

Abstract
Background & Aims
Tumors that develop in patients with Crohn's disease tend be multifocal, so field cancerization (the replacement of normal cells with non-dysplastic but tumorigenic clones) might contribute to intestinal carcinogenesis. We investigated patterns of tumor development from pretumor intestinal cell clones.
Methods
We performed genetic analyses of multiple areas of intestine from 10 patients with Crohn's disease and intestinal neoplasia. Two patients had multifocal neoplasia; longitudinal sections were collected from 3 patients. Individual crypts were microdissected and genotyped; clonal dependency analysis was used to determine the order and timing of mutations that led to tumor development.
Results
The same mutations in KRAS, CDKN2A(p16), and TP53 that were observed in neoplasias were also present in nontumor, nondysplastic, and dysplastic epithelium. In 2 patients, carcinogenic mutations were detected in nontumor epithelium 4 years before tumors developed. The same mutation (TP53 p.R248W) was detected at multiple sites along the entire length of the colon from 1 patient; it was the apparent founder mutation for synchronous tumors and multiple dysplas-tic areas. Disruption of TP53, CDKN2A, and KRAS were all seen as possible initial events in tumorigenesis; the sequence of mutations (the tumor development pathway) differed among lesions.
Conclusions
Pretumor clones can grow extensively in the intestinal epithelium of patients with Crohn's disease. Segmental resections for neoplasia in patients with Crohn's disease might therefore leave residual pretumor disease, and dysplasia might be an unreliable biomarker for cancer risk. Characterization of the behavior of pretumor clones might be used to predict the development of intestinal neoplasia.
Keywords: Inflammatory Bowel Disease, Tumor Progression, Clonal Ordering, Oncogene, CRC
Long-standing Crohn's disease (CD), an inflammatory bowel disease, is associated with an increased risk of developing intestinal cancer that is between 2 and 3 times greater than that of the healthy population.1–4 Compared with non–inflammatory bowel disease tumors, synchronous and multifocal neoplasia occurs in as many as 30% of patients with CD with neoplastic disease.5–7 This suggests that field cancerization, the widespread replacement of the normal cell population by a histologically nondysplastic mutant clone that is predisposed to tumor development,8–10 could underlie tumorigenesis in CD. The identification and characterization of pretumor mutant fields in patients with CD could provide better predictors of a patient's risk of neoplasia.
The widely accepted paradigm for tumorigenesis, the somatic mutation theory, is that tumorigenesis begins with rate-limiting mutations in a key growth control gene, resulting in immediate lesion growth, and then subsequent alteration of other genes drives the evolution of subclones within the tumor, leading eventually to the development of a malignant clone.11,12 However, the identification of field cancerization in the lung13 and colon14,15 and skin16 makes it increasingly clear that tumor development can begin long before any overt tumor growth.17 The dynamics of such pretumor clones remain singularly unknown. Determining parameters such as the clone growth rate would permit more precise estimations of a patient's risk and timescale for neoplastic development, while also revealing the basic biology of stem cell clone growth in the human intestine. Given their risk of malignancy, patients with CD are often subjected to routine endoscopic surveillance with concomitant surgery.18–20 Thus, tissue samples are collected in some patients over many years, providing a fortuitous but uncommon means to study the longitudinal dynamics of pretumor clones in the human intestine.
The genetic pathways of tumor development in CD have not been conclusively determined; however, the etiologic similarities between CD and ulcerative colitis (UC) suggest that tumorigenesis in the 2 diseases may share genetic pathways. UC-associated neoplasia frequently shows TP53 or KRAS mutations,14,21–23 whereas inactivating mutations of the adenomatous polyposis coli (APC) gene, found in most sporadic colorectal tumors,24 are rarely observed.14,22 Loss of heterozygosity (LOH) of chromosome arm 18q, possibly targeting the SMAD4 gene, is also relatively frequent in UC cancer.21,25
Here, we investigate pretumor clonal dynamics in the inflamed human CD bowel. Inferring clonal dynamics in the human bowel is technically challenging, because invasive labeling studies are impractical. Instead, we used clonal ordering techniques that exploit the relative spatial location of mutations to infer the order in which they were acquired.26,27 Rare point mutations mark clones, because 2 cells are very unlikely to bear the same point mutation unless they share a clonal origin. Longitudinally collected tissue samples allow the extent and rate of clonal expansion to be determined, indicating the relative fitness of each clone.28
Patients and Methods
See Supplementary Materials and Methods for full methods.
Patient and Clinical Material
Paraffin-embedded biopsy and resection specimens were obtained from 10 patients with CD-associated intestinal neoplasia (Table 1). Specimens from multiple areas of intestine were available from each patient; 3 patients had longitudinally collected samples, and 2 patients had multifocal neoplasia. The case of patient 1 is particularly enlightening because this patient had tissue collected over an 8-year period (1996–2004), during which he developed multifocal and metachronous tumors.
Table 1. Patient Details.
| Patient | Age (y) CD diagnosed |
Age (y) neoplasm diagnosed |
Family history of inflammatory bowel disease |
Family history of colorectal cancer |
Location of CD | Immunomodulator use |
No. of neoplasms |
Location of neoplasm |
Type of neoplasm(s) | Mutant genes detected |
TP53 | Kras | p16 | Tissues examined over time |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 65 | 73 | No | No | Pancolonic and terminal ileum | No | 3 | Sigmoid Sigmoid Rectum |
Moderately differentiated adenocarcinoma High-grade dysplasia Poorly differentiated adenocarcinoma |
TP53 | Exon 7 c.731G>A G244D Exon 7 c.742C>T R248W Exon 7 c.742C>T R248W |
None | None | 1996 1998 2000 2001 2003 2004 |
| 2 | 39 | 39 | No | Yes | Ileocolic | No | 1 | Sigmoid | Moderately differentiated adenocarcinoma | TP53 | Exon 6 c.637C>T R213* | None | None | No |
| 3 | 44 | 70 | No | No | Small bowel and ileocolic | Yes, azathioprine | 1 | Jejunum | Moderately differentiated colloid adenocarcinoma | Kras p16 | None | c.35G>A G12D | c.442C>A A148T | No |
| 4 | 29 | 30 | Yes | Yes | Ileocolic | No | 1 | Right colon | Moderately differentiated adenocarcinoma | TP53 | Exon 6 c.637C>T R213* | None | None | No |
| 5 | 37 | 65 | No | No | Pancolonic terminal ileum, jejunum perianal | Yes, azathioprine | 1 | Perineal wound in fistula tract | Well-differentiated mucinous adenocarcinoma | TP53 Kras p16 | Exon 7 c.733G>A G245S | c.37G>A G13D | c.238C>T R80* | 2004 2006 2008 |
| 6 | 37 | 52 | Yes | No | Colonic | No | 1 | Rectal | Moderately differentiated adenocarcinoma | TP53 | Exon 8 c.817C>T R273C | None | None | No |
| 7 | 71 | 86 | No | No | Colonic | No | 2 | Transverse colon | Adenomas (DALM) | Kras | None | c.35G>A G12D | None | 2009 2010 |
| 8 | 10 | 32 | Yes | Yes | Ileocolic | No | 1 | Ileum | Poorly differentiated mucinous adenocarcinoma | None | None | None | None | No |
| 9 | 45 | 46 | No | No | Ileocolic perianal | No | 1 | Right colon | Poorly differentiated adenocarcinoma | None | None | None | None | No |
| 10 | 78 | 79 | No | No | Ileocolic | No | 1 | Right colon | Mucinous moderately differentiated adenocarcinoma | None | None | None | None | No |
DALM, dysplasia-associated lesion or mass as adenoma was in area of colitis.
