

Abstract
Cancers progress through a series of events that can be characterized as “somatic evolution”. A central premise of Darwinian evolutionary theory is that the environment imparts pressure to select for species that are most fit within that particular microenvironmental context. Further, the rate of evolution is proportional to both 1) the strength of the environmental selection and 2) the phenotypic variance of the selected population. It is notable that, during the progression of cancers from carcinogenesis to local invasion to metastasis, the selective landscape continuously changes and throughout this process there is increased selection for cells that have altered metabolic phenotypes: implying that these phenotypes impart a selective advantage during the process of environmental selection. One of the most prevalent selected phenotypes is that of aerobic glycolysis, i.e. the continued fermentation of glucose even in the presence of adequate oxygen. The mechanisms of this so-called “Warburg Effect” have been well studied and there are multiple models to explain how this occurs and the molecular level. Herein, we propose that unifying insights can be gained by evaluating the environmental context within which this phenotype arises. In other words, we focus not on the “how”, but the “why” do cancer cells exhibit high aerobic glycolysis. This is best approached by examining the sequelae of aerobic glycolysis that may impart a selective advantage. Many of these have been considered, including: generation of anabolic substrates, response rates of glycolysis via-a-vis respiration, and generation of anti-oxidants. A further sequeala considered here is that aerobic glycolysis results in a high rate of lactic acid production; resulting in acidification of the extracellular space. Indeed, it has been shown that a low pHe promotes local invasion, promotes metastasis and inhibits anti-tumor immunity. In naturally occurring cancers, low pHe is a strong negative prognostic indicator of metastasis free survival. Further, it has been shown that inhibition of extracellular acidosis can inhibit metastasis and promote anti-tumor immunity. Hence, we propose that excess acid production confers a selective advantage for cells during the somatic evolution of cancers.
Introduction
Cancers are open, complex, and adaptive systems. They are open because there is free interaction between the tumor and the host. They are complex because they have many components that typically interact through non-linear dynamics. They are adaptive because these components can change over time and space. As non-linear dynamics are non-intuitive, complex adaptive systems are difficult to understand without mathematical models. Deep understanding of such systems can be obtained through computational models based on first principles. This is best illustrated by the most well-studied complex non-linear system – the weather. Modern meteorology has achieved unparalleled success in predicting weather (hurricane models, for example) by combining three key elements: 1. Data that are spatially and temporally explicit; 2. Sophisticated computational models); and 3. Well-defined first principles (e.g. the Navier-Stokes equations). We contend that similar understanding of Cancer Dynamics can be achieved by these same three elements: Spatially and temporally explicit data are primarily generated from imaging, computational models have been derived that accommodate some level of stochastics, and we believe that a first principle of carcinogenesis and behavior is rooted in Ecology and Evolutionary biology. Importantly, as we and others have described, Darwinian evolution of cancer requires phenotypically diverse cells being selected by the local Ecology, which is described by physical and biochemical microenvironment. Table 1 lists some of these fundamental evolutionary principles as applied to cancer.
Table 1.
Principles of Evolutionary Dynamics (as applied to Cancer)
|
The physical microenvironment is a prominent component and excellent example of the dynamics that exist within the complex cancer system. Tumor O2, glucose, and H+ levels influence both the survival and proliferation of cancer cells and, in turn, are influenced by the metabolism of tumor cells and their complex interactions with blood vessels and other normal tissue. Furthermore, these parameters and interactions are dynamic in that they change over time and space within the tumor. Clearly, understanding the role of the physical microenvironment requires extensive empirical data. However, data alone are insufficient for understanding complex systems in the absence of clearly defined first principles, which we propose must come from evolutionary biology. The conceptual model of carcinogenesis as a manifestation of evolutionary dynamics is now widely accepted and can be traced to pioneering work by Knudson (1) and Nowell (2). However, somatic evolution is commonly viewed as simply a consequence of accumulating random mutations. Darwinian dynamics are actually far more complex as they require a microenvironment selecting against a phenotypically diverse population, as described in the deceptively simple equation summarizing evolutionary dynamics:
| (1) |
This equation shows that μ̇ (the evolutionary rate, or the temporal change in strategy [∂μ/∂t]), is a function of δ (the heritable phenotypic variance), and G (the fitness of each individual). ∂G/∂μ is the slope of the fitness landscape, which describes the increase in fitness provided by an incremental increase in strategy. ∂G/∂μ and, hence, the rate of evolution, increases as environments become more stressful and more selective.
For the first term, note that genetic mutations are not explicitly included in evolutionary dynamics because evolution selects for phenotypes and not genotypes. That is, Darwinian evolution requires heritable variations in phenotype. Mutations, chromosomal rearrangements, insertion/deletions (INDELs) and the whole host of epigenetic changes are all “mechanisms of inheritance” that can contribute to the cellular phenotype (3, 4). Diversity of these genetic alterations can contribute to phenotypic heterogeneity, although the relationship is non-linear. For example, phenotypic variance can be buffered against mutations through the actions of cellular chaperones such as heat shock proteins, e.g. HSP90 or GRPs (5, 6). Conversely, multiple genetic changes can give rise to common phenotypes, commonly referred to as “convergent evolution” or “functional equivalence” (7). Figure 1 shows some of the genetic changes that can underlie some of the well-known hallmarks of cancer. For example, resistance to apoptosis can be accomplished through dozens of different genetic perturbations, such as Bcl-2, Bad, Bax, Akt, death receptors, etc. (8). Notable is that the microenvironment selects for the presence of the phenotype, and not the mechanism by which it is acquired.
Figure 1. Functional Equivalence.

In evolutionary dynamics, environments select for phenotypes that confer a proliferative or survival advantage. In cancer, the most prevalent phenotypes are described as cancer “hallmarks”, shown here. Each of these phenotypes can be expressed in response to multiple genetic alterations.
The slope of the fitness landscape in the second term on the right side of the equation describes the environmental selection pressure. It explicitly includes the role of the environment in evolution, demonstrating that the fitness of any phenotype is entirely contextual (9, 10). The fitness benefit of any given phenotypic property is dependent on the current and extant environmental selection forces. For example, resistance to apoptosis will improve fitness only in the presence of cytotoxic stress, and may actually reduce fitness in a non-stressful environment (11). Additionally, in a non-selecting environment with an abundance of nutrients and a lack of stress, cells can be at their maximal fitness (G) with an abundance of strategies (μ) and hence, ∂G/∂μ is essentially zero, leading to no evolution.
