

Abstract
NF1 encodes a RAS GTPase-Activating Protein. Accordingly, aberrant RAS activation underlies the pathogenesis of NF1-mutant cancers. Nevertheless, it is unclear which RAS pathway components represent optimal therapeutic targets. Here we identify mTORC1 as the key PI3K effector in NF1-mutant nervous system malignancies and conversely show that mTORC2 and AKT are dispensable. However, we find that tumor regression requires sustained inhibition of both mTORC1 and MEK. Transcriptional profiling studies were therefore used to establish a signature of effective mTORC1/MEK inhibition in vivo. We unexpectedly found that the glucose transporter, GLUT1, was potently suppressed but only when both pathways were inhibited. Moreover, unlike VHL and LKB1 mutant cancers, reduction of 18F-FDG uptake required the suppression of both mTORC1 and MEK. Together these studies identify optimal and sub-optimal therapeutic targets in NF1-mutant malignancies and define a non-invasive means of measuring combined mTORC1/MEK inhibition in vivo, which can be readily incorporated into clinical trials.
Keywords: NF1, RAS, mouse models, targeted therapies, MEK, PI3K, mTOR
Introduction
The NF1 tumor suppressor is mutated or suppressed in a variety of sporadic cancers including glioblastoma, neuroblastoma, melanoma and non-small cell lung cancer (1-5). NF1 mutations also underlie the familial cancer syndrome, neurofibromatosis type 1 (NF1) (6,7). NF1 patients exhibit a variety of tumorigenic and non-tumorigenic manifestations but the most common cause of death is malignant peripheral nerve sheath tumors (MPNSTs). These highly aggressive tumors are lethal in approximately 70% of patients, and conventional chemotherapy and radiation do not reduce mortality in individuals with inoperable tumors (8-10). Therefore, developing effective targeted therapies for these individuals represents an important and unmet clinical need. Moreover, an effective therapy for this tumor type may be more broadly applicable to other sporadic NF1-mutant cancers.
The NF1 tumor suppressor gene encodes a RAS GAP, which inactivates RAS by catalyzing the hydrolysis of RAS-GTP (6,7). As such, when NF1 is mutated or suppressed, RAS and downstream effectors become hyperactivated (11). Both the PI3K/mTOR and MEK/ERK pathways have been shown to be important in various NF1-mutant tumors and therefore components of these pathways represent potential therapeutic targets (12-15). However, given the plethora of available drugs that target these pathways we set out to genetically and chemically deconstruct the most important signaling nodes in NF1-mutant MPNSTs. Together with preclinical studies in a genetically engineered mouse tumor model, we found that mTORC1 is the key PI3K pathway component in these NF1-mutant malignancies, AKT and TORC2 are dispensable, and only sustained mTORC1 and MEK inhibition promotes tumor regression.
Several combined PI3K/MEK pathway trials are in development or are being considered for other cancers (16). However, the clinical challenge will be to identify a drug combination and dose that effectively suppresses both pathways, while minimizing toxicity. It is currently unclear how dosing can be adjusted while confirming that both targets are sufficiently inhibited in real time, especially given that the duration of inhibition appears to be an important determinant of efficacy. Thus, establishing a tractable biomarker for effective, combined target inhibition would greatly facilitate this effort. By performing transcriptional profiling and imaging studies we unexpectedly identified GLUT1, which mediates 18F-fluorodeoxyglucose (18F-FDG) uptake, as a key gene that is suppressed prior to tumor regression but only when both pathways are effectively inhibited. Moreover, we show that 18F-FDG uptake is a reliable readout of combined target inhibition. This insight can be directly applied to the design of clinical trials in NF1 mutant cancers and may also have broader utility in other RAS-driven tumors.
Results
p110α and mTORC1 are the key effectors in NF1-mutant nervous system malignancies
We previously showed that loss or inactivation of NF1 triggers the aberrant activation of PI3K/mTORC1 signaling in human and mouse MPNSTs (17). However, it is currently unclear which specific components within this pathway represent the best therapeutic targets. Such insight would reveal which drugs should be preferentially evaluated or excluded in clinical trials. Therefore, we sought to genetically and chemically deconstruct this pathway in NF1-mutant MPNSTs. There are three Class 1A catalytic PI3K isoforms: p110α, p110β, and p110δ. While p110α is frequently mutated in human cancer, p110β has been shown to play an essential role in PTEN mutant cancers and p110δ is critical in chronic lymphocytic leukemia (18-20). To identify which catalytic isoform(s) are essential in NF1-mutant nervous system malignancies, we first assessed the biological effects of isoform-specific ablation in human MPNST cells derived from NF1 patients. While all three isoforms were present in MPNSTs, genetic ablation of p110α, but not p110β or p110δ, dramatically impaired the proliferation of both tumor lines (Fig 1A). Similarly, NF1-mutant glioblastoma (GBM) cells were exclusively sensitive to siRNA-mediated depletion of p110α, but not p110β or p110δ, suggesting that p110α may play a more general role in NF1-deficient cancers (Fig 1A). To complement these findings, we utilized PI3K isoform-specific inhibitors: the p110α- specific inhibitor A66-(S), the p110β-specific inhibitor AZD-6284, and the p110δ-specific inhibitor CAL-101, as well as GDC-0941, a pan-PI3K inhibitor (21-24). The reported specificities of each drug are outlined in Supplementary Table S1. In human MPNST cell lines, the p110α-specific inhibitor A66-(s) and GDC-0941 potently inhibited the phosphorylation of AKT and S6; however, the p110β- or p110δ-specific inhibitors, AZD-6284 and CAL-101 respectively, did not suppress the phosphorylation of either protein (Fig 1B). Accordingly, A66-(S) was the only isoform-specific inhibitor that suppressed proliferation in these cells (Fig 1C; p=0.039 in 90-8TLs and p=0.0006 in S462s). Together, these observations suggest that p110α is the primary catalytic subunit responsible for pro-proliferative PI3K signaling in NF1-mutant nervous system malignancies.
Figure 1. p110α and mTORC1 are Critical for the Proliferation of NF1-Deficient Tumor Cells.