Mutation Screening of Cancers
Initially, DNA was needle macrodissected from each neoplastic lesion (surgical resection specimens) and screened for somatic mutations using polymerase chain reaction (PCR) sequencing at loci frequently mutated in inflammatory bowel disease cancers: TP53 exon 5–9, KRAS exon 1 (codons 12–13), and CDKN2A (p16INK4a) exon 2. Primer details are listed in Supplementary Table 1.
Laser Capture Microdissection
Mutations identified in the screening phase were then investigated on a crypt-by-crypt basis in multiple tissue specimens from each patient. A total of 450 crypts were microdissected from serial sections using a PALM microdissection system (Zeiss, Munich, Germany); in poorly differentiated tumors, small areas of epithelial cells were microdissected. Pathology was assessed using a serial H&E-stained slide. Following DNA extraction by digestion in PicoPure proteinase K buffer (Applied Bio-systems, Warrington, United Kingdom), DNA lysates were PCR sequenced. Tubes containing digestion buffer but no microdissection material were included as negative controls.
LOH Analysis
LOH analysis was performed on individual crypt lysates at up to 19 informative markers close to the FHIT, APC, CDKN2A, SMAD4, and TP53 genes, respectively, using a multiplexed microsatellite PCR kit (Qiagen, Crawley, United Kingdom). LOH was then considered present if the area under one allelic peak was more than twice that of the other, after normalizing peak areas relative to constitutional DNA. Multiple informative markers located close to APC, TP53, and CDKN2A were available for some patients; in these cases, LOH was called if it was shown by at least 2 of the 3 available markers. Microsatellite instability was assessed using multiplexed assay for the BAT-25 and BAT-26 mononucleotide repeats29; the presence of microsatellite instability in a sample precluded reliable LOH analysis. Primer details are listed in Supplementary Table 2.
Image Cytometry
Areas of approximately 1 mm2 of epithelium were needle dissected from waxed 40-μm sections, using a serial H&E section as a guide. Image cytometry was performed using the Fairfield DNA Ploidy System (Fairfield Imaging, Kent, United Kingdom) as previously described.14
Immunohistochemistry
Immunohistochemistry was performed for p53 (Dako, Ely, United Kingdom), β-catenin (Transduction Labs, Oxford, United Kingdom), and lysozyme (Dako) using standard protocols.
Results
Somatic mutations in TP53, CDKN2A, or KRAS were detected in neoplastic tissue from 7 of 10 patients (Table 1). Individual histologically normal, inflamed, or dysplastic crypts, cancer glands, or small areas of poorly differentiated tumor (∼500 cells) were then microdissected from multiple tissue samples from each patient with detected mutations and genotyped for the somatic mutation(s) detected in the patient's neoplasia (Supplementary Table 3).
Clonality of Tumor and Nontumor Tissue
Individual crypts from nontumor tissue were examined in the 7 of 10 patients who had a detected mutation in their neoplasia. In 5 of 7 of these patients, the mutation present within the tumor could also be detected in nontumor tissue (Figure 1 and Supplementary Table 3), suggesting in each case a clonal relationship between the tumor and nontumor mucosa.
Figure 1.
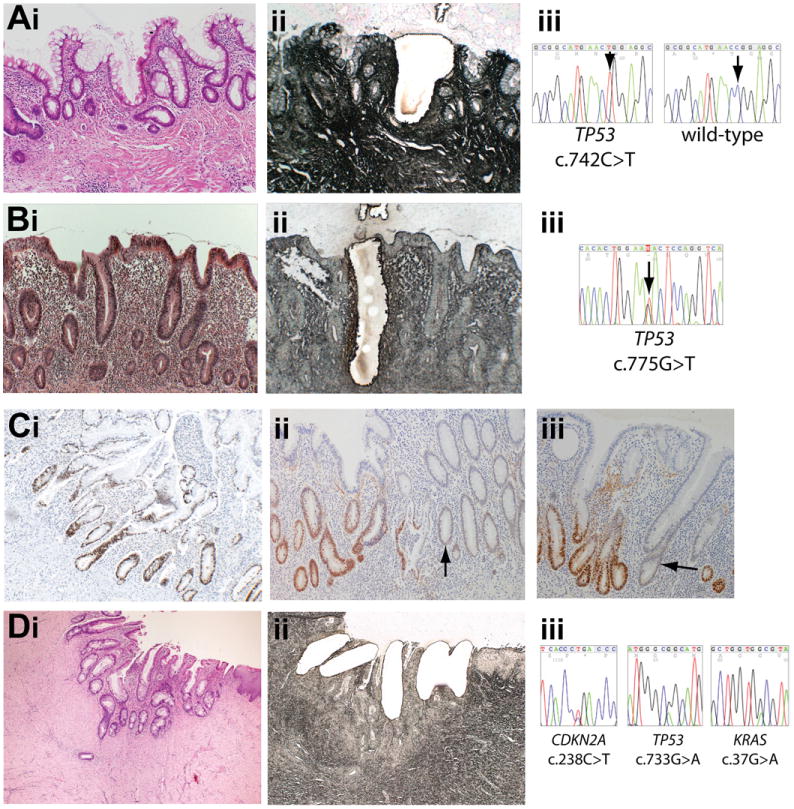
Mutations present in nontumor tissue. (A) (i) H&E stain (original magnification 100×) of nondysplastic (hyperplastic) mucosa showing crypt distortion with increased inflammatory cells and intraepithelial neutrophils in the transverse colon in patient 1 in 2004 and (ii) serial methylene green–stained PALM laser capture slide showing microdissected crypt. (iii) Sequencing shows the crypt contains a TP53 c.742C>T mutation. (B) (i) H&E stain (original magnification 100×) of nondysplastic (hyperplastic) mucosa with a marked increase in the inflammatory cells in the lamina propria and cryptitis in the resection margin of the sigmoid cancer resected from patient 1 in 2000 with (ii) serial PALM slide showing microdissection. (iii) Sequencing shows the crypt contains a TP53 c.775G>T mutation. (C) p53 immunohistochemistry on a nondysplastic colon resection specimen from patient 1 (original magnification 100×). (i) Patches of crypts with nuclear accumulation of p53 protein were observed, frequently demarked by odd p53-negative or low-expressing crypts (ii and iii) (arrows). (D) (i) H&E stain (original magnification 40×) of inflammatory atypia within cells in a perianal fistula from patient 5 four years before tumor growth. (ii) Laser capture slide (original magnification 100×). (iii) TP53, CDKN2A, and KRAS mutations in each crypt.
