

Abstract
Background
The phosphatonins FGF-23 and FRP-4 are inhibitors of tubular phosphate reabsorption that may play a role in the hyperphosphatemia associated with chronic kidney disease (CKD) or in the hypophosphatemia associated with renal transplants.
Methods
Plasma FGF-23, FRP-4, phosphorus and PTH were measured in patients at all stages of CKD. Phosphate regulation of FGF-23 and sFRP-4 was examined in ESRD patients in the presence and absence of therapeutic phosphate binder usage. In renal transplant patients, plasma FGF-23, sFRP-4 and phosphorus concentrations were determined before and 4-5 days after transplantation.
Results
Plasma FGF-23 correlated with creatinine clearance (r2 = - 0.584, p < 0.0001) and plasma phosphorus (r2 = 0.347, p < 0.001) in CKD patients and with plasma phosphorus (r2 = 0.448, p < 0.001) in ESRD patients. Phosphate binder withdrawal increased FGF-23 levels. In kidney transplant patients, dramatic decreases in FGF-23 (-88.8 ± 5.4%) and phosphorus (-64 ± 10.2%) were observed by 4-5 days post-transplantation. In patients with post-transplant hypophosphatemia, FGF-23 levels correlated inversely with plasma phosphorus (r2 = 0.661, p < 0.05). sFRP-4 levels did not change with creatinine clearance or hyperphosphatemia in CKD or ESRD patients, and no relation was noted between post-transplant sFRP-4 levels and hypophosphatemia.
Conclusions
In CKD, FGF-23 levels rose with decreasing creatinine clearance rates and increasing plasma phosphorus levels, and rapidly decreased post-transplantation suggesting FGF-23 is cleared by the kidney. Residual FGF-23 may contribute to the hypophosphatemia in post-transplant patients
Introduction
Phosphate homeostasis is complex and is influenced by phosphate intake, intestinal absorption, bone deposition/resorption and renal excretion. In addition, circulating levels of parathyroid hormone (PTH) and 1,25(OH)2D3 are important endocrine regulators of plasma phosphate (1-4). The impaired renal function in chronic kidney disease is associated with decreased 1,25(OH)2D3 and increased phosphate levels leading to a subsequent elevation in PTH (5). One of the key goals for CKD therapy is to normalize plasma phosphate since hyperphosphatemia is correlated with significant cardiovascular morbidity and mortality (6-10). Therefore, it is critical to understand the relationship between plasma phosphate and the phosphate regulating hormones, 1,25(OH)2D3 and PTH in order to optimize the treatment of CKD patients.
Over the past few years, new phosphate regulating proteins have been identified that induce phosphaturia by decreasing renal phosphate reabsorption. The two best characterized phosphatonins are fibroblast growth factor-23 (FGF-23) and secreted frizzled related protein-4 (sFRP-4) (11-20). In addition to its phosphaturic effects, FGF-23 has also been shown to decrease active vitamin D levels (21-23). Athymic nude mice implanted with Chinese hamster ovary cells expressing FGF-23 or injected with recombinant FGF-23 had a reduction in both plasma phosphorus and 1,25(OH)2D levels. FGF-23 null mice exhibited hyperphosphatemia and elevated 1,25(OH)2D levels (24,25). A role in normal phosphate and vitamin D physiology is suggested by the hyperphosphatemia and elevated 1,25(OH)2D3 levels observed in normal adult mice injected with neutralizing antibodies to FGF-23 (Shimada T, J Bone Miner Res, 18, S 97, 2003 Sep 2003, abstract). Phosphate loading of rats increases FGF-23 levels (26-28), but studies in healthy adults have produced mixed effects of dietary phosphate on FGF-23. Escalating phosphate diets were reported to increase (29)(Allen HC, J Bone Miner Res 17, S159, 2002 abstract) or to have no effect (30) on FGF-23 levels in humans.
sFRP-4 is measurable in normal adults. Like FGF-23, sFRP-4 is phosphaturic, and blunts the compensatory increase in 1,25(OH)2D3 in response to hypophosphatemia (31), but its role in phosphate homeostasis and regulation by phosphate is not clear. A third protein, MEPE (matrix extracellular phosphoglycoprotein) is expressed by phosphate wasting tumors, and has been reported to be phosphaturic (32), although this remains controversial since MEPE knockout mice do not display altered plasma phosphate levels (33,34).
Thus, the phosphatonins are known to cause phosphaturia (12-16,31) and are implicated in several hereditary and acquired hypophosphatemic disorders (21,26,32-52), but their roles in normal physiology or in kidney disease are less well understood. Although FGF-23 levels are detectable in normal individuals (11) and elevated in patients with chronic kidney disease (30,53-55), it is not known if this change is a consequence of reduced renal clearance and/or elevated phosphorus. If plasma phosphorus concentrations control phosphatonin expression, it is likely that their expression is also altered in other disease entities where phosphate dysregulation is evident. Post renal transplantation hypophosphatemia is seen commonly, and has clinical importance due to its linkage with post transplant bone disease (56-60). The present study examines the impact of hyperphosphatemia and chronic kidney disease on FGF-23 and sFRP-4, and the potential role of these phosphatonins in post transplantation hypophosphatemia.
Materials and Methods
After approval by the Washington University School of Medicine Human Studies Committee, informed and written consent was taken from 86 patients; 40 were chronic kidney disease (CKD) patients not on dialysis, 34 were patients on hemodialysis (ESRD) and 12 were pre-renal transplant patients. There were no inclusion or exclusion criteria used in patient recruitment. The CKD patients (n = 40) were 13 females and 27 males. The etiology of their disease was predominantly from hypertension (30%), diabetes (20%), focal segmental glomerulosclerosis (20%), and other glomerular disorders (7.5%). The remaining 22.5% was from miscellaneous causes. A control group was comprised of 12 healthy subjects with normal renal function.
Blood was collected for measurement of plasma levels of phosphorus, calcium, intact parathyroid hormone (iPTH), 1,25(OH)2D3, albumin, sFRP-4 and FGF-23. All the biochemistries except 1,25(OH)2(D)3, sFRP-4 and FGF-23 were measured at the chemistry laboratory of Barnes-Jewish Hospital, St Louis. 1,25(OH)2D was measured using the calf thymus receptor assay of Reinhardt et al (61) on the 1,25(OH)2D3 fraction extracted by the method of Hollis (62). The FGF-23 was measured using a kit that measures the C-terminal fragment of FGF-23 (Immutopics Corporation, San Clemente, CA). Plasma sFRP-4 measurements were performed using a two-antibody sandwich ELISA as described previously (31).