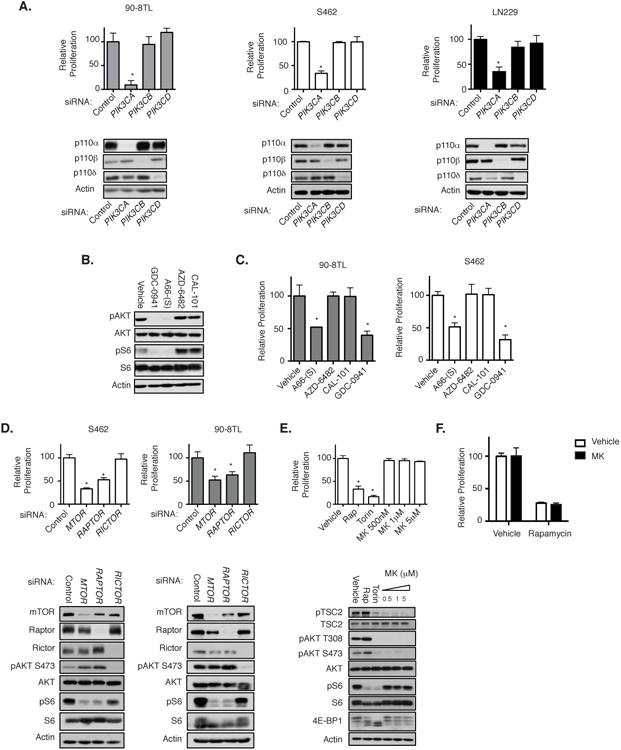
A, S462, 90-8TL, and LN-229 cells were transfected with pooled siRNAs targeting PIK3CA, PIK3CB, PIK3CD, or non-targeting siRNA. Bar graphs, relative change in cell number from day 0 to 96 hours as compared with control cells transfected with the non-targeting siRNA. Data points, triplicate averages ± SD. Immunoblots depict p110α, p110β, and p110δ protein levels 72 hours after transfection with the indicated siRNA. Actin serves as a loading control. *, P < 0.002. B, Immunoblots showing pAKT and pS6 levels in S462 cells following treatment with indicated inhibitors (4 hours; 500 nmol/L). AKT, S6, and actin serve as controls. C, Bar graphs of S462 and 90-8TL cells treated inhibitors as specified. Numbers represent the relative change in cell number from day 0 to 96 hours as compared with vehicle-treated control cells. Data points, triplicate averages ± SD. *, P < 0.04. D, S462 and 90-8TL cells were transfected with pooled siRNAs targeting MTOR, RAPTOR, RICTOR, or non-targeting siRNA. Bar graphs, relative change in cell number from day 0 to 96 hours as compared with control cells transfected with the non-targeting siRNA. Data points, triplicate averages ± SD. Immunoblots show mTOR, Raptor, Rictor, pAKT, and pS6 levels 72 hours after transfection with the indicated siRNA. AKT, S6, and actin levels serve as controls. *, P < 0.02. E, S462 cells were treated with the rapamcyin (Rap) at 100 nmol/L, Torin1 at 250 nmol/L, or MK-2206 (concentration indicated). Bar graph, relative change in cell number from day 0 to 96 hours as compared with vehicle-treated control cells. Data points, triplicate averages ± SD. Immunoblots show pAKT, pAKT, pTSC2, pS6, and 4E-BP1 levels in the presence of the specified inhibitors. AKT, TSC2, S6, and actin serve as controls. *, P < 0.0001. F, S462 cells treated with either rapamycin at 100 nmol/L, MK-2206 at 5 μmol/L, or both drugs together. Bar graphs, relative change in cell number from day 0 to 96 hours as compared with vehicle-treated control cells. Data points, triplicate averages ± SD. p, phosphorylated.
mTOR functions in two distinct complexes: the rapamycin-sensitive complex mTORC1, which phosphorylates 4E-BP1 and S6 kinase, and the relatively rapamycin-insensitive complex mTORC2, which phosphorylates AKT at serine 473 (25,26). NF1-deficient MPNSTs have been shown to be sensitive to rapamycin, indicating that mTORC1 plays a role in this tumor type; however, the contribution of mTORC2 activity, if any, to MPNST growth is unknown (13,17). We genetically targeted essential component proteins of each complex in order to evaluate the relative contribution of these two complexes. RAPTOR, is an essential component of mTORC1, but is not present in mTORC2, while RICTOR, a primary component protein of mTORC2, is not a member of the mTORC1 complex (27,28) As expected, siRNA-mediated-loss of RAPTOR or mTOR suppressed S6 phosphorylation and led to impaired proliferation of MPNST cell lines (Fig 1D). However, loss of RICTOR had no effect on MPNST proliferation, despite the effective suppression of phosphorylation of the mTORC2 target AKT (Fig 1D).
To further evaluate a role for AKT, or lack thereof, tumors cells were treated with the allosteric AKT inhibitor MK-2206 (29). MK-2206 suppressed the phosphorylation of AKT at S473 and T308, and effectively inhibited AKT kinase activity as confirmed by the loss of TSC2 phosphorylation on T1462 (Fig 1E and Supp Fig. S1). However, unlike rapamycin, MK-2206 had no effect on the proliferation of NF1-mutant MPNST cells (Fig 1E). The mTOR kinase inhibitor Torin1 inhibits both the mTORC1 and mTORC2 complexes. Notably, Torin1 has been reported to more effectively inhibit mTORC1, as compared to rapamycin, and in particular more potently suppresses 4E-BP1 phosphorylation, as observed in these studies (Fig 1E and Supp Fig. S1). Accordingly, Torin1 potently suppressed the proliferation of NF1 mutant cells and did so better than rapamycin (p< 0.02). As noted, both MK-2206 and Torin1 equivalently and potently suppressed AKT phosphorylation and activity, although only Torin1 suppressed MPNST cell proliferation. Moreover, MK-2206 did not enhance the anti-proliferative effects of rapamycin (Fig 1F). Taken together, these results suggest that mTORC1 is a critical effector in NF1-mutant cancers and that mTORC2 and AKT are dispensable in these tumor cells.
