

Abstract
The first synthesis of C5-Curcumin-Fatty Acid (C5-Curc-FA) conjugates was successfully performed. Through a two-step synthetic route, 10 analogs were synthesized for a structure-activity relationship (SAR) study. It was found that C5-Curc-FA conjugates containing either decanoic acid or palmitic acid moieties were cytotoxic against colorectal adenocarcinoma cell (CCL-229) at IC50s ranging from 22.5 to 56.1 µg/mL, being 5c the most active C5-Curc-FA conjugate. Our results strongly suggests that a decanoic acid moiety at the meta position in C5-Curc-FA conjugates is important for their anticancer activity effect. Possible mechanisms for the anticancer activity of C5-Curc-FA conjugates were also investigated including apoptosis induction, mitochondrial damage and caspases activation. It was shown that 5c inhibited the luminescence activity of NFκB, a key signaling molecule involved in cell apoptosis and cell proliferation, at IC50 = 18.2 µg/mL. In addition, it was demonstrated that 5c displayed significant apoptotic effect at GI50 = 46.0 µg/mL in colorectal adenocarcinoma cell line (ATCC CCL-222), which can be explained by the significant mitochondrial membrane permeabilization and caspases 3 and 7 activation effect of 5c. Finally, it was investigated that C5-Curc-FA conjugates can affect the replication process of cancer cells, since compounds 5c, 5e, and 6c inhibited the relaxing activity of the human DNA topoisomerase I at minimum inhibitory concentrations (MICs) that range from 50 to 500 µg/mL. Our results strongly support the hypothesis that the inhibition of both NFkB and DNA topoisomerase I by C5-Curc-FA conjugates is associated with their anticancer activity.
Keywords: C5-curcuminoids synthesis, anticancer agents, NFkB, apoptosis, DNA topoisomerase I

“Cancer is characterized by the uncontrolled growth and spread of abnormal cells”. This disease affects approximately 1,665,540 United States citizens, causing 585,720 deaths per year.1 In Puerto Rico, 51,805 persons have been diagnosed with cancer and the rate of death is 127 per 100,000 cases.2 One problem that is affecting the nation’s health is the high toxicity of the current drugs to treat patients with cancer that destroy blood cells such as T-cells, B-cells, macrophages, NK cells, and neutrophils. This situation is complicated when these treatments break down the tissue of natural infection barriers such as the skin, urogenic, and gastrointestinal tract covering thus allowing cancerous agents to travel through these openings and invade immunocompromised patients. Despite the fact that several compounds are being evaluated as anticancer agents, there is a critical need to develop new cancer chemotherapy treatments with low toxicity.
Among the compounds presently been evaluated as anticancer agents, curcumin has demonstrated to exhibit anticancer properties.3–7 Curcumin [1,7-bis (4-hydroxy-3-methoxyphenyl)-1,6- heptadiene-3,5-dione, 1, Figure 1] is a yellowish polyphenolic compound isolated from turmeric, a rhizome of the Indian plant Curcuma longa. Although preclinical and clinical studies have shown that 1 is not toxic towards normal human cells, several pharmacokinetic disadvantages, such as poor bioavailability, fast metabolism and requiring repetitive oral doses were reported, which limits its pharmacological applications.8–9 One of the reasons for the reduced bioavailabilty of 1 is its high rate of metabolism.10 Several studies performed in rats and in humans have suggested that orally and intravenously administered 1 is rapidly metabolized in the liver into hexahydrocurcumin and hexahydrocurcuminol, while curcumin glucuronide and curcumin sulfate are generated extrahepatically, probably in the gastrointestinal tract.11–13 The fast metabolism of 1 can be due to the presence of the β-diketone moiety, which appears to be a specific substrate for a series of aldo-keto reductases.14–16 Several approaches have been performed in order to improve the pharmacokinetics disadvantages mentioned above including the preparation of liposome-encapsulated 1 and the synthesis of mono-carbonyl analogs of 1 (C5-curcuminoids).17–20 Recently, the synthesis of (1E,4E)-1,5-bis(4-hydroxy-3-methoxyphenyl)penta-1,4-dien-3-one (C5-curcumin, 2, Figure 1) was performed with the objective of evaluating its anticancer properties.18–19 It was found that 2 was particularly active against both CNE (nasopharyngeal adenocarcinoma) and LS 174T (colorectal adenocarcinoma), thus displaying a good pharmacokinetic profile.19
Figure 1.