In the ESRD population, FRP-4, FGF-23 and plasma phosphorus levels were measured. Six patients were enrolled in a sub-study in which FGF-23 and sFRP-4 were measured when their phosphate binders were temporarily stopped. FGF-23 and sFRP-4 were measured in these patients after their plasma phosphorus rose to greater than 5 mg/dl. These individuals were then placed back on phosphorus binders and FGF-23 and FRP4 were measured again when once their plasma phosphorus fell to less than 4.5 mg/dl.
The patients on the transplant list had blood drawn within one day pre-transplantation and 4-5 days post-transplantation. Plasma FGF-23, sFRP-4, phosphorus, iPTH and creatinine were measured in these samples. Two patients with delayed graft function were excluded from the study.
Correlations between the levels of FGF-23, sFRP-4 and other blood chemistries were analyzed by ANOVA linear regression and by multivariable regression analysis (GraphPad Instat).
Results
FGF-23 in Chronic Kidney Disease(CKD) and End-Stage Renal Disease (ESRD)
Patients with chronic kidney disease, not on dialysis, had creatinine clearances ranging from 8 ml/min to 114ml/min. The dominant etiologies included hypertension, diabetes and focal segmental glomerulosclerosis. Plasma phosphorus ranged from 2.0 to 6.5 mg/dl and PTH from 11 pg/ml to 445 pg/ml. The FGF-23 levels in our control population were 67 ± 12 RU/ml. In the CKD population, there was a significant inverse relation between creatinine clearance and log FGF-23 levels (r2 =0.584, p < 0.0001) (fig. 1), although the relationship is curvilinear even when plotted on a semilog scale, suggesting multiple regulatory factors. FGF-23 levels were in the normal range in patients with creatinine clearances greater than 60-70 ml/min. In the ESRD population, all the patients were on hemodialysis from one to sixteen years. FGF-23 levels in this subset were much higher than the CKD patients (fig. 2). There was no relation between FGF-23 concentrations and years on dialysis (fig. 3).
Figure 1. Correlation of FGF-23 with creatinine clearance in CKD patients.
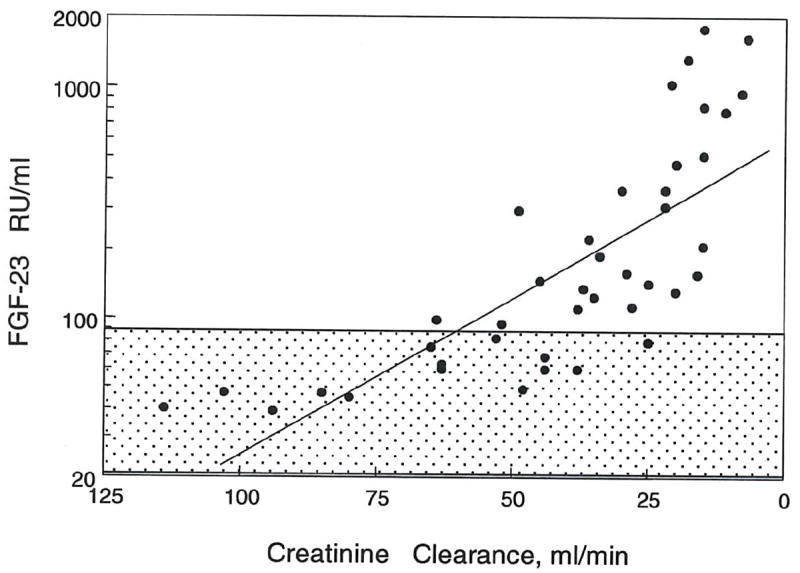
A negative correlation was observed between log FGF-23 and creatinine clearance in CKD patients (n = 40, r2 = - 0.584, p < 0.0001).
Figure 2. Mean FGF-23 levels in defined stages of CKD.
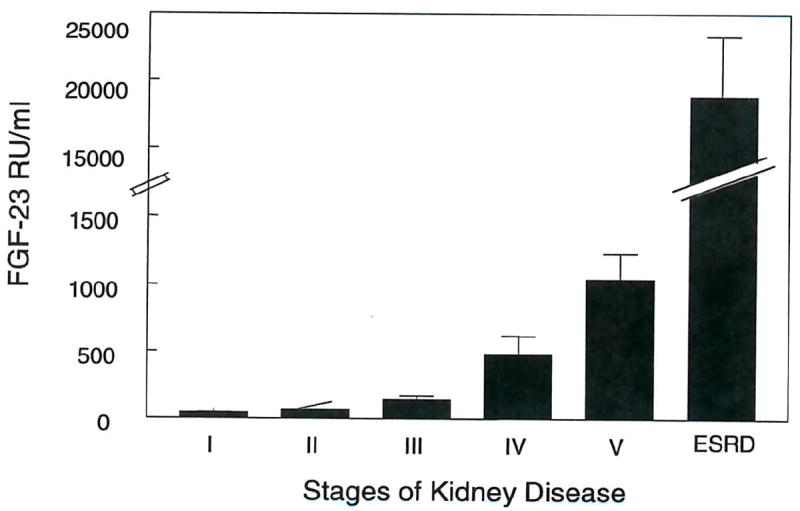
FGF-23 levels (mean ± S.D.) are given for each stage of CKD defined by K DOQI guidelines and for ESRD patients.
Figure 3. Lack of relationship between FGF-23 and years on dialysis.

FGF-23 correlated positively with plasma phosphorus in both CKD patients (r2 = 0.347, p < 0.001) (fig. 4) and in ESRD patients (r2 = 0.448, p < 0.001) (fig. 5). Multiple regression analysis indicated that both creatinine clearance and plasma phosphate independently correlated with FGF-23 levels. As illustrated in figure 6, the increment in FGF-23 with increasing phosphorus is similar when comparing patients grouped by renal function. The slopes for the correlation between FGF-23 and phosphorus are similar for CKD stages 1-3, CKD stages 4-5 and ESRD. However, the regression lines shift upward with decreasing renal function. These findings indicate independent effects of phosphate and renal insufficiency on FGF-23 levels.
Figure 4. Correlation between FGF-23 and plasma phosphorus in CKD patients.
