

Abstract
Insulin resistance is an underlying mechanism of type 2 diabetes and its vascular complications. Recent evidence suggests that crosstalk between angiotensin II (Ang II) and the insulin signaling in vascular smooth muscle cell (VSMC) may contribute to cellular insulin resistance. We hypothesized that Ang II inhibits the anti-mitogenic pathways while enhancing the mitogenic pathways stimulated by insulin via activation of Protein Tyrosine Phosphatase-1B (PTP-1B) in VSMC. We found that Ang II significantly inhibited insulin-induced phosphorylation of tyrosine 608 of IRS-1 and serine 473 of Akt, a downstream member of anti-mitogenic pathway of insulin. In contrast, Ang II increased the serine phosphorylation of IRS-1 which was not affected by the presence of insulin. Activation of p42/p44 MAPK (a mitogenic pathway) induced by insulin was further enhanced by Ang II. Transfection of VSMC with PTP-1B antisense oligonucleotide markedly reduced the effects of Ang II on insulin signaling. Furthermore, an increase in VSMC growth was attenuated by PTP-1B antisense only in the presence of both Ang II and insulin. Finally, we also showed that Ang II -induced activation of PTP-1B in VSMC was PKA/JAK2 dependent. We conclude that Ang II modulates both anti-mitogenic and mitogenic pathways of insulin via the activation of PTP-1B.
Keywords: insulin resistance, Angiotensin II, PTP-1B, growth
Graphical Abstract
1. Introduction
Insulin resistance is generally referred to reduced biological effects of insulin (Biddinger and Kahn, 2006). It is closely associated with the development of type2 diabetes and cardiovascular diseases (Hsueh et al., 2004; Saltiel and Kahn, 2001). Despite the metabolic function, previous studies have consistently shown that insulin can also regulate cell growth and differentiation, known as the non-metabolic functions of insulin (Biddinger and Kahn, 2006). Although vasculature is not considered as “a classic insulin sensitive tissue”, insulin resistance in blood vessel can occur and has been suggested to be a cause of vascular diseases during insulin resistant condition (Nigro et al., 2006). In vasculature, insulin mediates glucose uptake (Izawa et al., 2005) and acts as an anti-atherosclerotic factor by increasing nitric oxide (NO) release by endothelial cells via activation of phosphatidylinositol 3 kinase (PI3K)/Akt (Zeng et al., 2000; Zeng and Quon, 1996). Moreover, insulin regulates VSMC growth, proliferation and migration via activation of the mitogen-activated protein kinase (MAPK) (Wang et al., 2003). Similar to other tissues, when insulin resistance takes place, not all of the insulin regulated pathways become equally resistant to insulin (Biddinger and Kahn, 2006; Nigro et al., 2006). Attenuated activation of the insulin receptor substrate-1 (IRS-1)/PI3K/Akt pathway while maintained or enhanced the activation of the MAPK pathway is observed in different tissues during insulin resistant condition (Nigro et al., 2006). However, the underlying mechanisms leading to alterations of insulin signaling pathways in the vasculature are not yet clearly understood.
Previous studies have shown that Ang II infusion induces insulin resistance in vivo (Ogihara et al., 2002). and inhibiting the Ang II actions (by Ang II converting enzyme inhibitors and AT1 receptor blockers) can reduce the development of type 2 diabetes in hypertensive patients by improving insulin sensitivity (Carvalho et al., 1997; Folli et al., 1999). Studies in different insulin resistance models e.g. obese zucker rats (Henriksen et al., 2001) and fructose-fed hypertensive rats (Navarro-Cid et al., 1995) demonstrated that angiotensin receptor (AT1R) antagonists reduced insulin resistance. All of these data suggest that Ang II –mediated development of insulin resistance is associated with activation of AT1R (Igarashi et al., 2007). However, the mechanism by which Ang II causes alterations of insulin signaling pathways especially in the vasculature has not been fully understood.
