

Abstract
Mutated KRAS (KRAS*) is a fundamental driver in the majority of pancreatic ductal adenocarcinomas (PDAC). Using an inducible mouse model of KRAS*-driven PDAC, we compared KRAS* genetic extinction to pharmacological inhibition of MEK1 in tumor spheres and in vivo. KRAS* ablation blocked proliferation and induced apoptosis while MEK1 inhibition exerted cytostatic effects. Proteomic analysis evidenced that MEK1 inhibition was accompanied by a sustained activation of the PI3K-AKT-MTOR pathway and by the activation of AXL, PDGFRa, and HER1-2 receptor tyrosine kinases (RTKs) expressed in a large proportion of human PDAC samples analyzed. While single inhibition of each RTK alone or plus MEK1 inhibitors was ineffective, a combination of inhibitors targeting all three co-activated RTKs and MEK1 was needed to inhibit proliferation and induce apoptosis in both mouse and human low-passage PDAC cultures. Importantly, constitutive AKT activation, which may mimic the fraction of AKT2-amplified PDAC, was able to bypass the induction of apoptosis caused by KRAS* ablation, highlighting a potential inherent resistance mechanism that may inform the clinical application of MEK inhibitor therapy. This study suggests that combinatorial targeted therapies for pancreatic cancer must be informed by the activation state of each putative driver in a given treatment context. Additionally, our work may offer explanative and predictive power in understanding why inhibitors of EGFR signaling fail in PDAC treatment and how drug resistance mechanisms may arise in strategies to directly target KRAS.
Keywords: KRAS, receptor tyrosine kinases, pancreatic ductal adenocarcinoma, MEK inhibitor, AKT2 amplification, targeted therapy
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is among the deadliest tumors, and fewer than 5% of newly diagnosed patients survive more than five years (1). Only a small fraction of cases present with local disease and are eligible for potentially curative surgical resection; whereas, the remaining patients possess locally advanced or metastatic disease and receive systemic and palliative therapies. The treatment of locally advanced or metastatic PDAC with gemcitabine and radiation provides modest survival benefit, and recent clinical trials show further marginal improvement with combination of 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) or with nab-paclitaxel (2,3).
Oncogenic mutation of KRAS is a signature molecular event in PDAC and functions as a key driver of disease initiation and maintenance in approximately 90% of cases (4). Targeting oncogenic KRAS represents an ideal therapeutic strategy, although attempts to target KRAS directly have thus far failed, while farnesyltransferase inhibitors have shown no clinical benefit (5). Indeed targeting KRAS (or other structurally related family members) remains one of the biggest challenges in oncology (5) and has inspired a large scale NCI-directed effort (6,7). Inhibition of KRAS downstream effectors might in principle achieve similar results, and attempts have been made to target the MAPK signaling cascade and the PI3K-AKT-MTOR signaling pathway in multiple cancer subtypes. Combinatorial inhibition of these two critical effector pathways has resulted in favorable therapeutic outcomes in multiple preclinical studies in mutant RAS-driven tumor models (8,9); however, recent clinical data showed that these combination therapies are associated with toxicities that might ultimately limit the feasibility of this approach (10). A multitude of alternative targeted therapy regimens have been clinically tested for the treatment of PDAC and, apart from the statistically significant albeit minimal effects seen with epidermal growth factor (EGFR) inhibitors, none has provided a clear clinical benefits (11). Moreover, despite the frequent overexpression of EGFR in KRAS-driven PDAC and the essentiality of EGFR for tumor establishment (12), the therapeutic benefits of EGFR inhibition alone or in combination with gemcitabine remain very modest (13).
Here we use a conditional genetically engineered mouse model (GEMM) of PDAC engineered with pancreas-specific inducible mutant KRAS (KRASG12D) allele to evaluate signaling pathway patterns emerging with either genetic abrogation of KRASG12D or pharmacological inhibition of MEK1 in vivo or in vitro. As 2D-cultured cancer cell lines are poorly predictive of in vivo responses to pharmacologic inhibition (14), we employ a 3D-culture system of primary tumor-derived spheres as well as in in vivo tumor testing. We show that MEK1 inhibition activates a network of receptor tyrosine kinases (RTKs) that may limit the efficacy of single anti-RTK-based therapies alone or in combination with MEK1 inhibitors. Furthermore, activation of AXL, which is highly expressed in our inducible KRASG12D GEMM as well as in a significant number of clinical PDAC samples, might limit the benefits of MEK1-EGFR inhibitor combination treatments currently undergoing clinical testing.
Materials and Methods
Cell Culture
iKRAS cells were derived from P48 CRE_TetO-LSL-KRAS G12D_Rosa-rtTA_p53fl/+, tumor-bearing mice (15). Tumors were minced and digested in collagenase IV and dispase (4 mg/ml) for 1 h at 37 °C on an orbital shaker and subsequently filtered through a 40-μM nylon cell strainer. For conventional tissue culture, cells were maintained in RPMI1640 supplemented with 10% FBS, 2 mM glutamine, and 1% pen-strep. For 3D tissue culture, cells were maintained in low-attachment plates in stem cell medium (MEBM, Lonza) supplemented with 2mM glutamine (Invitrogen), B27 (Invitrogen), 20 ng/ml hEGF (PeproTech), 20 ng/ml hFGF (PeproTech), 5 μg/ml h-Insulin (Roche), 0.5 μM hydrocortisone (Sigma Aldrich), 100 μM β-mercaptoethanol (Sigma Aldrich), 4 μg/ml heparin (Sigma Aldrich), Methocult M3134. (StemCell Technologies) was added to stem cell medium (final concentration 0.8%) to keep tumor cells growing as clonal spheres (16). MIA PaCa-2 and Panc1 cells were obtained from ATCC.