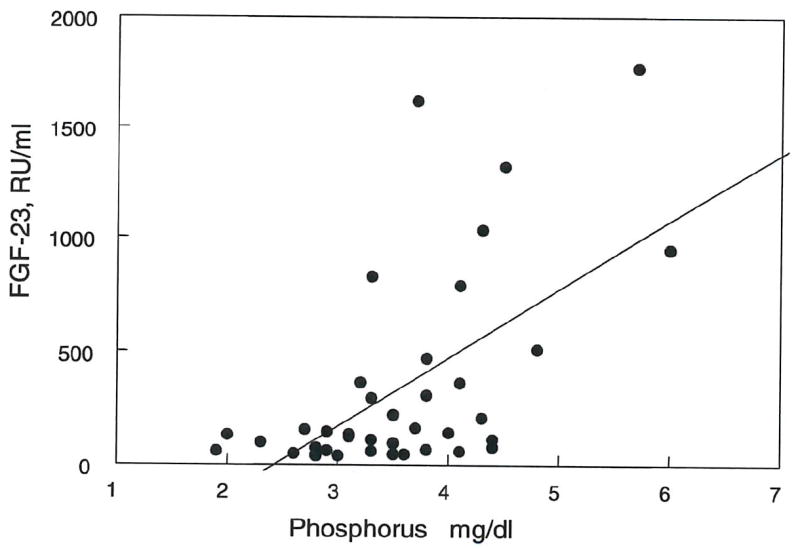
A positive correlation was seen between FGF-23 levels and plasma phosphorus (r2 = 0.448, p < 0.001).
Figure 5. Correlation between FGF-23 and plasma phosphorus in ESRD patients.
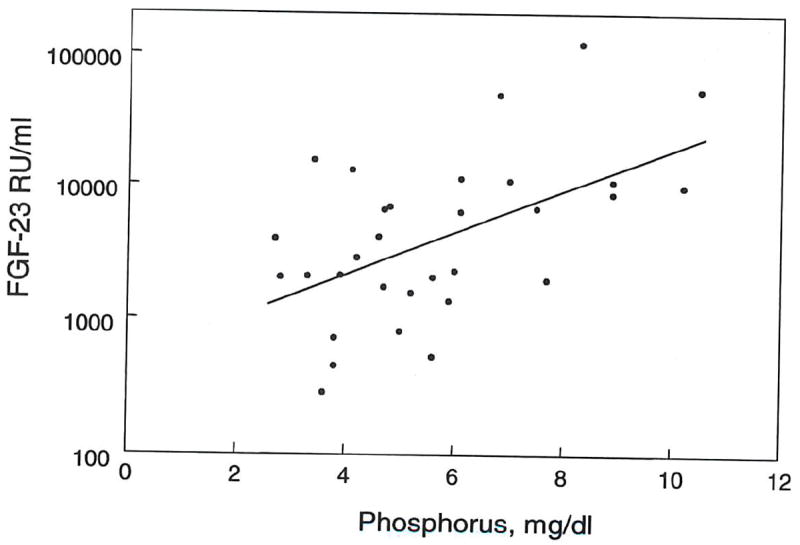
Log FGF-23 levels correlated positively with plasma phosphorus in CKD patients (r2 = 0.346, p < 0.0001, n = 30).
Figure 6. Correlations of FGF-23 with plasma phosphorus and stages of chronic kidney disease.
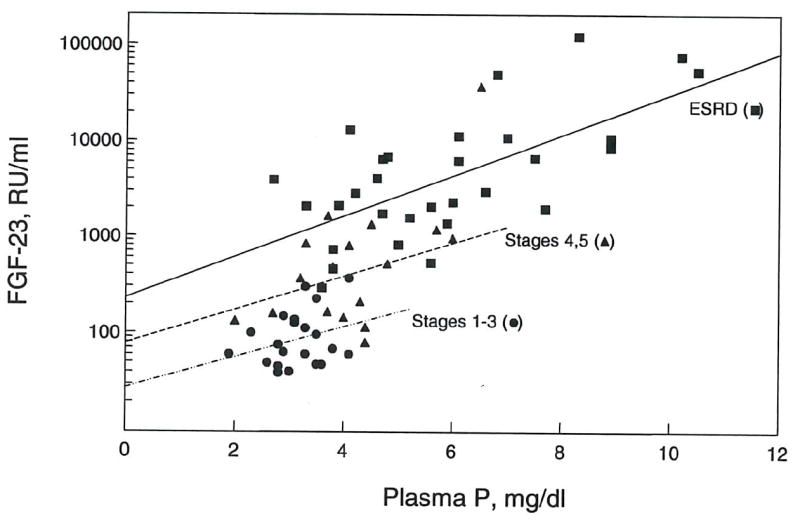
Correlations between FGF-23 and plasma phosphorus are shown for CKD stages 1-3 (r2 = 0.089, NS, n = 21), CKD stages 4-5 (r2 = 0.376, p < 0.01, n = 17) and ESRD (r2 = 0.448, p < 0.001, n = 30).
Most of the CKD patients had secondary hyperparathyroidism and were on phosphate binders and vitamin D analogs. Despite this, a positive correlation was seen between PTH and FGF-23 levels (r2 = 0.128, p = 0.02) (fig. 7). There was no correlation between plasma calcium and FGF-23, or between FGF-23 and 1,25(OH)2(D)3 concentrations (data not shown).
Figure 7. Correlation between plasma FGF-23 and PTH.
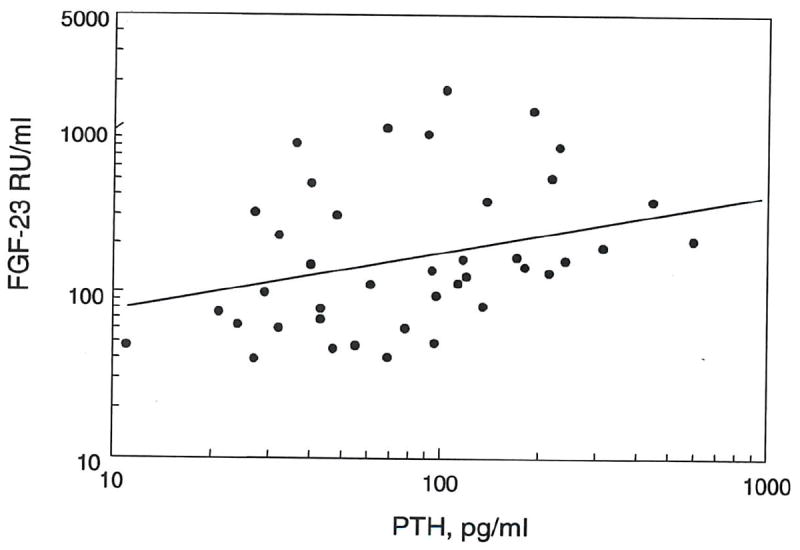
Log FGF-23 levels correlated positively with PTH levels in CKD patients (r2 = 0.128, p = 0.02).