Within nontumor tissue, the mutations found in the cancer were frequently present in both dysplastic and nondysplastic crypts, although always in areas of active disease (Figure 1 and Supplementary Table 3). Patients 3 and 4 had cancer in the small intestine, whereas patients 1, 5, and 6 had colon cancer, indicating that pretumor clone growth occurred in both intestinal compartments.
The remaining 2 patients (patients 2 and 7) had detected mutations only within the cancer and dysplasia-associated lesion or mass, respectively. Although an undetected pretumor clone may have preceded tumor growth, it is conceivable that these neoplasia followed a sporadic pattern of tumorigenesis.
Detection of Mutant Clones Many Years Before Cancer Growth and at Sites Distinct From the Cancer
Longitudinally collected samples, available from 2 informative patients (1 and 5), provided a means to study the dynamic behavior of putative pretumor clones (Figure 2 and Supplementary Figure 1). Patient 5 developed an adenocarcinoma arising within a perineal proctectomy scar that was resected in 2008. Both the cancer and the nearby nondysplastic crypts in the inflamed resection margin contained KRAS c.37G>A, TP53 c.733G>A, and CDKN2A mutations, and the same 3 mutations could also be detected in a proctectomy specimen collected 4 years earlier (Supplementary Table 3). However, a nearby perianal fistula tract in the earlier proctectomy specimen showed the same KRAS c.37G>A mutation, a different TP53 mutation, and no CDKN2A mutation, indicating that KRAS c.37G>A was the first pretumor mutation. This second TP53 mutation was not detected in later samples, suggesting the clone had died out (Supplementary Figure 1). Interestingly, a small bowel biopsy specimen from 2004 showed a different KRAS c.35G>A mutant clone in nondysplastic crypts; however, a subsequent biopsy specimen from the same area in 2006 did not contain this clone, suggesting this second KRAS clone had undergone only limited clonal expansion or died out.
Figure 2.
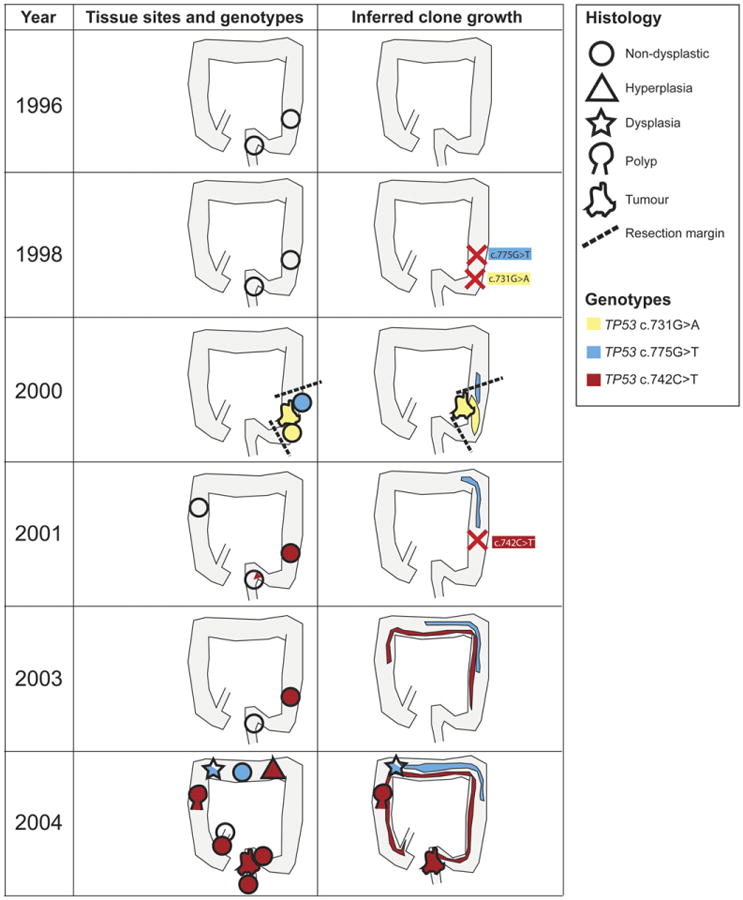
Longitudinal analysis showing clone spread in patient 1. Tissue collected before 2000 contained no widespread detected genetic abnormalities. A c.731G>A TP53 mutation was first detected throughout a sigmoid cancer in 2000 and within the resection margins, but not in later samples, suggesting the mutant arose in the sigmoid after the biopsy in 1998 and was completely removed by the cancer resection. A second c.775G>T TP53 was also found in the resection margin of the sigmoid cancer. This second clone had spread to the transverse colon by 2004. A third TP53 c.742C>T mutation was first detected in sigmoid and rectal biopsy specimens from 2001. By 2004, the mutant TP53 c.742C>T clone was present in every major segment of the colon and also in the terminal ileum and in multifocal neoplasia. Shapes indicate the predominant pathological diagnosis at the site, and color indicates the predominant TP53 genotype (see key).
An extensive tissue archive was available from patient 1 (Figure 2 and Supplementary Table 3). The sigmoid adenocarcinoma resected from this patient in 2000 contained a TP53 c.731G>A mutation throughout (Supplementary Figure 2), which was also present in morphologically nondysplastic crypts in the resection margin. Crypts from earlier rectal and sigmoid biopsy specimens (1996 and 1998) contained no detected TP53 mutations, suggesting that the TP53 c.731G>A mutation occurred in the 2 years before cancer growth. A follow-up biopsy around the anastomosis area did not contain the c.731G>A mutation, suggesting the resection successfully removed the mutant clone.
A second TP53 mutant, c.775G>T, was detected in the inflamed but nondysplastic resection margin of the sigmoid cancer in 2000. The same mutation was later detected in the transverse colon mucosa resected in 2004.