Cell viability
To determine viability of cells grown in 3D conditions, cells embedded in methylcellulose-based semisolid media were exposed to 1 μM calcein (life technology), incubated for 30 minutes, and quantified through ImageXpress velos (Molecular Devices) apparatus. Alternatively, cells were collected through centrifugation, trypsinized, stained in 1X Annexin V buffer with Annexin V-PE and 7AAD (BD Bioscience) for 5 minutes at room temperature and analyzed by flow cytometry.
Animal Studies
Animal studies were conducted according to IACUC guidelines. For allograft and xenograft establishment, 5 × 104 iKRAS cells or 2 × 106 PATC cells were suspended in 200 μl of 50% HBSS, 50% growth factor reduced matrigel and injected subcutaneously in the right flank of nude mice. Tumor volume was assessed using caliper measurements and calculated according to the standard formula: length/2 x width2. AZD6244, BEZ235, lapatinib, and imatinib were administered through oral gavage, whereas AXLi was administered by intraperitoneal injection. The drugs were dissolved in the following vehicles: i) AZD6244 and lapatinib, 10% methylcellulose, 2% tween 20; ii) BEZ235, 50% 2 methylpyrrolidone, 50% PEG300; iii) imatinib, sterile PBS; and iv) AXLi, 10% DMSO, 90% PEG300.
Reverse Phase Protein Array (RPPA)
The RPPA protein expression profiles were generated by the MD Anderson RPPA core facility following standard protocols (17). More information can be found at http://www.mdanderson.org/education-and-research/resources-for-professionals/scientific-resources/core-facilities-and-services/functional-proteomics-rppa-core/index.html. The RPPA dilution curves were fitted with a logistic model from the SuperCurve R-package (18,19), and RPPA data were normalized by protein loading. Normalized log2 transformed data were used for further statistical analyses. Differential expression between two conditions was calculated using Student’s t-test and multiple conditions with one-way analysis of variance with custom R-scripts. Raw p-values were corrected for multiple hypothesis testing using the Benjamini-Hochberg correction (false discovery rate; FDR) and protein changes were considered significant when FDR was less than 10%.
Western blot
Whole cell extracts were electrophoresed by SDS-PAGE and transferred to a nitrocellulose membrane using a semi-dry transfer apparatus according to the manufacturer’s instructions (Bio-Rad). After incubation with 5% nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) for 60 minutes, the membranes were incubated with primary antibodies (see supporting online material for detailed antibodies list) at 4° C overnight. Membranes were washed three times for 10 minutes and incubated with a 1:5000 dilution of horseradish peroxidase-conjugated secondary antibodies. Blots were washed with TBST three times and developed after ECL-based chemiluminescence reaction through film exposure.
Immunohistochemistry
Formalin-fixed tumors were dehydrated and paraffin embedded according to standard procedures. 5-μm slices were cut using a microtome, rehydrated, and subjected to antigen unmasking by heating at 95° C for 30 minutes with a commercially available antigen unmasking solution (Citra Plus - Biogenex). Slices were subsequently incubated with 3% hydrogen peroxide for 15 minutes, incubated with primary antibodies, washed, incubated with HRP-conjugated secondary antibodies, washed, and developed through DAB incubation. Slices were counterstained with haematoxilin, dehydrated, and mounted.
Real Time PCR
cDNAs were synthesized from RNA through reverse transcription with a commercially available kit following manufacturer’s instructions (Invitrogen). Real time PCR was performed by a TAQMAN-based reaction by utilizing the following sets of Invitrogen primers and probes: GAS6 (Mm00490378_m1), PDGFA (Mm01205760_m1), PDGFB (Mm00440677_m1), PDGFC (Mm00480205_m1). Relative mRNA quantities were calculated using the comparative ΔΔCT method.
Lentivirus Production
A semiconfluent 15-cm plate of HEK293T was transfected with 30 μg of pHAGE plasmid and a mixture of the packaging–encoding vectors PMD2.G (9 μg) and pCMVΔR-8.74 (19.5 μg). Virus-containing media was collected 72 hours post-transfection and filtered; the virus was concentrated through ultracentrifugation.
RESULTS
Pharmacological inhibition of MAPK signaling provides limited antitumor impact relative to genetic extinction of KRASG12D
To evaluate changes in cell signaling upon genetic ablation of KRASG12D, we derived tumor cell lines from our previously characterized GEMM model harboring an inducible KRASG12D allele (P48 CRE_TetO-LSL-KRAS G12D_Rosa-rtTA_ p53fl/+, hereafter referred to as iKRAS cells) (15). In this system, a doxycycline-inducible promoter controls the expression of the constitutively active KRASG12D allele such that addition or removal of doxycycline induces or represses gene expression, respectively, with the consequent induction and subsequent regression of very aggressive established tumors. In addition, cells isolated from explanted tumors readily reformed tumors upon transplantation in recipient mice in the presence of doxycycline. As previously described (15), genetic extinction of KRASG12D upon doxycycline removal resulted in rapid tumor regression that was accompanied by dramatic induction of apoptosis and arrest of cell proliferation, demonstrating that KRASG12D is required for tumor maintenance (Figures 1, S1). In contrast, extinction of KRASG12D upon doxycyline withdrawal in iKRAS cells maintained in standard 2D-culture conditions did not exhibit anti-proliferative or apoptotic phenotypes (Figure S2). The poor correlation of 2D models with the in vivo setting prompted the development of a 3D-culture model in which iKRAS cells clonally proliferate and form tumor spheres only in the presence of doxycycline, and removal of doxycycline precipitates robust proliferative arrest and apoptosis as observed in vivo (Figure 1C, D). We speculated that this 3D-culture system may better predict antitumor responses to various drug treatments.
FIGURE 1.