Effect of Modulation of Phosphorus in ESRD patients
In ESRD patients receiving phosphate binders to control serum phosphorus levels, cessation of binder therapy for one to two weeks increased plasma phosphorus and produced a significant increase in FGF-23 levels (fig. 8). The decrease in plasma phosphorus with reinstatement of phosphate binders was accompanied by a decline in FGF-23 concentrations by two weeks in 4 of the 6 patients studied, but the reduction did not reach statistical significance (fig. 8). There were no changes in sFRP-4 in response with binder therapy (data not shown).
Figure 8. Effects of stopping and reinstating phosphate binders on plasma FGF-23 levels in ESRD patients.
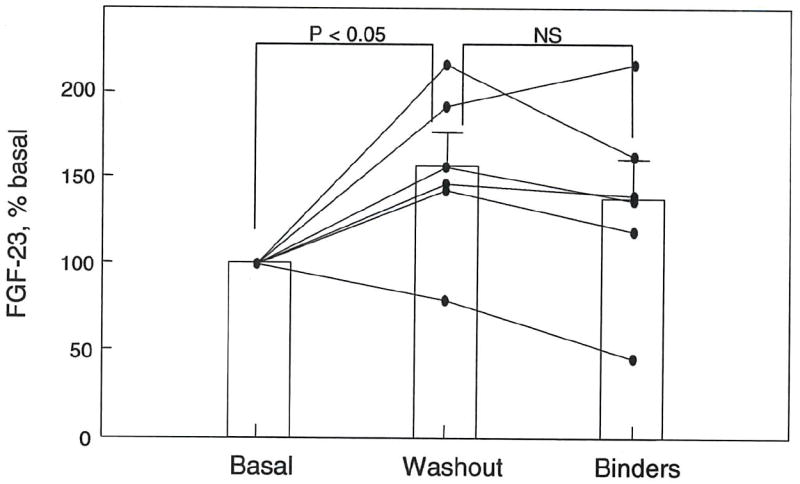
Basal levels of FGF-23 and plasma phosphorus were determined in hemodialysis patients (n = 6) taking phosphate binders to control hyperphosphatemia. Binder therapy was stopped, and FGF-23 levels were measured when plasma phosphorus levels increased to greater than 5.0 mg/dl (1-2 weeks; Washout). Binder therapy was then resumed, and FGF-23 levels were measured when plasma phosphorus fell to less than 4.5 mg/dl (1-2 weeks; Binders). All data are expressed as % basal and plotted as mean ± S.E.M.
FGF-23 in Renal Transplantation
In the transplant patients, 2 patients had delayed graft function and were excluded from the analysis. In the patients with functioning grafts, plasma creatinine decreased by an average of 76.6% and FGF-23 concentrations declined by 89% (fig. 9A). A 66% reduction in plasma phosphorus was seen in the 10 patients with functioning grafts (fig. 9B). Those patients with post transplant hypophosphatemia (defined as plasma P ≤ 2.0 mg/dl) (n = 7) were analyzed further. It should be noted that patients were given phosphorus supplements if their plasma phosphorus fell below 1.5 mg/dl. In these hypophosphatemic patients, plasma phosphorous correlated negatively with FGF-23 concentrations (r2 = 0.661, p <0.05) (fig. 10A). No correlation was observed between post-transplant PTH and post-transplant hypophosphatemia (fig. 10B).
Figure 9. Changes in FGF-23 and plasma phosphorus with kidney transplantation.
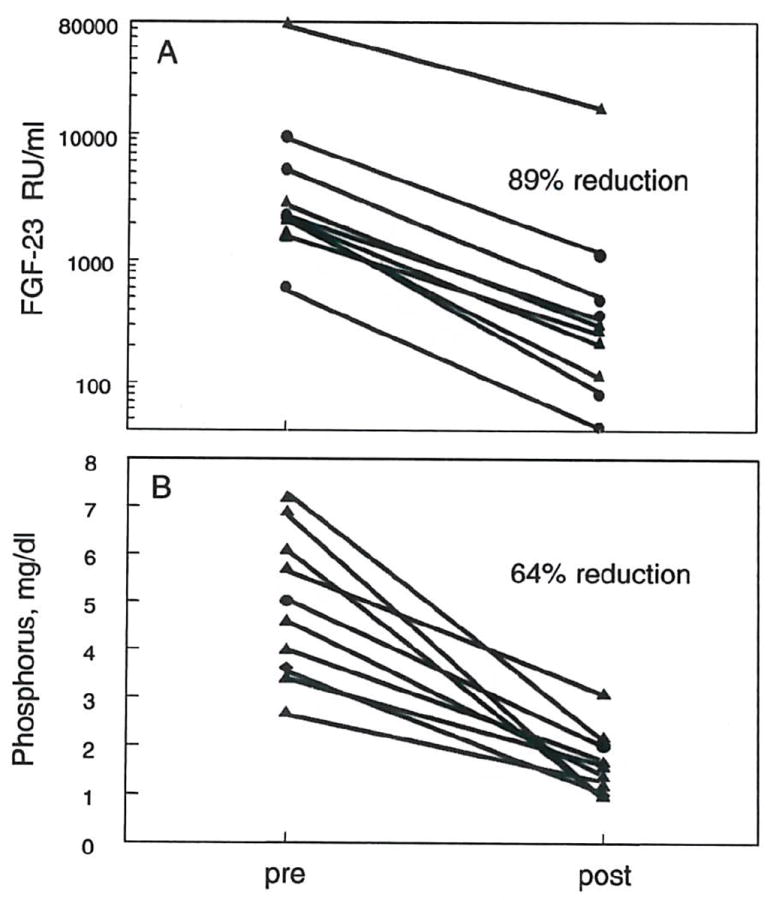
Plasma levels of FGF-23 (A) and phosphorus (B) were measured within one day of renal transplant (pre) and 4 to 5 days post-transplant (post). Data are shown for individual patients. FGF-23 and plasma phosphorus decreased by 89% and 64%, respectively.
Figure 10. Plasma phosphorus in post-transplant hypophosphatemia correlates with FGF-23, but not PTH.