Darwinian dynamics govern cancer biology throughout the course of tumor development and treatment. While this evolutionary arc can be measured through genetic and epigenetic changes, it also fundamentally reflects alterations in the physical microenvironment that both affect and are effected by the phenotypic properties of the cancer cells. Figure 2 shows that the microenvironment changes during the course of breast cancer development from a thin layer of cells in normal epithelium to a thicker mass as cells progress to ductal carcinoma in situ, DCIS. After the basement membrane is breached during local invasion the previously avascular disease now has unfettered access to blood supplies. In the following sections, we will describe the role of the physical microenvironment in the changing adaptive landscape through the lifetime of a cancer, from Carcinogenesis to Therapy, and highlight some of the critical vacancies in our knowledge of these dynamics that require further investigation.
Figure 2. The changing environment during carcinogenesis.

As cancers progress from normal epithelia to metastatic disease, the microenvironment housing the cancer cells also changes. In normal epithelia, secretory- and myo-epithelial cells are no more than two layers off the basement membrane and no cell is more than 100 microns away from its blood supply. Hyperplasia can result in neoplasia, wherein cells can grow many layers thick so that they are far from their blood supplies and hence, are nutrient deprived. In carcinoma in situ, cells have been selected for adaptation to these nutrient deprived conditions. Following breach of the basement membrane, the microenvironment is radically different with mesenchymal and immune cells, and an abundant blood supply. Cells are selected for adaptation to this new environment and can result in a locally invasive cancer. In metastasis, cells are subjected to many of the same selection pressures, in addition to shear forces in blood and a new set of host antigens.
Carcinogenesis
The cellular transition from normal to cancer is a multistep, multi-pathway process that is often described as “somatic evolution.” These Darwinian dynamics are typically viewed as a sequence of mutations as illustrated by the “Vogelgram”. We note, however, that by equation 1, the genetic changes in a population are deeply connected to the underlying environmental properties of the tissue in which the tumor develops. In breast cancers and other ductal tumors, for example, the cells of origin line the basement membrane but have a large empty space (i.e. the ducal lumen) into which they can potentially proliferate (see Figure 2). This spatial context then dictates the order of mutation as initial selection forces are dominated by “anoikis”. In other words, cell proliferation cannot occur until the cells are able to survive when not in contact with the basement membrane. Resistance to anoikis has been associated with constitutive upregulation of EGF (e.g. ERBB2) receptors, and/or inflammation mediated activation of NFkB (12, 13). However, it is likely that there are multiple other mechanisms involved, as in Figure 1. Once the anoikis constraint is released, epithelial hyperplasia is observed as the cells are able to grow into the ductal lumens. Eventually, these hyperplastic lesions evolve into Ductal Carcinoma in Situ (DCIS), a nascent cancer that is not necessarily clinically significant because it is confined to the ductal lumen.
The spatial organization of ducts dictates subsequent evolution through altered microenvironmental selection. While abrogation of anoikis permits the new population to grow into the lumen, other growth constraints emerge due to the anatomy and physiology of ducts. That is, while the tumor grows into the duct, the blood vessels remain within the surrounding stroma and the consequent diffusion-reaction dynamics result in regional variations in growth factors, substrate, and metabolites. Figure 3 shows a late-stage comedo-type DCIS lesion. Note that the cell layer can exceed 160 microns thick. This is important because, both mathematical modeling and empirical measurements demonstrate that the oxygen concentrations approach 0 in tissue that is more than 160–200 microns from a blood vessel (14). Thus, as the peri-luminal cells in the duct grow further from the basement membrane, they must adapt to increasingly severe hypoxic conditions (15). This can be assessed by staining for HIF clients, such as carbonic anhydrase-9 (CA-IX) and we, and others, have demonstrated strong CA-IX staining in the peri-luminal region of late-stage DCIS (16). Other HIF clients, such as the glucose transporter-1 (GLUT-1), have similar spatial staining patterns (Figure 4) demonstrating that, in the absence of oxygen, glycolysis is upregulated to meet cellular energy requirement and the consequent reduction in efficiency of ATP production requires increased glucose flux. Importantly, the end-product of glycolytic metabolism is lactic acid, and hence, the peri-luminal regions of late-stage DCIS are also expected to be profoundly acidic.
Figure 3. Ductal Carcinoma in situ (DCIS).

Section of late stage comedo-type DCIS stained with hematoxylin and eosin (H&E). Notable landmarks are: a blood supply that is limited to the stroma; the reactive fibrotic stroma surrounding DCIS-involved ducts. The pleiomorphic cells exist in layers more than 160 microns thick, which is the diffusion distance of O2 in tissues. Because of oxygen diffusion, the peri-luminal cells are expected to be hypoxic and acidic. The presence of central necrosis indicates strong micro-environmental selection of peri-luminal cells, which is spatially coincident with presence of hypoxia and acidosis.
Figure 4. Ductal Carcinoma in situ (DCIS).

Section of late stage comedo-type DCIS with central necrosis stained immunohistochemically with antibody against the glucose transporter, GLUT-1. Notable is the strong staining of peri-luminal cells which are expected to be hypoxic (see figure 3). GLUT-1 is a client of the hypoxia inducible factor, HIF-1α, and can be a biomarker for high rates of glycolysis.
This hypoxic/acidic environment is, we propose, critical for understanding the later stages of carcinogenesis. In the context of equation 1, this results in a relatively large slope in the fitness landscape (e.g. high values for [∂G∂u]) which will promote rapid evolution to cellular adaptive strategies that permit survival and proliferation in both hypoxic and acidotic environments. Furthermore, both hypoxia and acidosis induce chromosomal instability and hence, this adaptive landscape favors emergence of genetically diverse clades of cells, increasing the variance (δ), and further accelerating the rate of evolution toward cancer (17).
The consequence of these selection forces are, to some extent, predictable. For example, hypoxia has been shown by Giaccia and others to select for p53-deficiency presumably to increase survival in the harsh physical microenvironment (18). We have observed spontaneous loss of p53 via chromosomal deletions in two different breast epithelial cell lines flowing selection by intermittent hypoxia (19). Acquisition of a p53-null phenotype results in further genomic instability, and can induce even higher glycolysis via reduction of the TP53-indicuble glycolysis and apoptosis regulator, TIGAR, which acts to inhibit glycolysis in response to genotoxic stress (20). Thus, regional variations in the physical microenvironment of in-situ cancers will generally select for molecular pathways that increase glycolysis to maintain energy supplies in hypoxia and increase tolerance to the acidosis that results from glycolysis. We have developed models over the last decade to describe these adaptations, (15, 21). In the latest of these, shown in Figure 5 (17), we explicitly account for the fact that hypoxia and acidosis induce both genomic instability (phenotypic variance) and environmental selection. Hence, these microenvironmental factors affect both components of the evolutionary dynamic equation 1. The evolutionary sequence described in these models confers a powerful combination of phenotypic properties on local cell populations allowing them to create an acidic environment through constitutively activated glycolysis (i.e. aerobic glycolysis or the Warburg Effect) that produces an acidic environment to which they are well adapted. Further, in all of these models we have attempted to explicitly couple the emergent phenotypes to the microenvironmental selection pressures that would favor them. Among these, the most important is emergence of constitutively activated glycolysis and, despite the fact that we proposed that this emerges following hypoxic selection, the microenvironmental conditions that select for this phenotype are unknown. We do know, after dozens of attempts in multiple cells lines, that cyclic or chronic hypoxia does not select for cells with a Warburg Effect. As an alternative, there is evidence that highly glycolytic cells with mutant k-ras are selected under conditions of nutrient deprivation (22), and periluminal cells in DCIS are expected to be glucose-deprived. This hypothesis remains to be tested.