A: Phospho-ERK levels of iKRAS cells cultured in 3D conditions 48 hours after KRASG12D ablation or AZD6244 2 μM treatment;
B: Representative image of spheres derived from iKRAS cells cultured for 7 days with continuous KRASG12D expression, 7 days after KRASG12D extinction (doxycycline withdrawal), or in KRASG12D-expressing cells treated for 7 days with 2 μM AZD6244;
C: Proliferation and D: apoptosis of iKRAS cells with continuous KRASG12D expression, after KRASG12D extinction, or in KRASG12D-expressing cells treated with 2 μM AZD6244;
E: Tumor volumes of iKRAS allografts. Arrow = time point where doxycycline was withdrawn to extinguish KRASG12D expression or 100 mg/kg/day AZD6244 administration was initiated in mice receiving doxycycline for continuous KRASG12D expression in tumor cells;
F: representative IHC of iKRAS-derived allografts stained for Ki67 (proliferation), cleaved caspase 3 (apoptosis), or phospho-ERK.
As KRASG12D is undruggable, significant effort has focused on dampening its signaling cascade. To determine which pharmacologic treatment(s) most closely recapitulate the signaling events and the phenotypic consequences of KRASG12D ablation observed in our iKRAS model, we treated iKRAS cells kept in the presence of doxycycline with the MEK1 inhibitor AZD6244 at a dose (2 μM) capable of inhibiting ERK phosphorylation to an extent comparable to what is observed upon 48 hours of doxycycline withdrawal (Figure 1A). While AZD6244 treatment significantly inhibited cell proliferation, there was a notable lack of cell death (Figure 1C, D). To conduct parallel in vivo studies, mice carrying iKRAS-derived subcutaneous allograft were divided into three groups: continued doxycycline administration, doxycycline withdrawal, or continued doxycycline administration plus AZD6244 at 100 mg/kg/day, a dose which achieves the highest pharmacological pathway repression. Similar to the 3D model, we observed complete tumor regression upon doxycycline withdrawal but only blunted tumor growth with AZD6244 treatment (Figure 1E). Correspondingly, immunohistochemical analysis of tumors following doxycycline withdrawal confirmed robust inhibition of cell proliferation (Ki67) and induction of apoptosis (cleaved caspase 3) compared to reduced cell proliferation and no apoptosis with AZD6244 treatment. Of note, inhibition of ERK phosphorylation was similar in AZD6244-treated and KRAS ablated tumors (Figure 1F).
KRASG12D ablation results in co-extinction of MAPK and PI3K-AKT-MTOR signaling, whereas MEK inhibition results in extinction of MAPK signaling with concomitant activation of AKT
We next audited signaling pathways to potential identify differentially regulated signaling events that may underlie the distinct biological phenotypes of KRASG12D extinction versus AZD6244 treatment. To that end, we performed a reverse-phase protein array (RPPA) analysis of lysates derived from three independent iKRAS lines cultured in the presence of doxycyline, after doxycycline withdrawal, or doxycycline plus AZD6244 treatment (2 μM). The heatmap (Figure 2A) reveals that multiple effectors of the PI3K-AKT-MTOR signaling pathway showed differential regulation in the setting of KRASG12D extinction versus MEK inhibition. Specifically, KRASG12D extinction showed dramatic repression of PI3K-AKT-MTOR targets: phospho-PRAS40, phospho-GSK alpha/beta, and phospho MTOR relative to KRASG12D–expressing cells, whereas MEK inhibition resulted in a partial attenuation of these signaling components. Conversely KRASG12D-ablation or MEK inhibitor treatment induced robust and minor accumulations of FOXO3a, respectively. Notably, ERK phosphorylation on Thr202 and Tyr204 and RAS-MAPK regulated phosphorylation of ribosomal protein S6 on Ser235 and Ser236 (20) were similarly inhibited by KRASG12D ablation or MEK inhibitor treatment. Immunohistochemistry analysis confirmed comparable signaling changes in treated tumors (Figure 2B). Moreover, western blot analysis of iKRAS cells cultured with doxycyline, after doxycycline withdrawal, or doxycycline plus AZD6244 treatment (2 μM) evidenced a robust increment in phospho-AKT at Thr308 and Ser473 in AZD6244-treated samples whereas KRASG12D ablation caused a time dependent reduction of phospho AKT levels (Figure 2C).
FIGURE 2.

A: Heatmap generated from RPPA analysis (mean values from 3 independent lines) of PI3K-AKT effector proteins from iKRAS cells cultured with doxycycline (KRASG12D expressed), 48 hours after doxycycline withdrawal (KRASG12D ablated), or in the presence of doxycycline after 48-hour exposure to 2 μM AZD6244
B: representative IHC of AKT-mTOR effector proteins P-GSKα/β, FOXO3a, P6S, and P-4EBP1 in iKRAS-derived allografts. When tumors became palpable mice were maintained with doxycyline (KRASG12D expressed) with or without 100 mg/kg AZD6244 daily treatment or removed from doxycycline (KRASG12D ablated ). For immunohistochemical evaluation, tumors were collected five days after the beginning of treatment or doxycycline withdrawal;
C: time course of AKT phosphorylation levels on residues T308 and S473 in iKRAS cells with continuous KRASG12D expression, after KRASG12D ablation, or in KRASG12D-expressing cells treated with 2 μM AZD6244,
D: Macroscopic tumor appearance (right) and tumor volumes (left) from iKRAS allografts from mice treated with vehicle, 100 mg/kg QD AZD6244, 40 mg/kg QD BEZ235, or combined AZD6244 plus BEZ235. Treatments were started when tumors become palpable and continued until excessive tumor burden or ulcerations occurred.
To assess whether the differential PI3K-AKT-MTOR activation status observed upon KRASG12D ablation versus MEK inhibition may be responsible for the different tumor response, we established iKRAS-derived allografts and treated them with AZD6244 (100 mg/kg/day) and/or BEZ235 (40 mg/kg/day) (a dual PI3K-MTOR inhibitor). Inhibition of the MAPK signaling arm or PI3K-AKT-MTOR arms alone provided minimal antitumor benefit, while concurrent inhibition of both effector pathways exerted robust antitumor efficacy mimicking the phenotypic changes observed upon KRASG12D ablation (Figure 2D).
