

Abstract
Combined deficiency of factor V (FV) and FVIII (F5F8D) is an autosomal recessive bleeding disorder characterized by simultaneous decreases of both coagulation factors. This review summarizes recent reports on the clinical presentations, treatments, and molecular mechanism of F5F8D. Genetic studies identified LMAN1 and MCFD2 as causative genes for this disorder, revealing a previously unknown intracellular transport pathway shared by the two important blood coagulation factors. LMAN1 and MCFD2 form a Ca2+-dependent cargo receptor complex that functions in the transport of FV/FVIII from the endoplasmic reticulum (ER) to the Golgi. Disrupting the LMAN1-MCFD2 receptor, complex formation is the primary molecular defect of missense mutations leading to F5F8D. The EF-hand domains of MCFD2 are necessary and sufficient for the interactions with both LMAN1 and FV/FVIII. Similarly, the carbohydrate recognition domain of LMAN1 contains distinct and separable binding sites for both MCFD2 and FV/FVIII. Therefore, FV and FVIII likely carry duel sorting signals that are separately recognized by LMAN1 and MCFD2 and necessary for the efficient ER-to-Golgi transport. FV and FVIII likely bind LMAN1 through the high-mannose N-linked glycans under the higher Ca2+ conditions in the ER and dissociate in the lower Ca2+ environment of the ER–Golgi intermediate compartment.
Keywords: hemorrhagic disorders, ERGIC, protein trafficking, carbohydrate binding, calcium binding
Combined deficiency of factor V (FV) and FVIII (F5F8D, OMIM 227300) is a rare bleeding disorder first reported in a pair of Swiss siblings in 1954.1 Since then, there have been over 200 cases reported in the literature. As per other rare autosomal recessive disorders, F5F8D is often associated with consanguinity. Although found all over the world, F5F8D cases are most concentrated in the Mediterranean, Middle Eastern, and South Asian countries, likely due to the prevalence of consanguineous marriages in these regions. F5F8D is characterized by simultaneous decreases of plasma FV and FVIII to 5 to 30% of normal and mild-to-moderate bleeding symptoms. The molecular basis of F5F8D was discovered at the turn of the last century to be mutations in either LMAN1 (lectin mannose binding, 1) or MCFD2 (multiple coagulation factor deficiency gene 2).2,3 Proteins encoded by these two genes function in a novel intracellular trafficking pathway required for the efficient secretion of FV and FVIII. This review provides an update on recent progress toward the clinical and mechanistic understanding of F5F8D since the last comprehensive reviews on this topic.4,5
Symptoms and Diagnosis
Symptoms of F5F8D are generally mild. Comparison of relatively large cohorts of F5F8D in India, Iran, and Israel indicates that bleeding from trauma/surgery is the most frequently reported clinical manifestation.6–9 This observation likely reflects the fact that often F5F8D is brought to the attention of physicians following excessive bleeding during and after trauma, surgery, and labor. Common spontaneous bleeding symptoms include epistaxis, gum bleeding, easy bruising, and menorrhagia. Less frequently reported are hemarthroses, gastrointestinal bleeding, hematuria, and intracranial bleeding. In males, bleeding due to circumcision is frequently reported in regions where it is commonly practiced. In females, menorrhagia is found in a majority of patients in all reported studies. There appears to be some differences in the prevalence of the types of spontaneous bleeding symptoms between different regions.6 The reasons for this discrepancy are unclear. Recent reports also confirmed the previous observation that defects of two factors do not lead to more severe bleeding symptoms than single defect of FV or FVIII of similar degree.6,10
F5F8D is suspected in bleeding patients with prolonged prothrombin time and activated partial thromboplastin time. Diagnosis is made on laboratory findings of decreased FV and FVIII levels in plasma, usually in the range of 5 to 30% of normal (typically 10–20%). No correlations of bleeding severity and the levels of FV and FVIII have been observed in F5F8D patients. Although most F5F8D diagnosis occurred in children and adults, a successful diagnosis of F5F8D was reported in a 2-day-old infant with spontaneous cephalhematoma.11 The 9-year-old sibling of the proband had been symptom free since birth, despite laboratory findings of F5F8D,11 highlighting the variability of bleeding symptoms and possibly contributing to the underdiagnosis of this disorder. FV deficiency (parahemophilia) combined with type 1 von Willebrand disease can also present with decreases in FV and FVIII levels, due to the indirect effect of von Willebrand factor deficiency on FVIII stability. Although chance coinheritance of parahemophilia and hemophilia A is a possibility, it is extremely unlikely due to the low frequency of both disorders in general population (1/1,000,000 for parahemophilia and 1/5,000 in males for hemophilia A). Ultimate confirmation of F5F8D comes from the identification of mutations in either LMAN1 or MCFD2. However, no routine genetic testing is currently available for F5F8D. Mutation analysis is done on a research basis in several medical centers. Fortunately, clinical management of F5F8D does not rely on molecular diagnosis.