Several protein tyrosine phosphatases (PTPases) such as PTP-1B, LAR, SHIP2 and PTEN are implicated in the development of insulin resistance (Asante-Appiah and Kennedy, 2003; Byon et al., 1998). However, the most convincing data support a critical role of PTP-1B as a modulator of insulin signaling (Asante-Appiah and Kennedy, 2003; Byon et al., 1998). For example, PTP-1B knockout mice are insulin sensitive and maintain euglycemia with one half the insulin level found in the wild type controls (Elchebly et al., 1999). However, the role of PTP-1B was only investigated from a metabolic aspect. Little is known about the functions of PTP-1B in regulating insulin signaling pathways in the vasculature. Interestingly, our group has recently shown that Ang II inhibits insulin-induced tyrosine phosphorylation of insulin receptor (IR) in VSMC by activation of protein kinase A (PKA) (Marrero et al., 2004). Based on our recent findings and others, we speculate that activation of PTP-1B through PKA is an underlying mechanism of Ang II-induced blockade of insulin-induced IRS-1/PI3K/Akt pathway in VSMC (Marrero et al., 2004). Furthermore, Dube et al. demonstrated that PTP-1B acts as a positive regulator for Ras and thus enhances MAPK activity (Dube et al., 2004), suggesting a growth promoting effect of PTP-1B via MAPK. Taken together, activation of PTP-1B by Ang II in VSMC may potentially alter insulin signaling and facilitates VSMC growth and proliferation. While the role of Ang II in hypertension is well established, not much is known regarding the effect of Ang II on insulin signaling pathways in VSMC. Therefore, we tested the hypothesis that Ang II modulated insulin signaling pathways in VSMC via activation of PTP-1B. Our study sheds light on a novel molecular mechanism of insulin resistance in VSMC.
2. Materials and Methods
2.1. Culture of rat vascular smooth muscle cells (VSMC)
VSMC were obtained from the aorta of male Sprague-Dawley rats using an explant method as previously described (Florian and Watts, 1998). Subcultures 3–8 were used in these studies. 80%–90% confluent VSMC was placed into serum-free media with normal (5.5 mM) glucose 24 hours prior to experimentation. Cells were stimulated with insulin (100 nM) (Sigma) for indicated time with or without pre-incubation of Ang II (100 nM) (Sigma) for 1 hr. We chose to pre-incubate VSMC with Ang II for 1 hour because our preliminary studies showed a maximal inhibitory effect of Ang II on insulin signaling molecules (IRS-1 and Akt) compared to other pre-incubation time points (15 min or 30 min; data not shown).
In some experiments, VSMC were treated with specific inhibitors before stimulation of Ang II, H-89 (PKA inhibitor, 0.05μM, 30 min), Calphostin C (PKC inhibitor, 0.05 μM, 30 min), KT-5827 (PKG inhibitor, 10 μM, 30 min), PP2 (Src inhibitor, 0.1 μM, 30 min) and AG-490 (JAK2 inhibitor, 0.1 μM, 1 hour). After the experiments were carried out, VSMC were washed with an ice-cold phosphate buffered saline (1xPBS) solution containing sodium orthovanadate (1 mM). VSMC were lysed in 500 μL of ice cold RIPA lysis buffer. Cells were then harvested and sonicated. Lysates were centrifuged at 14000 rpm for 10 min. The supernatant was collected and total protein was measured by Lowry high assay, using BSA standard (BioRad) and then samples were frozen at −80°C until used.