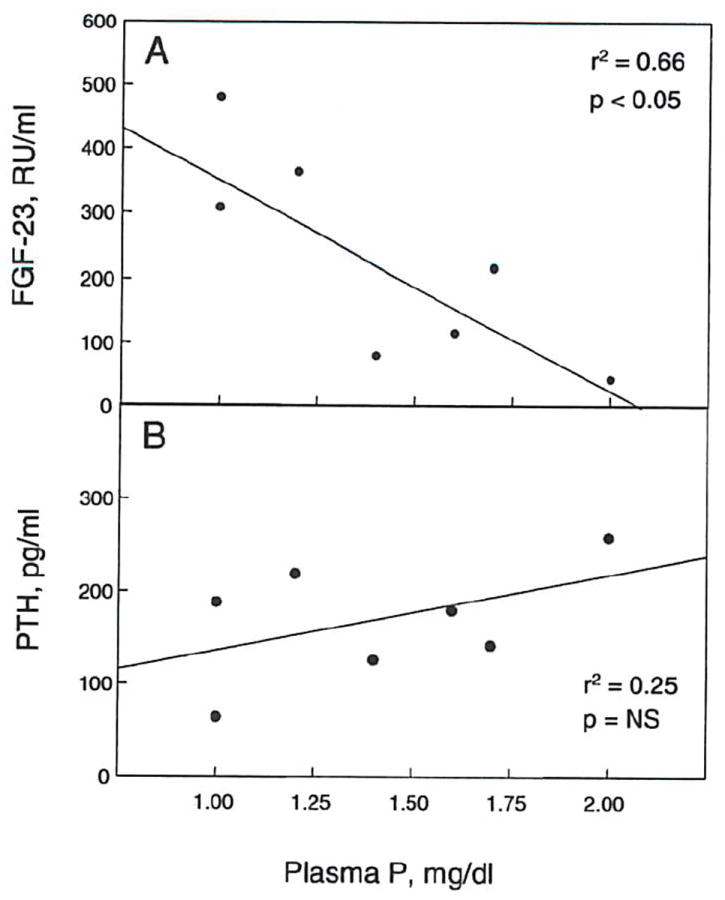
In patients with post transplantation hypophosphatemia (plasma phosphorus ≤ 2 mg/dl), a correlation was observed between plasma phosphorus and FGF-23 levels (r2 = 0.66, P < 0.05), but not PTH (r2 = 0.250, p = NS) (n = 7).
sFRP-4 in Renal Disease and Transplantation
In the control group, sFRP-4 levels were 22.3 ± 10.5 ng/ml. In patients with chronic kidney disease, sFRP-4 levels did not correlate with creatinine clearance (r2 = 0.0026, NS) or with plasma phosphorus (r2 = 0.046, NS), FGF-23 (r2 = 0.0097, NS) or calcium (r2 = 0.0018, NS) (fig. 11). When patients were stratified according to different stages of chronic kidney disease, based on kidney disease outcomes quality initiative (K DOQI) guidelines, sFRP-4 did not increase with worsening renal function. In stages 4-5 of CKD (creatinine clearance less than 30 ml/min) many patients had sFRP-4 concentrations that fell within the normal range. Additionally, there was no statistical difference in the pre- and post-transplant sFRP-4 concentrations.
Figure 11. Lack of correlation of sFRP4 levels with phosphorus, creatinine clearance, FGF-23 or calcium in CKD patients.
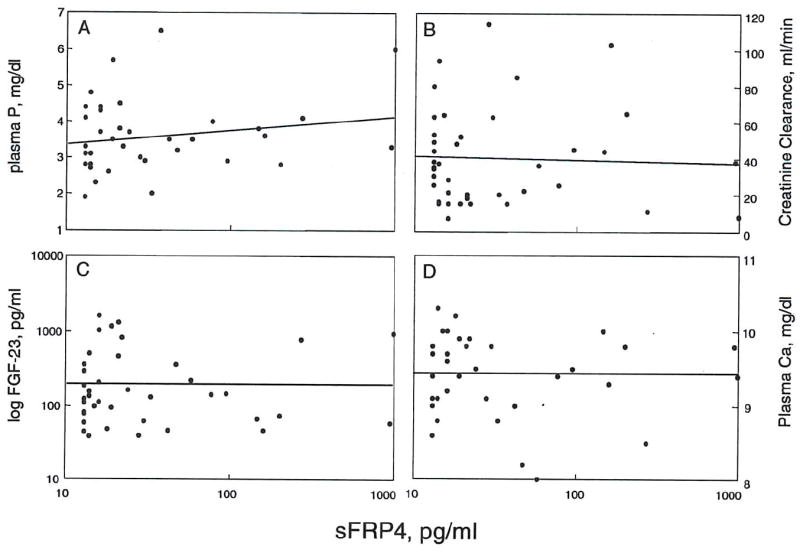
Regression analysis revealed no statisticallv significant relationships of FRP-4 levels with (A) plasma phosphorus (r2 = -0.003, NS). (B) creatinine clearance (r2 = .046, NS). (C) FGF-23 (r2 = 0.010, NS) or (D) calcium (r2 = -0.002, NS). (n = 41).
DISCUSSION
Secondary hyperparathyroidism is commonly associated with chronic kidney disease and develops in response to a combination of hypocalcemia, hyperphosphatemia and decreased 1,25(OH)2D3. The elevated PTH levels drive an increase in renal phosphorus excretion, thereby countering the rising levels of plasma phosphate. Our data show that FGF-23 has a positive correlation with plasma phosphate in chronic kidney disease and ESRD (figs. 4-6), consistent with recruitment of this phosphatonin to aid in phosphate excretion. Studies in experimental animals and in some, but not all, studies in humans indicate that phosphate loading and hyperphosphatemia can increase FGF-23 levels.
A positive correlation was also observed between FGF-23 and PTH concentrations (fig. 7). Interestingly FGF-23 has been shown to impair synthesis of 1,25(OH)2D3 through inhibition of 1α-hydroxylase (24). It is therefore possible that rising FGF-23 levels are responsible for the decline of 1,25(OH)2D3, and the ensuing hypocalcemia and hyperparathyroidism. Our study, however, could not establish any correlation between FGF-23 levels and 1,25(OH)2D levels, likely attributable to the many variables that can influence 1,25(OH)2D in renal patients.
The functional significance of the correlation between PTH and FGF-23 is not clear. A role for PTH in the regulation of FGF-23 has been suggested by a study demonstrating that parathyroidectomy of uremic rats blocks the increase in FGF-23 in response to a high phosphorus diet, but, curiously, infusion of the uremic rats with PTH could not restore the response, suggesting that perhaps other secreted factors from the parathyroid glands may be involved (Hasegawa et al, J Am Soc Nephrol 14:208A 2003, abstract). Parathyroidectomy has recently been reported to reduce FGF-23 in patients with kidney disease (55).