Disease Management
Because of the mild-to-moderate bleeding symptoms, treatment is on demand depending on the severity of bleeding. According to the guideline for rare coagulation disorders from the United Kingdom Haemophilia Centre Doctors’ Organization,12 the recommended therapy includes fresh frozen plasma (FFP), which provides FV, and FVIII concentrate, which compensates for the shorter half-life of plasma FVIII. This treatment regimen has been demonstrated to be effective in controlling bleeding during dental extraction,13 male circumcision,10 and preparation for labor in pregnant women.14 Desmopressin (DDAVP), a synthetic derivative of the antidiuretic hormone arginine vasopressin, was shown to increase the FVIII level by more than twofold over 60 to 120 minutes in a patient with F5F8D.15 In patients whose FVIII levels are at the higher end of the range (> 15% of normal), DDAVP could be used in lieu of FVIII concentrate for preventing bleeding in tooth extraction, labor, or other minor surgeries.14,15 FVIIa has been successfully used as a bypass therapy to treat hemophilia patients with inhibitors. In a case of F5F8D with persistent abdominal hematoma following surgery that combined FFP and FVIII concentrate infusions failed to control, FVIIa was successfully used to stop the bleeding complications.16 FVIIa treatment has the advantage over FFP infusion of avoiding fluid overload. However, the safety of off-label use of FVIIa in treating F5F8D remains to be established.
Percutaneous coronary intervention (PCI) in patients with congenital coagulation factor deficiencies presents a unique challenge. They are not only at increased risk of perioperative bleeding but can also suffer thrombosis of the stent. It has been postulated that in patients with classic hemophilia, platelet adhesion and aggregation are generally intact and therefore these patients suffer similar risks of rethrombosis.17,18 The use of antiplatelet agents during and after PCI is essential, given that thrombosis of stents is a significant source of mortality especially in the first few months following the procedure. Recently, successful PCI with bare metal stent placement was reported in a patient with F5F8D.19 Hemostasis was managed perioperatively with FFP and recombinant FVIII. Unfractionated heparin was used during the procedure for anticoagulation. Long-term prevention of thrombosis was achieved with antiplatelet agents, aspirin and clopidogrel, with good outcome. This suggests that patients with F5F8D can safely undergo PCI for coronary artery disease, with treatment individualized to the specific patient.
Mutations that Cause F5F8D
Mutations in LMAN1 and MCFD2 account for nearly all cases of F5F8D.20,21 LMAN1 is a 13-exon gene localized to chromosome 18q21. MCFD2 is a 4-exon gene localized to chromosome 2p21. At least 36 LMAN1 mutations and 18 MCFD2 mutations have been reported.19,20,22–25 Most of the mutations are insertion/deletion, nonsense, and splice site mutations that completely abolish the protein function.20 Mutations in the regulatory regions of the genes have also been reported, including a promoter deletion in LMAN1.26 Some recurrent mutations appear to arise independently in multiple ethnic backgrounds, for example, the c.149 + 5G > A mutation in MCFD2 and the p.R202X mutation in LMAN1, possibly due to mutation hotspots. Another notable mutation hotspot is a microsatellite repeat of 9-adenine in exon 8 of LMAN1. Three different mutations (c.912delA, c.912-913insA, and c.904A > T) have been identified at this location in patients of distinct origins.19,20,23 Some mutations are exclusively associated with certain genetic backgrounds, suggesting a founder effect. For example, the M1T (c.2T > C) mutation, which abolishes the initiation codon, is exclusively found in families of Italian origin. Likewise, two mutations in LMAN1 (c.89-90insG and c.1149 + 2T > G) account for all Jewish F5F8D patients. An unusual LMAN1 mutation was recently reported in a US patient; this is a homozygous single nucleotide transition (c.1083A > G) in exon 9 that does not change the underlying amino acid residue, but creates a new splice donor site.25 Analysis of RNA extracted from the immortalized lymphocytes indicates that this ectopic splice donor site completely displaces the weak natural splice donor site at the exon 9-intron 9 junction. The resulting alternatively spliced mRNA introduces a frameshift following exon 9 and a premature stop codon shortly after. Interestingly, the premature stop codon does not lead to nonsense-mediated decay (NMD), suggesting that LMAN1 belongs to a group of genes that are resistant to NMD.
Molecular Mechanism of F5F8D
F5F8D is among the growing number of genetic disorders caused by defects in the early secretory pathway,27 which refers to protein transport from the endoplasmic reticulum (ER) to the Golgi. Molecular studies of F5F8D provide insight into FV and FVIII trafficking in the early secretory pathway. FV and FVIII are two large plasma glycoproteins that share similar domain structures (A1-A2-B-A3-C1-C2), with high sequence identities between the A and C domains. Like other secreted proteins, FV and FVIII are synthesized on the rough ER and translocated into the ER lumen, where both proteins undergo N-linked glycosylation, proper folding, and quality control. FVIII is more prone to misfolding than FV and hence engages in more extensive interactions with chaperones in the ER. Proteins that have passed the quality control are packaged into coat protein complex-II (COPII) vesicles that bud from distinct regions of the ER.28 Upon budding, COPII vesicles undergo homotypic fusion to form the ER–Golgi intermediate compartment (ERGIC), a structure located between the ER and Golgi that is unique to higher eukaryotic cells.29 Accessory transport factors and proteins that display ER-retrieval signals are returned to the ER via coat protein complex-I (COPI) vesicles, where they can participate in subsequent rounds of vesicle formation.