A third TP53 c.742C>T mutation was also detected in this patient, in multiple neoplastic lesions, and at multiple sites spanning the length of the colon (Figure 2). The mutation was first identified in crypts from sigmoid and rectum collected in 2001 and was not present in earlier samples, making it likely the mutation was first acquired in this area around this time. The c.742C>T mutation was again detected in sigmoid crypts collected in 2003 but not in a simultaneous rectal biopsy specimen. By 2004, the c.742C>T mutation was detected at sites along the entire length of the colon: a rectal cancer (Supplementary Figure 3), a transverse colon hyperplastic mucosa (Figure 1), a right colon adenoma, and in the nondysplastic but inflamed mucosa surrounding each of these lesions. This mutation was a founding mutation for areas of dysplasia, an adenoma, and a cancer. Immunohistochemical analysis of the patient's colectomy specimen in 2004 revealed patchy and infrequent p53 overexpression along the colon length (Figure 1), suggesting that p53 mutant clones were present only in a minority of crypts.
Noninflamed areas of terminal ileum were available from patients 3 and 4. Crypts microdissected from these nondiseased areas did not contain the mutations found in these patients' cancer and inflamed mucosa (Supplementary Figure 4).
Crypts on Either Side of the Ileal-Cecal Valve Have a Common Somatic Mutation
Individual crypts from inflamed terminal ileum tissue from patient 1 collected in 2004 contained the same TP53 c.742C>T mutation present in this patient's colon. Separate microdissection of crypts and villi showed that the mutation was also present on the villus fed by a mutant crypt (Figure 3). Immunohistochemistry for lysozyme staining highlighted the presence of Paneth cells within mutated crypts (Figure 3).
Figure 3.
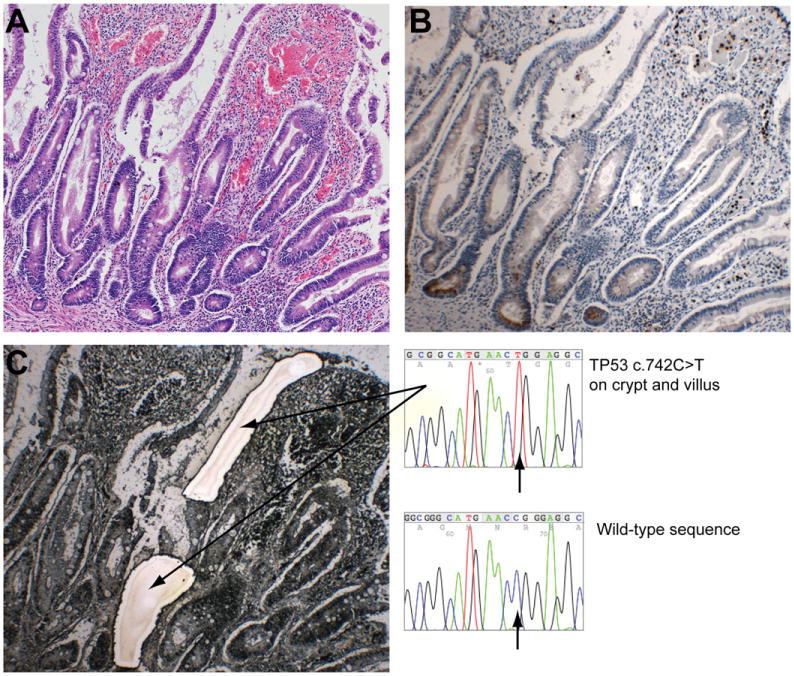
Presence of the same TP53 mutation in functional small intestinal and colon crypts. (A) H&E section showing inflamed terminal ileum mucosa with crypt/villous distortion and an increase in inflammatory cells and intraepithelial neutrophils. (B) Serial section stained for lysozyme indicating the presence of functional Paneth cells at the small intestinal crypt base. (C) Serial laser capture slide stained with methylene green showing separately microdissected crypt and villus. Genotyping revealed the same TP53 c.742C>T was present in both the crypt and villus. This same mutation was found in colon tissue collected at the same and previous times. Original magnification 100× for all micrographs.
Interchangeable Order of Mutations in Tumorigenesis
The order of mutations in tumorigenesis differed between tumors. Mutation order was inferred from the relative spatial localization of mutants; genetic heterogeneity was a hallmark of most tumors. In all 3 polypoid lesions detected in patient 1, a TP53 point mutation was present throughout the lesion and within the resection margins, suggesting TP53 was the founder mutation for each lesion. Tumor development involved sequential 17p, 9p and 18q LOH, and aneuploidy (Figure 4 and Supplementary Figure 3). The sigmoid cancer resected in patient 1 in 2000 also showed widespread 5q LOH and nuclear β-catenin (Supplementary Figure 2), implicating APC in early tumorigenesis of this lesion.
Figure 4.
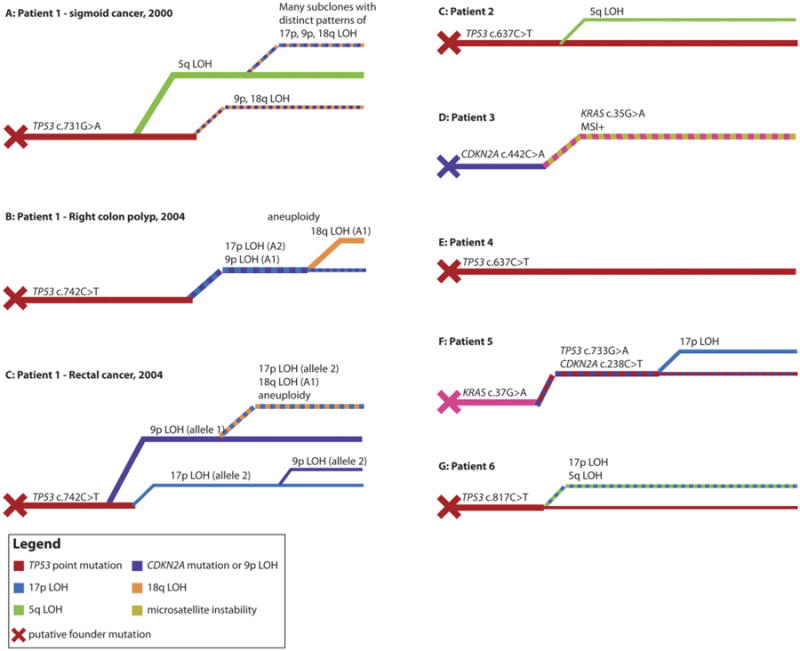
Phylogenetic trees of neoplasia development. (A) Phylogenetic tree of sigmoid cancer resected in patient 1 in 2000. Tumor growth was preceded by the acquisition of a TP53 c.731G>A mutation, because this mutation was found in the cancer and its resection margin. A clone with 5q LOH was dominant in the cancer, and a single crypt showed 9p and 18q LOH but no 5q LOH. Numerous small subclones with distinct patterns of 9p, 18q, and 17p LOH were found within the 5q subclone. This frequent LOH could be attributable to the cancer containing an aneuploid clone, which was not tested for in this cancer. (B) Phylogenetic tree of the ascending colon adenoma resected in patient 1 in 2004. The cancer developed from the preneoplastic clone distinguished by the TP53 c.742C>T mutation, and subsequent carcinogenesis involved the sequential acquisition of 17p, 9p, and then 18q LOH. Unlike the previous cancer in patient 1, no 5q LOH was detected in the lesion in 2004. (C–G) Phylogenetic trees for tumors from patients 1–6. Most lesions were initiated by a TP53 mutant clone, with the exceptions of patient 3, where the putative founder clone was a CDKN2A (p16) mutant, and patient 5, where the founder clone was a KRAS mutant. The inferred sequence of subsequent mutations and LOH events was different in every lesion. Colors indicate genotypes (see key), and line thicknesses are indicative of relative clone abundance.
