., Wakayama T., Nakada D., Matsumoto K., Sugimoto K. Association of Rad9 with double-strand breaks through a Mec1-dependent mechanism. Mol. Cell. Biol. 2004;24:3277–3285. doi: 10.1128/MCB.24.8.3277-3285.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X., Cui D., Papusha A., Zhang X., Chu C.D., Tang J., Chen K., Pan X., Ira G. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature. 2012;489:576–580. doi: 10.1038/nature11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J.A., Kruhlak M., Dotiwala F., Nussenzweig A., Haber J.E. Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J. Cell. Biol. 2007;178:209–218. doi: 10.1083/jcb.200612031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi Y.H., Schulze J.M., Jackson J., Hentrich T., Seidel C., Jaspersen S.L., Kobor M.S., Shilatifard A. Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol. Cell. 2011;42:118–126. doi: 10.1016/j.molcel.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szilard R.K., Jacques P.E., Laramee L., Cheng B., Galicia S., Bataille A.R., Yeung M., Mendez M., Bergeron M., Robert F., et al. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat. Struct. Mol. Biol. 2010;17:299–305. doi: 10.1038/nsmb.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L., et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mantiero D., Clerici M., Lucchini G., Longhese M.P. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007;8:380–387. doi: 10.1038/sj.embor.7400911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell. Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 52.Carneiro T., Khair L., Reis C.C., Borges V., Moser B.A., Nakamura T.M., Ferreira M.G. Telomeres avoid end detection by severing the checkpoint signal transduction pathway. Nature. 2010;467:228–232. doi: 10.1038/nature09353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonetti D., Villa M., Gobbini E., Cassani C., Tedeschi G., Longhese M.P. Escape of Sgs1 from Rad9 inhibition reduces the requirement for Sae2 and functional MRX in DNA end resection. EMBO Rep. 2015;16:351–361. doi: 10.15252/embr.201439764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shim E.Y., Chung W.H., Nicolette M.L., Zhang Y., Davis M., Zhu Z., Paull T.T., Ira G., Lee S.E. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010;29:3370–3380. doi: 10.1038/emboj.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen H., Lisby M., Symington L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lisby M., Barlow J.H., Burgess R.C., Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 57.Puddu F., Granata M., Di Nola L., Balestrini A., Piergiovanni G., Lazzaro F., Giannattasio M., Plevani P., Muzi-Falconi M. Phosphorylation of the budding yeast 9-1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol. Cell. Biol. 2008;28:4782–4793. doi: 10.1128/MCB.00330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navadgi-Patil V.M., Burgers P.M. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol. Cell. 2009;36:743–753. doi: 10.1016/j.molcel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andreadis C., Nikolaou C., Fragiadakis G.S., Tsiliki G., Alexandraki D. Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Res. 2014;42:12650–12667. doi: 10.1093/nar/gku915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pandita R.K., Sharma G.G., Laszlo A., Hopkins K.M., Davey S., Chakhparonian M., Gupta A., Wellinger R.J., Zhang J., Powell S.N., et al. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol. Cell. Biol. 2006;26:1850–1864. doi: 10.1128/MCB.26.5.1850-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.An L., Wang Y., Liu Y., Yang X., Liu C., Hu Z., He W., Song W., Hang H. Rad9 is required for B cell proliferation and immunoglobulin class switch recombination. J. Biol. Chem. 2010;285:35267–35273. doi: 10.1074/jbc.M110.161208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Broustas C.G., Lieberman H.B. Contributions of Rad9 to tumorigenesis. J. Cell. Biochem. 2012;113:742–751. doi: 10.1002/jcb.23424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.