Both KRASG12D ablation and MEK inhibition activate EGFR, HER2, PDGFRa, and AXL
It has been shown that MEK inhibition results in hyperactivation of the erythroblastic leukemia viral oncogene homologue (ERBB) receptor tyrosine kinases (21), which could explain the increase in AKT phosphorylation observed upon MEK1 inhibitor treatment. Phospho-RTK analyses performed on resected iKRAS-derived tumors showed detectable basal levels of phosphorylated platelet-derived growth factor receptor alpha (PDGFRa) in KRASG12D-expressing tumors, as well as a robust increase in phosphorylation of EGFR, human epidermal growth factor receptor 2 (HER2), and P-AXL upon either AZD6244 treatment or doxycycline withdrawal (Figure 3A). To determine whether increased RTK activation resulted from increased expression of the RTKs or their ligands, we analyzed protein lysates and cDNA from iKRAS cells. We found that RTK activation was the result of both, as KRAS ablation increased expression of PDGFRa and AXL, as well as the PDGFR ligands PDGFa, PDGFb, PDGFc, and the AXL ligand growth arrest-specific 6 (GAS6) (Figures 3B, C and S4). In contrast, none of the EGFR family ligands assessed showed an increased expression (data not shown).
FIGURE 3.

A: Phospho-RTK array of iKRAS-derived allografts from mice receiving doxycycline (KRASG12D expressed), after doxycycline withdrawal (KRASG12D ablated), or treated with 100 mg/kg AZD6244 daily in the presence of doxycycline. Tumors were collected five days after the beginning of the treatment or doxycycline withdrawal;
B: Expression levels of GAS6 (AXL ligand), PDGFa, PDGFb, and PDGFc (PDGFR ligands) from iKRAS cells cultured in the presence (KRASG12D expressed) or absence (KRASG12D ablated) of doxycycline or treated with 2 μM AZD6244 in the presence of doxycycline;
C: Western blot of AXL, HER2, and PDGFRa from iKRAS cells cultured in the presence (KRASG12D expressed) or absence (KRASG12D ablated) of doxycycline for 48 hours. Phospho-ERK levels manifest effective abrogation of KRASG12D expression, whereas total ERK serves as a loading control;
D: C-myc expression in iKRAS cells cultured in the presence of doxycycline (KRASG12D expressed), upon doxycycline widthdrawal (KRASG12D ablated) or in the presence of doxycycline and 2 μM AZD6244 for 48 hours;
E: AXL and c-myc expression in iKRAS cells transduced with either scramble or c-myc shRNA and cultured in the presence of doxycycline (KRASG12D expressed).
Diverse mechanisms may be responsible for the RTK activation observed in response to MEK inhibition. In particular, it has been shown that AZD6244 treatment inhibits a MAPK-mediated phosphorylation of EGFR and HER2 that prevents receptor dimerization and activation (21), whereas a more broad transcriptional RTK activation occurs due to inhibition of c-myc (22). Because AXL levels were elevated upon KRASG12D ablation, we postulated that c-myc may negatively regulate AXL expression. As expected, both ablation of KRASG12D and MEK inhibitor treatment repressed c-myc expression, and knockdown of c-myc in iKRAS lines resulted in increased AXL expression (Figure 3D, E).
RTKs drive redundant signaling to the PI3K-AKT MTOR pathway
To assess the effect on signaling modulation of each RTK activated upon MEK inhibition, we performed RPPA analysis on iKRAS cells exposed to single RTK inhibitors as well as their different combinations with or without AZD6244 treatment. To inhibit each RTK, we used the dual EGFR-HER2 inhibitor lapatinib, the PDGFRa inhibitor imatinib, and the novel AXL inhibitor CH5451098 (23) (hereafter referred to as AXLi). AXLi showed superior selectivity and potency over other available AXL inhibitors, and we extensively evaluated its in vivo safety and pharmacokinetics (Figures S6, S7, and S8). RPPA profiles (Figure 4A) from single or combined RTK inhibitor-treated iKRAS cells showed no significant impact on protein or phosphoprotein abundance of members of the PI3K-AKT-MTOR signaling pathway compared to untreated controls. This is consistent with our observations that in basal, untreated conditions, iKRAS cells show minimal RTK activation and supports the view that the inhibitors used in the present study are “on target” and are delivered at appropriate doses to extinguish MEK inhibitor-mediated RTK activation (Figure S9). Moreover, no off-pathway effects were detected in cells lacking RTK activation. In contrast, combined treatment with RTK inhibitors and AZD6244 showed a significant impact on proteins and phosphoproteins of the PI3K-AKT-MTOR signaling pathway only when AZD6244 was combined with all three RTK inhibitors, which produced pathway inhibition analogous to that observed upon KRASG12D ablation.
FIGURE 4.

A: Heatmap generated from RPPA analysis (mean values from 3 independent lines) of PI3K-AKT effector proteins from iKRAS cells cultured with doxycycline (KRASG12D expressed) and treated for 48 hours with one or all three (RTKi) RTK inhibitors (1 μM AXLi, 1 μM imatinib or 1 μM lapatinib) alone or in combination with 2 μM AZD6244;
B: Representative IHC of phospho-ERK and phospho-S6 of tumors derived from iKRAS allografts treated with AXLi 30 mg/kg QD, imatinib 100 mg/kg QD, or lapatinib 100 mg/kg QD, or the combination of each RTK inhibitor with 100 mg/kg QD AZD6244;
C: AKT phosphorylation levels on residues T308 and S473 in iKRAS cells cultured in doxycycline (KRASG12D expressed) and treated for 48 hours with a single RTK inhibitor (1 μM AXLi, 1 μM imatinib or 1 μM lapatinib) alone or in combination with 2μM AZD6244;
D: AKT phosphorylation levels on residues T308 and S473 in iKRAS cells cultured in doxycycline and treated for 48 hours with 3 RTK inhibitors (RTKi) (1 μM AXLi, 1 μM imatinib or 1 μM lapatinib), alone or in combination with 2 μM AZD6244. iKRAS cells were also analyzed 48 hours after doxycycline withdrawal (KRASG12D ablated).