Biallelic mutation of TP53, presumed on the detection of both a TP53 point mutation and LOH, was not requisite for tumor growth; the ascending colon adenoma in patient 1 in 2004 had a TP53 point mutation throughout the adenoma but only a subclone had 17p LOH; furthermore, biallelic TP53 mutations were infrequently observed in nondysplastic crypts in this patient (Supplementary Table 3). Similarly, a putative biallelic TP53 mutation was observed in the dysplastic margins of the cancers from both patients 5 and 6.
The presence of a CDKN2A mutation in both tumor and nondysplastic tissue in patient 3 implicates CDKN2A disruption in the initiation of this patient's tumor; tumor progression involved a subsequent KRAS mutation and the development of microsatellite instability (Figure 4). The initial mutation in patient 5 was in KRAS, with later CDKN2A and TP53 mutations implicated in subsequent tumor development.
Discussion
The genetic and histologic mechanisms driving the development of CD-associated cancers have not been conclusively determined. The data presented herein are strong evidence that field cancerization, the replacement of the normal epithelium with a protumorigenic clone,8,9 before any dysplastic histologic change contributes to carcinogenesis in patients with CD. In 5 of 7 informative patients with neoplasia, the same point mutation in KRAS, CDKN2A, or TP53 could be detected within the tumor, neighboring resection margins that showed active disease, and even in more distant diseased areas, strongly suggesting that this mutant tissue was clonal in origin.
In 1 patient, a TP53 c.742C>T mutation was first detected focally in the sigmoid colon and then 4 years later at sites along the entire colon length. These data suggest that the mutant clone had arisen in the left colon and appeared to have spread both proximally to the right colon and distally to the rectum within 4 years (Figure 2), suggesting pretumor growth can occur at a substantial scale and rate. This conclusion rests on the strength of the TP53 c.742C>T point mutation to uniquely identify a clonal population of cells in this patient. If this particular mutation occurred at high frequency, it is conceivable that 2 unrelated cells could independently develop the mutation; if this were true, then the data suggest pretumor clone growth of more restricted spatial extent. The International Agency for Research on Cancer p53 mutation database30 suggests that the c.742C>T mutation comprises approximately 7% of the total detected somatic TP53 mutations in the human intestine. Given 2 crypts that both have a TP53 mutation, the odds that crypts will both have the c.742C>T mutation are small (approximately 200:1). This calculation assumes that the distribution of TP53 mutations selected in the CD bowel mirrors that reported in the International Agency for Research on Cancer database for the intestine at large. However, given that other gastrointestinal cancers arising in an inflammatory environment show a broad range of TP53 mutations, this is likely a reasonable assumption.14,31–33 The notion that the TP53 c.742C>T mutation marks a single clone is further supported by LOH data; both the rectal and ascending colon lesions in patient 1, which were clonal for the TP53 c.742C>T point mutation, showed loss of the same alleles of markers on chromosome arms 9p, 18q, and 17p. Because the same allele of 17p was consistently lost around the entire colon, this suggests that the universally retained allele bore the c.742C>T point mutation. The odds of finding 2 cells, which both have LOH at these 3 loci and have lost the same alleles, are 1:8; the odds of 2 mutated cells both bearing these same patterns of LOH and the c.742C>T point mutation are at most 1:1600. This probability must also be considered in light of the presumably very small likelihood of a crypt acquiring a fixed genetic lesion at all. Therefore, it is likely that the widely dispersed TP53 c.742C>T mutant cells, frequently marked by an additional 3 genetic lesions, in patient 1 comprised a single clone. Furthermore, evidence of long-range clone spread in this patient is clearer still when the TP53 c.775G>T mutant is considered. This mutation is reported to be relatively rare, comprising approximately 0.1% of the reported somatic TP53 variants in the International Agency for Research on Cancer database, and so the odds of this mutant arising more than once in the bowel are minimal. The TP53 c.775G>T mutation was detected in the descending colon at baseline and the transverse colon 4 years later, indicating extensive clone migration during this time.
How does a mutant clone grow in the CD intestine? Crypt fission is the primary mechanism of clonal expansion in the intestinal epithelium.34,35 Although fission is rare in the normal intestine, patients with colitis have an elevated fission rate,36 and tumorigenic mutations may further increase the crypt division rate. Chronic inflammation, resulting in cycles of crypt atrophy and mucosal healing by crypt fission, likely provides a major growth stimulus; indeed, mutant clones were only identified in areas of active disease. Despite this, it is unlikely that a mutant clone could sustain exponential growth,37 raising the possibility that extensive clonal expansion may occur through a noncanonical mechanism such as stem cell migration between crypts, perhaps analogous to the migration between niches observed in the Drosophila ovary.38
p53 and p16 mutants were observed to have undergone extensive clonal expansion in the colitic bowel, indicating that these mutated clones have a selective advantage in the CD intestine. Mutations that impair the ability of p53 and p16 to regulate the apoptosis and senescence responses presumably provide a survival advantage, because such mutant clones are likely more able to withstand the inflammatory stress of the colitic bowel. Specific clone fitness may be determined by the mutation type; indeed, the c.742C>T, p.R248W mutant, which underwent the most extensive clonal expansion, is associated with severe down-regulation of p53 activity.30,39 Differential effects of each mutation may also regulate the ability of a mutant cell to compete with wild-type or other mutant cells for a place in the bowel.40 Further selective advantage for TP53 mutants may be due to their tolerance of genetic instability,41,42 seemingly an early event in colitis-associated cancers.43 Supporting this hypothesis, an aneuploid population was observed in transverse colon nondysplastic but p53-mutant tissue in patient 1 (Supplementary Table 3 and Supplementary Figure 5).
Notably, mutations were only detected in diseased tissue; consequently, we would suggest that there is interdependence between the persistence of mutant clones and inflammatory disease. This relationship gives further credence to the suggestion that tumor suppressor gene mutations could confer a survival advantage in the inflamed bowel; indeed, 4 of the 5 patients who had identified field cancerization were also recorded as having long-standing active CD. Further, it is tempting to speculate that persistent inflammation may be necessary for significant clone growth. Fission rates are markedly increased in inflamed mucosa,36 and potentially this is the direct cause of clone spread. Future analyses of isolated “skip lesions” and their histologically normal margins, and also of pathology archives that fully catalog nondiseased bowel, will provide a means to fully investigate any relation between active disease and clone spread. Our data are insufficient to investigate any potential relationship between mucosal injury from an endoscopic biopsy and clone growth.