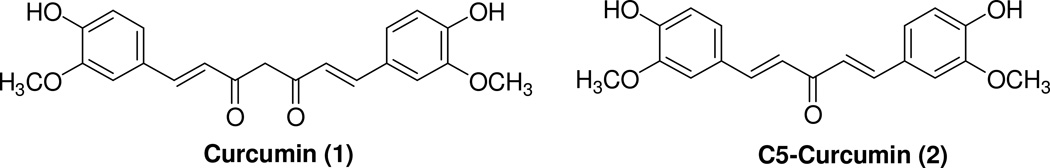
Chemical structures of Curcumin (1) and C5-Curcumin (2).
In 2010, Singh and collaborators reported the first total synthesis of curcumin bioconjugates containing fatty acids aimed at studying both its antibacterial and antiviral properties.21 Results from this study revealed that the conjugation of both decanoic acid and palmitic acid to curcumin improved the antimicrobial activity of 1. These authors reasoned that the improved biological properties of curcumin-fatty acid (Curc-FA) conjugates is due to the presence of the fatty acid moiety, which decreases the rate of metabolism of 1 needing an additional hydrolysis of ester bonds by carboesterases present in cells; provides structural similarity to the cell wall allowing the uptake of 1 by enhancing its effective concentration; and improves the biological activity of 1 after enzymatic hydrolysis of the ester group in Curc-FA conjugates.21 However, to the best of our knowledge, the chemical connection of a fatty acid to a monocarbonyl analog of 1 and evaluation of its anticancer activity has not been reported.
In the present study the Singh approach was used to synthesize, for the first time, C5-curcumin-fatty acid (C5-Curc-FA) conjugates in order to determine structure-activity relationship (SAR) using the CCL-229 (colorectal adenocarcinoma) cell line. We chose to use the CCL-229 cell line because colorectal cancer is one of the cancers that mostly affects the adult population in both the United States and Puerto Rico.2,22 Our approach of connecting FAs to 2 is totally new, since to the best of our knowledge, only one study has been reported addressing the chemical connection of FAs to 1, which contains a β-diketone moiety in its structure. In this study, we decided to conjugate 2 to FAs because the presence of only one carbonyl moiety in 2 enhances its in vitro stability profile, its in vivo pharmacokinetic profile, and its cytotoxic activity against several cancer cell lines including CNE and LS 174T adenocarcinomas in vitro.19
The synthesis of C5-curcumin-fatty acid conjugate 5 started with the conjugation of the substituted phenol 3 with either decanoic acid or palmitic acid through Steglich esterification conditions described in the literature23, which afforded 4a–g in 43–80% yields. Compounds 4a–g were subsequently reacted with acetone and lithium hydroxide monohydrate in ethanol obtaining the desired C5-Curc-FA conjugates 5a–g in 47–69% yields. The synthetic strategy displayed in Scheme 1 was also used for preparing C5-Curc-FA conjugates containing cyclohexanone moieties. In this approach, compounds 4e–g were reacted with cyclohexane and lithium hydroxide monohydrate in ethanol for 24 h. This reaction afforded the desired 6a–c in 42–62% yields (Scheme 2). Melting points for 5a–g and 6a–c were determined with a Melt-Temp apparatus and were uncorrected.
Scheme 1.

Synthetic route towards the C5-Curcumin-Fatty acid conjugates 5a–g. i) Decanoic acid or palmitic acid, DIC, DMAP, CH2Cl2/THF (1:1 v/v), 0°C (for 1 hr), 24 h and ii) acetone, LiOH-H2O, EtOH, rt, 24 h.
Scheme 2.

Synthesis of C5-Curc-FA conjugates containing the cyclohexanone moiety. i) Cyclohexanone, LiOH-H2O, EtOH, rt, 24 h.
The NMR analyses were performed in a Bruker Avance AV-500 spectrometer. Both 1H-NMR and 13C-NMR spectroscopic data are shown in Table 1. In 1H-NMR two doublets at 7.5 and 6.5 ppm were observed for 5a–5g. These signals have a coupling constant (J) of 16 Hz, which are characteristic of compounds containing a trans alkene moiety.24 In the case of 6a–6c, a singlet at 7.6 ppm was also observed in 1H-NMR, which is in agreement with spectroscopic data from other C5-curcuminoids containing cyclohexanone moieties previously reported in the literature.18 The purity of 5 and 6 was determined to be > 95% by 13C-NNR and confirmed by a melting point analysis. For 5 and 6, two signals at 191 and 170 ppm were observed. These signals are evidence of the presence of the α,β-unsaturated carbonyl and ester carbonyl groups, respectively.24
Table 1.