Figure 5. Evolutionary dynamics in carcinogenesis (redrawn from Gillies et al., ref. 17).
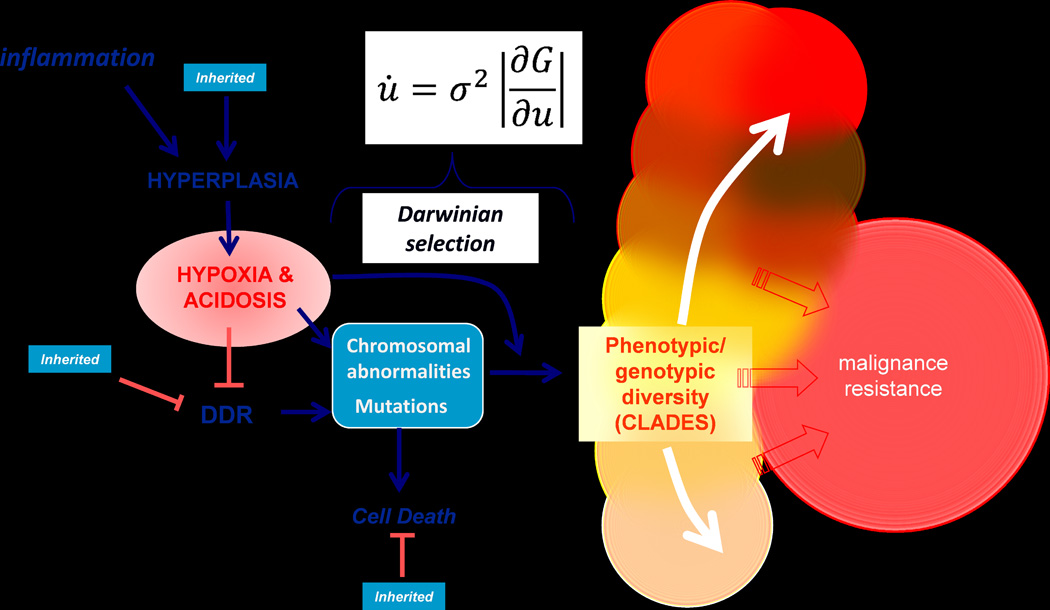
Hyperplasia can be induced by chronic inflammation, and this results in volumes that are hypoxic and acidic (see figures 3 and 4). Through mechanisms that include reactive oxygen (ROS) generated genotoxicity and inhibition of DNA damage response (DDR) pathways, hypoxia and acidosis are known to induce mutations and chromosomal abnormalities, which normally result in apoptotic cell death. With loss of apoptosis, these genomic changes are expected to generate increased phenotypic variance (σ in equation). In addition, hypoxia and acidosis impart strong selection pressure on the emerging phenotypes (∂G/∂μ in equation). These processes combine to generate a high rate of evolution (∂μ/∂t), generating multiple successful clades of cells, each adapted to be maximally fit in their unique microenvironmental niches. The preponderance of these clades is synonymous with malignance and resistance. Notably, many components of this process can be inherited, such as inflammation-independent hyperplasia, reduced DDR, or resistance to apoptosis.
Micro-invasive Cancer
To progress to a potentially life threatening cancer, the tumor cells must breach the basement membrane and gain access to the underlying stroma. Figure 6 captures this process (9) and, importantly, demonstrates that locally invasive cells can take their glycolytic phenotype with them. What are the physical and biological mechanisms that are necessary and sufficient for this transition? Note, in figure 3, that the stroma surrounding late stage DCIS is acellular and fibrotic; characteristics typical of reactive stroma. Collagen re-organization can be revealed by second harmonic microscopy (2HM) of unstained tissue sections (23) and Bhujwalla et al. have shown an induction of 2HM changes induced by hypoxia (24). Notably, the acid secreted by growing tumors induces the reactivity of stroma to secrete inflammatory mediators such as TNFα and nitric oxide, via p38 MAP-K pathway (25). We thus propose, yet it remains to be proved, that the normal stroma provides a physical barrier to local invasion and that the glycolytic, acid-adapted tumor cells that arise within DCIS induce stromal remodeling, possibly via an acid-mediated mechanism, which is necessary and permissive for neoplastic epithelial cells to penetrate the basement membrane and invade locally into the stroma.
Figure 6. Locally Invasive Breast Cancer.

Section of breast duct with comedo-type DCIS lesion wherein cells have broken through the basement membrane and invaded local stroma. Section is stained immunohistochemically with antibody against GLUT-1. Similar to Figure 4, strong peri-luminal staining is observed. Notably, some of the invasive cells are also strongly positive, even though in the stroma these cells have access to vasculature and are not expected to be hypoxic (see figure 3). Hence, we suspect that these cells are “pseudo-hypoxic” and likely exhibit aerobic glycolysis (Warburg Effect).
Furthermore, this phenotype persists as the tumor progresses because it continues to provide an adaptive advantage as, upon breeching the membrane, a novel adaptive landscape emerges (see Figure 2). That is, prior to the formation of a microinvasive tumor focus, mesenchymal cells in the stroma have never been in contact with epithelial cell and, similarly, the epithelially-derived cancer cells have never directly interacted with the mesenchyma. Furthermore, this adaptive landscape also includes immune cells, which are only rarely observed in in-situ tumors. This requires significant adaptations in the invading cancer that is derived from epithelia. Immune evasion strategies can involve mimicry such as epithelial-to-mesenchymal transition (EMT) or epithelial-to-leukocyte transition (ELT) (26), expression of checkpoint ligands, release of kyneuranines (27, 28), or simply producing an acidic environment (29, 30).