2.2. Western Blotting
VSMC lysates were placed in denaturing SDS buffer and boiled for 5 minutes, then resolved by SDS-PAGE gel electrophoresis. Proteins were transferred to nitrocellulose membranes and blocked (60 min, 22 °C) by 5% skim milk in T-TBS (TBS with 0.0.5% Tween-20, pH 7.4). Membranes were incubated overnight (4°C) with affinity purified anti-phosphospecificanti-Tyr608 IRS-1 (1:500 dilution) from Biosource, Anti-Ser307-IRS-1 (1:1000 dilution), anti-Ser612-IRS-1 (1:1000 dilution), anti-Ser473-Akt (1:1000 dilution) and anti-Thr202/Tyr204-p42/44 MAPK (1:1000 dilution) from Cell Signaling. Membranes were washed with T-TBS and then incubated with the appropriate secondary antibody (60 min, 22 °C). After washing with T-TBS the bands were visualized using Pierce Supersignal substrate chemiluminescence and Kodax Biomax film. Membranes were stripped with 37°C stripping buffer (Pierce) for 30 minutes and the complete removal of the primary antibody was ensured. Membranes were washed with T-TBS and re-blocked with 5% skim milk before re-probing with the following antibodies: total IRS-1 (1:1000 dilution) (BD Biosciences), Akt and p42/44 MAPK (1:1000 dilution) (Cell Signaling). At the end of experiment, membranes were also incubated with smooth muscle alpha actin antibody (Oncogene, Boston, MA) and the appropriate secondary antibody to ensure equal loading of total protein. Molecular weight markers assessed specificity of the bands.
2.3. Immunoprecipitation Studies
Aliquots of 500 μg of VSMC lysate were used for immunoprecipitation. Anti-PTP-1B antibody (1 μg) (BD Biosciences) were added to the VSMC lysate and samples were incubated for 2 hours at 4 °C with rocking. 50 μl of protein A/G agarose beads (Santacruz Biotechnology, Santacruz, CA, U.S.A.) were added, followed by incubation overnight at 4 °C with rocking. The agarose beads then were pelleted by centrifugation at 2,000 rpm for 3 minutes. The beads were washed three times with ice-cold rinse buffer containing 1x PBS and 1 mM Sodium Vanadate. Immunoprecipitated proteins were dissolved by boiling for 5 minutes at 95 °C in SDS sample buffer and analyzed by SDS-PAGE. Proteins then were transferred to a nitrocellulose membrane and blotted with either anti-phosphotyrosine (1:1000 dilution) (PY4G10, Upstate) or anti-phosphoserine (1:1000 dilution) antibodies (Zymed). Finally, proteins were visualized using horseradish-peroxidase conjugated with goat anti-mouse or donkey anti-rabbit IgG and enhanced chemiluminescence kit and Kodax Biomax film. Memebranes were stripped and re-probed with anti-PTP–1B antibody.
2.4. PTP-1B Activity Assay
PTP-1B phosphatase assays were performed on the PTP-1B immunoprecipitates as previously described (Marrero et al., 1998). Briefly, VSMCs were starved for 24 hours prior to experimentation and then stimulated by 100 nM Ang II for the indicated time. Cells were lysed and total proteins were immunoprecipitated with PTP-1B antibodies. Immunocomplexes were washed three times with ice-cold wash buffer and then three times with phosphatase buffer. Immunocomplex pellets were then resuspended in 100 μl of phosphatase buffer containing 1 mg/ml BSA, 5 mM EDTA, and 10 mM dithiothreitol. The reaction was initiated by the addition of p-nitrophenyl phosphate (10 mM final concentration). After 30-min incubation at room temperature, the reaction was stopped by the addition of 1 M NaOH and absorbance of the sample was determined at 410 nm in a spectrophotometer.
2.5. PTP-1B sense and antisense
The experiments were carried out as described in a previous study by Duff et al. (Duff et al., 1995). Oligonucleotides (ISIS-113715, Isis Pharmaceuticals, Carlsbad, CA) were added to the cells in Opti-MEM containing Lipofectin. After indicated times, the medium was removed, 0.1% calf serum/DMEM was added, and cells were allowed to recover for 30 min prior to stimulation by Ang II or insulin.
2.6 Akt activity assay
This experiment was done using PathScan Phospho-Akt and PathScan Total Akt1 Sandwich ELISA Kit (Cell Signaling) according to the manufacturer’s instructions.