| Compound |  |
||||||
|---|---|---|---|---|---|---|---|
| 13C-NMR chemical shift (ppm) |
1H-NMR chemical shift (ppm), multiplicity |
H2–H3 coupling constant (Hz) |
|||||
| C1 | C2 | C3 | C4 | H2 | H3 | ||
| 5a | 190.0 | 121.8 | 148.3 | 168.6 | 6.45, d | 7.60, d | 15.8 |
| 5b | 191.9 | 122.2 | 147.6 | 170.8 | 6.56, d | 7.53, d | 16.1 |
| 5c | 190.8 | 122.0 | 147.2 | 167.0 | 6.67, d | 7.56, d | 16.2 |
| 5d | 190.3 | 122.6 | 148.0 | 173.0 | 6.45, d | 7.60, d | 15.8 |
| 5e | 190.0 | 122.3 | 147.9 | 168.9 | 6.44, d | 7.60, d | 15.8 |
| 5f | 192.1 | 120.9 | 141.1 | 172.9 | 6.52, d | 7.48, d | 15.8 |
| 5g | 188.9 | 121.1 | 147.1 | 171.6 | 6.48, d | 7.37, d | 16.3 |
| 6a | 190.0 | 137.4 | 139.7 | 168.9 | None | 7.53, s | None |
| 6b | 192.6 | 136.4 | 139.8 | 174.4 | None | 7.62, s | None |
| 6c | 190.7 | 136.9 | 139.5 | 173.4 | None | 7.52, s | None |
Signals from FA moieties were observed at 0.85–2.44 ppm in 1H-NMR and 13.1–34.0 ppm in 13C-NMR. Signals from cyclohexanone moieties were observed at 1.62–2.28 ppm and 2.95–3.33 ppm in 1H-NMR and 22.1–23.8 ppm and 25.6–29.4 ppm in 13C-NMR.
Signals from methoxy groups in compounds 5a, 5b, 5e, 5f, 6a, and 6b were observed at 3.78–3.83 ppm in 1H-NMR and 54.1–56.0 ppm in 13C-NMR.
Signals from aromatic groups were observed at 6.59–7.80 ppm in 1H-NMR and 107.2–157.3 ppm in 13C-NMR.
Once 5a–g and 6a–c were synthesized, these compounds were tested against colon cancer cells (CCL-229). These tests were performed through the MTT (methyl thiazolyl tetrazolium) assay for determining cell viability. CCL-229 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Kaighn's Modification of Ham's F-12 Medium containing 2 mM L-glutamine and 1500 mg/L sodium bicarbonate (F12K, Mediatech, Inc., Manassas, VA, USA). A 10,000 cells/200 µL/well were seeded into a Corning 96-well microplate (Corning, NY, USA) and compounds 5a–g and 6a–c were added to the cell cultures. The final concentrations of C5-Curc-FA conjugates 5a–g and 6a–c ranged from 5 to 500 µg/mL. The cells were incubated at 37°C for three days in a humidified 5% CO2 incubator. Results from MTT assays are displayed in Table 2.
Table 2.
Cytotoxic activity C5-Curc-FA conjugates 5a–g and 6a–c against colorectal cancer cells (CCL-229).a
| Compound | IC50 ± SEM, µg/mLb |
|---|---|
| 5a | 143.1 ± 14.6 |
| 5b | 66.0 ± 6.5 |
| 5c | 22.5 ± 3.0 |
| 5d | 179.7 ± 31.1 |
| 5e | 56.1 ± 1.5 |
| 5f | 70.5 ± 16.9 |
| 5g | 112.9 ± 7.1 |
| 6a | 139.0 ± 23.3 |
| 6b | 129.9 ± 24.1 |
| 6c | 29.3 ± 0.7 |
| 1c | 26.8 ± 1.6 |
Experiments were performed in triplicate (N =3).
Results were reported as mean ± SEM from three determinations. IC50 represents the sample concentration that is required to achieve 50% cell viability and this value was determined from dose-response curves that were generated by using Graph-Pad Prism v. 5.01.
Curcumin was used as positive control.