In one patient, the same TP53 mutation was detected in crypts on both sides of the ileal-cecal valve. These data therefore suggest that a colon-derived cell can cross into the small intestine and form functional crypt-villus units. The strength of this conclusion depends on the reliability of the TP53 mutation as a clone marker; its merits have been discussed previously. There are a number of possible mechanisms for this apparent cell migration. First, an impaired ileocecal valve, perhaps compromised by inflammation, could permit retrograde clone expansion. Second, the relatively common fissures in patients with CD44 could provide a migratory route for a cryptogenic cell. Third, it is conceivable that endoscopy could seed colon-derived cryptogenic cells into the small intestine; denuded epithelium in areas of active disease may provide a fertile ground for migrating cells. Once in the small intestine, mesenchymal signaling may then induce the progeny of the colon-derived stem cell(s) to follow a small intestinal pattern of differentiation.45,46
The clinical implications of our findings could be considerable. First, we have shown that tumorigenic mutations may be present in large sections of morphologically nondysplastic mucosa. This questions the adequacy of dysplasia as a biomarker for neoplasia risk, because nondysplastic crypts can carry what may prove to be a biologically significant mutation burden. Molecular profiling of the nondysplastic epithelium could potentially better identify patients at risk for neoplasia, with the detection of particular mutants predicting enhanced risk. A controlled study comparing the mutant-clone burden and dynamics between patients with CD cancer and patients without cancer over time is required. Second, it questions the adequacy of performing endoscopic or limited resections for colonic neoplasms in patients with CD, but close surveillance in patients with long-standing Crohn's colitis is still needed until more is understood about the process. It is conceivable that patients who have developed a prolific mutant clone may be best treated by colectomy rather than limited resection, whereas a less prolific clone could be dealt with by localized treatment, although an assessment of both the efficacy and cost-effectiveness of these suggestions would need to be performed. Definite conclusions here are prohibited by the absence of data from patients who never acquired cancers; this is a limitation of our study. The remarkable motility of pretumor clones may be a hallmark of other inflammation-associated cancers such as hepatocellular carcinoma, cholangiocarcinoma, and Barrett's-associated adenocarcinoma, highlighting the need for further investigation into the dynamics of pretumor clones to understand patterns of disease occurrence.
We have shown frequent and occasionally very extensive field cancerization in the chronically inflamed bowel of a few patients with CD. These data provide a precedent for further study of pretumor clones as biomarkers in inflammatory diseases. Our observation of nondysplastic crypts harboring tumorigenic clones questions the utility of dysplasia as the sole biomarker of neoplastic risk. Foremost, we have highlighted that pretumor clonal dynamics can contribute to patterns of tumor occurrence.
Supplementary Material
Supplementary Figure 1. Longitudinal analysis of clone spread over time in patient 5. Tissue was collected for patient 5 between 2004 and 2008. A nondysplastic small intestine biopsy specimen in 2004 showed a KRAS codon 12 mutation; a biopsy specimen 2 years later was genetically wild type. A KRAS codon 13 mutation was detected within intestinalized areas of the anal canal (fistula tracts) and was found with 2 spatially distinct TP53 mutants, suggesting that the KRAS mutation was the founder mutation for the lesion. Four years later, this same KRAS mutation was found throughout a cancer located within the perineal proctectomy scar, indicating that this pretumor clone had grown into a cancer. Clonality between the cancer and the precursor lesion confirmed the presence of the same CDKN2A (p16) and TP53 mutations within both lesions.
Supplementary Figure 2. Genetic and protein analysis of the sigmoid cancer resected in patient 1 in 2000. (A) H&E stain of cancer biopsy specimen showing moderately differentiated adenocarcinoma (original magnification 25×). (B) p53 immunohistochemistry (original magnification 25×). (C) β-catenin immunohistochemistry (original magnification 25×). (D) Methylene green–stained PALM membrane slide showing microdissected areas of the cancer (original magnification 25×). (E) H&E stain showing moderately differentiated adenocarcinoma (original magnification 100×). (F) p53 immunohistochemistry (original magnification 100×). Strong nuclear expression of p53 is visible. (G) β-catenin immunohistochemistry (original magnification 100×). The majority of epithelial cells show nuclear accumulation of β-catenin. (H) Sequencing showing the TP53 c.731G>A mutation present throughout the cancer. (I) LOH data for marker D5S346 indicating 5q LOH.
Supplementary Figure 3. Genetic and protein analysis of the rectal cancer resected in patient 1 in 2004. (A) H&E stain of cancer biopsy specimen showing moderately differentiated adenocarcinoma (original magnification 25×). (B) H&E stain of dysplastic crypt (original magnification 100×). (C) β-catenin immunohistochemistry shows little nuclear accumulation of β-catenin (original magnification 100×). (D) p53 immunohistochemistry shows strong nuclear expression of p53 (original magnification 100×). (E) Sequencing showing the TP53 c.742C>T mutation that was present throughout the cancer. (F) LOH data for marker D17S1881 indicating 17p LOH in cancer crypts.
Supplementary Figure 4. Noninflamed mucosa did not contain mutant clones. (A and B) Jejunum resection margin from patient 3 showing no significant inflammation. The crypts microdissected from this area were wild-type for the CDKN2A and KRAS mutations found in this patient's jejunal cancer and its inflamed resection margins. (A) H&E stain (original magnification 100×) and (B) methylene green–stained laser capture slide (original magnification 100×) showing microdissected crypt. (C and D) Noninflamed terminal ileum tissue from patient 4. Crypts microdissected from this area were all wild type for the TP53 mutation found in the patient's cecal cancer. (C) H&E stain (original magnification 100×) and (D) methylene green–stained laser capture slide (original magnification 100×).
Supplementary Figure 5. Image cytometry. (A) Patient 1 had transverse colon dysplasia resected in 2004. The dysplasia contained a c.775G>T TP53 mutation and was diploid. (B) Patient 1 had transverse colon nondysplastic tissue resected in 2004. The tissue contained a c.742C>T TP53 mutation and was aneuploid. (C and D) Moderately and poorly differentiated regions, respectively, of the rectal cancer in patient 1 in 2004 containing c.742C>T TP53 mutation. (C) The moderately differentiated region was diploid. (D) The poorly differentiated region had developed aneuploidy.
Supplementary Table 1. Primer Details and Reaction Conditions for Sequenced Loci
Supplementary Table 2. Microsatellite LOH Analysis Primers and Reaction Details
Acknowledgments
The authors thank members of the Equipment Park and Experimental Histopathology Laboratories at the Cancer Research UK London Research Institute for technical assistance.