How cargo proteins are recruited into COPII vesicles is not entirely clear. Although ER export by bulk flow (passive diffusion into budding vesicles) might be sufficient for the transport of some highly abundant cargo proteins, many other proteins are thought to be selectively packaged into export vesicles. This selective model of export envisions that cargo proteins display specific sorting signals that promote their incorporation into COPII vesicles.30,31 The cytoplasmic side of many transmembrane cargo proteins can directly bind components of the COPII coat. Some of these transmembrane proteins in turn serve as cargo receptors for both soluble and transmembrane proteins. However, cargo receptors have proven elusive to identify, with only a few characterized to date, mostly in yeast.30,31 In mammals, the LMAN1-MCFD2 complex is the first known example of a specific ER-to-Golgi cargo receptor, discovered largely through the studies of F5F8D. The requirement of both a transmembrane component (LMAN1) and a soluble cofactor (MCFD2) suggests a more sophisticated mechanism for cargo trafficking in higher eukaryotes. In addition to FV and FVIII, LMAN1 has also been shown to transport other cargo proteins such as cathepsin C, cathepsin Z, and α1-antitrypsin.32,33 However, MCFD2 appears to be dispensable for the transport of cathepsins C and Z, suggesting that it is specifically required for FV/FVIII transport.34
Organization of the LMAN1-MCFD2 Receptor Complex
LMAN1 (also called ERGIC-53) is a homohexameric 53-kD type-1 transmembrane protein cycling between the ER and the ERGIC/cis-Golgi. The short C-terminal cytosolic tail contains diphenylalanine ER exit motif that interacts with the COPII, and a dilysine ER retrieval motif that interacts with the COPI (Fig. 1). The ER luminal domain contains a carbohydrate recognition domain (CRD) responsible for ligand binding and a putative coiled-coiled domain required for oligomerization (Fig. 1). All three domains contribute to LMAN1 subcellular localization.35 MCFD2 is a 16-kDa soluble protein with an N-terminal sequence of unknown function and two calmodulin-like EF-hand domains at the C terminus (Fig. 1).3 LMAN1 and MCFD2 form a Ca2+-dependent complex with 1:1 stoichiometry.36 Localization of MCFD2 is dependent on the interaction with LMAN1 because it lacks ER retrieve signal and overexpression of MCFD2 results in MCFD2 secretion.36 The LMAN1-MCFD2 complex can directly interact with FV/FVIII, indicating LMAN1-MCFD2 is a cargo receptor for FV/FVIII.36 LMAN1 level is normal in patients with MCFD2 mutations,3 indicating that, despite meeting all the requirements for a cargo receptor, LMAN1 alone is not sufficient for FV/FVIII transport. In fact, patients with MCFD2 mutations tend to have lower FV/FIII levels,20 suggesting that MCFD2 is more directly involved in the cargo receptor function.
Fig. 1.
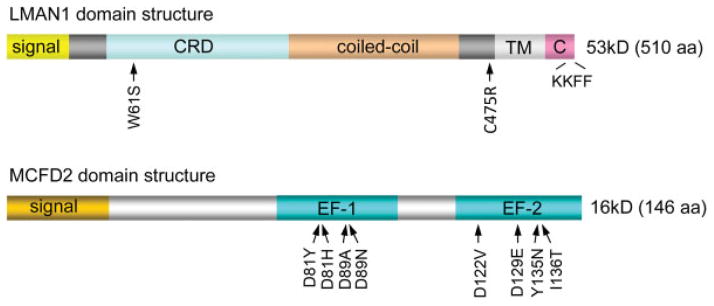
Domain structures of LMAN1 and MCFD2 and the locations of missense mutations identified in F5F8D patients. F5F8D, combined deficiency of FV and FVIII; TM, transmembrane domain. CRD, carbohydrate recognition domain.
All missense mutations of LMAN1 and MCFD2 found in F5F8D patients disrupt the interaction of the two proteins, indicating that the LMAN1-MCFD2 complex formation is critical for the receptor function. Recently, significant progress has been made in understanding the organization of this receptor complex, which is central to understanding this disease mechanism. We showed that the EF-hand domains of MCFD2 are necessary and sufficient to bind LMAN1.37 Deletion of all the N-terminal non-EF-hand sequence has no effect on LMAN1 binding. However, further deletions that disturb the EF-hand structure abolish LMAN1 binding. Domain deletion mutagenesis studies showed that the CRD of LMAN1 (LMAN1-CRD) is responsible for MCFD2 binding.38 The LMAN1-CRD has an overall globular shape that predominantly consists of several β sheets that form a β-sandwich. Deletion of the first β sheet disrupts MCFD2 binding without affecting mannose binding, indicating that the first β sheet of the CRD is the MCFD2-binding motif. On the contrary, point mutations in the ligand-binding pocket that abolish mannose and FV/FVIII binding have no effect on MCFD2 binding.38 Therefore, the LMAN1-CRD contains distinct, separable binding sites for both its partner protein (MCFD2) and the cargo proteins (FV/FVIII). These results were confirmed by the crystal structures of the LMAN1-CRD in complex with MCFD2 (Fig. 2A), reported by two independent groups.39,40 Consistent with the biochemical analysis, the crystal structures demonstrate the interaction of the first β sheet of the CRD with the EF-hand domains of MCFD2. The ligand-binding pocket is on the opposite side of the protein and distal to the LMAN1-MCFD2–binding interface (Fig. 2A). In addition, the LMAN1-CRD structure undergoes no significant changes after MCFD2 binding, whereas MCFD2 experiences considerable conformational changes after binding to LMAN1.39,40
Fig. 2.