Clearly, despite these obstacles, some cancer cells succeed in evading immune surveillance and setting up a successful colony within the stroma, which has an abundance of oxygen, nutrients, and a neutral pH. While these conditions would appear to be ideal, they are not evolutionarily optimal because they favor proliferation of stromal cells as well as tumor cells; creating a subtle competition. In fact, proliferation of mesenchymal cells (e.g. fibroblasts) with extinction of local tumor cells could be a specific tumor defense mechanism of the host. We propose that, in fact, tumor cells can gain an advantage by recapitulating the intra-ductal conditions (i.e. acidic, hypoxic and nutrient deprived) to which they were adapted but are unfavorable for the normal cells. Notably, this can be achieved by expression of glycolytic phenotype even in the presence of oxygen (aerobic glycolysis). The success of this adaptive strategy is readily apparent in the exceptionally high sensitivity and specificity of FDG PET scans to identify malignant disease (9). It is likely that the strategy also results in subtle dysangiogenesis, which may be initially induced by hypoxia but also increases hypoxia in a slippery slope of feed-forward dynamics (31) as well as metabolic co-option of stromal cells to form a metabolic network wherein the stromal cells eat what the cancer produces, and vice-versa, as illustrated in Figure 7, drawn after (32). This figure shows that cells can be either fermentative, wherein they consume glucose and export lactic acid via monocarboxylate transporter-4 (MCT-4), or they can be oxidative and consume lactate to produce CO2. While oxidative metabolism has an absolute requirement for oxygen, fermentative metabolism can occur in the absence or presence of oxygen (the Warburg Effect). Figure 8 shows differential staining for MCT-4 in well-differentiated pancreatic ductal adenocarcinoma, PDAC, as well as in parts of the stroma, suggestive of metabolic multi-compartmentation. Notably, this metabolic cooperation need not be limited to tumor-stromal interactions as different components of the stroma may cooperate with each other. Hence, we propose that successful transition from in-situ to invasive cancer can be characterized as a failure of the stroma to adequately compete with the invading cancer cells.
Figure 7. Metabolic symbiosis in cancer, re-drawn after Sonveaux (ref 32).

Oxidative cells (A) consume lactic acid, which is converted to pyruvate and subsequently oxidized in the TCA cycle to produce ATP and CO2. The CO2 is hydrated on the outside of the cell by carbonic anhydrases 9 or 12 (CA9/12) to produce HCO3− + H+. Additional H+ are exported by the sodium/hydrogen exchanger, NHE. In fermentative cells (B), glucose is imported via GLUT-1 or GLUT-3, converted to G6P, whereupon it enters glycolysis. The oxidative step catalyzed by GAPDH results in acid production and release of the H+. These H+ are removed by NHE1 and the sodium dependent bicarbonate transporter, NBC, working in concert with CA9. The lactate produced by reduction of pyr to lactate by LDH-5 (a.k.a. LDH-A) is exported by MCT4. Notably, while oxidative cells have an absolute requirement for oxygenation, fermentative cells can exist in anaerobic (Pasteur Effect) or aerobic (Warburg Effect) environments.
Figure 8. Metabolic compartmentation.

Figure shows a 3 mm core from a pancreatic ductal adenocarcinoma tissue microarray stained for MCT-4. Note strong staining in the ductal cancer cells and the high heterogeneity in stromal staining.
Primary Cancers
Formation of a clinically apparent cancer requires expansion of the micro-invasive foci. This requires new adaptations to promote angiogenesis, suppress proliferation of normal mesenchymal cells, and defeat the negative effects of the immune system. We note, however, that FdG PET imaging has clearly demonstrated that the vast majority of primary and metastatic tumors exhibit increased glucose flux indicating that upregulated glycolysis that emerges in carcinogenesis continues to provide an adaptive advantage. Thus, it is not surprising that studies in preclinical models and primary cancers have demonstrated that solid tumors are acidic and actually export acid into the surrounding normal tissue. This tumor-induced perturbation of the pH in adjacent host tissue was predicted first by reaction-diffusion modeling (33) and subsequently confirmed with imaging of tumors in dorsal window chambers (33, 34). These have led to the hypothesis that aerobic glycolysis provides an adaptive advantage despite its inefficient energy production by promoting “acid mediated invasion.” Specifically, acid that diffuses along concentration gradients into peri-tumoral normal tissue promotes tumor growth and invasion because it reduces proliferation of surrounding stromal cells, induces breakdown of normal extracellular matrix (ECM), promotes angiogenesis, and blunts the normal immune response to tumor antigens (35, 36).
Clearly, the mechanisms by which tumor cells invade into adjacent tissue are complex and can be modified in response to environmental conditions. The acid-mediated invasion hypothesis is supported by evidence that tissue pH affects many of these molecular and cellular events. There is clear evidence that increased acid production, combined with poor perfusion, results in an acidic extracellular pH (pHe) in malignant tumors (pH = 6.5 – 6.9) compared to normal tissue under physiological conditions (pHe= 7.2 – 7.4) (37–39). Indeed, it has been convincingly been shown that the pHe in canine sarcomas ranged from 6.37–7.42; and that, furthermore, an acidic pHe was a strong negative predictor of metastasis free survival (HR, 0.29; p+0.005)(40). Furthermore, window chamber studies in vivo have demonstrated that H+ ions flow along concentration gradients from tumor into adjacent normal tissue causing significant acidosis. A number of studies have demonstrated acidic pHe can induce release of (cysteine or aspartyl) cathepsin proteinases (41–43), which promote local invasion and tissue remodeling (44–46). In vivo studies have demonstrated that the acidic peri-tumoral normal tissue undergoes significant tissue remodeling with reduction of the ECM density (47). This can be seen in the far right panel in Figure 2. Furthermore, cells exposed to in vitro low pH demonstrate increased invasion both in vitro and in vivo (43, 48, 49). An acidic environment can also increase angiogenesis through the release of VEGF, and inhibits the immune response to tumor antigens (35, 36, 50). Importantly, cancer cells, because of the evolutionary dynamics of in situ tumors, enter the micro-invasive stage of tumor growth with already-developed adaptive mechanisms that allow them to survive and even proliferate in acidic environments. We have shown, via modified cellular automaton models, that acquisition of acid resistance is a pre-requisite to acquiring a glycolytic phenotype (51). These adaptations can involve, inter alia, chronically activated autophagy as a survival mechanism (52), upregulation of the sodium-hydrogen exchange (NHE-1) or carbonic anhydrase (CA-IX) (53–55) to maintain a neutral intracellular pH, and decoration of the plasma membrane with lysosomally-associated proteins (56, 57), coupled to release of lysosomal proteases, discussed above. As normal cells die and the extracellular matrix is degraded, cancer cells continue to proliferate and invade this open space. This “niche engineering” strategy promotes local invasion and subsequent in vivo growth of malignant tumors.