2.7. Thymidine incorporation
The experiments were done as previously described (Marrero et al., 1997). Briefly, VSMC were plated in 96-well plates and maintained in DMEM supplemented with 10% fetal bovine serum. 24 and 48 h after transfection with PTP-1B sense or antisense, some groups were stimulated with insulin and/or pre-incubation with Ang II (100 nM) for 1 hour. VSMC were pulsed with 1 mCi/ml [3H] thymidine (New England Nuclear, Boston, MA) and then harvested into trichloroacetic acid-precipitable material. Cells were washed with phosphate-buffered saline, incubated in 10% trichloroacetic acid at 4 °C, dissolved at room temperature in 1 mol/liter, and dried on filter paper. The paper was washed three times with phosphate-buffered saline, and then the samples were placed in scintillation liquid and counted on a scintillation counter (Beckman Inc., Palo Alto, CA). Data were plotted as the number of cpm/well. Each experimental data point represents duplicate wells from at least four different experiments.
2.8. PKA activity assay
The assay was performed with the PKA assay kit from Calbiochem (Promega) according to the manufacturer’s instructions.
2.9. Data analysis and statistics
Data are presented as means ± standard error of the mean (SEM) for the number of animals in parentheses. Statistical analysis was carried out with the Sigma Stat program (Jandel Scientific, San Rafael, CA). Unpaired Student t-test was used to compare between two groups. One-way ANOVA followed by a Student-Newman-Keuls post hoc test was used to compare three or more groups. P < 0.05 values were considered to be significant. Band density was quantified using the program NIH Image.
3. Results
3.1. Ang II inhibits insulin-induced activation of anti-mitogenic pathways
We first investigate the effect of Ang II on insulin-induced activation of IRS-1 and Akt in culture rat aortic VSMC. It is well documented that tyrosine phosphorylation of IRS-1 is required for insulin-stimulated PI3K/Akt (Gual et al., 2005). Moreover, the specific tyrosine phosphorylation at 608 is important for the docking between IRS-1 and PI3K (Esposito et al., 2003). As shown in Figure 1A, insulin alone significantly increased tyrosine 608 phosphorylation of IRS-1 in a time-dependent manner (% phosphorylation: 84.78±5.9 at 5 min and 82.79±4.67 at 10 min vs. 18.87±5.93 in control group,* p<0.05). However, the phosphorylation of tyrosine 608 of IRS-1-induced by insulin was significantly inhibited by Ang II (% phosphorylation from insulin alone: 84.78±5.9 at 5 min and 82.79±4.67 at 10 min vs. % phosphorylation from insulin+Ang II: 48.4±3.3 at 5 min and 64.79±4.67 at 10 min, *p<0.05).
Figure 1.

Ang II (100 nM) altered the anti-mitogenic pathways of insulin. Phosphorylation of tyrosine 608 of IRS-1 (A), serine 307 IRS-1 (B), serine 612 IRS-1 (C) and serine 473 Akt (D). Representative blots are shown and the results represent the mean ± SEM of 3–6 independent experiments. # p<0.05 vs. control, * p<0.05 vs. insulin alone.
IRS-1 also can be phosphorylated at specific serine sites which negatively regulate IRS functions (Gual et al., 2005). Previous studies have shown that phosphorylation of serine 307 and serine 612 of IRS-1 results in impairment of insulin signaling transmission (Gual et al., 2005). Therefore, we next examined the effect of Ang II on the phosphorylation of serine 307 and 612 of IRS-1 in order to delineate further the mechanism of Ang II-induced impairment of insulin signaling in VSMC. Ang II significantly increased phosphorylation of serine 307 and 612 of IRS-1 (*p<0.05 vs. insulin alone) in VSMC but this activation was not affected by insulin (Figure 1B and 1C).
It has been suggested that Akt is required for insulin-stimulated glucose uptake and it is important for insulin signal activation (Saltiel and Kahn, 2001). Accumulating data also show that decreases in insulin-induced Akt activation is found in various models of insulin resistance (Krook et al., 1997; Shao et al., 2000). As shown in Figure 1D, insulin alone significantly increased phosphoryaltion of serine 473 of Akt maximally at 10 min (% phosphorylation: 124.82±15.9 at 10 min vs. 21±8.8 in control group, *p<0.05) and this effect was attenuated by pre-incubation with Ang II (*p<0.05 vs. insulin alone). Therefore, these data suggest that Ang II inhibited the ability of insulin to activate the anti-mitogenic pathway in VSMC, as shown by decreased tyrosine phosphorylation of IRS-1, increased serine phosphorylation of IRS-1 and decreased serine phosphorylation of Akt.