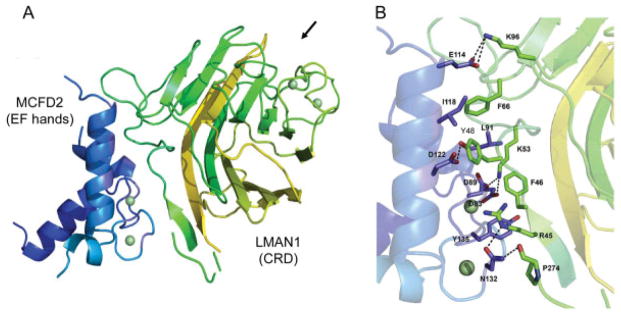
Crystal structure of MCFD2 (blue) in complex with the CRD of LMAN1 (green). (A) Overview of the LMAN1 (CRD)-MCFD2 complex. Arrow points to the mannose-binding pocket. (B) Details of the binding interface between MCFD2 and the CRD of LMAN1. Interacting residues are labeled. Hydrogen bonds are shown as dashed lines. Green spheres represent Ca2+. CRD, carbohydrate recognition domain.
Ca2+ plays a pivotal role in the interaction of LMAN1 with MCFD2.3,36 However, the structural effects of Ca2+ on LMAN1 and MCFD2 are different. Crystal structures of the LMAN1-CRD in the presence or absence of Ca2+ indicate that Ca2+ binding only causes localized conformational changes around the ligand-binding pocket.41 In contrast, Ca2+ is critical for overall MCFD2 tertiary structure formation and in the absence of Ca2+, MCFD2 assumes a disordered structure.42 The LMAN1-MCFD2 complex formation needs intact EF-hand domains of MCFD2. Therefore, the importance of Ca2+ in cargo receptor formation is manifested mainly through its role in maintaining the MCFD2 structure.
Missense mutations often provide clues to the important functional domains of proteins. Eight of the 18 MCFD2 mutations identified to date are missense mutations (Fig. 1). Combining biochemical and structural analyses, we can now explain the molecular defects of reported mis-sense mutations in MCFD2. As stated above, the EF-hand domains of MCFD2 are necessary and sufficient for LMAN1 binding. Two Ca2+ ions are the linchpins that hold the structure of the EF-hand domains. Most MCFD2 missense mutations (D81H, D81Y, D89A, D89N, D129E, and Y135N) change amino acid residues that are directly involved in coordinating Ca2+ binding and result in the collapse of MCFD2 tertiary structure.40,42 I136 is not directly involved in coordinating Ca2+, but it is located in the second EF-hand loop and involved in the formation of a hydrophobic core. Mutation of a hydrophobic isoleucine to a polar amino acid residue (I136T) will disrupt the EF-hand structure.42 The circular dichroism (CD) analysis demonstrated that all mutations located in the EF-hand loops disrupt the tertiary structure of MCFD2 to the extent that is similar to the Ca2+-free state.40,43 The D122V mutation is the only mutation located outside the Ca2+-binding loops, replacing a charged amino acid with a hydrophobic one on the first helix of the second EF-hand domain. Its CD spectrum is distinct from other mutants, suggesting minor structural disruptions.37,40 Crystal structures revealed details of the binding interface of LMAN1 with MCFD2 (Fig. 2B). Three amino acid residues of MCFD2 mutated in F5F8D are among those that are in direct contact with LMAN1. D122 forms a hydrogen bond with Y48 in LMAN1, which is an amino acid critical for the interaction because the Y48A mutation of LMAN1 completely abolishes MCFD2 binding.38 D89 forms a salt bridge with K53 in LMAN1 and Y135 packs with F46 in LMAN1.39,40
Only two missense mutations have been identified in LMAN1, including W67S and C475R. The W67S (c.200G > C) mutation was recently identified in a Japanese patient.44 This mutation is located in the second β sheet of the CRD of LMAN1, which is distal to the sugar-binding pocket of the protein. Biochemical analysis of W67S demonstrated that this mutant is unable to bind mannose or interact with MCFD2,38,44 suggesting that it compromises the structural integrity of the protein. The C475R mutation, which has been reported in compound heterozygous state in an Argentinian patient21 and homozygosity in an Italian patient,16 disrupts one of two membrane-proximal cysteines. A previous mutagenesis study showed that LMAN1 with the C475A mutation can dimerize but cannot form hexamers.35 However, LMAN1 in lymphoblasts derived from a patient carrying the C475R mutation failed to accumulate in cells, suggesting that this mutation destabilizes LMAN1.21 Replacing the cysteine 475 with a bulky arginine near the transmembrane domain may be more detrimental to the protein structure. Oligomerization of LMAN1 is essential for its function, as monomeric LMAN1 is defective in ER exit and unable to bind MCFD2 in vivo.38 Structural determination of the complex formed by full-length LMAN1 hexamers with MCFD2 promises to yield a better functional understanding of this cargo receptor in vivo. In summary, all missense mutations identified in LMAN1 and MCFD2 to date in F5F8D either destabilize protein structures or disrupt the LMAN1-MCFD2 interaction, emphasizing the functional importance of receptor complex formation in the secretion of FV and FVIII.
Mechanism of FV/FVIII Binding and Release by the LMAN1-MCFD2 Receptor Complex
The EF-hand domains of MCFD2 not only bind to LMAN1 but also interact with FV and FVIII.37 Missense mutations in MCFD2 that disrupt the tertiary structure and abolish LMAN1 binding still retain the FV/FVIII-binding activities, suggesting that this interaction is independent of Ca2+-induced folding of the protein.37 However, the precise FV/FVIII-binding site on MCFD2 remains to be determined. LMAN1 with a deletion of the coiled-coil domain can still interact with FV/FVIII.38 Deletion of the CRD and mutations in the ligand-binding pocket of the CRD that disrupt mannose binding abolish the interaction with FV/FVIII.38 These results suggest that LMAN1 mainly interacts through its carbohydrate-binding site with the high-mannose glycan side chains on FV/FVIII. Therefore, FV and FVIII likely carry duel sorting signals that are separately recognized by LMAN1 and MCFD2. Both sorting signals are necessary for the efficient transport from the ER to the Golgi.