Altering the physical environment for cancer therapy and prevention
In the prior sections we argued that the physical microenvironment generally and pH specifically are both a cause and a consequence of tumor cell evolution and subsequent tumor growth and invasion. This model suggests that neutralization of the acidity could under, some circumstances, “tip the balance of power” thus delaying carcinogenesis and reducing tumor invasive growth. We, and others, have demonstrated that oral buffers could neutralize the acidic pH of tumors (58–60). Neutralizing tumor acidity can increase the effectiveness of weak base chemotherapeutics through ion trapping (61, 62). Further, in multiple systems, we have observed that chronic ingestion of oral buffers increases tumor pH and inhibits experimental or spontaneous metastases (Fig. 9) (63). That this was a buffer effect was confirmed by observations that many buffers work, including bicarbonate, imidazoles and TRIS (64). Furthermore, lysine HCl (pH=8.0) had no effect, whereas lysine free base (pH=10.6) completely inhibited formation of experimental PC3m prostate cancer metastases (65). The effects have been most pronounced in prostate cancer models and, hence, we have investigated the effects of buffer therapy on the behavior of TRAMP (Transgenic Adenoma of Mouse Prostate) mice, which express middle T antigen under control of the probasin promoter. We have observed that, if buffer therapy is initiated prior to 4 weeks, cancers do not develop in these mice (66). If buffer therapy is initiated after 10 weeks (after the cancers are extracapsular), it has no effect on the primary tumor, but completely inhibits formation of metastases (Ibrahim-Hashim, in review). The results of buffer therapy have been so promising, that a phase I clinical trial to determine tolerability has been initiated (NCT01846429). A practical question going forward for clinical trials is if buffer therapy can be improved upon, as it required chronic ingestion of alkaline buffers and some grade 2 gastrointenstinal events have been observed.
Figure 9. Inhibition of metastasis with buffer therapy, from Robey et al. (ref. 41).

Lung metastases of mice bearing GFP-expressing MDA-mb-231 breast cancer xenografts treated with 200 mM ad lib bicarbonate. Primary tumors were allowed to grow to a volume of 1.0 cc after which they were resected. Animals remained until metastases in draining lymph nodes reached volume of 0.5 cc. Image show lungs of mice under white light and fluorescence.
Finally, an important advantage of an acidic micoenvironment is that it tends to render the immune response less effective (29). This suggests that systemic buffers would improve tumor response to immunotherapy, and this is currently under investigation (Pilon-Thomas, in review).
The problem of heterogeneity
As primary cancers grow, they usually develop substantial spatial heterogeneity that is governed primarily by blood flow. Peter Nowell is credited for proposing the clonal basis of tumors (2), and this has been well-validated with modern molecular techniques that have sequenced genomes from different regions of the same tumor (67–69). In fact, the existence of intratumoral chromosomal heterogeneity has been known since the 1930’s (70). Nonetheless, the clinical practice of medical oncology continues to ignore heterogeneity and treat the most visible genetic lesion, without accommodating minor populations that surely exist. Hence, tumors are often described as well mixed homogeneous systems. For example, a breast cancer can be classified as “estrogen receptor (ER) positive” even when as few as 10% of the tumor cells express ER. Notably, such patients often respond initially to anti-estrogen therapy as well as those with high levels of ER-positive cells (71). However, most patients eventually fail due to acquisition of resistance, which is fundamentally due to heterogeneity and its relevance to treatment. Planned emergence of resistance is not readily accommodated in today’s clinical practice. Such regional variations, as well as temporal changes with or without therapy, clearly limit the role of genomic/proteomic data in long-term tumor therapy, especially since these data are obtained once prior to the beginning of therapy and rarely during active treatment. We have proposed that regional variation can be characterized and quantified by defining “habitats” that can be measured by combining physiological parameters derived from clinical images; including perfusion, cell density, interstitial edema, glucose uptake, etc (72). In a strict analogy to ecology, these habitats are defined by the availability of water, giving rise to well-perfused rain forests, constantly perfused riparian zones, intermittently perfused savannahs, and rarely perfused deserts (73). In turn, these regional variations in physiological conditions select for local cell populations that are best-adapted to these specific habitats. These spatial variations will be apparent in regional heterogeneity of the molecular properties of the cells. Yet cancers are complex dynamical systems and spatiotemporal changes in blood, cell density and cellular metabolism undoubtedly alter the local physical microenvironment, thus influencing regional variations in tumor growth and invasion. Recently, we examined these dynamics in growth of tumors in window chambers and observed that the acidic pHe of peritumoral tissues around the circumference of the tumor was predictive of subsequent tumor invasion (34).
Clearly it is not possible to continuously sample regional tumor populations to assess spatial and temporal variability in the genotypic and phenotypic tumor populations. We have proposed that non-invasive imaging must play a critical in defining the heterogeneity and the presence of habitats within individual tumors. This approach, called “radiomics” defines heterogeneity through quantitative analysis of radiological images, and these are being shown to have high prognostic value, especially in combination with genomic analyses, or “radiogenomics” (74, 75).
Conclusion
The physiological microenvironment of nascent and clinically apparent primary and metastatic tumors is hostile: the pH is acidic, oxygen is variable, substrates are in short supply and there is an abundance of toxic reactive oxygen and reactive nitrogen species. These factors indisputably play a role in carcinogenesis and tumor progression, yet their exact roles and the mechanisms involved are only now beginning to be defined. As expressed in equation 1, the Darwinian dynamics that govern cancer biology include complex interactions between the changing tumor genome/phenome and local environmental conditions that include normal mesenchymal cells, immune cells, as well as underlying concentrations of substrate, metabolites, and cell-products. The physical environment within the tumor is, thus, both a force that selects for optimal tumor phenotypes and a strategy of the tumor that can be manipulated to enhance its own fitness – an evolutionary strategy termed “niche engineering”. Defining these complex dynamics must include: (i) the cancer cells; (ii) the host stromal and immune cells; and (iii) the bidirectional interactions between these populations. This is particularly evident in the physical microenvironment that is governed by blood flow and tumor metabolism as well as the tumor effects on blood flow and vice-versa. Understanding these interactions at a mechanistic level permits novel therapeutic perturbations that can delay or even prevent the evolutionary dynamics that govern carcinogenesis and tumor invasion.