3.2. Ang II enhances insulin-induced activation of mitogenic pathways
Another main signaling cascade stimulated by insulin is the MAPK. The p42/44 MAPK pathway is specifically involved in growth and proliferation of VSMC (mitogenic effect of insulin) (Nigro et al., 2006). Therefore, we next investigated the effect of insulin-induced phosphorylation of p42/44 MAPK in the presence or absence of Ang II in VSMC. Insulin itself only slightly increased p42/44 MAPK phosphorylation and this activation did not reach statistically significance (p>0.05 vs. control group). As expected, Ang II markedly increased activation of p42/44 MAPK (% phosphorylation: 49.95±5.8 from Ang II alone vs. 0±0 from control group, *p<0.05). Interestingly, we observed a slight decrease in p42/44 MAPK phosphorylation after pre-incubation of Ang II for 1 hour followed by insulin stimulation at 5 and 10 min. It is possible that insulin induces some effects to oppose Ang II actions at early time point. However, Ang II significantly enhanced p42/44 MAPK activation induced by insulin at 30 min (*p<0.05 vs. insulin alone and #p<0.05 vs. Ang II alone) (Figure 2). Therefore, these data suggest that Ang II enhanced insulin-induced activation of the mitogenic pathway. In this experiment, we chose to utilize the lower concentration of Ang II at 1 nM in this experiment because Ang II at 100 nM maximally activates MAPK. Therefore, it was not possible to detect differences in the levels of p42/44 MAPK activation comparing between Ang II alone and Ang II with insulin (data not shown) when Ang II 100 nM was used. This limitation is due to the sensitivity of western blot analysis with ECL and film detection methods.
Figure 2.

Ang II (1 nM) enhanced insulin-induced activation of mitogenic pathway. Representative blots are shown and the results represent the mean ± SEM of 3–5 independent experiments. * p<0.05 vs. insulin alone. # p<0.05 compared to Ang II alone.
3.3. Ang II impairs insulin-induced activation of anti-mitogenic pathways by activation of PTP-1B
Accumulating data support a critical role of PTP-1B in insulin signaling (Asante-Appiah and Kennedy, 2003) in the classic insulin sensitive tissues but little is known about the role of PTP-1B in VSMC. Therefore, we set forth to investigate whether PTP-1B is activated by Ang II in VSMC. As shown in Figure 4B, Ang II increased PTP-1B activity in VSMC, which was blocked by the tyrosine phosphatase inhibitor, sodium orthovanadate (*p<0.05 vs. Ang II alone). We then hypothesized that Ang II modulated insulin signaling molecules by activation of PTP-1B. We utilized a PTP-1B antisense oligonucleotide to deplete the expression of PTP-1B. We found that PTP-1B antisense oligonucleotide markedly down-regulated PTP-1B expression in VSMC after 6 hours of treatment compared to treatment with sense (Figure 3A). Importantly, reduction of PTP-1B expression by this antisense inhibited the blockade action of Ang II on insulin-induced tyrosine phosphorylation of IRS-1 (Figure 3B). We further investigated the activation of Akt in the presence or absence of the PTP-1B antisense (Figure 3C) and consistent with the tyrosine phosphorylation of IRS-1, the inhibitory effect of Ang II on the insulin-induced Akt activation also was significantly inhibited by the PTP-1B antisense (Figure 3C) (* p<0.05 vs. Ang II+insulin, Ang II+insulin+PTP-1B sense). Taken together, these data suggest that the activation of PTP-1B is involved in Ang II-induced insulin resistance in VSMC.
Figure 4.