A recent article reports the crystal structures of the LMAN1-CRD bound to α-1,2-mannobiose, the terminal carbohydrate moiety of high-mannose glycans.45 In the crystal structures, α-1,2-mannobiose interacts with both side-chain and main-chain atoms of several amino acid residues in the ligand-binding pocket. Interaction of FVIII with the H178A or G251/252A mutants, which disrupt sugar binding without affecting Ca2+ binding, was significantly reduced compared with wild type LMAN1. These results show that specific mannose recognition at the sugar-binding site is required for proper LMAN1-cargo interactions in cells.
LMAN1-MCFD2 cargo receptor captures cargo in the ER and releases them in the ERGIC/cis-Golgi before recycling back to the ER. The half-lives of LMAN1 and MCFD2 are similar,36 suggesting that the receptor complex does not dissociate during multiple cycles of shuttling between the ER and the ERGIC/cis-Golgi. How is cargo release regulated? Decreasing pH during trafficking from the ER to the ERGIC/cis-Golgi has been proposed to deprotonate H178 and trigger Ca2+ dissociation from LMAN1, thus inducing release of glycoprotein cargo.46 The pH of the ER is estimated to be approximately 7.2 to 7.4 while that of the cis-Golgi is approximately 6.4 to 6.6.47 However, in vitro measurements showed that binding of the LMAN1-CRD with either Ca2+ or α-1, 2-mannobiose is insensitive to pH in this range.45 Furthermore, the H178A mutation of LMAN1-CRD protein mutant does not affect Ca2+ binding, arguing against its proposed role as a pH sensor. The Ca2+ concentration of the ER is estimated to be in the low-millimolar range with heterogeneous distribution.48 Although the precise luminal Ca2+ concentration of the ERGIC is unknown, evidence suggests that it is much lower than in the ER.49 Quantitative analysis showed that the interaction of the LMAN1-CRD with MCFD2 is strong even at low Ca2+ concentrations in the μM range,39,50 while the binding affinity of the LMAN1-CRD with α-1, 2-mannobiose is relatively weak and decreases as Ca2+ concentration decreases.45 These results suggest that the LMAN1-CRD interaction with Man-α-1,2-Man is sensitive to low Ca2+ concentrations at which the LMAN1-MCFD2 interaction is maintained. A drop in compartmental Ca2+ during ER-to-ERGIC traffic may thus trigger the dissociation of glycoprotein cargo from the LMAN1-CRD in the ERGIC, without disrupting the LMAN1-MCFD2 interaction.
Phenotype of LMAN1-Deficient Mice
LMAN1-deficient mice were established from a gene-trap ES cell line, which contains a lacZ cassette under the control of the endogenous Lman1 promoter.51 β-gal staining showed that although LMAN1 is widely expressed, the expression levels vary considerably in different tissues and cell types. LMAN1 deficient mice mimic the phenotypes of F5F8D patients, though with the average factor levels at the high-end range of human patients. Plasma FV and FVIII, and platelet FV in Lman1 −/− mice are all reduced to 40 to 50% of the wild type level,51 compared with 5 to 30% levels typically observed in human F5F8D patients. LMAN1 deficiency had no apparent effect on COPII-coated vesicle formation in an in vitro assay. However, mild ER stress was detected in Lman1 −/− hepatocytes, perhaps due to the accumulation of LMAN1 cargo in the ER lumen.51 Despite previous reports identifying lysosomal proteins cathepsins C and Z, and plasma protein α1-antitrypsin as additional potential cargoes for LMAN1, no differences were observed between the wild type and Lman1−/− mice in the levels of cathepsins C and Z in liver lysates or α1-antitrypsin levels in plasma. Furthermore, plasma α1-antitrypsin level is also not reduced in F5F8D patients compared with normal and obligate carrier controls.51 However, α1-antitrypsin accumulates in the ER of hepatocytes, suggesting its dependence on LMAN1 for ER exit. It is unclear why the effects of LMAN1 deficiency on the steady-state levels of α1-antitrypsin and FV/FVIII are different. An unexpected, partially penetrant, perinatal lethality was observed for Lman1 −/− mice, dependent on the specific inbred strain genetic background, suggesting a potential role for other, as yet unidentified LMAN1-dependent cargo proteins.