References
- 1.Knudson AG., Jr Mutation and cancer in man. Cancer. 1977;39(4 Suppl):1882–1886. doi: 10.1002/1097-0142(197704)39:4+<1882::aid-cncr2820390821>3.0.co;2-#. PubMed PMID: 851956. [DOI] [PubMed] [Google Scholar]
- 2.Nowell PC. The clonal nature of neoplasia. Cancer cells. 1989;1(1):29–30. PubMed PMID: 2701359. [PubMed] [Google Scholar]
- 3.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–1570. doi: 10.1126/science.1230184. PubMed PMID: 23539597; PubMed Central PMCID: PMC3821556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. PubMed PMID: 22770212. [DOI] [PubMed] [Google Scholar]
- 5.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nature reviews Cancer. 2005;5(10):761–772. doi: 10.1038/nrc1716. PubMed PMID: 16175177. [DOI] [PubMed] [Google Scholar]
- 6.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nature reviews Cancer. 2014;14(4):263–276. doi: 10.1038/nrc3701. PubMed PMID: 24658275; PubMed Central PMCID: PMC4158750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gatenby RA, Gillies RJ, Brown JS. Of cancer and cave fish. Nature reviews Cancer. 2011;11(4):237–238. doi: 10.1038/nrc3036. PubMed PMID: 21548400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nature reviews Cancer. 2002;2(4):277–288. doi: 10.1038/nrc776. PubMed PMID: 12001989. [DOI] [PubMed] [Google Scholar]
- 9.Gillies RJ, Gatenby RA. Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? Journal of bioenergetics and biomembranes. 2007;39(3):251–257. doi: 10.1007/s10863-007-9085-y. PubMed PMID: 17624581. [DOI] [PubMed] [Google Scholar]
- 10.Gillies RJ, Gatenby RA. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer metastasis reviews. 2007;26(2):311–317. doi: 10.1007/s10555-007-9065-z. PubMed PMID: 17404691. [DOI] [PubMed] [Google Scholar]
- 11.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, Wang L, Amato KR, Arcila M, Sos ML, Socci ND, Viale A, de Stanchina E, Ginsberg MS, Thomas RK, Kris MG, Inoue A, Ladanyi M, Miller VA, Michor F, Pao W. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Science translational medicine. 2011;3(90):90ra59. doi: 10.1126/scitranslmed.3002356. PubMed PMID: 21734175; PubMed Central PMCID: PMC3500629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whelan KA, Schwab LP, Karakashev SV, Franchetti L, Johannes GJ, Seagroves TN, Reginato MJ. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. The Journal of biological chemistry. 2013;288(22):15865–15877. doi: 10.1074/jbc.M112.426999. PubMed PMID: 23585570; PubMed Central PMCID: PMC3668743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SH, Riley Pt, Frisch SM. Regulation of anoikis by deleted in breast cancer-1 (DBC1) through NF-kappaB. Apoptosis : an international journal on programmed cell death. 2013;18(8):949–62. doi: 10.1007/s10495-013-0847-1. PubMed PMID: 23588592, PubMed Central PMCID: PMC3691317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–257. doi: 10.1038/35025220. PubMed PMID: 11001068. [DOI] [PubMed] [Google Scholar]
- 15.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature reviews Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. Epub 2004/11/02. PubMed PMID: 15516961. [DOI] [PubMed] [Google Scholar]
- 16.Wykoff CC, Beasley NJP, Watson PH, Turner KJ, Pastorek J, Amen Sibtain, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. Hypoxia-inducible Expression of Tumor-associated Carbonic Anhydrases. Can Res. 2000;60:7075–7083. [PubMed] [Google Scholar]
- 17.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nature reviews Cancer. 2012;12(7):487–93. doi: 10.1038/nrc3298. Epub 2012/06/15. PubMed PMID: 22695393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379(6560):88–91. doi: 10.1038/379088a0. Epub 1996/01/04. PubMed PMID: 8538748. [DOI] [PubMed] [Google Scholar]
- 19.Verduzco D, Xu L, lloyd MC, Ibrahim Hashim A, Balagurunathan Y, Gatenby RA, Gillies RJ. Intermittent hypoxia selects for genotypes and phenotypes that increase survival, invasion, and therapy resistance. PloS one. 2015 doi: 10.1371/journal.pone.0120958. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. PubMed PMID: 16839880. [DOI] [PubMed] [Google Scholar]
- 21.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nature reviews Cancer. 2008;8(1):56–61. doi: 10.1038/nrc2255. Epub 2007/12/07. PubMed PMID: 18059462. [DOI] [PubMed] [Google Scholar]
- 22.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA, Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325(5947):1555–1559. doi: 10.1126/science.1174229. PubMed PMID: 19661383; PubMed Central PMCID: PMC2820374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burke K, Tang P, Brown E. Second harmonic generation reveals matrix alterations during breast tumor progression. Journal of biomedical optics. 2013;18(3):31106. doi: 10.1117/1.JBO.18.3.031106. PubMed PMID: 23172133; PubMed Central PMCID: PMC3595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kakkad SM, Penet MF, Akhbardeh A, Pathak AP, Solaiyappan M, Raman V, Leibfritz D, Glunde K, Bhujwalla ZM. Hypoxic tumor environments exhibit disrupted collagen I fibers and low macromolecular transport. PloS one. 2013;8(12):e81869. doi: 10.1371/journal.pone.0081869. PubMed PMID: 24349142; PubMed Central PMCID: PMC3861360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riemann A, Ihling A, Thomas J, Schneider B, Thews O, Gekle M. Acidic environment activates inflammatory programs in fibroblasts via a cAMP-MAPK pathway. Biochimica et biophysica acta. 2014;1853(2):299–307. doi: 10.1016/j.bbamcr.2014.11.022. PubMed PMID: 25461841. [DOI] [PubMed] [Google Scholar]
- 26.Luddy KA, Robertson-Tessi M, Tafreshi NK, Soliman H, Morse DL. The role of toll-like receptors in colorectal cancer progression: evidence for epithelial to leucocytic transition. Frontiers in immunology. 2014;5:429. doi: 10.3389/fimmu.2014.00429. PubMed PMID: 25368611; PubMed Central PMCID: PMC4202790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chow MT, Moller A, Smyth MJ. Inflammation and immune surveillance in cancer. Seminars in cancer biology. 2012;22(1):23–32. doi: 10.1016/j.semcancer.2011.12.004. PubMed PMID: 22210181. [DOI] [PubMed] [Google Scholar]