Our CKD population spanned all the stages of kidney disease (per K DOQI guidelines) and was representative of the most common causes of kidney disease. FGF-23 levels were found to vary inversely with creatinine clearance in chronic kidney disease with levels rising as kidney disease progressed from stages one to five (figs.1 and 2). At a creatinine clearance of more than 60ml/min, which equates to stages one and two, FGF-23 levels were normal. These findings are in agreement with those in several recently-published studies (53-55).
FGF-23 is an approximately 26-kDa protein, and so would not be cleared by dialysis. It would be expected then that increasing years on dialysis should lead to accumulation of FGF-23. Although our data showed a trend to for increased FGF-23 levels with years on dialysis, the relationship did not reach significance (fig. 3). On the other hand, such this relationship was found to be significant in the recent study of Imanishi et al (53) using a larger patient population. Our observation that FGF-23 levels fall rapidly following renal transplantation would be consistent with renal clearance of this phosphatonin, but this remains to be directly established. Other mechanisms for FGF-23 turnover are also possible.
The accumulation of FGF-23 with duration of dialysis may be prevented by the medications being used to control phosphate absorption. We found that stopping phosphate binders significantly increased FGF-23 within two weeks. Five of six patients showed an increase in FGF-23 when binder administration was halted and plasma phosphorus rose to greater than 5 mg/dl. The mean increase was 55 ± 19% (p < 0.05). Reinstating binder therapy until plasma phosphorus fell to less than 4.5 mg/dl led to a reduction in FGF-23 levels that was not statistically different from either the washout values or from the basal values (fig. 8). A larger patient population may be necessary to observe these changes, but our results indicate regulation of FGF-23 in dialysis patients. This finding is in agreement with the animal data showing regulation of FGF-23 by dietary phosphate (26-28), although studies in humans have produced disparate results (29,30). It is expected that the impaired phosphate handling in patients with kidney disease would accentuate the effects of phosphate loading.
Renal osteodystrophy seen in chronic kidney disease persists after kidney transplantation. Post-transplant bone disease has been attributed to a variety of factors, including hyperparathyroidism, pre-transplant bone disease, steroids, immunosuppressive medications and hypophosphatemia (56-59).
Hypophosphatemia is often observed following renal transplantation; its etiology is attributed to PTH-dependent and independent mechanisms. The PTH-independent mechanisms have been ascribed to corticosteroids, cyclosporine, and tubular dysfunction. Green et al demonstrated the presence of an unknown humoral factor in the plasma of transplanted patients, which leads to decreased NaPi activity in OK cells (59). We hypothesized that the large amount of FGF-23 that accumulates pre-transplantation is not cleared immediately post transplantation, and may drive the phosphate leak. Our results showed that all transplanted patients with good renal function (plasma creatinine less than 2.0 mg/dl), became hypophosphatemic. The hypophosphatemia was seen as early as 2 or 3 days post-transplant, and many of these patients were on phosphate supplements by the time their FGF-23 was measured on the 5th post-transplant day. After transplantation, there was an 89% and 64% drop in the FGF-23 and phosphorus levels, respectively (figs. 9A and 9B). Post-transplantation hypophosphatemia (plasma P < 2 mg/dl) correlated inversely with FGF-23 levels (fig. 10A), indicating that elevated FGF-23 likely contributes to the post-transplant hypophosphatemia. Other factors, including PTH, may also be involved, although we did not observe a correlation of PTH with post-transplantation phosphorus levels in our study (fig 10B).
Secreted frizzled-related protein-4 (sFRP-4) belongs to a family of soluble proteins that have a frizzled-like cysteine-rich domain and function as modulators of Wnt-Fz signals. Recent studies have shown that sFRP-4 is a phosphatonin (16,31). Infusion of recombinant sFRP-4 led to phosphaturia in rats, but did not produce an expected rise in 1,25(OH)2D3 levels (31).
sFRP-4 is phosphaturic, but its role in phosphate homeostasis is unclear. We were unable to demonstrate any relationship between plasma sFRP-4 levels and plasma phosphate, and, unlike FGF-23, sFRP-4 did not increase with progressive renal failure, suggesting that sFRP-4 is not cleared by the kidney. Modulating plasma phosphate by ceasing and reinstating phosphate binders also did not alter sFRP-4. Further studies will be required to determine the factors that regulate sFRP-4.
In conclusion, plasma FGF-23 appears to be cleared by the kidney, with levels increasing with progressive renal failure. However, the lack of correlation with years on dialysis would indicate a complex control of FGF-23 synthesis and degradation. Our findings show a correlation of FGF-23 with plasma phosphate in patients with kidney disease and an elevation in FGF-23 when phosphate levels are allowed to increase by withdrawal of phosphate binders, supporting the notion that FGF-23 plays a key role in phosphate homeostasis. Recovery of renal function post-transplantation leads to a rapid clearance of the FGF-23, but correlation with plasma phosphate suggests that residual FGF-23 may contribute to the post-transplant hypophosphatemia and bone disease.
Acknowledgments
This research was supported by a grant from the National Institutes of Health grants DK53774 and 5P30DK56341 (AJB).