Other Potential Functions of LMAN1
LMAN1 is highly expressed in multiple tissues that are not involved in FV/FVIII production,51 suggesting additional functions. Because F5F8D patients do not present any obvious clinical symptoms other than mild-to-moderate bleeding, phenotypes associated with other potential functions of LMAN1 may be masked by functional redundancy and/or require additional challenges to manifest. LMAN1 mutations are frequently found in colorectal cancer52 and gastric cancer patients53 with microsatellite instability. Mutations occur at the 9-adenine microsatellite in exon 8 of LMAN1. Whether LMAN1 mutations are relevant to cancer development remains to be determined. LMAN1 is upregulated after gammaherpesvirus infection of cells and knockdown of LMAN1 resulted in a significant reduction in virus production, suggesting that the LMAN1-associated secretory pathway may be rate limiting for virus production.54 Retina is one of the tissues with a high level of LMAN1 expression.51 A recent report identified Lman1 as a direct target of the transcription factor neural retina leucine zipper in mouse retina.55 LMAN1 is subject to regulation by other proteins. Proteomic studies of the ERGIC identified UBXD1 as a cytoplasmic protein that interacts with the C-terminus of LMAN1 and modulates its subcellular trafficking.56 Another protein that may regulate the subcellular localization of LMAN1 is VIPL, a lectin with homology to LMAN1 that is also localized to the early secretory pathway. It was reported that LMAN1 changes to a compact distribution pattern overlapping with Golgi markers and interact with VIPL under ER stress conditions.57
Concluding Remarks
Studies of the molecular mechanism of F5F8D have provided important basic biological insight into the early secretory pathway. Better understanding of the unique roles of LMAN1 and MCFD2 in the secretion of FV/FVIII may also provide clinical benefits in improved treatments of bleeding disorders, such as improving FVIII expression that may expedite the eventual goal of somatic gene therapy for hemophilia A, as well as suggesting new approaches for boosting FVIII production in vivo and in vitro. F5F8D patients present no obvious clinical symptoms other than mild-to-moderate bleeding. Therefore, inhibition of this pathway offers an attractive alternative to the oral anticoagulants, such as warfarin, in treatment of venous thrombosis associated with FV Leiden and elevated FVIII levels.
Acknowledgments
Work in the authors’ laboratory is supported in part by a grant from the National Institutes of Health (HL094505 to B.Z.).
References
- 1.Oeri J, Matter M, Isenschmid H, Hauser F, Koller F. Angeborener mangel an faktor V (parahaemophilie) verbunden mit echter haemophilie A bein zwei brudern. Med Probl Paediatr. 1954;1:575–588. [PubMed] [Google Scholar]
- 2.Nichols WC, Seligsohn U, Zivelin A, et al. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93(1):61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 3.Zhang B, Cunningham MA, Nichols WC, et al. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nat Genet. 2003;34(2):220–225. doi: 10.1038/ng1153. [DOI] [PubMed] [Google Scholar]
- 4.Spreafico M, Peyvandi F. Combined Factor V and Factor VIII Deficiency. Semin Thromb Hemost. 2009;35(4):390–399. doi: 10.1055/s-0029-1225761. [DOI] [PubMed] [Google Scholar]
- 5.Zhang B. Recent developments in the understanding of the combined deficiency of FV and FVIII. Br J Haematol. 2009;145 (1):15–23. doi: 10.1111/j.1365-2141.2008.07559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viswabandya A, Baidya S, Nair SC, et al. Clinical manifestations of combined factor V and VIII deficiency: a series of 37 cases from a single center in India. Am J Hematol. 2010;85(7):538–539. doi: 10.1002/ajh.21741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mansouritorgabeh H, Rezaieyazdi Z, Pourfathollah AA, Rezai J, Esamaili H. Haemorrhagic symptoms in patients with combined factors V and VIII deficiency in north-eastern Iran. Haemophilia. 2004;10(3):271–275. doi: 10.1111/j.1365-2516.2004.00890.x. [DOI] [PubMed] [Google Scholar]
- 8.Peyvandi F, Tuddenham EG, Akhtari AM, Lak M, Mannucci PM. Bleeding symptoms in 27 Iranian patients with the combined deficiency of factor V and factor VIII. Br J Haematol. 1998;100 (4):773–776. doi: 10.1046/j.1365-2141.1998.00620.x. [DOI] [PubMed] [Google Scholar]
- 9.Seligsohn U, Zivelin A, Zwang E. Combined factor V and factor VIII deficiency among non-Ashkenazi Jews. N Engl J Med. 1982;307 (19):1191–1195. doi: 10.1056/NEJM198211043071907. [DOI] [PubMed] [Google Scholar]
- 10.Mansouritorghabeh H, Banihashem A, Modaresi A, Manavifar L. Circumcision in males with bleeding disorders. Mediterr J Hematol Infect Dis. 2013;5(1):e2013004. doi: 10.4084/MJHID.2013.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdullah WZ, Ismail R, Nasir A, Mohamad N, Hassan R. Developmental haemostasis for factor V and factor VIII levels in neonates: a case report of spontaneous cephalhaematoma. Fetal Pediatr Pathol. 2013;32(2):77–81. doi: 10.3109/15513815.2012.671447. [DOI] [PubMed] [Google Scholar]
- 12.Bolton-Maggs PH, Perry DJ, Chalmers EA, et al. The rare coagulation disorders—review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004;10(5):593–628. doi: 10.1111/j.1365-2516.2004.00944.x. [DOI] [PubMed] [Google Scholar]