Ang II-induced PTP-1B activation in VSMC was a PKA dependent mechanism. Only H-89 inhibited serine phosphorylation of PTP-1B (A). Representative blots are shown from 5 independent experiments. Ang II-induced PTP-1B activation was significantly attenuated by H-89 and Sodium Vanadate (B). *p<0.05 compared to Ang II alone (control) (n=5).
Figure 3.

PTP-1B antisense inhibited the effects of Ang II on insulin signaling. PTP-1B expression was attenuated by PTP-1B antisense after 6 hours of transfection (A). Ang II failed to inhibit tyrosine phosphorylation of IRS-1(B) and activity of Akt - induced by insulin after VSMC was transfected with PTP-1B antisense (C) * p<0.05 vs. Ang II+insulin, Ang II+insulin+PTP-1B sense. PTP-1B antisense blocked the combined effect of insulin and Ang II on DNA synthesis (D) #p<0.05 vs. control group without Ang II, *p<0.05 vs. Ang II alone, &p<0.05 vs. Ang II+insulin+PTP-1B antisense. Representative blots are shown and the results represent the mean ± SEM of 5 independent experiments.
3.4. Ang II enhances insulin-promoted VSMC growth by activation of PTP-1B
We next determined the effect of PTP-1B antisense on VSMC growth by measuring DNA synthesis. As shown in Figure 3D, insulin alone had no effect on DNA synthesis. However, Ang II itself significantly increased VSMC growth (#p<0.05 vs. control group without Ang II). We observed that DNA synthesis of VSMC was markedly enhanced by pre-incubation of VSMC with Ang II followed by insulin exposure compared to those stimulated with either insulin or Ang II alone (*p<0.05 vs. Ang II alone). Although PTP-1B antisense oligonucleotide had no effect on Ang II’s ability to increase DNA synthesis, it markedly blocked the combined effect of insulin and Ang II (&p<0.05 vs. Ang II+insulin+PTP-1B antisense).
3.5. Ang II induces activation of PTP-1B via a PKA dependent mechanism
It has been reported that PTP-1B can be activated via a serine phosphorylation (Tao et al., 2001). Two potential serine kinases have been reported to be involved in the regulation of PTP-1B activity: PKA (Tao et al., 2001) and PKC (Flint et al., 1993). To determine the potential involvement of these two kinases in our system, we used specific pharmacological inhibitors, H-89 and Calphostin C. VSMC were treated with either H-89 or Calphostin C prior to Ang II exposure and PTP-1B then was immunoprecipitated with an anti-PTP-1B antibody and immunoblotted with either anti- phosphoserine or anti- phosphotyrosine antibody. H-89 markedly inhibited the serine phosphorylation of PTP-1B, whereas Calphostin C had no effect (Figure 4A). These data suggest that activation of PTP-1B in VSMC by Ang II occurs via a PKA dependent serine phosphorylation and is independent of PKC. We also confirmed our findings by investigating the PTP-1B activity in the presence or absence of H-89. As shown in Figure 4B, the Ang II induced PTP-1B activation was attenuated significantly by the PKA inhibitor (* p<0.05 vs. Ang II alone). All of these data indicate that activation of PTP-1B by Ang II occurs through PKA dependent mechanism.
3.6. JAK2 is an upstream kinase of Ang II-induced PKA activation in VSMC
To further examine the up-stream molecule of PKA in our system, we then utilized various pharmacological inhibitors and examined the effects of these inhibitors on PKA activity. We found that PKC (Calphosin C), PKG (KT5823) and Src kinase (PP2) specific inhibitors did not affect the Ang II-induced activation of PKA, whereas a JAK2 specific inhibitor (AG-490) significantly (*p<0.01 vs. Ang II alone) reduced the Ang II-induced PKA activation (Figure 5). We conclude that Ang II increases activity of PKA in VSMC through JAK2.
Figure 5.

Effects of various inhibitors on the Ang II-induced activation of PKA in VSMC. AG-490 markedly inhibited the Ang II-induced PKA activity. #p<0.05 vs. control, *p<0.01 vs. Ang II alone (n=5).