References
- 1.Pullman TN, Lavender AR, Aho I, Rasmussen H. Direct renal action of a purified parathyroid extract. Endocrinology. 1960;67:570–582. doi: 10.1210/endo-67-5-570. [DOI] [PubMed] [Google Scholar]
- 2.Kurnik BR, Hruska KA. Effects of 1,25 dihydroxycholecalciferol on phosphate transport in Vitamin D deprived rats. Am J Physiol. 1984;247:F 177–184. doi: 10.1152/ajprenal.1984.247.1.F177. [DOI] [PubMed] [Google Scholar]
- 3.Kumar R. Vitamin D and the kidney. In: Feldman D, Glorieux FH, Pike JW, editors. Vitamin D (eds) 1. San Diego: Academic Press; 1997. pp. 275–292. [Google Scholar]
- 4.Murer H, Hernando N, Forster I, Biber J. Molecular mechanisms in proximal tubular and small intestinal phosphate reabsorption. Mol Membrane Biol. 2001;18:3–11. doi: 10.1080/09687680010019357. [DOI] [PubMed] [Google Scholar]
- 5.Martinez I, Saracho R, Montenegro J, Llach F. The importance of dietary calcium and phosphorus in the secondary hyperparathyroidism of patients with early renal failure. Am J Kidney Dis. 1997;29:496–502. doi: 10.1016/s0272-6386(97)90330-9. [DOI] [PubMed] [Google Scholar]
- 6.Qunibi WY, Nolan CA, Ayus JC. Cardiovascular calcification in patients with end-stage renal disease: A century-old phenomenon. Kidney Int. 2002;82(Suppl):73–80. doi: 10.1046/j.1523-1755.62.s82.15.x. [DOI] [PubMed] [Google Scholar]
- 7.Lindner A, Charra B, Sherrard DJ, Scribner BH. Accelerated atherosclerosis in prolonged maintenance dialysis. N Eng J Med. 1974;290:697–701. doi: 10.1056/NEJM197403282901301. [DOI] [PubMed] [Google Scholar]
- 8.Budisavljevic MN, Cheek D, Ploth DW. Calciphylaxis in chronic renal failure. J Am Soc Nephrol. 1996;7:978–82. doi: 10.1681/ASN.V77978. [DOI] [PubMed] [Google Scholar]
- 9.Coates T, Kirkland GS, Dymock RB, Murphy BF, Brealey JK, Mathew TH, Disney AP. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:514–518. doi: 10.1053/ajkd.1998.v32.pm9740153. [DOI] [PubMed] [Google Scholar]
- 10.Slatapolsky E, Finch J, Denda M, Ritter C, Zhong M, Dusso A, MacDonald PN, Brown AJ. Phosphorus restriction prevents parathyroid gland growth. High phosphorus directly stimulates PTH secretion growth in vitro. J Clin Invest. 1996;97:2534–2540. doi: 10.1172/JCI118701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonnson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. Fibroblast growth factor 23 in oncogenic osteomalcia and X linked hypophosphatemia. N Eng J Med. 2003;348:1656–1663. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 12.Schiavi SC, Kumar R. The phosphatonin pathway: New insights in phosphate homeostasis. Kidney Int. 2004;65:1–14. doi: 10.1111/j.1523-1755.2004.00355.x. [DOI] [PubMed] [Google Scholar]
- 13.Silve C, Beck L. Is FGF-23 the long sought after phosphaturic factor phosphatonin? Nephrol Dial Transplant. 2002;17:958–961. doi: 10.1093/ndt/17.6.958. [DOI] [PubMed] [Google Scholar]
- 14.Fukomoto S, Yamashita T. Fibroblast growth factor 23 is the phosphaturic factor in tumor induced osteomalacia and may be phosphatonin. Curr Opin Nephrol Hypertens. 2002;11:385–389. doi: 10.1097/00041552-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Schiavi SC, Moe OW. Phosphatonins: a new class of phosphate regulating proteins. Curr Opin Nephrol Hypertens. 2002;11:423–430. doi: 10.1097/00041552-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Kumar R. New insights into phosphate homeostasis: fibroblast growth factor 23 and frizzled-related protein-4 are phosphaturic factors derived from tumors associated with osteomalacia. Curr Opin Nephrol Hypertens. 2002;11:547–553. doi: 10.1097/00041552-200209000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Tenenhouse HS, Murer H. Disorders of renal tubular phosphate transport. J Am Soc Nephrol. 2003;14:240–248. doi: 10.1097/01.asn.0000045045.47494.71. [DOI] [PubMed] [Google Scholar]
- 18.Kumar R. Phosphatonin-a new phosphaturetic hormone? (lessons from tumor induced osteomalacia and X-linked hypophosphatemia) Nephrol Dial Transplant. 1997;12:11–13. doi: 10.1093/ndt/12.1.11. [DOI] [PubMed] [Google Scholar]
- 19.Econs MJ, Drezner MK. Tumor-induced osteomalacia-unveiling a new hormone. N Engl J Med. 1994;330:1679–1681. doi: 10.1056/NEJM199406093302310. [DOI] [PubMed] [Google Scholar]
- 20.Drezner M. PHEX gene and hypophosphatemia. Kidney Int. 2000;57:9–18. doi: 10.1046/j.1523-1755.2000.00807.x. [DOI] [PubMed] [Google Scholar]
- 21.Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto K, Fukushima N. Human fibroblast growth factor-23 mutants suppress Na+- dependent phosphate co-transport activity and 1α, 25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278:2206–2211. doi: 10.1074/jbc.M207872200. [DOI] [PubMed] [Google Scholar]
- 22.Larsson T, Marsell R, Shipani E, Ohisson C, Ljunggren O, Tenenhouse HS, Juppner H, Honsson KB. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1 (I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 23.Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004;314:409–414. doi: 10.1016/j.bbrc.2003.12.102. [DOI] [PubMed] [Google Scholar]
- 24.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sitara D, Razzaque MS, Hesse M, Yaganathan S, Taguchi T, Erben RG, Juppner H, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2003;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 27.Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N. Circulating FGF-23 is regulated by 1α,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280:2543–2549. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 28.Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto KI. Vitamin D and phosphate regulate fibroblast growth factor-23 in K562 cells. Am J Physiol Endocrinol Metab. doi: 10.1152/ajpendo.00502.2004. in press. [DOI] [PubMed] [Google Scholar]
- 29.Gupta A, Winer K, Econs MJ, Marx SJ, Collins MT. FGF-23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492. doi: 10.1210/jc.2004-0724. [DOI] [PubMed] [Google Scholar]
- 30.Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 31.Theresa Berndt T, Craig T, Bowe A, Vassiliadis J. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112:785–794. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rowe PS, Kumagai Y, Guttierez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR. MEPE has properties of an osteoblastic phosphatonin and minhibin. Bone. 2004;34:303–319. doi: 10.1016/j.bone.2003.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson DN, Tkalcevic GT, Mansolf AL, Rivera-Gonzalez R, Brown TA. Identification of osteoblast/osteocyte factor 45 (OF45), a bone-specific cDNA encoding an RGD-containing protein that is highly expressed in osteoblasts and osteocytes. J Biol Chem. 2000;275:36172–36180. doi: 10.1074/jbc.M003622200. [DOI] [PubMed] [Google Scholar]