Results from Table 2 show that 5a–g and 6a–c exert direct cytotoxic effect on CCL-229 cells and inhibit their growth in vitro in a dose dependent manner. Among the ten C5-Curc-FA tested, compounds 5c, 5e, and 6c were the most active conjugates against CCL-229 displaying IC50 values ranging from 22.5–56.1 µg/mL. It can be appreciated from Table 2 that the anticancer activity of C5-Curc-FA increases when decreasing the number of carbon atoms in the fatty acyl chain. This is the case of the pairs of compounds 5f/5b and 5g/5f whose IC50 values increase 1.1 and 5.0 times, respectively. With the exception of 5g and 6c, the cyclohexanone moiety in a C5-Curc-FA conjugates decrease their anticancer activity against CCL-229 cells. For example, when the anticancer activity of 5e and 6a are compared, it can be observed that the IC50 value of 6a is 2.5-fold higher than 5e. A similar pattern was observed when the activity of 5f and 6b are compared. It is interesting to observe that 5c showed similar IC50 values than 1, which demonstrate its efficacy as anticancer agent. Results in Table 2 also suggest that the presence of a methoxy group in a C5-Curc-FA conjugate affects its anticancer activity towards CCL-229. In curcuminoids 5f and 5g, which contain 16-carbon chain length FA moieties, the presence of a methoxy group at the para position increases their anticancer activity. On the other hand, the methoxy group at the para position in curcuminoid 5b, decreases its anticancer activity. These results strongly support that the FA moiety plays an important role in the anticancer activity of C5-Curc-FA conjugates towards colorectal cancer cells.
The toxicities of the most active C5-Curc-FA conjugates were determined against peripheral blood mononuclear cells (PBMCs) from healthy volunteers (Table 3). PBMCs were cultured in culture medium supplemented with interleukin-2 (IL-2) and seeded into a 96-well microplate (4 × 105–5 × 104 cells/ 200 µL/ well). C5-Curc-FA conjugates were added to the cell cultures obtaining final concentrations that ranged from 2 to 100 µg/mL. The cells were incubated at 37°C for 3 days in humidified 5% CO2 incubator. The cytotoxicity of the cells was evaluated by the MTT assay. Results in Table 3 show that 5c, 5e and 6c were 2.9–3.3-fold less toxic than 1 suggesting that the presence of a fatty acid moiety and removal of a carbonyl group in 1 decreases the cytotoxic activity of C5-Curc-FA conjugates against PBMC. Singh et al. explained that fatty acids are natural components of cell membranes, thus by conjugating fatty acids to 2 it is expected that this connection enhances the cellular uptake of 2, increases its lipophilicity, improves its half-life, and reduces its rate of metabolism by carboesterases inside the cell, which ensures its low toxicity and high bioavailability.21 In fact, it can be noted from Table 3 that the cytotoxicity of C5-Curc-FA conjugates against PBMC decreases when their logP values increase.
Table 3.
Toxicity of C5-Curc-FA conjugates against PBMC from healthy volunteers. a
| Compound | IC50 ± SEM, µg/mL | logPb |
|---|---|---|
| 5c | 35.9 ± 0.4 | 11.66 |
| 5e | 40.6 ± 0.9 | 16.68 |
| 6c | 36.3 ± 0.4 | 17.77 |
| 1c | 12.3 ± 0.4 | 4.12 |
Experiments were performed in triplicate (N =3). Results were reported as mean ± SEM from three determinations. IC50 represents the sample concentration that is required to achieve 50% cell viability and this value was determined from dose-response curves that were generated by using Graph-Pad Prism v. 5.01 (GraphPad Software).
LogP values were predicted by using MarvinSketch v. 6.2 (ChemAxon).
Curcumin was used as positive control.
One of the mechanisms that has been widely studied for 1, and its derivatives, is the inhibition of the transcription factor NFκB.3,25–26 The NfkB is a key signaling molecule in the elaboration of the inflammatory response and apoptosis of cancer cell lines.27 In order to further investigate the mechanism of action of C5-Curc-FA conjugates, we carried out several NFkB luciferase reporter assays aimed at determining the inhibitory effects of these conjugates on the activity of NFkB. For these assays, we selected 5c because 5c displayed the best anticancer activity against the CCL-229 cells. Prior to these assays, NFkB Reporter-HEK293 (BPS bioscience, CA) cells were cultured in MEM (Hyclone, UT) medium with 10% FBS (Life technologies, CA), 1% non-essential amino acid, 1mM Na-pyruvate (Hyclone, UT), 1% Penn-strep (Hyclone, UT), and 100 µg/mL of Hygromycin B (Hyclone, UT). The NFkB luciferase reporter assays were performed by seeding 30,000 cells per well into a white clear-bottom 96-well microplate in 40 µL of assay medium (growth medium without Hygromycin B). Cells were incubated at 37 °C and 5% CO2 overnight to allow them to recover and reattach. After 24 h, compounds 5c and 1 were diluted in the assay medium and 5 µL of dilution were added to each well. Also, 5 µL of assay medium with DMSO were added to the untreated control wells and cell-free control wells. Cells were treated with the tested compounds for 16 h. Then, the cells were treated with mouse TNFα (5 ng/mL, Sigma-Aldrich, MO) for an additional 6 h. After treatment, cells were lysed and luciferase assays were performed by adding 50 µL of One-Step Luciferase reagent per well and shaking at room temperature for approximately 15 min. Luminescence was measured using a luminometer (BioTek SynergyTM 2 microplate reader).