- 28.Hamai A, Benlalam H, Meslin F, Hasmim M, Carre T, Akalay I, Janji B, Berchem G, Noman MZ, Chouaib S. Immune surveillance of human cancer: if the cytotoxic T-lymphocytes play the music, does the tumoral system call the tune? Tissue antigens. 2010;75(1):1–8. doi: 10.1111/j.1399-0039.2009.01401.x. PubMed PMID: 20196816. [DOI] [PubMed] [Google Scholar]
- 29.Lardner A. The effects of extracellular pH on immune function. Journal of leukocyte biology. 2001;69(4):522–530. PubMed PMID: 11310837. [PubMed] [Google Scholar]
- 30.Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, Huber V, Parmiani G, Generoso L, Santinami M, Borghi M, Fais S, Bellone M, Rivoltini L. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer research. 2012;72(11):2746–2756. doi: 10.1158/0008-5472.CAN-11-1272. PubMed PMID: 22593198. [DOI] [PubMed] [Google Scholar]
- 31.Gillies RJ, Schornack PA, Secomb TW, Raghunand N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia. 1999;1(3):197–207. doi: 10.1038/sj.neo.7900037. Epub 2000/08/10. PubMed PMID: 10935474; PubMed Central PMCID: PMC1508079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Frontiers in pharmacology. 2011;2:49. doi: 10.3389/fphar.2011.00049. Epub 2011/09/10. PubMed PMID: 21904528; PubMed Central PMCID: PMC3161244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer research. 2006;66(10):5216–5223. doi: 10.1158/0008-5472.CAN-05-4193. PubMed PMID: 16707446. [DOI] [PubMed] [Google Scholar]
- 34.Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ. Acidity generated by the tumor microenvironment drives local invasion. Cancer research. 2013 doi: 10.1158/0008-5472.CAN-12-2796. Epub 2013/01/05. PubMed PMID: 23288510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer research. 2001;61(16):6020–6024. Epub 2001/08/17. PubMed PMID: 11507045. [PubMed] [Google Scholar]
- 36.Xu L, Fukumura D, Jain RK. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. The Journal of biological chemistry. 2002;277(13):11368–11374. doi: 10.1074/jbc.M108347200. Epub 2001/12/14. PubMed PMID: 11741977. [DOI] [PubMed] [Google Scholar]
- 37.Stubbs M, McSheehy PM, Griffiths JR, Bashford CL. Causes and consequences of tumour acidity and implications for treatment. Molecular medicine today. 2000;6(1):15–19. doi: 10.1016/s1357-4310(99)01615-9. PubMed PMID: 10637570. [DOI] [PubMed] [Google Scholar]
- 38.Gillies RJ, Liu Z, Bhujwalla Z. 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. The American journal of physiology. 1994;267(1 Pt 1):C195–C203. doi: 10.1152/ajpcell.1994.267.1.C195. PubMed PMID: 8048479. [DOI] [PubMed] [Google Scholar]
- 39.van Sluis R, Bhujwalla ZM, Raghunand N, Ballesteros P, Alvarez J, Cerdan S, Galons JP, Gillies RJ. In vivo imaging of extracellular pH using 1H MRSI. Magn Reson Med. 1999;41(4):743–750. doi: 10.1002/(sici)1522-2594(199904)41:4<743::aid-mrm13>3.0.co;2-z. Epub 1999/05/20. PubMed PMID: 10332850. [DOI] [PubMed] [Google Scholar]
- 40.Lora-Michiels M, Yu D, Sanders L, Poulson JM, Azuma C, Case B, Vujaskovic Z, Thrall DE, Charles HC, Dewhirst MW. Extracellular pH and P-31 magnetic resonance spectroscopic variables are related to outcome in canine soft tissue sarcomas treated with thermoradiotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(19):5733–5740. doi: 10.1158/1078-0432.CCR-05-2669. PubMed PMID: 17020978. [DOI] [PubMed] [Google Scholar]
- 41.Rozhin J, Sameni M, Ziegler G, Sloane BF. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer research. 1994;54(24):6517–6525. PubMed PMID: 7987851. [PubMed] [Google Scholar]
- 42.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer research. 2009;69(6):2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. PubMed PMID: 19276390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer research. 2006;66(13):6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. PubMed PMID: 16818644. [DOI] [PubMed] [Google Scholar]
- 44.Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. Journal of the National Cancer Institute. 1997;89(17):1260–1270. doi: 10.1093/jnci/89.17.1260. PubMed PMID: 9293916. [DOI] [PubMed] [Google Scholar]
- 45.Sloane BF, Moin K, Krepela E, Rozhin J. Cathepsin B and its endogenous inhibitors: the role in tumor malignancy. Cancer metastasis reviews. 1990;9(4) doi: 10.1007/BF00049523. PubMed PMID: 5824. [DOI] [PubMed] [Google Scholar]
- 46.Rochefort H, Liaudet-Coopman E. Cathepsin D in cancer metastasis: a protease and a ligand. Apmis. 1999;107(1):86–95. doi: 10.1111/j.1699-0463.1999.tb01530.x. Epub 1999/04/06. PubMed PMID: 10190284. [DOI] [PubMed] [Google Scholar]
- 47.Gatenby RA, Gawlinski ET. A reaction-diffusion model of cancer invasion. Cancer research. 1996;56(24):5745–5753. Epub 1996/12/15. PubMed PMID: 8971186. [PubMed] [Google Scholar]
- 48.Moellering RE, Black KC, Krishnamurty C, Baggett BK, Stafford P, Rain M, Gatenby RA, Gillies RJ. Acid treatment of melanoma cells selects for invasive phenotypes. Clinical & experimental metastasis. 2008;25(4):411–425. doi: 10.1007/s10585-008-9145-7. PubMed PMID: 18301995. [DOI] [PubMed] [Google Scholar]
- 49.Schlappack OK, Zimmermann A, Hill RP. Glucose starvation and acidosis: effect on experimental metastatic potential, DNA content and MTX resistance of murine tumour cells. Br J Cancer. 1991;64(4):663–670. doi: 10.1038/bjc.1991.378. Epub 1991/10/01. PubMed PMID: 1911214; PubMed Central PMCID: PMC1977701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lardner A. The effects of extracellular pH on immune function. Journal of leukocyte biology. 2001;69(4):522–530. Epub 2001/04/20. PubMed PMID: 11310837. [PubMed] [Google Scholar]
- 51.Robertson-Tessi M, Gillies RJ, Gatenby RA, Anderson AR. The impact of heterogeneity on tumor growth, invasion and treatment outcomes. PloS one. 2015 doi: 10.1158/0008-5472.CAN-14-1428. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jonathan Wojtkowiak JMR, Kumar Virendra, Schramm Karla J, Haller Edward, Proemsey Joshua B, Lloyd Mark C, Sloane Bonnie F, Gillies Robert J. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer research. 2012 doi: 10.1158/0008-5472.CAN-11-3881. Published OnlineFirst June 19, 2012; Epub Published OnlineFirst June 19, 2012; doi:10.1158/0008-5472.CAN-11-3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouyssegur J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer research. 2009;69(1):358–368. doi: 10.1158/0008-5472.CAN-08-2470. Epub 2009/01/02. PubMed PMID: 19118021. [DOI] [PubMed] [Google Scholar]