- 13.Mansouritorghabeh H, Rezaieyazdi Z, Bagheri M. Successful use of factor VIII concentrate and fresh frozen plasma for four dental extractions in an individual with combined factor V and VIII deficiency. Transfus Med Hemother. 2009;36(2):138–139. doi: 10.1159/000205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oukkache B, El Graoui O, Zafad S. Combined factor V and VIII deficiency and pregnancy. Int J Hematol. 2012;96(6):786–788. doi: 10.1007/s12185-012-1201-z. [DOI] [PubMed] [Google Scholar]
- 15.Guglielmone H, Minoldo S, Jarchum G. Response to the DDAVP test in a patient with combined deficiency of factor V and factor VIII. Haemophilia. 2009;15(3):838–839. doi: 10.1111/j.1365-2516.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 16.Di Marzio I, Iuliani O, Malizia R, et al. Successful use of recombinant FVIIa in combined factor V and FVIII deficiency with surgical bleeding resistant to substitutive treatment. A case report Haemophilia. 2011;17(1):160–161. doi: 10.1111/j.1365-2516.2010.02368.x. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura M, Yamashita T, Yajima J, et al. Long-term safety and efficacy of sirolimus-eluting stents in Japanese patients: a single-center cohort study. J Invasive Cardiol. 2009;21(10):526–531. [PubMed] [Google Scholar]
- 18.Wenaweser P, Dörffler-Melly J, Imboden K, et al. Stent thrombosis is associated with an impaired response to antiplatelet therapy. J Am Coll Cardiol. 2005;45(11):1748–1752. doi: 10.1016/j.jacc.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 19.Patel AJ, Liu H-H, Lager RA, Malkovska V, Zhang B. Successful percutaneous coronary intervention in a patient with combined deficiency of FV and FVIII due to novel compound heterozygous mutations in LMAN1. Haemophilia. 2013;19:607–610. doi: 10.1111/hae.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang B, Spreafico M, Zheng C, et al. Genotype-phenotype correlation in combined deficiency of factor V and factor VIII. Blood. 2008;111(12):5592–5600. doi: 10.1182/blood-2007-10-113951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang B, McGee B, Yamaoka JS, et al. Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2. Blood. 2006;107(5):1903–1907. doi: 10.1182/blood-2005-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdallah HE, Gouider E, Amor MB, Jlizi A, Meddeb B, Elgaaied A. Molecular analysis in twoTunisian families with combined factor V and factor VIII deficiency. Haemophilia. 2010;16(5):801–804. doi: 10.1111/j.1365-2516.2010.02268.x. [DOI] [PubMed] [Google Scholar]
- 23.Ge J, Xue F, Gu DS, et al. Combined deficiency of factors V and VIII caused by a novel compound heterozygous mutation of gene Lman1. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2010;18(1):185–190. [PubMed] [Google Scholar]
- 24.Hejer E, Adnen LM, Asma J, Ibtihel M, Benammar-Elgaaied A, Gouider E. Identification of a novel mutation in the MCFD2 gene in a Tunisian family with combined factor V and VIII deficiency. Tunis Med. 2012;90(4):343–344. [PubMed] [Google Scholar]
- 25.Zhu M, Das V, Zheng C, Majumdar S, Zhang B. A synonymous mutation in LMAN1 creates an ectopic splice donor site and causes combined deficiency of FV and FVIII. J Thromb Haemost. 2012;10:2407–2409. doi: 10.1111/jth.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nyfeler B, Kamiya Y, Boehlen F, et al. Deletion of 3 residues from the C-terminus of MCFD2 affects binding to ERGIC-53 and causes combined factor V and factor VIII deficiency. Blood. 2008;111 (3):1299–1301. doi: 10.1182/blood-2007-09-112854. [DOI] [PubMed] [Google Scholar]
- 27.Khoriaty R, Vasievich MP, Ginsburg D. The COPII pathway and hematologic disease. Blood. 2012;120(1):31–38. doi: 10.1182/blood-2012-01-292086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122(4):605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 29.Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119(Pt 11):2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- 30.Baines AC, Zhang B. Receptor-mediated protein transport in the early secretory pathway. Trends Biochem Sci. 2007;32(8):381–388. doi: 10.1016/j.tibs.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Dancourt J, Barlowe C. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem. 2010;79:777–802. doi: 10.1146/annurev-biochem-061608-091319. [DOI] [PubMed] [Google Scholar]
- 32.Nyfeler B, Reiterer V, Wendeler MW, et al. Identification of ERGIC-53 as an intracellular transport receptor of alpha1-antitrypsin. J Cell Biol. 2008;180(4):705–712. doi: 10.1083/jcb.200709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Appenzeller C, Andersson H, Kappeler F, Hauri HP. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat Cell Biol. 1999;1(6):330–334. doi: 10.1038/14020. [DOI] [PubMed] [Google Scholar]
- 34.Nyfeler B, Zhang B, Ginsburg D, Kaufman RJ, Hauri HP. Cargo selectivity of the ERGIC-53/MCFD2 transport receptor complex. Traffic. 2006;7(11):1473–1481. doi: 10.1111/j.1600-0854.2006.00483.x. [DOI] [PubMed] [Google Scholar]
- 35.Nufer O, Kappeler F, Guldbrandsen S, Hauri HP. ER export of ERGIC-53 is controlled by cooperation of targeting determinants in all three of its domains. J Cell Sci. 2003;116(Pt 21):4429–4440. doi: 10.1242/jcs.00759. [DOI] [PubMed] [Google Scholar]
- 36.Zhang B, Kaufman RJ, Ginsburg D. LMAN1 and MCFD2 form a cargo receptor complex and interact with coagulation factor VIII in the early secretory pathway. J Biol Chem. 2005;280(27):25881–25886. doi: 10.1074/jbc.M502160200. [DOI] [PubMed] [Google Scholar]