Results from the NFkB inhibitory assays are displayed in Figure 2. Our results demonstrate that 5c has the ability of inhibiting the activity of NFkB since its luminescence properties, after treatment with 5c, exhibited a sigmoidal behavior with the logarithm of the concentration of the conjugate. A similar dose-response behavior was observed for 1, since its inhibitory activity against NFkB is known in the literature.3,25–26,28 Also, it was observed that the luminescence in the presence of 1 is 2.3-fold higher than in the presence of 5c, which suggests that the lipophilic nature of 5c is affecting the NFkB inhibitory activity. The fact that 5c was able to inhibit the NFkB luminescence activity implies that the disruption of the NFkB pathway is taking place. The NFkB pathway is a key mediator of the control of genes that are highly involved in apoptosis and cell proliferation such as c-IAP1, c-IAP2, and IXAP (antiapoptotic genes), the TNF receptor-associated factors (TRAF1 and TRAF2), the Bcl-2 homologue A1/Bfl-1, and IEX-IL.27 Results from proliferation of CCL-229 cells and NFkB inhibitory assays strongly support the hypothesis that the disruption of the NFkB pathway can be a possible mechanism for 5c, since its activity as an anticancer agent and an NFkB inhibitor was observed at IC50s values of 22.5 and 18.2 µg/mL, respectively.
Figure 2.

Dose-response curves for the NFkB inhibitory activity for both 1 (A) and 5c (B).a,b
aExperiments were performed in triplicate (N =3). Results were reported as mean ± SEM from three determinations.
b Non-linear regression analysis was performed by using GraphPad Prism v. 5.01 (GraphPad Software). The IC50 values were determined by the concentration causing a half-maximal percent activity.
Due to the fact that 1 and 5c displayed NFkB inhibitory activity, we hypothesize that these compounds might be able to induce apoptosis in colorectal cancer cells. For this reason, we performed the Annexin V assay in order to determine whether compounds 1 and 5c induce apoptosis. For this assay the human COLO-205 colorectal adenocarcinoma (ATCC CCL-222) was used and cultures were maintained in RPMI 1640 media (ATCC, Manassas VA) containing 10% fetal bovine serum (ATCC) and incubated at 37°C with humidified atmosphere of 95% air and 5% CO2. Apoptosis endpoint was measured after 24 h of exposure. The Annexin V assay has been used as an indicator of apoptosis. We used Annexin V as the apoptosis indicator because this assay allows for the detection of phosphatidylserine (PS) on the exterior surface of cells, a hallmark in apoptotic cells.29 For this assay, approximately 2.5 × 106 cells were treated for 24 h with the GI50 dose of each test compound 46.0 µg/mL of 5c and 7.4 µg/mL of 1. Controls used in all assays were camptothecin (3.5 µg/mL), and the vehicle (DMSO). After a 24 h exposure, cells were stained with Annexin V conjugate, and propidium iodide (Biotium, Hayward, CA). Samples were analyzed using the Nucleo Counter NC3000 system (Chemometec, Allerod, Denmark). A one way ANOVA with post hoc Tukey test was also performed for determining statistical significance. Results obtained from the Annexin V (Figure 3) assay showed a statistically significant apoptosis induction by 5c (P <0.05). The experimental compound presented an average of 27.5% apoptotic cells, which contrasts significantly to the negative control that presented an average of 14.3% (Figure 3). Positive controls camptothecin and 1 presented averages of 29.0% and 20.7%, respectively, indicating that all test compounds induced apoptosis when comparing to the negative compound.
Figure 3.

(A) Annexin V assay results showing the apoptotic effect of compounds 5c and curcumin (1). Statistical distribution showing the percentage of apoptotic cells per compound tested.a
aExperiments were performed in triplicate (N =3). Negative control presented an average of 14.3% apoptotic cells, positive controls camptothecin and 1 (curc) presented averages of 29.0 and 27.5% respectively. Compound 5c presented an average of 27.5% which is statistically significant (P<0.05) when comparing to the negative control.