- 54.Ivanov S, Liao SY, Ivanova A, Danilkovitch-Miagkova A, Tarasova N, Weirich G, Merrill MJ, Proescholdt MA, Oldfield EH, Lee J, Zavada J, Waheed A, Sly W, Lerman MI, Stanbridge EJ. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol. 2001;158(3):905–919. doi: 10.1016/S0002-9440(10)64038-2. Epub 2001/03/10. PubMed PMID: 11238039; PubMed Central PMCID: PMC1850340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gatenby RA. The potential role of transformation-induced metabolic changes in tumor-host interaction. Cancer research. 1995;55(18):4151–4156. Epub 1995/09/15. PubMed PMID: 7664293. [PubMed] [Google Scholar]
- 56.Steffan JJ, Snider JL, Skalli O, Welbourne T, Cardelli JA. Na+/H+ exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic. 2009;10(6):737–753. doi: 10.1111/j.1600-0854.2009.00904.x. PubMed PMID: 19302267. [DOI] [PubMed] [Google Scholar]
- 57.Glunde K, Guggino SE, Solaiyappan M, Pathak AP, Ichikawa Y, Bhujwalla ZM. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia. 2003;5(6):533–545. doi: 10.1016/s1476-5586(03)80037-4. PubMed PMID: 14965446; PubMed Central PMCID: PMC1502575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW, Paine-Murrieta G, Roe D, Bhujwalla ZM, Gillies RJ. Enhancement of chemotherapy by manipulation of tumour pH. British journal of cancer. 1999;80(7):1005–1011. doi: 10.1038/sj.bjc.6690455. Epub 1999/06/11. PubMed PMID: 10362108; PubMed Central PMCID: PMC2363059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen LQ, Howison CM, Jeffery JJ, Robey IF, Kuo PH, Pagel MD. Evaluations of extracellular pH within in vivo tumors using acidoCEST MRI. Magn Reson Med. 2014;72(5):1408–1417. doi: 10.1002/mrm.25053. PubMed PMID: 24281951; PubMed Central PMCID: PMC4033731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andreev OA, Dupuy AD, Segala M, Sandugu S, Serra DA, Chichester CO, Engelman DM, Reshetnyak YK. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(19):7893–7898. doi: 10.1073/pnas.0702439104. PubMed PMID: 17483464; PubMed Central PMCID: PMC1861852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahoney BP, Raghunand N, Baggett B, Gillies RJ. Tumor acidity, ion trapping and chemotherapeutics. I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochemical pharmacology. 2003;66(7):1207–1218. doi: 10.1016/s0006-2952(03)00467-2. Epub 2003/09/25. PubMed PMID: 14505800. [DOI] [PubMed] [Google Scholar]
- 62.Raghunand N, Mahoney BP, Gillies RJ. Tumor acidity, ion trapping and chemotherapeutics. II. pH-dependent partition coefficients predict importance of ion trapping on pharmacokinetics of weakly basic chemotherapeutic agents. Biochemical pharmacology. 2003;66(7):1219–1229. doi: 10.1016/s0006-2952(03)00468-4. Epub 2003/09/25. PubMed PMID: 14505801. [DOI] [PubMed] [Google Scholar]
- 63.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer research. 2009;69(6):2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. Epub 2009/03/12. PubMed PMID: 19276390; PubMed Central PMCID: PMC2834485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ibrahim Hashim A, Cornnell HH, Coelho Ribeiro Mde L, Abrahams D, Cunningham J, Lloyd M, Martinez GV, Gatenby RA, Gillies RJ. Reduction of metastasis using a non-volatile buffer. Clinical & experimental metastasis. 2011;28(8):841–849. doi: 10.1007/s10585-011-9415-7. Epub 2011/08/24. PubMed PMID: 21861189; PubMed Central PMCID: PMC3213349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ribeiro M, Silva AS, Bailey K, Kumar NB, Sellers TA, Gatenby RA, Ibrahim Hashim A, Gillies RJ. Buffer Therapy for Cancer. J Nutr Food Sci. 2012;S2:1–7. [PMC free article] [PubMed] [Google Scholar]
- 66.Ibrahim-Hashim A, Cornnell HH, Abrahams D, Lloyd M, Bui M, Gillies RJ, Gatenby RA. Systemic buffers inhibit carcinogenesis in TRAMP mice. The Journal of urology. 2012;188(2):624–631. doi: 10.1016/j.juro.2012.03.113. Epub 2012/06/19. PubMed PMID: 22704445; PubMed Central PMCID: PMC3694604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. PubMed PMID: 20981102; PubMed Central PMCID: PMC3148940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. Epub 2012/03/09. PubMed PMID: 22397650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(10):4009–4014. doi: 10.1073/pnas.1219747110. Epub 2013/02/16. PubMed PMID: 23412337; PubMed Central PMCID: PMC3593922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winge O. Zytologische untersuchungen uber die natur maligner tumoren. II. Teerkarzinome bei mausen. Z Zellforsch Mikrosk Anat. 1930;10:683–735. [Google Scholar]
- 71.Allred DC, Brown P, Medina D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast cancer research : BCR. 2004;6(6):240–245. doi: 10.1186/bcr938. PubMed PMID: 15535853l; PubMed Central PMCID: PMC1064085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gatenby RA, Grove O, Gillies RJ. Quantitative imaging in cancer evolution and ecology. Radiology. 2013;269(1):8–15. doi: 10.1148/radiol.13122697. PubMed PMID: 24062559; PubMed Central PMCID: PMC3781355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alfarouk KO, Ibrahim ME, Gatenby RA, Brown JS. Riparian ecosystems in human cancers. Evolutionary applications. 2013;6(1):46–53. doi: 10.1111/eva.12015. PubMed PMID: 23396634; PubMed Central PMCID: PMC3567470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aerts HJ, Velazquez ER, Leijenaar RT, Parmar C, Grossmann P, Cavalho S, Bussink J, Monshouwer R, Haibe-Kains B, Rietveld D, Hoebers F, Rietbergen MM, Leemans CR, Dekker A, Quackenbush J, Gillies RJ, Lambin P. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nature communications. 2014;5:4006. doi: 10.1038/ncomms5006. PubMed PMID: 24892406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lambin P, Rios-Velazquez E, Leijenaar R, Carvalho S, van Stiphout RG, Granton P, Zegers CM, Gillies R, Boellard R, Dekker A, Aerts HJ. Radiomics: extracting more information from medical images using advanced feature analysis. European journal of cancer. 2012;48(4):441–446. doi: 10.1016/j.ejca.2011.11.036. PubMed PMID: 22257792. [DOI] [PMC free article] [PubMed] [Google Scholar]