Selection of an effective PI3K/mTOR pathway inhibitor
These in vitro studies suggested that pan-PI3K inhibitors, p110α-specific inhibitors or mTORC1 inhibitors should suppress the growth of NF1-mutant MPNSTs. Therefore, we first evaluated the in vivo effects of GDC-0941 and rapamycin in a genetically engineered mouse MPNST model. Like human MPNSTs, tumors from these animals harbor compound mutations in Nf1 and p53, and develop with an average latency of five months. These MPNSTs are highly aggressive, and mice survive for an average of 10.7 days after tumors are detected, thus recapitulating the aggressive nature of human tumors (30). As previously shown, rapamycin suppressed the growth of Nf1/p53 mutant MPNSTs (p<0.0001) (13); however, GDC-0941 did so significantly less well (p=0.0021) (Fig 2A). Notably, the maximum tolerated dose of GDC-0941 (150mg/kg) inhibited the phosphorylation of AKT, S6 and 4E-BP1 in tumors within 1 hour, however these pathways were reactivated within 4 hours after treatment (Fig 2B). In contrast, rapamycin suppressed S6 and 4E-BP1 phosphorylation for at least 18 hours, consistent with the observed enhanced efficacy and the demonstrated importance of mTORC1 is these tumors. It should be noted that AKT is not activated by relief of feedback mechanisms in this model, as we have previously shown (Fig 2B) (13,31). Several other PI3K/mTOR pathway inhibitors including BEZ-235, Torin2, and INK-128 were evaluated in these animals (data not shown); however we were unable to identify an inhibitor that exhibited better pharmacodynamics or growth inhibition than rapamycin at tolerable doses in these animals. Therefore, rapamycin was selected for further studies.
Figure 2. Therapeutic effects of PI3K and MEK pathway inhibitors in vivo.
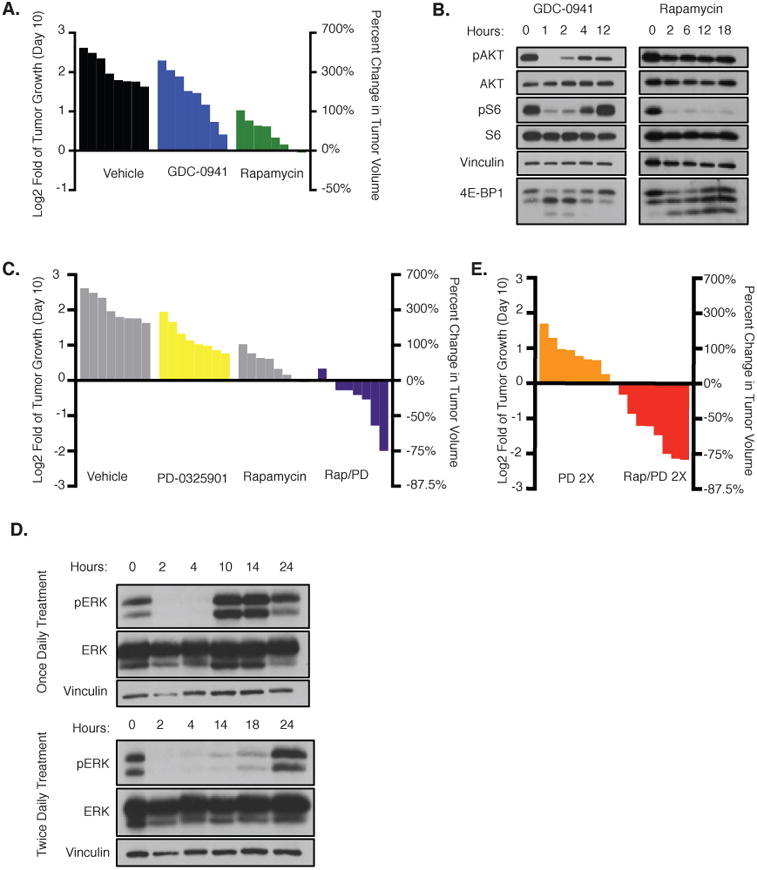
A, Waterfall plot depicting change in tumor volume in NPcis mice after 10 days of treatment with vehicle (black), GDC-0941 (blue), or rapamycin (green). The left y-axis indicates the log2 of the fold change in volume after 10 days. The right y-axis indicates the percent change in tumor volume relative to day 0. B, pAKT/pS6/4E-BP1 immunoblots of tissue from animals exposed to GDC-0941 or rapamycin for the indicated amount of time. AKT, S6, and vinculin serve as controls. C, Waterfall plot depicting change in tumor volume after 10 days of treatment with PD-0325901 (yellow) or rapamycin and PD-0325901 (Rap–PD) in combination (purple). Vehicle and rapamycin (gray) are reprinted from A for reference. The left y-axis indicates the log2 of the fold change in volume after 10 days. The right y-axis indicates the percent change in tumor volume relative to day 0. D, Immunoblots showing pERK levels in tissue after treatment with PD-0325901 once daily (top) or twice daily (bottom). Hours, number of hours from initial treatment. Representative samples from three biologic replicates are shown. Vinculin and ERK serve as controls. E, Waterfall plot depicting change in tumor volume after 10 days of treatment with PD-0325901 twice daily or PD-0325901 twice daily in combination with rapamycin. The left y-axis indicates the log2 of the fold change in tumor volume relative to day 0 and the right y-axis indicates the percent change in tumor volume.
Combined, sustained inhibition of mTORC1 and MEK promotes MPNST regression in vivo
Although mTORC1 is a critical signaling node in NF1 mutant tumors, mTORC1 inhibition exerted only cytostatic effects on MPNSTs in vitro and in vivo (Fig 1D, E, 2A) (13). Therefore, we evaluated the effects of rapamycin combined with a MEK inhibitor, which targets a second critical RAS effector pathway. Tumor-bearing mice were treated with vehicle, the MEK inhibitor PD-0325901, rapamycin, or the combination of rapamycin and PD-0325901. As a monotherapy, PD-0325901 slightly attenuated the growth of MPNSTs, but did so less than rapamycin (Fig 2C). However, combined PD-0325901 and rapamycin treatment induced tumor regression in these mice (Fig 2C). Interestingly, these observations differ from effects observed in benign NF1-deficient peripheral nervous system tumors and myeloid malignancies, where MEK appears to function as the dominant RAS-effector pathway and MEK inhibitors exert cytotoxic effects alone, suggesting that different tumor types harboring the same initial driving genetic lesion may rely on different downstream signals (32,33). Nevertheless, upon examining the pharmacodynamics of PD-0325901 at this dose, we found that ERK phosphorylation was inhibited for only 4-6 hours, whereas sustained inhibition could be achieved by dosing with PD-0325901 twice daily (Fig 2D). As such, we hypothesized that a revised dosing schedule might exert more potent therapeutic effects. Twice-daily PD-0325901 treatment did not promote tumor regression as a monotherapy, however when combined with rapamycin, twice daily PD-0325901 treatment improved the therapeutic response (Fig. 2E). All mice treated with this combination responded, and more than half of the tumors regressed 50% or more, with several shrinking 75% or more. Together, these observations indicate that the duration of both MEK and mTORC1 inhibition is a critical determinant of the therapeutic response.