After determining apoptosis activity with the annexin assay, several hallmarks of this process were analyzed. Among these endpoints is mitochondrial membrane permeabilization (MMP) and effector caspase activation. Mitochondrial membrane permeabilization (MMP) is one of the hallmarks of the apoptotic process.30 A total of 1 ×106 cells were exposed to compounds and the controls camptothecin and 1 at the previously described doses, the samples were labeled with 200 µg/ml of 5, 5’, 6, 6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolocarbocyanineiodide (JC-1). An additional counter stain with 1 µg/ml DAPI in PBS was also applied, then analyzed using the Nucleo Counter NC3000 system. A one way ANOVA with post hoc Tukey test was also performed. Our results present a statistically significant permeabilization of the experimental compound comparable to the Camptothecin control. All compounds except the negative control (14.0%) presented average statistically significant (P<0.05) depolarization. The test compounds show varied activation rates; Camptothecin presented 48.5% and 1 23.0% average depolarization (Figure 4a and 4b). The experimental compound 5c presented and average of 68.0% depolarized cells.
Figure 4.

(A) Mitochondrial membrane permeabilization effects of 5c.a (B) Diagram showing the distribution cells with permeabilized mitochondrial membrane among all tested compounds
a Experiments were performed in triplicate (N =3).Control sample presented an average of 14% permeabilized cells whereas the Camptothecin positive control 48.5% and Curcumin analog 23.0%. Compound 5c presented 68% of cells with permeabilized mitochondrial membrane which is statistically significant (P<0.05).
The activation of effector caspases 3 and 7 is a key event in the apoptosis process.31 These enzymes activate the nucleases tasked with degradation of nuclear material. Cell cultures were exposed to the test compounds as described previously in the annexin assay. Cells were harvested then stained with the Fluorescent Labeled Inhibitors of Caspases (FLICA). These markers bind to the active caspase enzymes, the green FAM FLICA kit (Immunochemistry Technologies, Bloomington Min.) was utilized as per manufacturer’s specification then analyzed using the Nucleocounter instrument. Data obtained presents statistically significant (P<0.05) effector caspase activation in the experimental 5c compound and positive controls (Figure 5). The background negative samples presented an average of 8% cell with active caspase 3 whereas the positive camptothecin presented 30% activation. Compound 5c and its analog 1 presented 19.5% and 23.3% respectively demonstrating statistically significant caspase activation.
Figure 5.

Caspase 3 and 7 activation effect of 5c.a
a Experiments were performed in triplicate (N =3). Results present a statistically significant (P<0.05) activation in the experimental compound 5c and positive controls. Negative control presented a mean of 8% of cells with active caspase 3. Camptothecin, curcumin and the experimental 5c compound presented averages of 30%, 23.3% and 19.5% respectively.
Other studies have demonstrated that curcumin and other curcuminoids displayed inhibitory activity against DNA topoisomerase I and II.32–34 DNA topoisomerases are enzymes involved in generating the necessary topological and conformational changes in DNA, which are essential for several cellular processes such as replication, recombination, and transcription.35 In addition to their normal cellular functions, DNA topoisomerases are known to be important molecular targets for some anticancer drugs.36 In an effort to further understand the mechanism of action of 5 and 6, we studied the inhibitory activity of 5c, 5e and 6c against the human topoisomerase I (Figure 6). The inhibitory activity of these curcuminoids towards topoisomerase I was studied because they were the most active anticancer conjugates based on their IC50 values (Table 2).
Figure 6.

Representative ethidium bromide stained gels showing the inhibitory activity of C5-Curc-FA conjugates 5c, 5e, and 6c.a
a Experiments were performed in triplicate (N =3).
The enzyme activity of topoisomerase I was assessed by using human DNA topoioserase I kit (TopoGen, Port Orange, FL, USA, 10 unit of enzyme relaxes 0.06 µg of pHOT1 supercoiled DNA in 30 min at 37 °C). Compounds 5c, 5e, and 6c were dissolved in 50% DMSO and were tested at different concentrations that range from 10 to 1,500 µg/mL. Reactions (final volume 20 µL) were carried out for 30 min at 37 °C. Reactions were stopped by adding 2 µL of 10% sodium dodecyl sulfate (SDS). Bound proteins were digested by incubation with proteinase K (0.05 mg/mL) for 15 min at 37 °C. After an incubation period, 2 µL of electrophoresis universal loading buffer were added. Samples were loaded followed by electrophoresis in a 1% agarose gel (72V) until the dye front of bromophenol blue is about 70% down gel. To visualize the reaction products, the gel was stained with ethidium bromide for 1 h and distained for 30 min in distilled water. DNA bands were detected and quantified in a Min BIS bioimaging system (model 241016P1, Israel).