Funding: S.G. received funding from the Price Institute of Surgical Research, University of Louisville, and Sarah Shallenberger Brown and support in part from National Institutes of Health/National Institute of Environmental Health Sciences grant 1P30ES014443-01A1. T.A.G., R.J., S.J.L., and N.A.W. were supported by Cancer Research UK. M.R.-J. is supported by UCLH/UCL Comprehensive Biomedical Research Centre. T.A.G. and M.R.-J. received funding for this study from the University College London Hospitals Charities - Fast Track Grant.
Abbreviations used in this paper
- CD
Crohn's disease
- LOH
loss of heterozygosity
- PCR
polymerase chain reaction
- UC
ulcerative colitis
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Supplementary Material: Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2011.12.004.
References
- 1.Laukoetter MG, Mennigen R, Hannig CM, et al. Intestinal cancer risk in Crohn's disease: a meta-analysis. J Gastrointest Surg. 2011;15:576–583. doi: 10.1007/s11605-010-1402-9. [DOI] [PubMed] [Google Scholar]
- 2.Hemminki K, Li X, Sundquist J, et al. Cancer risks in Crohn disease patients. Ann Oncol. 2009;20:574–580. doi: 10.1093/annonc/mdn595. [DOI] [PubMed] [Google Scholar]
- 3.von Roon AC, Reese G, Teare J, et al. The risk of cancer in patients with Crohn's disease. Dis Colon Rectum. 2007;50:839–855. doi: 10.1007/s10350-006-0848-z. [DOI] [PubMed] [Google Scholar]
- 4.Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn's disease. Aliment Pharmacol Ther. 2006;23:1097–1104. doi: 10.1111/j.1365-2036.2006.02854.x. [DOI] [PubMed] [Google Scholar]
- 5.Gyde SN, Prior P, Macartney JC, et al. Malignancy in Crohn's disease. Gut. 1980;21:1024–1029. doi: 10.1136/gut.21.12.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connell WR, Sheffield JP, Kamm MA, et al. Lower gastrointestinal malignancy in Crohn's disease. Gut. 1994;35:347–352. doi: 10.1136/gut.35.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ribeiro MB, Greenstein AJ, Sachar DB, et al. Colorectal adenocarcinoma in Crohn's disease. Ann Surg. 1996;223:186–193. doi: 10.1097/00000658-199602000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–968. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 9.Braakhuis BJM, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727–1730. [PubMed] [Google Scholar]
- 10.Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40:142–150. doi: 10.5483/bmbrep.2007.40.2.142. [DOI] [PubMed] [Google Scholar]
- 11.McCombs R. A hypothesis on the causation of cancer. Science. 1930;72:423–424. doi: 10.1126/science.72.1869.423. [DOI] [PubMed] [Google Scholar]
- 12.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 13.Franklin WA, Gazdar AF, Haney J, et al. Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest. 1997;100:2133–2137. doi: 10.1172/JCI119748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leedham SJ, Graham TA, Oukrif D, et al. Clonality, founder mutations, and field cancerization in human ulcerative colitis-associated neoplasia. Gastroenterology. 2009;136:542–550.e6. doi: 10.1053/j.gastro.2008.10.086. [DOI] [PubMed] [Google Scholar]
- 15.Nosho K, Kure S, Irahara N, et al. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology. 2009;137:1609–1620. e1–3. doi: 10.1053/j.gastro.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hafner C, Toll A, Fernandez-Casado A, et al. Multiple oncogenic mutations and clonal relationship in spatially distinct benign human epidermal tumors. Proc Natl Acad Sci U S A. 2010;107:20780–20785. doi: 10.1073/pnas.1008365107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung GT, Sundaresan V, Hasleton P, et al. Clonal evolution of lung tumors. Cancer Res. 1996;56:1609–1614. [PubMed] [Google Scholar]
- 18.Farraye FA, Odze RD, Eaden J, et al. AGA medical position statement on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:738–745. doi: 10.1053/j.gastro.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 19.Cairns SR, Scholefield JH, Steele RJ, et al. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002) Gut. 2010;59:666–689. doi: 10.1136/gut.2009.179804. [DOI] [PubMed] [Google Scholar]
- 20.Ullman T, Odze R, Farraye FA. Diagnosis and management of dysplasia in patients with ulcerative colitis and Crohn's disease of the colon. Inflamm Bowel Dis. 2009;15:630–638. doi: 10.1002/ibd.20766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kern SE, Redston M, Seymour AB, et al. Molecular genetic profiles of colitis-associated neoplasms. Gastroenterology. 1994;107:420–428. doi: 10.1016/0016-5085(94)90167-8. [DOI] [PubMed] [Google Scholar]
- 22.Rashid A, Hamilton SR. Genetic alterations in sporadic and Crohn's-associated adenocarcinomas of the small intestine. Gastroenterology. 1997;113:127–135. doi: 10.1016/s0016-5085(97)70087-8. [DOI] [PubMed] [Google Scholar]
- 23.Hussain SP, Amstad P, Raja K, et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancerprone chronic inflammatory disease. Cancer Res. 2000;60:3333–3337. [PubMed] [Google Scholar]
- 24.Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci U S A. 2000;97:3352–3357. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoque AT, Hahn SA, Schutte M, et al. DPC4 gene mutation in colitis associated neoplasia. Gut. 1997;40:120–122. doi: 10.1136/gut.40.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham TA, Wright NA. Investigating the fixation and spread of mutations in the gastrointestinal epithelium. Future Oncol. 2008;4:825–839. doi: 10.2217/14796694.4.6.825. [DOI] [PubMed] [Google Scholar]
- 27.Thirlwell C, Will OCC, Domingo E, et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology. 2010;138:1441–1454. 1454.e1–7. doi: 10.1053/j.gastro.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 28.Maley CC, Galipeau PC, Li X, et al. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett's esophagus. Cancer Res. 2004;64:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 29.Stone JG, Tomlinson IP, Houlston RS. Optimising methods for determining RER status in colorectal cancers. Cancer Lett. 2000;149:15–20. doi: 10.1016/s0304-3835(99)00324-9. [DOI] [PubMed] [Google Scholar]
- 30.Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 31.Prevo LJ, Sanchez CA, Galipeau PC, et al. p53-mutant clones and field effects in Barrett's esophagus. Cancer Res. 1999;59:4784–4787. [PubMed] [Google Scholar]
- 32.Flejou JF, Gratio V, Muzeau F, et al. p53 abnormalities in adenocarcinoma of the gastric cardia and antrum. Mol Pathol. 1999;52:263–268. doi: 10.1136/mp.52.5.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida T, Mikami T, Mitomi H, et al. Diverse p53 alterations in ulcerative colitis-associated low-grade dysplasia: full-length gene sequencing in microdissected single crypts. J Pathol. 2003;199:166–175. doi: 10.1002/path.1264. [DOI] [PubMed] [Google Scholar]