Identifying GLUT1 as a component of the therapeutic signature that is suppressed prior to tumor regression
Pharmocodynamic markers in tumors are often not examined during clinical trials, and when they are, the kinetics of suppression are difficult to evaluate. Therefore, if a treatment does not show efficacy, especially in cases of dose de-escalation, it is often unclear whether the target or targets were sufficiently inhibited. Therefore, we sought to identify a molecular change that might serve as a functional biomarker of effective, combined inhibition of mTORC1 and MEK pathways. The transcriptional profiles of tumors from animals treated with vehicle, rapamycin, PD-0325901 (twice daily), or the combination of rapamycin and PD-0325901 were evaluated. Importantly, tissues were collected after 14 hours of treatment: a time point that would capture transcriptional changes caused by sustained target inhibition but occurring prior to tumor regression. Using a gene expression class comparison, we identified a gene set that was exclusively regulated by combined rapamycin and PD-0325901 treatment (Fig 3A). Interestingly Slc2a1, which encodes a glucose transporter and is commonly referred to as Glut1, was identified as one of the uniquely suppressed genes in rapamycin/PD-0325901 treated tumors (Fig 3A) Effective mTORC1 and MEK target inhibition in tumor tissue was verified (Fig 3B). Q-PCR analysis confirmed that Glut1 levels were reduced 64% after only 14 hours of treatment compared to vehicle treated tumors and that neither rapamycin nor PD-0325901 exerted suppressive effects alone (Fig 3C). A dramatic decrease in GLUT1 protein levels was further confirmed by evaluating its expression in tumor biopsies taken before and 3 days after treatment (Fig 3D). These findings differ from observations in VHL and LKB1 mutant tumors, where GLUT1 mRNA and consequently protein expression is primarily regulated by mTOR and HIF1α, and its expression can be suppressed by mTORC1 inhibitors alone (34,35). However in these Nf1-mutant MPNSTs, suppression of both mTORC1 and the MEK/ERK pathways are required. This finding resolves a longstanding observation that rapamycin is not sufficient to suppress the expression of GLUT1, or other HIF-1α target genes in vitro or in vivo in this tumor type (13). Together, these results demonstrate that GLUT1 is suppressed in MPNSTs only after combined mTORC1 and MEK inhibition, which could be exploited for developing an imaging biomarker of combined target inhibition.
Figure 3. GLUT1 is an early biomarker of effective combined MEK/mTORC1 inhibition.
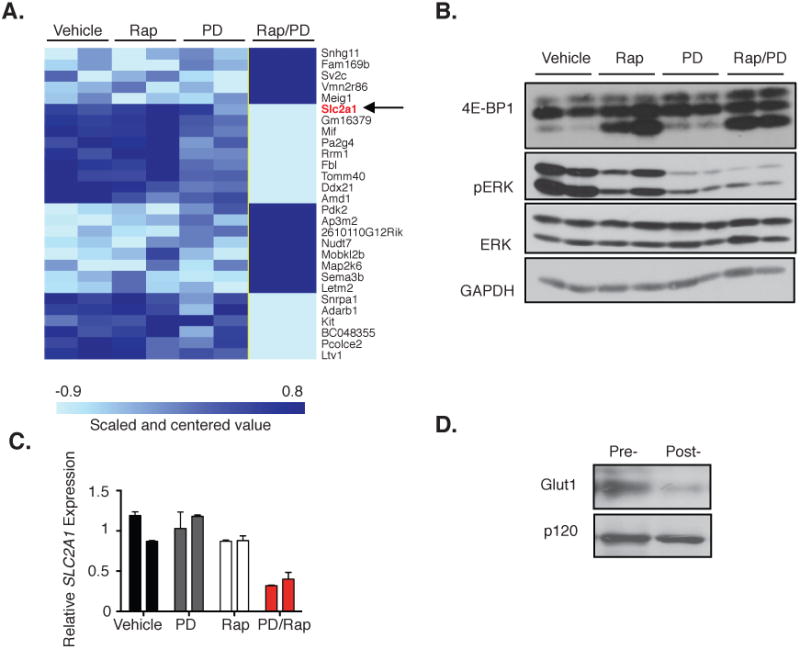
A, Microarray analysis of mouse tumors 14 hours after treatment with vehicle, rapamycin, PD-0325901 (PD), or both rapamycin and PD-0325901 (Rap–PD). Heat map depicts the uniquely upregulated genes (dark blue) or downregulated genes (light blue) from tumors in mice treated with Rap–PD as compared with all other groups reaching a significance level of P = 0.001. The arrow denotes Slc2a1(encoding GLUT1), highlighted in red, as one gene of particular interest within this signature. B, Immunoblot of pERK and 4E-BP1 levels in individual tumors as described in A. ERK and GAPDH serve as a control. C, Quantitative PCR showingSlc2a1 transcript levels in individual tumors described in A. D, Immunoblot showing GLUT1 levels in a pretreatment biopsy sample, and in the same tumor after treatment with PD-0325901 (twice daily) and rapamycin. p120 serves as a control.