To confirm our results in vivo we evaluated the levels of phospho-S6 kinase as a marker of the activity of the PI3K-AKT-MTOR pathway in tumors treated with AXLi (30 mg/kg/day), imatinib (100 mg/kg/day), or lapatinib (100 mg/kg/day) alone or in combination with AZD6244 (100 mg/kg/day) for 5 days. Consistent with the RPPA data, neither treatment with a single RTK inhibitor nor with combined single RTK and MEK inhibitors affected P-S6 levels (Figure 4B), thus confirming our earlier conclusion that diverse RTKs drive redundant inputs to the PI3K-AKT-MTOR signaling pathway. Ultimately, an analysis of AKT phosphorylation on Thr308 and Ser473 (Figure 4C, D) further validated our findings that single RTK inhibition does not affect the MEK1-mediated feedback loop of activation of the PI3K signaling pathway and, conversely, this feedback is abrogated only when co-extinction of multiple RTKs is enabled.
Inhibition of multiple RTKs is required to synergize with MEK inhibition and to induce antitumor activity
Because each RTK independently sustains the activity of the PI3K-AKT-MTOR signaling pathway, we sought to evaluate the therapeutic potential of anti RTK therapy alone or in combination with MEK inhibition. In vitro treatment of iKRAS cells evidenced that none of the RTK inhibitors alone or in combination significantly inhibited growth of iKRAS tumor spheres (Figure 5A), suggesting that KRASG12D-driven tumors may not require RTK signaling for their growth and survival. Moreover, concomitant treatment of iKRAS cells with AZD6244 and any single RTK inhibitor did not potentiate the activity of AZD6244. Conversely, a robust inhibitory effect was observed when AZD6244 was combined with lapatinib and AXLi, imatinib and AXLi, or even more effectively when AZD6244 was combined with all three RTK inhibitors concurrently (Figure 5A). To validate these findings in vivo, we treated iKRAS-tumor-bearing mice with combinations of AXLi (30 mg/kg/day), imatinib (100 mg/kg/day), or lapatinib (100 mg/kg/day) and AZD6244 (100 mg/kg/day). These experiments confirmed our in vitro findings that co-extinction of MEK and of the RTKs activated by MEK inhibition was required to achieve substantial tumor growth inhibition (Figure 5B).
FIGURE 5.
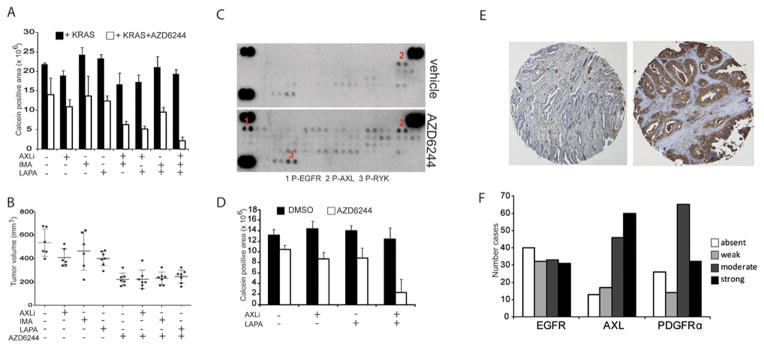
A: Viability (calcein staining) of iKRAS cells with continuous KRASG12D expression (cultured in the presence of doxycycline) cultured for 7 days in the presence or absence of 2 μM AZD6244 and treated with one or more RTK inhibitors: 1 μM AXLi, 1 μM imatinib or 1 μM lapatinib;
B: Tumor volumes of iKRAS-derived allografts with continuous KRASG12D expression and treated with either AXLi 30 mg/kg QD, imatinib 100 mg/kg QD, or lapatinib 100 mg/kg QD, or the combination of each RTK inhibitor with 100 mg/kg QD AZD6244. Treatments were started when tumors become palpable and continued until excessive tumor burden or ulcerations occurred;
C: Phospho-RTK array of lysates derived from xenografts from the human primary PDAC-derived line PATC. Mice received either vehicle or AZD6244 100 mg/kg daily and were sacrificed five days after treatment;
D: Viability (Calcein staining) of PATC cells cultured for 7 days in the presence or absence of 2 μM AZD6244 and treated with either 1 μM AXLi, 1 μM lapatinib or their combination;
E: Representative staining of human PDAC strongly positive or negative for AXL;
F: Number of PDAC cases presenting absent, weak, moderate and strong staining for either EGFR, AXL, or PDGFRa in a TMA series representative of 136 cases;
To address the human relevance of our findings, we treated xenografts derived from a human primary PDAC harboring mutant KRAS (hereafter named PATC) with vehicle or AZD6244 (100 mg/kg/day) for five days and profiled RTK activation status in tumor-derived lysates. MEK inhibition induced a modest increase in P-AXL levels compared to vehicle-treated tumors, and a robust increase of EGFR phosphorylation (Figure 5C). We then treated cells with lapatinib, AXLi, or their combination alone or in addition to AZD6244 2 μM to determine whether co-extinction of multiple RTKs is necessary to induce anti-spherogenic responses. Consistent with previous findings in our murine model of PDAC, only co-inhibition of AXL and EGFR along with MEK inhibitor treatment significantly repressed the spherogenic activity of PATC cells (Figure 5D). These results suggest that inhibiting the RTKs whose activation is induced by MEK inhibition may be required for the development of a MEK inhibitor treatment regimen.