- 37.Zheng C, Liu HH, Zhou J, Zhang B. EF-hand domains of MCFD2 mediate interactions with both LMAN1 and coagulation factor V or VIII. Blood. 2010;115(5):1081–1087. doi: 10.1182/blood-2009-09-241877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng C, Liu HH, Yuan S, Zhou J, Zhang B. Molecular basis of LMAN1 in coordinating LMAN1-MCFD2 cargo receptor formation and ER-to-Golgi transport of FV/FVIII. Blood. 2010;116(25):5698–5706. doi: 10.1182/blood-2010-04-278325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishio M, Kamiya Y, Mizushima T, et al. Structural basis for the cooperative interplay between the two causative gene products of combined factor V and factor VIII deficiency. Proc Natl Acad Sci U S A. 2010;107(9):4034–4039. doi: 10.1073/pnas.0908526107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wigren E, Bourhis JM, Kursula I, Guy JE, Lindqvist Y. Crystal structure of the LMAN1-CRD/MCFD2 transport receptor complex provides insight into combined deficiency of factor V and factor VIII. FEBS Lett. 2010;584(5):878–882. doi: 10.1016/j.febslet.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Velloso LM, Svensson K, Pettersson RF, Lindqvist Y. The crystal structure of the carbohydrate-recognition domain of the glycoprotein sorting receptor p58/ERGIC-53 reveals an unpredicted metal-binding site and conformational changes associated with calcium ion binding. J Mol Biol. 2003;334(5):845–851. doi: 10.1016/j.jmb.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 42.Guy JE, Wigren E, Svärd M, Härd T, Lindqvist Y. New insights into multiple coagulation factor deficiency from the solution structure of human MCFD2. J Mol Biol. 2008;381(4):941–955. doi: 10.1016/j.jmb.2008.06.042. [DOI] [PubMed] [Google Scholar]
- 43.Elmahmoudi H, Wigren E, Laatiri A, et al. Analysis of newly detected mutations in the MCFD2 gene giving rise to combined deficiency of coagulation factors V and VIII. Haemophilia. 2011;17 (5):e923–e927. doi: 10.1111/j.1365-2516.2011.02529.x. [DOI] [PubMed] [Google Scholar]
- 44.Yamada T, Fujimori Y, Suzuki A, et al. A novel missense mutation causing abnormal LMAN1 in a Japanese patient with combined deficiency of factor V and factor VIII. Am J Hematol. 2009;84 (11):738–742. doi: 10.1002/ajh.21532. [DOI] [PubMed] [Google Scholar]
- 45.Zheng C, Richard CP, Das V, et al. Structural characterization of carbohydrate binding by LMAN1 provides new insight into the endoplasmic reticulum export of FV and FVIII. J Biol Chem. 2013 doi: 10.1074/jbc.M113.461434. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Appenzeller-Herzog C, Roche AC, Nufer O, Hauri HP. pH-induced conversion of the transport lectin ERGIC-53 triggers glycoprotein release. J Biol Chem. 2004;279(13):12943–12950. doi: 10.1074/jbc.M313245200. [DOI] [PubMed] [Google Scholar]
- 47.Paroutis P, Touret N, Grinstein S. The pH of the secretory pathway: measurement, determinants, and regulation. Physiology (Bethesda) 2004;19:207–215. doi: 10.1152/physiol.00005.2004. [DOI] [PubMed] [Google Scholar]
- 48.Montero M, Alvarez J, Scheenen WJ, Rizzuto R, Meldolesi J, Pozzan T. Ca2+ homeostasis in the endoplasmic reticulum: coexistence of high and low [Ca2+] subcompartments in intact HeLa cells. J Cell Biol. 1997;139(3):601–611. doi: 10.1083/jcb.139.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pezzati R, Bossi M, Podini P, Meldolesi J, Grohovaz F. High-resolution calcium mapping of the endoplasmic reticulum-Golgi-exocytic membrane system. Electron energy loss imaging analysis of quick frozen-freeze dried PC12 cells. Mol Biol Cell. 1997;8(8):1501–1512. doi: 10.1091/mbc.8.8.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawasaki N, Ichikawa Y, Matsuo I, et al. The sugar-binding ability of ERGIC-53 is enhanced by its interaction with MCFD2. Blood. 2008;111(4):1972–1979. doi: 10.1182/blood-2007-06-097022. [DOI] [PubMed] [Google Scholar]
- 51.Zhang B, Zheng C, Zhu M, et al. Mice deficient in LMAN1 exhibit FV and FVIII deficiencies and liver accumulation of α1-antitrypsin. Blood. 2011;118(12):3384–3391. doi: 10.1182/blood-2011-05-352815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roeckel N, Woerner SM, Kloor M, et al. High frequency of LMAN1 abnormalities in colorectal tumors with microsatellite instability. Cancer Res. 2009;69(1):292–299. doi: 10.1158/0008-5472.CAN-08-3314. [DOI] [PubMed] [Google Scholar]
- 53.Nagarajan N, Bertrand D, Hillmer AM, et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012;13(12):R115. doi: 10.1186/gb-2012-13-12-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mages J, Freimüller K, Lang R, et al. Proteins of the secretory pathway govern virus productivity during lytic gammaherpesvirus infection. J Cell Mol Med. 2008;12(5B):1974–1989. doi: 10.1111/j.1582-4934.2008.00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hao H, Kim DS, Klocke B, et al. Transcriptional regulation of rod photoreceptor homeostasis revealed by in vivo NRL targetome analysis. PLoS Genet. 2012;8(4):e1002649. doi: 10.1371/journal.pgen.1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haines DS, Lee JE, Beauparlant SL, et al. Protein interaction profiling of the p97 adaptor UBXD1 points to a role for the complex in modulating ERGIC-53 trafficking. Mol Cell Proteomics. 2012;11 (6):016444. doi: 10.1074/mcp.M111.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin SY, Kawasaki N, Hu D, Tozawa H, Matsumoto N, Yamamoto K. Subcellular localization of ERGIC-53 under endoplasmic reticulum stress condition. Glycobiology. 2012;22(12):1709–1720. doi: 10.1093/glycob/cws114. [DOI] [PubMed] [Google Scholar]