Only combined, effective suppression of mTORC1 and MEK inhibits 18F-FDG uptake
GLUT1 is a membrane bound glucose transporter that is frequently over-expressed in tumors, in part, because altered tumor metabolism requires increased glucose uptake (36-39). This metabolic activity can be measured by positron emission tomography (PET) scans designed to quantify 18F-FDG uptake (40). GLUT1 has been shown to regulate 18F-FDG uptake in a variety of tumor types (41,42). MPNSTs are generally FDG-PET positive, and enhanced 18F-FDG uptake is used to diagnose a conversion to malignancy, as MPNSTS often arise from benign precursor lesions (43). Because human MPNSTs exhibit a strong FDG-PET signal, and because GLUT1 was specifically suppressed in tumors treated with combined rapamycin and PD-0325901, we hypothesized that the substantial reduction in GLUT1 mRNA and protein might inhibit 18F-FDG uptake in these tumors. To evaluate this possibility, FDG-PET imaging was performed on tumor bearing mice. As expected, MPNSTs were FDG-PET positive at baseline, mirroring the behavior of human MPNSTs (Fig 4A). Mice were then treated with vehicle, PD-0325901, rapamycin or PD-0325901/rapamycin and PET analysis was performed a second time, 40 hours after the baseline scan. This time point was selected because it represents a time before detectable regression occurs, in order to avoid any confounding change in the FDG-PET signal due to a reduction in tumor size. It should be noted that the initial (64%) decrease in Glut1 mRNA levels can be detected 14 hours after treatment, however given the dramatic decrease in GLUT1 protein after 72 hours this repression is sustained and perhaps enhanced, Animals treated with vehicle, PD-0325901, or rapamycin, did not have a significant change in 18F-FDG uptake after treatment (Fig 4A,B); however, animals treated with both PD-0325901 and rapamycin exhibited a significant decrease in SUVmax values (p<0.004) (Fig 4A,B). Importantly, while hexokinase and other GLUT genes can regulate glucose uptake in some settings (41,44-46), rapamycin/PD-0325901 treatment did not affect the expression of any of these genes suggesting that GLUT1 may be the rate limiting step for FDG-PET uptake in MPNSTs (Supplemental Table S2).
Figure 4. 18F-FDG uptake is a non-invasive biomarker of combined MEK/mTORC1 inhibition.
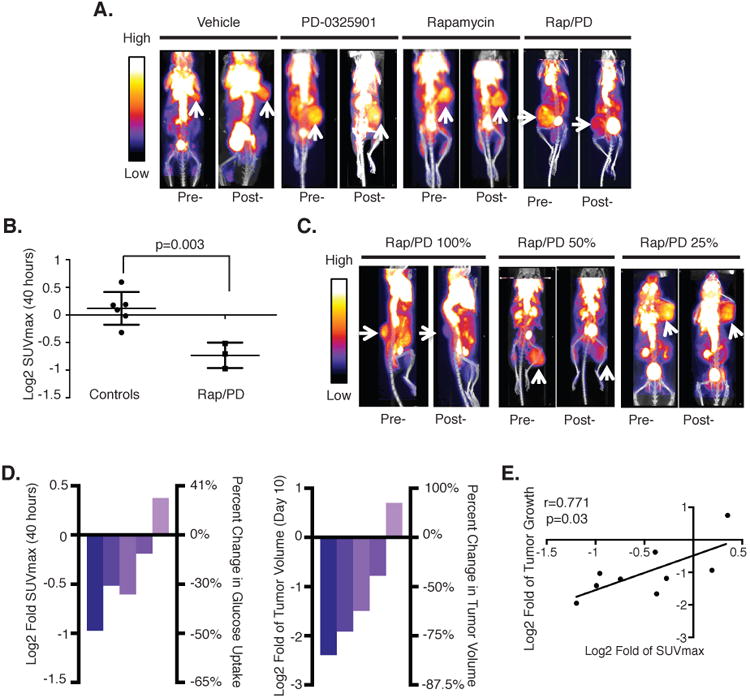
A, Representative images of FDG–PET scans of animals treated with vehicle, PD-0325901, rapamycin, or both (Rap–PD). PD-0325901 was dosed twice daily. Baseline scans are shown at left (pre-), and the right image shows the same view of the same animal 40 hours after treatment with the indicated compound (post-). Arrow, MPNST. Scale bar, relative 18F-FDG uptake, with low uptake in blue and highest uptake in white. B, The 18F-FDG uptake by tumors was quantified using SUVmax, and the log2 of the fold change of this number was calculated to indicate the change in 18F-FDG uptake 40 hours after treatment relative to baseline for each animal. The change in combination-treated animals (Rap–PD) is compared with monotherapy and vehicle-treated animals (controls). The y-axis indicates the log2 of the fold change in SUVmax relative to baseline. C, Representative images of FDG–PET scans of animals treated with 100% PD-0324901–rapamycin, 50%PD-0325901–rapamycin, or 25% PD-0325901–rapamycin. The left image (pre-) shows the baseline scan for each animal and the right (post-) shows the scan 40 hours after treatment with the indicated dose. Again, the tumors are indicated with arrows. Scale bar, relative FDG uptake, with low uptake in blue and highest uptake in white. D, Left, a waterfall plot depicting the log2 fold change of SUVmax, a quantification of tumor FDG–PET activity, of selected tumors at 40 hours after treatment with PD-0325901 (25%, 50%, or 100% of the dose) and rapamycin. Right, a waterfall plot depicting the log2 of fold change in tumor volume of the same mice after 10 days. Individual tumors are graphed in the same order on each plot. E, Regression analysis of nine combination-treated mice (100%, n = 3; 50%, n = 3; 25%, n = 3). The x-axis represents the log2 of fold change in tumor volume at 10 days, and they-axis represents the log2 of fold change in SUVmax levels after 40 hours of treatment. Both numbers are relative to day 0 measurements. Line, best-fit linear correlation, and the Pearson coefficient (r) was calculated.
These observations suggested that FDG-PET imaging could be used as a biomarker of effective combined mTORC1/MEK inhibition in NF1-mutant tumors. Such a biomarker would be invaluable in the course of evaluating similar therapies in the clinic and in the course of dose de-escalation/escalation studies. This biomarker would be particularly useful if the early change in FDG-PET imaging were predictive of a later change in tumor size. To experimentally evaluate this possibility, we performed a dose de-escalation study in mice. Mice were treated with rapamycin in combination with 100%, 50%, or 25% of the PD-0325901 dose. As expected, this produced a range of responses in FDG-PET uptake at 40 hours and tumor regression after 10 days (Fig. 4C,D). Importantly, the suppression of FDG-PET activity at 40 hours, as measured by change in SUVmax, correlated with the ultimate decrease in tumor size after 10 days (Pearson R=0.711, p=0.03) (Fig 4D,E). These results suggest that early changes in the FDG-PET signal are indicative of the degree of target inhibition and correlate with eventual tumor regression in MPNSTs treated with combined mTORC1/MEK inhibitors.