There have been several reports of RTK overexpression in human PDAC tissues (12,24–26), prompting analysis of EGFR, AXL, and PDGFRa levels in a tissue microarray (TMA) containing 136 untreated PDAC cases. All three RTKs were moderately or strongly expressed in a significant number of cases (24.2% moderate and 22.8% strong for EGFR, 33.8% moderate and 44.0% strong for AXL and 47.8% moderate and 23.3% strong for PDGFRa; Figure 5F), supporting the possibility that a relevant fraction of PDAC tumors basally express determinants of resistance to MEK1 inhibition.
Constitutive AKT activation prevents tumor regression upon KRASG12D ablation
Our data demonstrate that a number of RTKs are upregulated upon MEK inhibition, and that each is able to sustain PI3K-AKT-MTOR activation. To determine whether constitutive activation of PI3K-AKT-MTOR signaling can rescue KRASG12D dependence, we derived allografts from iKRAS cells transduced with either GFP or a constitutively active myristylated AKT (myr-AKT) (Figure 6A). Both GFP- and myr-AKT-expressing iKRAS cells formed tumors with analogous kinetics, indicating that AKT hyperactivation does not provide an additional growth advantage. However, upon doxycycline withdrawal, GFP-transduced tumors underwent rapid and robust regression, whereas AKT-transduced tumors showed arrested growth but no significant reduction in tumor volume (Figure 6C). Immunohistochemical analysis of the phosphorylation status of S6 and eIF4E-binding protein 1 (4EBP1) in tumors upon KRASG12D ablation confirmed the maintenance of pathway activity in Myr-AKT expressing tumors (Figure 6B). These data demonstrate that AKT activation, while unable to overcome dependency on mutated KRAS for tumor growth, is sufficient to promote tumor maintenance, thereby facilitating eventual escape mechanisms. Our data further support the conclusion that the limited antitumor activity observed upon MAPK inhibition is likely caused by MEK inhibition-mediated AKT activation.
FIGURE 6.
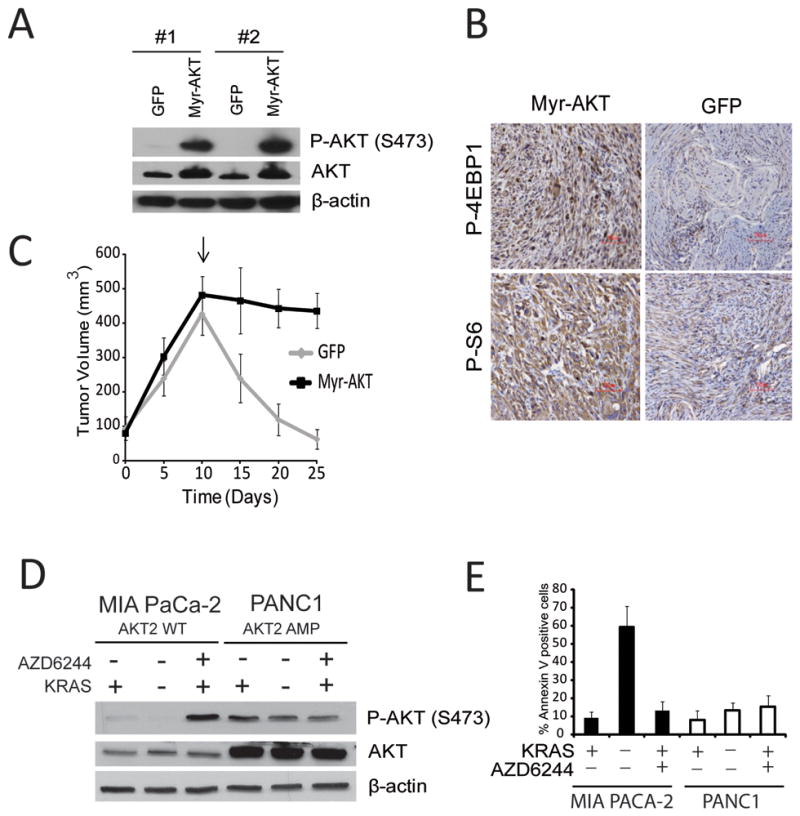
A: Phospho-AKT and AKT levels of iKRAS cells transduced with lentivirus encoding myristilated-AKT (Myr-AKT);
B: Representative IHC of P-S6 or P-4EBP1 in allografts of iKRAS cells expressing GFP or Myr-AKT collected 5 days after doxycycline widthdrawal (KRASG12D ablated);
C: Tumor volumes from allografts of iKRAS cells expressing GFP or Myr-AKT. Arrows = time point of doxycycline withdrawal;
D: Phospho-AKT and AKT levels of MIA PaCa-2 (KRASG12C, AKT2wt) and PANC1 (KRASG12D AKT2amp) cells transduced with a doxycycline-inducible shRNA for KRAS and cultured for 72 hours in the absence of doxycycline (KRAS+) with our without treatment with AZD6244 2 μM, or in the presence of doxycycline (KRAS−);
E: Quantification of apoptotic cells in MIA PaCa-2 and PANC1 cells transduced with doxycycline-inducible shRNA for KRAS cultured for 72 hours in the absence of doxycycline (KRAS+) with our without treatment with AZD6244 2 μM, or in the presence of doxycycline (KRAS−).