Results from Figure 6 show that 5c inhibits the relaxing activity of the human DNA topoisomerase I at 50 µg/mL, while 5e and 6c completely inhibit the activity of the enzyme at minimum inhibitory concentrations (MICs) of 100 and 250 µg/mL, respectively. These results are comparable with those findings described by Roth et al., where they reported that curcumin inhibits the relaxing activity of DNA topoisomerase I at 50 µg/mL.32 It is very interesting to observe that the inhibitory activity of 5c is 5–10-fold higher than 5e and 6c implying that the decanoic acid moiety at the meta position is important for the topoisomerase inhibitory activity of C5-Curc-FA conjugates. Also, it was observed that the methoxy group in 5e and the cyclohexanone moiety in 6c decrease their inhibitory activity against the DNA topoisomerase I. These topoisomerase inhibitory results are in agreement with the anticancer activity displayed by these compounds (Table 2) and they suggest that the inhibition of the relaxing activity of DNA topoisomerase I can be another mechanism of action for these C5-Curc-FA conjugates.
Our results clearly demonstrated that 5c, 5e and 6c warrant further investigation for the development of valuable anticancer agents. In the case of 5c, this compound displayed a similar anticancer activity as 1 towards CCL-229 cells, but resulted to be less toxic towards PBMC. Therefore, 5c displayed the best therapeutic index. We are in the process of synthesizing other C5-Curc-FA conjugates in order to further explore their anticancer properties as well as their mechanism of action.
Acknowledgements
This project was supported by the National Center for Research Resources and the National Institute of General Medical Sciences of the National Institutes of Health through Grant Number 8 P20 GM 103475. We thank Material Characterization Center (MCC) for the use of NMR. J. Rosario and C. Rios acknowledge the support of the PR-INBRE program for undergraduate fellowships. This project also was supported in part by the PRCTRC Grant through the Grant Numbers U54 RR026139 and 8U54MD 007587-03, and National Institute on Minority Health and Health Disparities of the National Institute of Health through the Grant Number 8G12MD007583-28.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.American Cancer Society. Cancer Facts & Figures 2014. Atlanta: American Cancer Society; 2014. pp. 1–2. [Google Scholar]
- 2.Figueroa-Vallés NR. Bulletin of Registry of Cancer v.1(2) San Juan: Department of Health of Puerto Rico; 2008. pp. 1–8. [Google Scholar]
- 3.Aggarwal BB, Kumar A, Bharti AC. Anticancer Res. 2003;23:363–398. [PubMed] [Google Scholar]
- 4.Aggarwal BB. Cancer Biology & Therapy. 2008;7:1436–1440. doi: 10.4161/cbt.7.9.6659. [DOI] [PubMed] [Google Scholar]
- 5.Bar-Sela G, Epelbaum R, Schaffer M. Current Medicinal Chemistry. 2010;17:190–197. doi: 10.2174/092986710790149738. [DOI] [PubMed] [Google Scholar]
- 6.Agrawal DK, Mishra PK. Med. Res. Rev. 2010;30:818–860. doi: 10.1002/med.20188. [DOI] [PubMed] [Google Scholar]
- 7.Basnet P, Skalko-Basnet N. Molecules. 2011;16:4567–4598. doi: 10.3390/molecules16064567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma RA, Steward WP, Gescher AJ. Adv. Exp. Med. Biol. 2007;595:453–470. doi: 10.1007/978-0-387-46401-5_20. [DOI] [PubMed] [Google Scholar]
- 9.Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Mol. Pharmacol. 2007;4:807–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 10.Sharma RA, Gescher AJ, Steward WP. Eur. J. Cancer. 2005;41:1955–1968. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Pan MH, Huang TM, Lin JK. Drug Metab. Dispos. 1999;27:486–494. [PubMed] [Google Scholar]
- 12.Ireson C, Orr S, Jones DJ, Verschoyle R, Lim CK, Luo JL, Howells L, Plummer S, Jukes R, Williams M, Steward WP, Gescher A. Cancer Res. 2001;61:1058–1064. [PubMed] [Google Scholar]