Discussion
Numerous PI3K pathway inhibitors have been developed and are being evaluated in clinical trials (16). However in many cancers it is not clear which specific component(s) within this pathway are most critical or to what degree they must be inhibited. Such information would undoubtedly facilitate the selection of the most appropriate drugs for clinical studies. In this study we used a genetic and chemical approach to systematically deconstruct the PI3K signaling pathway in NF1-mutant nervous system malignancies. Importantly, we found that mTORC1, which is regulated by p110α in these tumors, is the minimal, essential PI3K pathway component and that surprisingly AKT and mTORC2 are dispensable. However, while agents that inhibit mTORC1 promote cytostasis in human tumor cells and genetically engineered models, tumor regression requires concomitant suppression of the MEK/ERK pathway.
Notably, there are currently no effective therapies for MPNSTs. As such, these studies reveal a promising therapeutic approach as well as a mechanistic framework for selecting the most appropriate agents for clinical trials. For example, because p110β does not appear to contribute to the therapeutic response in these tumors perhaps p110α-specific, β-sparing PI3K inhibitors could be used with less toxicity (47). Alternatively, because mTORC1 appears to be the key PI3K effector in these tumors perhaps rapalogues, which exhibit excellent pharmacokinetic properties, may be suitable for combination therapies. The observation that AKT is not activated in these tumors by feedback inhibition and that AKT inhibitors do not enhance the effects of rapamycin, further alleviates the concern that AKT suppression may be required in this setting. Nevertheless, these studies suggest that successful agents must promote sustained inhibition of both ERK and mTORC1. Importantly, suppression of these same targets results in tumor regression in a mouse model of NF1-mutant melanoma, underscoring the importance of these pathways in NF1-deficient cancers (4). Nevertheless, establishing the sufficient degree/length of inhibition of both targets that will be required to mediate an efficacious response in patients represents a formidable challenge.
While mouse models are useful for identifying critical therapeutic targets in genetically defined cancers, the ultimate success of a therapy in humans depends on many factors. Certainly, species-specific differences in tumor complexity may limit efficacy or restrict therapeutic responses to a subset of patients. However, perhaps an even more important consideration relates to dosing. One of the primary obstacles in developing combination therapies, especially when targeting two major signaling pathways, is achieving efficacy while preventing toxicity. As such, even if the correct therapeutic targets have been identified, it may not be possible to sufficiently suppress these targets in humans. MEK inhibitors have been shown to exhibit toxicity in humans at high doses (48,49). Therefore in this study we used a dose of PD-0325901 that is comparable to the tolerable dose in humans. Similarly, the dose of rapamycin was selected based on a previous preclinical study that led to successful human clinical trial in a number of tuberous sclerosis complex related pathologies, although reported trough plasma levels were somewhat higher than what has been observed in humans (∼50ng/ml versus 3-20 ng/ml in humans) (50-53). However our preliminary observations suggest that lower doses and/or intermittent dosing of mTOR inhibitors/rapalogues are also effective when combined with MEK inhibitors. Given the differences in toxicity observed between mice and humans, only clinical trials will reveal whether an effective, non-toxic dose can be achieved. As such, another important goal of this study was to develop a biomarker that could be used to guide dosing in the clinic.
Current clinical trial strategies involve dosing up to the Maximum Tolerated Dose of one drug, and adding the second drug to the tolerable dose when possible. However, it is not always clear how dose escalation/de-escalation affects the degree or kinetics of target inhibition or if dosing at the MTD is necessary. As such, we set out to identify a biomarker(s) that would serve as an early downstream readout of effective, combined inhibition of MEK and TORC1. While several genes were identified in these tumors, GLUT1 stood out as an important and tractable molecular change. Consistent with the documented role of GLUT1 in regulating glucose uptake, we found that 18F-FDG uptake, as measured by FDG-PET, was a reliable readout of effective, combined target inhibition in vivo. Importantly, changes in GLUT1 expression and 18F-FDG uptake occurred prior to tumor regression, supporting its role as a molecular marker of TORC1/MEK suppression rather than a consequence of tumor shrinkage. Interestingly, neither GLUT1 expression nor glucose uptake were suppressed after treatment with either rapamycin or MEK inhibitors alone. This observation differs from findings in a subset of other mTOR-driven tumor types, where HIF1α-dependent GLUT1 expression is decreased after treatment with rapamycin, as is 18F-FDG uptake (34,35). We have previously shown that neither GLUT1 nor HIF1α levels are altered in MPNSTs when treated with rapamycin, marking an important distinction between NF1-deficient tumors and these other mTOR-driven tumors (13). Here, we provide an explanation for this difference, as simultaneous inhibition of both the mTORC1 and MEK are required to suppress GLUT1 and 8F-FDG uptake in MPNSTs. It will be interesting to determine whether inhibition of both pathways is required to alter glucose uptake in other tumors, in particular other RAS-driven tumors. Certainly, other factors, such as hexokinase activity or other members of the GLUT family, may contribute to glucose uptake in some cancers and in these instances 8F- FDG uptake might not be an effective biomarker. However our studies suggest that FDG-PET imaging represents a promising, non-invasive means of measuring combined mTORC1/MEK inhibition in vivo in these NF1-mutant tumors, which can be readily incorporated into clinical trials. Such a tool should help identify the most effective drugs, facilitate dosing, and its utility may extend beyond NF1-mutant cancers.
Methods
Cell Lines and Reagents
S462s and LN229s were purchased from ATCC. 90-8TLs were generously provided by Dr. Eric Legius (KULeuven). The authors performed no further authentication of the cell lines. Cell lines were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with fetal bovine serum (10%) and L-glutamine. Antibodies were obtained from the following sources: Cell Signaling Technologies: pAKT (4060), AKT (9272), pERK (4370), ERK (9102), pS6 (2211), S6 (2217), p110α (4255), p110β (3011), Vinculin (4650), 4E-BP1 (9452), mTOR (7C10), Rictor (53A2), Raptor (24C12) pTSC2 (3611), TSC2 (3612) GAPDH (2118); Santa Cruz Biotechnology: p110δ (sc-7176); Trans Labs: p120 (G12920), Sigma: Actin (A2066), Alpha Diagnostics: Glut1 (GT11-A). Torin1, A66-(S), AZD-6284, and CAL-101 were kindly provided by Nathanael Gray (Dana Farber Cancer Institute/Harvard Medical School). MK-2206 was generously provided by D. Wade Clapp (Indiana University). GDC-0941 was provided by Genentech (San Francisco, CA). PD-0325901 was a gift from Kevin Shannon (University of California, San Francisco). Rapamycin was purchased from LC Labs.