Ten to twenty percent of PDAC tumors carry gain/amplification of AKT2 (27,28), and we hypothesized that this subset of tumors may be insensitive to KRASG12D ablation. To test this hypothesis, we employed MIA PACA-2 and PANC1 cells, which both harbor mutant KRAS, but only the latter has amplification of 19q13 encompassing the AKT2 locus (29). Both cell lines were transduced with an inducible anti-KRAS shRNA to allow KRAS ablation, and MEK was inhibited with 2 μM AZD6244. In MIAPACA2 cells, KRAS knockdown did not increase phospho-AKT levels above the low basal levels observed, in contrast to a marked accumulation of phospho-AKT upon MEK inhibition (analogous to what we previously observed in our murine model; Figure 6D). Conversely, PANC1 cells presented with basally elevated levels of total AKT and phospho-AKT, neither of which was further increased upon either KRAS knockdown or MEK inhibition, suggesting that, in this cell line, AKT2 amplification drives constitutive activity of the pathway. KRAS knockdown induced apoptosis only in the MIAPACA2 cell line (Figure 6E), supporting the contention that tumors harboring AKT2 amplification are intrinsically independent from mutant KRAS for maintenance.
DISCUSSION
In contrast to other driver oncogenes such as mutated BRAF or EGFR, effective therapies that directly target mutated KRAS are still unavailable, therefore attempts to impact KRAS-driven tumors largely depend on an improved understanding of the key effector pathways required for tumor maintenance downstream of KRAS. Unfortunately, clinical studies have shown that abrogation of the MAPK effector pathway alone through MEK inhibitors in KRAS-driven tumors fails to achieve clinical responses (30,31). Accordingly, we failed to see a robust therapeutic response upon MEK inhibition in our murine model of PDAC, whereas ablation of KRASG12D resulted in visibly complete tumor regression, thus underscoring the utility of this model to deeply characterize KRAS effectors. Through comparative analyses of genetic KRASG12D ablation versus MEK inhibition, we identified differentially regulated key effectors. Specifically, in agreement with previous reports (32,33), the sole inhibition of MEK1 promoted the activation of the PI3K pathway whereas KRAS ablation profoundly inhibited both PI3K-AKT-MTOR and MAPK signaling, further emphasizing the need to simultaneously inhibit both pathways to yield a robust antitumor response such as was observed upon KRAS ablation in our iKRAS murine model. Despite numerous previous reports of promising therapeutic efficacy from various combinations of inhibitors of these two pathways (8,9,34), an emerging set of clinical data have shown that these combinations are poorly tolerated, thus limiting their utility (10).
It has been established in multiple tumor models that MEK inhibition engages feedback loops that promote hyperactivation of the PI3K-AKT-MTOR pathway through the recruitment of diverse RTKs including AXL and PDGFRa (21,22,35). Diverse feedback circuits have been described, including the MTORC1-mediated recruitment of insulin receptor kinase (36,37) and the MEK inhibition-mediated hyperactivation of the ERBB receptors EGFR, HER2, and ERBB3 (21,38). Moreover MEK inhibition-mediated RTK activation was observed in both RAS mutant and RAS wt tumor models indicating that such adaptive response is not unique to mutant RAS driven tumors (21,22,35). Both of these RTK-mediated feedback loops activate the PI3K pathway and thereby represent valuable “druggable” therapeutic opportunities that may afford lower toxicity than MEK/PI3K inhibitor combinations. However, while a variety of preclinical studies in diverse tumor models have reported promising antitumor activity by co-targeting of MEK and ERBB receptors (39–41), a recent clinical study of combined therapy with AZD6244 and the anti-EGFR drug erlotinib in patients with chemotherapy-refractory pancreatic cancer reported that antitumor activity was only achieved in a subset of patients (42).
We profiled RTK activation and therapeutic responses to targeting the RTK-mediated feedback loops in our murine model. Surprisingly, we observed that RTK activation occurs both upon KRASG12D ablation and MEK inhibition, although in the former this event fails to induce AKT hyperactivation or to promote tumor maintenance, suggesting that continuous KRASG12D signaling is required to promote RTK-mediated activation of the PI3K pathway. Importantly, the RTK activation we observed included not only HER1-2, but also the TAM receptor AXL as well as PDGFRa. Moreover, the mechanism of activation of AXL and PDGFRa involves both upregulation of the RTK itself as well as transcriptional upregulation of their ligands, suggesting that, upon MEK inhibition, tumor cells possess the ability to engage autonomous regulatory circuits rather than relying solely on the supply of growth factors from the microenvironment.
To evaluate the contribution of each RTK activated upon MEK inhibitor treatment, we tested whether inhibition of any one RTK exerted dominant control over pro-survival pathway activation, which may help elucidate strategies for targeted therapy combinations. These efforts included extensive characterization of a novel AXL inhibitor with unprecedented selectivity and potency compared to all others previously described. We showed that inhibition of any single RTK activated by MEK inhibitor treatment did not yield antitumor efficacy above that achieved with the MEK inhibitor alone. Rather, concomitant inhibition of all of the diverse RTKs activated upon MEK inhibition was required to yield synergistic effects with MEK inhibition in both our iKRAS murine model, as well as in a MEK inhibitor-treated model derived from human primary PDAC. This result has profound clinical relevance, as it underscores the need for personalized therapy combinations based on a rational co-extinction strategy versus blind enrollment into trials that combine a MEK inhibitor with a single RTK inhibitor. Moreover, consistent with previous reports, we found detectable AXL and PDGFRa expression in a number of human PDAC samples (24–26), suggesting that many patients present with tumors that may be easily resistant to MEK/HER inhibitor combinations. Furthermore, our data are in line with previous evidence that activation of AXL and PDGFRa can drive resistance to anti-EGFR treatment (43,44) and, in the case of AXL, to anti MEK1 therapy (45,46). Proteomic profiling of iKRAS cells upon MEK inhibition and treatment with single or multiple RTK inhibitors showed that each RTK can independently sustain hyperactivity of the PI3K pathway mediated by MEK inhibition.