- 34.Greaves LC, Preston SL, Tadrous PJ, et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci U S A. 2006;103:714–719. doi: 10.1073/pnas.0505903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez-Gonzalez L, Deheragoda M, Elia G, et al. Analysis of the clonal architecture of the human small intestinal epithelium establishes a common stem cell for all lineages and reveals a mechanism for the fixation and spread of mutations. J Pathol. 2009;217:489–496. doi: 10.1002/path.2502. [DOI] [PubMed] [Google Scholar]
- 36.Cheng H, Bjerknes M, Amar J, et al. Crypt production in normal and diseased human colonic epithelium. Anat Rec. 1986;216:44–48. doi: 10.1002/ar.1092160108. [DOI] [PubMed] [Google Scholar]
- 37.Chao DL, Eck JT, Brash DE, et al. Preneoplastic lesion growth driven by the death of adjacent normal stem cells. Proc Natl Acad Sci U S A. 2008;105:15034–15039. doi: 10.1073/pnas.0802211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nystul T, Spradling A. An epithelial niche in the Drosophila ovary undergoes long-range stem cell replacement. Cell Stem Cell. 2007;1:277–285. doi: 10.1016/j.stem.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 39.Kato S, Han SY, Liu W, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moreno E. Is cell competition relevant to cancer? Nat Rev Cancer. 2008;8:141–147. doi: 10.1038/nrc2252. [DOI] [PubMed] [Google Scholar]
- 41.Brentnall TA, Crispin DA, Rabinovitch PS, et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology. 1994;107:369–378. doi: 10.1016/0016-5085(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 42.Burmer GC, Rabinovitch PS, Haggitt RC, et al. Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterology. 1992;103:1602–1610. doi: 10.1016/0016-5085(92)91184-6. [DOI] [PubMed] [Google Scholar]
- 43.Chen R, Rabinovitch PS, Crispin DA, et al. DNA fingerprinting abnormalities can distinguish ulcerative colitis patients with dysplasia and cancer from those who are dysplasia/cancer-free. Am J Pathol. 2003;162:665–672. doi: 10.1016/S0002-9440(10)63860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams DR, Coller JA, Corman ML, et al. Anal complications in Crohn's disease. Dis Colon Rectum. 1981;24:22–24. doi: 10.1007/BF02603444. [DOI] [PubMed] [Google Scholar]
- 45.Kedinger M, Lefebvre O, Duluc I, et al. Cellular and molecular partners involved in gut morphogenesis and differentiation. Philos Trans R Soc Lond B Biol Sci. 1998;353:847–856. doi: 10.1098/rstb.1998.0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benahmed F, Gross I, Gaunt SJ, et al. Multiple regulatory regions control the complex expression pattern of the mouse Cdx2 homeobox gene. Gastroenterology. 2008;135:1238–1247. 1247 e1–3. doi: 10.1053/j.gastro.2008.06.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Longitudinal analysis of clone spread over time in patient 5. Tissue was collected for patient 5 between 2004 and 2008. A nondysplastic small intestine biopsy specimen in 2004 showed a KRAS codon 12 mutation; a biopsy specimen 2 years later was genetically wild type. A KRAS codon 13 mutation was detected within intestinalized areas of the anal canal (fistula tracts) and was found with 2 spatially distinct TP53 mutants, suggesting that the KRAS mutation was the founder mutation for the lesion. Four years later, this same KRAS mutation was found throughout a cancer located within the perineal proctectomy scar, indicating that this pretumor clone had grown into a cancer. Clonality between the cancer and the precursor lesion confirmed the presence of the same CDKN2A (p16) and TP53 mutations within both lesions.
Supplementary Figure 2. Genetic and protein analysis of the sigmoid cancer resected in patient 1 in 2000. (A) H&E stain of cancer biopsy specimen showing moderately differentiated adenocarcinoma (original magnification 25×). (B) p53 immunohistochemistry (original magnification 25×). (C) β-catenin immunohistochemistry (original magnification 25×). (D) Methylene green–stained PALM membrane slide showing microdissected areas of the cancer (original magnification 25×). (E) H&E stain showing moderately differentiated adenocarcinoma (original magnification 100×). (F) p53 immunohistochemistry (original magnification 100×). Strong nuclear expression of p53 is visible. (G) β-catenin immunohistochemistry (original magnification 100×). The majority of epithelial cells show nuclear accumulation of β-catenin. (H) Sequencing showing the TP53 c.731G>A mutation present throughout the cancer. (I) LOH data for marker D5S346 indicating 5q LOH.
Supplementary Figure 3. Genetic and protein analysis of the rectal cancer resected in patient 1 in 2004. (A) H&E stain of cancer biopsy specimen showing moderately differentiated adenocarcinoma (original magnification 25×). (B) H&E stain of dysplastic crypt (original magnification 100×). (C) β-catenin immunohistochemistry shows little nuclear accumulation of β-catenin (original magnification 100×). (D) p53 immunohistochemistry shows strong nuclear expression of p53 (original magnification 100×). (E) Sequencing showing the TP53 c.742C>T mutation that was present throughout the cancer. (F) LOH data for marker D17S1881 indicating 17p LOH in cancer crypts.
Supplementary Figure 4. Noninflamed mucosa did not contain mutant clones. (A and B) Jejunum resection margin from patient 3 showing no significant inflammation. The crypts microdissected from this area were wild-type for the CDKN2A and KRAS mutations found in this patient's jejunal cancer and its inflamed resection margins. (A) H&E stain (original magnification 100×) and (B) methylene green–stained laser capture slide (original magnification 100×) showing microdissected crypt. (C and D) Noninflamed terminal ileum tissue from patient 4. Crypts microdissected from this area were all wild type for the TP53 mutation found in the patient's cecal cancer. (C) H&E stain (original magnification 100×) and (D) methylene green–stained laser capture slide (original magnification 100×).
Supplementary Figure 5. Image cytometry. (A) Patient 1 had transverse colon dysplasia resected in 2004. The dysplasia contained a c.775G>T TP53 mutation and was diploid. (B) Patient 1 had transverse colon nondysplastic tissue resected in 2004. The tissue contained a c.742C>T TP53 mutation and was aneuploid. (C and D) Moderately and poorly differentiated regions, respectively, of the rectal cancer in patient 1 in 2004 containing c.742C>T TP53 mutation. (C) The moderately differentiated region was diploid. (D) The poorly differentiated region had developed aneuploidy.
Supplementary Table 1. Primer Details and Reaction Conditions for Sequenced Loci
Supplementary Table 2. Microsatellite LOH Analysis Primers and Reaction Details