RNAi
Non-Targeting and PIK3CA, PIK3CB, PIK3CD, Raptor, Rictor, mTOR siRNA pools were purchased from Dharmacon (D-001810-10, L-003018-00, L-003019-00, L-006775-00, L-004107-00, L-016984-00, L-003008-00, respectively). siRNAs were transfected overnight in antibiotic free medium using RNAiMax lipofectamine from Invitrogen.
Cellular Proliferation Studies
Approximately 125,000 cells per well were seeded in 6-well plates. For siRNA experiments, cells were seeded 12-16 hours after transfection. 24-hours after plating, day 0 counts were taken using a hemocytometer and trypan blue exclusion. For inhibitor experiments, drug treatments were started at this time. Inhibitors were changed once daily, except for PI3K isoform experiments where drugs were replenished twice daily. Final cell counts were taken 96 hours after day 0 counts.
Drug treatments and dosing schedule
Animal procedures were approved by the Center for Animal and Comparative Medicine in Harvard Medical School in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. C56/BL6 NPcis mice have been previously described (30). Mice were treated daily with rapamycin via IP injections at 5 mg/kg, which were prepared as previously described (13). PD-0325901 was administered at 1.5 mg/kg once or twice daily (10 hours apart) by oral gavage. PD-0325901 was prepared as previously described.(54). GDC-0941 was administered at 150mg/kg once daily by oral gavage. GDC-0941 was prepared as previously described (55). Compounds given in combination were administered sequentially.
Biopsy
Tumor biopsy was performed on mice prior to drug treatment using the wedge biopsy technique and snap-frozen. Drug treatment was started 8 hours after initial biopsy. The post-treatment biopsy was performed 3 days after treatment began. The mouse was anesthetized by isoflurane inhalation and given a local block with lidocaine and marcaine while the tumor biopsy was collected.
Tumor volume measurements
Mice were started on a treatment when tumor size reached 200–1000 mm3. Tumor size was measured every 2–3 days by Vernier calipers. Tumor volume was calculated using the standard formula L × W2 × 0.52. A mouse pathologist confirmed that all tumors in this study are MPNSTs.
18F-FDG-PET imaging and analysis
PET/CT scans were performed on the Bioscan NanoPET/CT at the Longwood Small Animal Imaging Facility. This PET scanner is equipped with a dedicated isoflurane anesthesia system, temperature controlled platform, cardiac gating, and respiratory gating. PET scanning was performed on anesthetized animals lying motionless on a table, after Retro-orbital IV injection of 0.1 to 10 mCi of F18-FDG PET radioisotope, while being imaged with a coincidence camera. The mice were imaged after a pre-determined “washout” period (30-60 min). Individual mice were first scanned pre-treatment and then 40 hours after the treatment regimen was initiated (see dosing schedule methods). For quantitative analysis, the standardized uptake value (SUV) normalized to body weight in the tumor was calculated using SUV= ACvoi (kBq/ml)/(FDGdose(MBq)/BW(kg)) where ACvoi is the average activity concentration in the tumor volume (or the maximum value); FDGdose is the dose of F18-FDG administered; and BW is the body weight. For evaluating tumors the highest SUV in the tumor was taken as the SUVmax.
Microarray
RNA isolated from MPNST tumor samples from NPcis mice treated for 14 hours with vehicle, rapamycin, PD-0325901, or the combination of rapamycin and PD-0325901 For all PD-0325901 treated samples, PD-0325901 was dosed a second time at 10 hours. RNA was isolated with Trizol following the manufactures protocol. RNA clean-up was then performed using Qiagen's RNA easy kit (#74104). The Partners HealthCare Center for Personalized Genetic Medicine core facility hybridized the RNA to the Affymetrics Mouse Gene 1.0 STS. To determine genes differentially expressed in the combination treatment, a class comparison between the combination treated samples and all other samples was performed. Analysis was completed using BRB-Array tools developed by Dr. Richard Simon (National Cancer Institute, NIH, Rockville, MD) and the BRB-ArrayTools Development team. Thresholds were set at p<0.001. Microarray data can be accessed in the GEO database (accession number: GSE57141).
Supplementary Material
Significance.
This work demonstrates that mTORC1 and MEK are key therapeutic targets in NF1-mutant cancers and establishes a non-invasive biomarker of effective, combined target inhibition that can be evaluated in clinical trials.
Acknowledgments
We would like to thank Haley Goodwill, John Frangioni, and the Longwood Small Animal Imaging facility at Beth Israel Hospital for performing the 18F-FDG PET imaging studies. We also thank Roderick T. Bronson for histologic confirmation of all MPNSTs.
Financial support: KC and this work were supported by a grant from the NCI (5R01CA111754-08) the Department of Defense (W81XWH-13-1-0044), the Children's Tumor Foundation (CTF), and the Ludwig Center at DF/HCC. CM was supported in part by the Pharmacological Sciences Training Program Grant from the National Institute of Health (T32 GM007306). JF was supported by the CTF.
Footnotes
There are no conflicts of interest to disclose.