- 13.Ravindranath V, Chandrasekhara N. Toxicology. 1981;20:251–257. doi: 10.1016/0300-483x(81)90056-1. [DOI] [PubMed] [Google Scholar]
- 14.Straganz GD, Glieder A, Brecker L, Ribbons DW, Steiner W. Biochem. J. 2003;369:573–581. doi: 10.1042/BJ20021047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosemond MJ, St John-Williams L, Yamaguchi T, Fujishita T, Walsh JS. Chem. Biol. Interact. 2004;147:129–139. doi: 10.1016/j.cbi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Grogan G. Biochem. J. 2005;388:721–730. doi: 10.1042/BJ20042038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Braiteh FS, Kurzrock R. Cancer. 2005;104:1322–1331. doi: 10.1002/cncr.21300. [DOI] [PubMed] [Google Scholar]
- 18.Liang G, Yang S, Jiang L, Zhao Y, Shao L, Xiao J, Ye F, Li Y, Li X. Chem. Pharm. Bull. 2008;56:162–167. doi: 10.1248/cpb.56.162. [DOI] [PubMed] [Google Scholar]
- 19.Liang G, Shao L, Wang Y, Zhao C, Chu Y, Xiao J, Zhao Y, Li X, Yang S. Bioorg. Med. Chem. 2009;17:2623–2631. doi: 10.1016/j.bmc.2008.10.044. [DOI] [PubMed] [Google Scholar]
- 20.Yamakoshi H, Ohori H, Kudo C, Sato A, Kanoh N, Ishioka C, Shibata H, Iwabuchi Y. Bioorg. Med. Chem. 2010;18:1083–1092. doi: 10.1016/j.bmc.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 21.Singh RK, Rai D, Yadav D, Bhargava A, Balzarini J, De Clercq E. Eur. J. Med. Chem. 2010;45:1078–1086. doi: 10.1016/j.ejmech.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.U.S. Cancer Statistics Working Group. Atlanta: Centers for Disease Control and Prevention and National Cancer Institute; [accessed 03/18/2015]. United States Cancer Statistics: 1999–2011 Incidence and Mortality Web-based Report U.S. Department of Health and Human Services. http://apps.nccd.cdc.gov/uscs/ [Google Scholar]
- 23.Higgins CA, Delbederi Z, McGarel K, Mills T, McGrath O, Feutren-Burton S, Watters W, Armstrong P, Johnston PG, Waugh D, van den Berg H. Bioconjugate Chem. 2009;20:1737–1751. doi: 10.1021/bc900122g. [DOI] [PubMed] [Google Scholar]
- 24.Lambert JB, Shurvell HF, Lightner DA, Graham Cooks R. Organic Structural Spectroscopy. New Jersey: Prentice Hall; 1998. [Google Scholar]
- 25.Aggarwal S, Ichikawa H, Takada Y, Sandur SK, Shishodia S, Aggarwal BB. Mol. Pharmacol. 2006;69:195–206. doi: 10.1124/mol.105.017400. [DOI] [PubMed] [Google Scholar]
- 26.Prakobwong S, Khoontawad J, Yongvanit P, Pairojkul C, Hiraku Y, Sithithaworn P, Pinlaor P, Aggarwal BB, Pinlaor S. Int. J. Cancer. 2011;129:88–100. doi: 10.1002/ijc.25656. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto Y, Gaynor RB. J. Clin. Invest. 2001;107:135–142. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tharakan ST, Inamoto T, Sung B, Aggarwal BB, Kamat AM. Biochem. Pharmacol. 2010;79:218–228. doi: 10.1016/j.bcp.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Schutters K, Reutelingsperger C. Apoptosis. 2010;15:1072–1082. doi: 10.1007/s10495-010-0503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wlodkowic D, Telford W, Skommer J, Darzynkiewicz Z. Methods in Cell Biology. 2011;103:55–98. doi: 10.1016/B978-0-12-385493-3.00004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyllie AH. Molecular Neurobiology. 2010;42:4–9. doi: 10.1007/s12035-010-8125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roth GN, Chandra A, Nair MG. J. Nat. Prod. 1998;61:542–545. doi: 10.1021/np970459f. [DOI] [PubMed] [Google Scholar]
- 33.Ketron AC, Gordon ON, Schneider C, Osheroff N. Biochemistry. 2013;52:221–227. doi: 10.1021/bi3014455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar A, Bora U. Interdisciplinary Sciences. 2014;6:285–291. doi: 10.1007/s12539-012-0048-6. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki K, Shono F, Kai H, Uno T, Uyeda M. J. Enzyme Inhib. 2000;15:357–366. doi: 10.1080/14756360009040693. [DOI] [PubMed] [Google Scholar]
- 36.Giovanella BC, Stehlin JS, Wall ME, Wani MC, Nicholas AW, Liu LF, Silber R, Potmesil M. Science. 1989;246:1046–1048. doi: 10.1126/science.2555920. [DOI] [PubMed] [Google Scholar]