Given the striking dominance of the PI3K signaling pathway activation in tumor maintenance, we asked whether a fraction of PDAC harboring genomic aberrations driving constitutive activation of this pathway such as AKT2 amplification which occurs in 10–20% of PDACs (27,28), may render this subset of patients insensitive not only to MEK/RTK inhibitors, but also to continuous KRAS signaling. Indeed, constitutive activation of the PI3K-AKT pathway prevented tumor regression upon KRAS ablation and, furthermore, cell lines harboring AKT2 amplification manifested resistance not only to MEK inhibition but also to genetic abrogation of KRAS.
Collectively, our findings suggest that combinatorial MEK/RTK targeted therapies may have limited impact without careful patients stratification to therapy combinations based on an individual’s oncogenome and on the activation state of signal transduction pathways at time of treatment. Based on this, a personalized therapy strategy that accounts for the genomic aberrations driving constitutive activity of the PI3K signaling pathway, as well as a deep analysis of feedback circuits engaged by MEK inhibition, is more likely to result in positive clinical outcomes than random assignment to combination therapies to inhibit MEK and a single RTK. An improved and comprehensive understanding of these and additional mechanisms that allow survival of tumors cells despite suppression of mutant KRAS-mediated signaling (16,47) will define possibly definitive therapeutic strategies.
Supplementary Material
Acknowledgments
Financial Support: This study was supported by grants from the Hirshberg Foundation for Pancreatic Cancer Research to A.V., Sheikh Ahmed Center for Pancreatic Cancer Research to G.F.D., T.P.H. and A.V, the American Italian Cancer Foundation to G.F.D., P01CA117969 to R.A.D, RPPA was support by CCSG grant P30 CA016672 and by an Italian American Cancer Foundation Post-Doctoral Research Fellowship to P.P.
We wish to acknowledge Shan Jiang for her expert management of the mouse colony and Dr. Lynda Chin for helpful suggestions.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker NM, Der CJ. Cancer: Drug for an ‘undruggable’ protein. Nature. 2013;497:577–8. doi: 10.1038/nature12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–42. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 7.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci USA. 2013;110:4015–20. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–25. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 11.Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144:1316–26. doi: 10.1053/j.gastro.2013.01.078. [DOI] [PubMed] [Google Scholar]
- 12.Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–30. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 14.Storch K, Eke I, Borgmann K, Krause M, Richter C, Becker K, et al. Three-dimensional cell growth confers radioresistance by chromatin density modification. Cancer Res. 2010;70:3925–34. doi: 10.1158/0008-5472.CAN-09-3848. [DOI] [PubMed] [Google Scholar]
- 15.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viale A, Pettazzoni P, Costas A, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014 doi: 10.1038/nature13611. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–21. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 18.Hu J, He X, Baggerly KA, Coombes KR, Hennessy BT, Mills GB. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–94. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- 19.Neeley ES, Kornblau SM, Coombes KR, Baggerly KA. Variable slope normalization of reverse phase protein arrays. Bioinformatics. 2009;25:1384–9. doi: 10.1093/bioinformatics/btp174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–64. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228–37. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–21. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono Y, Mizuguchi E, Harada T, Kimbara A, Tachibana K, Takano K, et al. Discovery of selective AXL kinase inhibitor CH5451098 and its biological activity. 244th ACS National meeting; 2012. p. 11501. [Google Scholar]
- 24.Koorstra JB, Karikari CA, Feldmann G, Bisht S, Rojas PL, Offerhaus GJ, et al. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol Ther. 2009;8:618–26. doi: 10.4161/cbt.8.7.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song X, Wang H, Logsdon CD, Rashid A, Fleming JB, Abbruzzese JL, et al. Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer. 2011;117:734–43. doi: 10.1002/cncr.25483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang RF, Yokoi K, Bucana CD, Tsan R, Killion JJ, Evans DB, et al. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin Cancer Res. 2003;9:6534–44. [PubMed] [Google Scholar]
- 27.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–64. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 28.Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 2011;1:158–69. doi: 10.1158/2159-8290.CD-11-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuuselo R, Savinainen K, Azorsa DO, Basu GD, Karhu R, Tuzmen S, et al. Intersex-like (IXL) is a cell survival regulator in pancreatic cancer with 19q13 amplification. Cancer Res. 2007;67:1943–9. doi: 10.1158/0008-5472.CAN-06-3387. [DOI] [PubMed] [Google Scholar]
- 30.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50:2072–81. doi: 10.1016/j.ejca.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 32.Parker R, Clifton-Bligh R, Molloy MP. Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol Cancer Ther. 2014;13:1894–906. doi: 10.1158/1535-7163.MCT-13-0938. [DOI] [PubMed] [Google Scholar]
- 33.Huang MH, Lee JH, Chang YJ, Tsai HH, Lin YL, Lin AM, et al. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol Oncol. 2013;7:112–20. doi: 10.1016/j.molonc.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts PJ, Usary JE, Darr DB, Dillon PM, Pfefferle AD, Whittle MC, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18:5290–303. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520–33. doi: 10.1158/2159-8290.CD-12-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248–59. doi: 10.1158/2159-8290.CD-11-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311–21. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abel EV, Basile KJ, Kugel CH, 3rd, Witkiewicz AK, Le K, Amaravadi RK, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. 2013;123:2155–68. doi: 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diep CH, Munoz RM, Choudhary A, Von Hoff DD, Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011;17:2744–56. doi: 10.1158/1078-0432.CCR-10-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med. 2014;6:224ra26. doi: 10.1126/scitranslmed.3007947. [DOI] [PubMed] [Google Scholar]
- 41.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ko AH, Tempero MA, Bekaii-Saab TB, Kuhn P, Courtin R, Ziyeh S, et al. Dual MEK/EGFR inhibition for advanced, chemotherapy-refractory pancreatic cancer: A multicenter phase II trial of selumetinib (AZD6244; ARRY-142886) plus erlotinib. J Clin Oncol. 2013;31(suppl):abstr 4014. [Google Scholar]
- 43.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–60. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–90. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:ra66. doi: 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–27. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158:185–97. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.