References
- 1.Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell. 2009;16:44–54. doi: 10.1016/j.ccr.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The I, Murthy AE, Hannigan GE, Jacoby LB, Menon AG, Gusella JF, et al. Neurofibromatosis type 1 gene mutations in neuroblastoma. Nat Genet. 1993;3:62–6. doi: 10.1038/ng0193-62. [DOI] [PubMed] [Google Scholar]
- 4.Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–49. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–9. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 7.Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 8.Zehou O, Fabre E, Zelek L, Sbidian E, Ortonne N, Banu E, et al. Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1: a 10-year institutional review. Orphanet J Rare Dis. 2013;8:127. doi: 10.1186/1750-1172-8-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–4. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DE, Prasad V, Foster L, Dall GF, Birch R, Grimer RJ. Survival in Malignant Peripheral Nerve Sheath Tumours: A Comparison between Sporadic and Neurofibromatosis Type 1-Associated Tumours. Sarcoma. 2009;2009:756395. doi: 10.1155/2009/756395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–73. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 12.See WL, Tan IL, Mukherjee J, Nicolaides T, Pieper RO. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res. 2012;72:3350–9. doi: 10.1158/0008-5472.CAN-12-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johannessen CM, Johnson BW, Williams SMG, Chan AW, Reczek EE, Lynch RC, et al. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 14.Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T, et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013;19:450–61. doi: 10.1158/1078-0432.CCR-12-1067. [DOI] [PubMed] [Google Scholar]
- 15.Parkin B, Ouillette P, Wang Y, Liu Y, Wright W, Roulston D, et al. NF1 inactivation in adult acute myelogenous leukemia. Clin Cancer Res. 2010;16:4135–47. doi: 10.1158/1078-0432.CCR-09-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71:1395–409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- 17.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102:8573–8. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 19.Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–9. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herman SEM, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–88. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, et al. A drug targeting only p110α can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J. 2011;438:53–62. doi: 10.1042/BJ20110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lannutti BJ, Meadows SA, Herman SEM, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–4. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nylander S, Kull B, Björkman JA, Ulvinge JC, Oakes N, Emanuelsson BM, et al. Human target validation of phosphoinositide 3-kinase (PI3K)β: effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J Thromb Haemost. 2012;10:2127–36. doi: 10.1111/j.1538-7836.2012.04898.x. [DOI] [PubMed] [Google Scholar]
- 24.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–32. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 25.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 26.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 27.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–22. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 29.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–67. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 30.Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–6. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 31.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–13. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340–7. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang T, Krisman K, Theobald EH, Xu J, Akutagawa J, Lauchle JO, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123:335–9. doi: 10.1172/JCI63193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 35.Shackelford DB, Vasquez DS, Corbeil J, Wu S, Leblanc M, Wu CL, et al. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci USA. 2009;106:11137–42. doi: 10.1073/pnas.0900465106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS, et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol. 2009;174:1544–52. doi: 10.2353/ajpath.2009.080596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Younes M, Ertan A, Lechago LV, Somoano J, Lechago J. Human erythrocyte glucose transporter (Glut1) is immunohistochemically detected as a late event during malignant progression in Barrett's metaplasia. Cancer Epidemiol Biomarkers Prev. 1997;6:303–5. [PubMed] [Google Scholar]
- 38.Sakashita M, Aoyama N, Minami R, Maekawa S, Kuroda K, Shirasaka D, et al. Glut1 expression in T1 and T2 stage colorectal carcinomas: its relationship to clinicopathological features. Eur J Cancer. 2001;37:204–9. doi: 10.1016/s0959-8049(00)00371-3. [DOI] [PubMed] [Google Scholar]
- 39.Grover-McKay M, Walsh SA, Seftor EA, Thomas PA, Hendrix MJ. Role for glucose transporter 1 protein in human breast cancer. Pathol Oncol Res. 1998;4:115–20. doi: 10.1007/BF02904704. [DOI] [PubMed] [Google Scholar]
- 40.Som P, Atkins HL, Bandoypadhyay D, Fowler JS, MacGregor RR, Matsui K, et al. A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med. 1980;21:670–5. [PubMed] [Google Scholar]
- 41.Smith TA. The rate-limiting step for tumor [18F]fluoro-2-deoxy-D-glucose (FDG) incorporation. Nucl Med Biol. 2001;28:1–4. doi: 10.1016/s0969-8051(00)00177-3. [DOI] [PubMed] [Google Scholar]
- 42.Avril N. GLUT1 expression in tissue and (18)F-FDG uptake. J Nucl Med. 2004;45:930–2. [PubMed] [Google Scholar]
- 43.Benz MR, Czernin J, Dry SM, Tap WD, Allen-Auerbach MS, Elashoff D, et al. Quantitative F18-fluorodeoxyglucose positron emission tomography accurately characterizes peripheral nerve sheath tumors as malignant or benign. Cancer. 2010;116:451–8. doi: 10.1002/cncr.24755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Younes M, Brown RW, Stephenson M, Gondo M, Cagle PT. Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer. 1997;80:1046–51. doi: 10.1002/(sici)1097-0142(19970915)80:6<1046::aid-cncr6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 45.Ito T, Noguchi Y, Satoh S, Hayashi H, Inayama Y, Kitamura H. Expression of facilitative glucose transporter isoforms in lung carcinomas: its relation to histologic type, differentiation grade, and tumor stage. Mod Pathol. 1998;11:437–43. [PubMed] [Google Scholar]
- 46.Aloj L, Caracó C, Jagoda E, Eckelman WC, Neumann RD. Glut-1 and hexokinase expression: relationship with 2-fluoro-2-deoxy-D-glucose uptake in A431 and T47D cells in culture. Cancer Res. 1999;59:4709–14. [PubMed] [Google Scholar]
- 47.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LoRusso PM, Krishnamurthi SS, Rinehart JJ, Nabell LM, Malburg L, Chapman PB, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–37. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 49.Renouf DJ, Velazquez-Martin JP, Simpson R, Siu LL, Bedard PL. Ocular toxicity of targeted therapies. J Clin Oncol. 2012;30:3277–86. doi: 10.1200/JCO.2011.41.5851. [DOI] [PubMed] [Google Scholar]
- 50.Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–32. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–87. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- 52.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125–32. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 53.Ando K, Kurihara M, Kataoka H, Ueyama M, Togo S, Sato T, et al. The efficacy and safety of low-dose sirolimus for treatment of lymphangioleiomyomatosis. Respir Investig. 2013;51:175–83. doi: 10.1016/j.resinv.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Brown AP, Carlson TCG, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59:671–9. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- 55.Wallin JJ, Guan J, Prior WW, Lee LB, Berry L, Belmont LD, et al. GDC-0941, a novel class I selective PI3K inhibitor, enhances the efficacy of docetaxel in human breast cancer models by increasing cell death in vitro and in vivo. Clin Cancer Res. 2012;18:3901–11. doi: 10.1158/1078-0432.CCR-11-2088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.