- 34.Gowen L, Peterson DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, McNeish JD, Brown TA. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278:1998–2007. doi: 10.1074/jbc.M203250200. [DOI] [PubMed] [Google Scholar]
- 35.Bowe AE, Finnegan R, Jan de Beur SM, Cho J, Levine MA, Kumar R, Schiavi SC. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun. 2001;284:977–981. doi: 10.1006/bbrc.2001.5084. [DOI] [PubMed] [Google Scholar]
- 36.Liu S, Guo R, Tu Q, Quarles D. Overexpression of Phex in osteoblasts fails to rescue the hyp mouse phenotype. J Biol Chem. 2002;277:3686–3697. doi: 10.1074/jbc.M107707200. [DOI] [PubMed] [Google Scholar]
- 37.Rowe PS. The molecular background to hypophosphatemic rickets. Arch Dis Child. 2000;83:192–194. doi: 10.1136/adc.83.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jan de Beur SM, Levine MA. Molecular pathogenesis of hypophosphatemic rickets. J Clin Endocrinol. 2002;87:2467–2473. doi: 10.1210/jcem.87.6.8688. [DOI] [PubMed] [Google Scholar]
- 39.Econs MJ, McEnery PT, Lennon F, Speer MC. Autosomal dominant hypophosphatemic rickets is linked to chromosome 12p13. J Clin Invest. 1997;100:2653–2657. doi: 10.1172/JCI119809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quarles LD, Drezner MK. Editorial: Pathophysiology of X-linked hypophosphatemia, tumor induced osteomalacia and autosomal dominant hypophosphatemia: A perPHEXing problem. J Clin Endocrinol Metab. 2000;86:494–496. doi: 10.1210/jcem.86.2.7302. [DOI] [PubMed] [Google Scholar]
- 41.Cai Q, Hodgson SF, Kao PC, Lennon VA, Klee GG, Zinsmiester AR, Kumar R. Brief report: inhibition of renal phosphate transport by a tumor product in a patient with oncogenic osteomalacia. N Engl J Med. 1994;330:1645–1649. doi: 10.1056/NEJM199406093302304. [DOI] [PubMed] [Google Scholar]
- 42.Wilkins GE, Granleese S, Hegele S, Holden J, Anderson DW, Bondy GP. Oncogenic osteomalacia: evidence for a humoral phosphaturic factor. J Clin Endocrinol Metab. 1995;80:1628–1634. doi: 10.1210/jcem.80.5.7745010. [DOI] [PubMed] [Google Scholar]
- 43.De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY, Kumar R, Levine MA, Schiavi SC. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res. 2002;17:1102–1110. doi: 10.1359/jbmr.2002.17.6.1102. [DOI] [PubMed] [Google Scholar]
- 44.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF-23 as a causative factor of tumor induced osteomalacia. Proc NatI Acad Sci USA. 2001;98:6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000;67:54–68. doi: 10.1006/geno.2000.6235. [DOI] [PubMed] [Google Scholar]
- 46.Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S. Increased circulatory level of biologically active full length FGF-23 in patients with hypophosphatemic rickets/osteomalcia. J Clin Endocrinol Metab. 2002;87:4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 47.Nelson AE, Bligh RC, Mirams M, Gill A, Au A, Clarkson A, Juppner H, Ruff S, Stalley P, Scolyer RA, Robinson BG, Mason RS, Bligh PC. Fibroblast growth factor-23: a new clinical marker for oncogenic osteomalcia. J Clin Endocrinol Metab. 2003;88:4088–4094. doi: 10.1210/jc.2002-021919. [DOI] [PubMed] [Google Scholar]
- 48.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23 preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophsy Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 49.The ADHR consortium. Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF-23. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 50.White KE, Jonsson KB, Cam G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren O, Meitinger T, Strom TM, Juppner H, Econs MJ. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86:497–500. doi: 10.1210/jcem.86.2.7408. [DOI] [PubMed] [Google Scholar]
- 51.Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Mutant FGF-23 is responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179–3182. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- 52.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imanishi Y, Inaba M, Nakatsuka K, Nagasue K, Okuno S, Yoshihara A, Miura M, Miyauchi A, Kobayashi K, Miki T, Shoji T, Ishimura E, Nishizawa Y. FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int. 2004;65:1943–1946. doi: 10.1111/j.1523-1755.2004.00604.x. [DOI] [PubMed] [Google Scholar]
- 54.Shigematsu T, Kazama JJ, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, Fukugawa M. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis. 2004;44:250–256. doi: 10.1053/j.ajkd.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 55.Sato T, Tominaga Y, Ueki T, Goto N, Matsuoka S, Katayama A, Haba T, Uchida K, Nakanishi S, Kazama JJ, Gejyo F, Yamashita T, Fukugawa M. Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am J Kidney Dis. 2004;44:481–487. [PubMed] [Google Scholar]
- 56.Rosenbaum RW, Hruska KA, Korkor A, Anderson C, Slatopolsky E. Decreased phosphate reabsorption after renal transplantation. Evidence for a mechanism independent of calcium and parathyroid hormone. Kidney Int. 1981;19:568–578. doi: 10.1038/ki.1981.54. [DOI] [PubMed] [Google Scholar]
- 57.Levi M. Post-transplant hypophosphatemia. Kidney Int. 2001;59:2377–2387. doi: 10.1046/j.1523-1755.2001.00755.x. [DOI] [PubMed] [Google Scholar]
- 58.Better OS. Tubular dysfunction following kidney transplantation. Nephron. 1980;25:209–213. doi: 10.1159/000181840. [DOI] [PubMed] [Google Scholar]
- 59.Green J, Debby H, Lederer E, Levi Moshe, Zajicek Hubert K, Bick Tova. Evidence for a PTH-independent humoral mechanism in post-transplant hypophosphatemia and phosphaturia. Kidney Int. 2001;60:1182. doi: 10.1046/j.1523-1755.2001.0600031182.x. [DOI] [PubMed] [Google Scholar]
- 60.Rojas E, Carlini R, Clesca P, Arminio Anabella, Suniaga Orlando, De Elguezabal Karen, Weisinger Jose R, Hruska Keith A, Bellorin-Font Ezequiel. The pathogenesis of osteodystrophy after renal transplantation as detected by early alterations in bone remodeling. Kidney Int. 2003;63:1915–1923. doi: 10.1046/j.1523-1755.2003.00938.x. [DOI] [PubMed] [Google Scholar]
- 61.Reinhardt TA, Horst RL, Orf JW, Holllis BW. A microassay for 1,25dihydroxyvitamin D not requiring high performance liquid chromatography application to clinical studies. J Clin Endocrinol Metab. 1984;58:91–98. doi: 10.1210/jcem-58-1-91. [DOI] [PubMed] [Google Scholar]
- 62.Hollis BW. Assay of circulating 1,25-dihydroxyvitamin D involving a novel cartridge extraction and purification procedure. Clinical Chemistry. 1986;32:2060–2063. [PubMed] [Google Scholar]