4. Discussion
Our present data demonstrated that activation of PTP-1B by Ang II resulted in aberration of insulin signaling pathways in VSMC. We observed a selective inhibition of insulin –induced activation of IRS-1/Akt pathway while enhanced activation of MAPK, which is consistent with other studies shown in various insulin resistant state (Jiang et al., 1999). Moreover, these studies demonstrated the activation of PTP1-B in VSMC by Ang II was via a JAK2/PKA-dependent mechanism. Our studies demonstrated the following novel findings: 1) the role of PTP-1B in smooth muscle insulin resistance and 2) JAK2/PKA is involved in PTP-1B activation in VSMC. These data suggest that alterations of insulin signaling in VSMC caused by Ang II may contribute to the vascular insulin resistance which is an important factor determined the cardiovascular complications.
Previous studies have shown that Ang II inhibits insulin signaling pathways in VSMC at multiple levels (Folli et al., 1997; Izawa et al., 2005). The specific inhibition of insulin-induced activation of IRS-1/PI3K/Akt by Ang II results in decreased GLUT4 translocation to the plasma membrane, followed by a reduction of glucose uptake in VSMC, suggesting insulin-induced glucose uptake in VSMC is dependent on IRS-1/PI3K/Akt, similar to other insulin-sensitive tissues (Izawa et al., 2005). Importantly, activation of IRS/PI3K/Akt signaling by insulin is also known as an anti-mitogenic pathway (non-metabolic effect) in both VSMC and endothelial cells (Nigro et al., 2006; Wang et al., 2003). Insulin maintains VSMC quiescence and counteracts platelet-derived growth factor (PDGF) –promoted dedifferentiation via a PI3K-dependent mechanism (Wang et al., 2003). Furthermore, in endothelium, insulin has been proposed to induce IRS-1/PI3K/Akt signaling, which stimulates the production of NO thereby providing an anti-mitogenic effects of insulin (Kuboki et al., 2000; Zeng et al., 2000). Our present studies support previous reported data which demonstrate that Ang II inhibited the insulin-induced activation of IRS-1 (Tyr608) and Akt (Ser473) in VSMC (Folli et al., 1997; Izawa et al., 2005). Moreover, in this study we showed that Ang II maintained the serine phosphorylation of IRS-1 at Ser307 and Ser612 even in the presence of insulin. It is well documented that serine phosphorylation of IRS-1 at these sites causes a reduction of insulin-induced activation of signaling downstream by a number of mechanisms such as decreasing tyrosine phosphorylation, promoting degradation and interrupting the association of IRS-1 with PI3K (Gual et al., 2005). We speculate that the reduction of insulin-induced tyrosine phosphorylation of IRS-1 and serine phosphorylation of Akt caused by Ang II in our experiments may be partly attributable to the serine phosphorylation of IRS-1. Furthermore, activation of MAPK by Ang II has been reported to increase serine phosphorylation of IRS (Izawa et al., 2005), leading to impairment of insulin-induced activation of IRS/PI3K/Akt signaling. Because we also observed an increase p42/44 MAPK activation and serine phosphorylation of IRS-1 in our system following stimulations of insulin and Ang II, we cannot rule out the possibility that p42/44 MAPK contributes to the decrease in insulin-induced activation of IRS/PI3K/Akt in VSMCs. However, our studies add another novel mechanism of Ang II- induced insulin resistance.
This study is the first to demonstrate that Ang II-induced alteration of insulin signaling in VSMC occurs via activation of PTP-1B. Although most of the previous studies have emphasized the critical roles of PTP-1B in insulin resistance in various tissues such as liver, skeletal muscle, adipose tissue (Elchebly et al., 1999; Wu et al., 2005; Zinker et al., 2002) and brain (Bence et al., 2006), the functions of PTP-1B in regulating insulin signaling in the vasculature have not been well investigated. We previously reported that Ang II blunted the insulin-induced tyrosine phosphorylation of IRβ in VSMC (Marrero et al., 2004). Taken together with current studies, we propose that PTP-1B is involved with inhibition of insulin signaling in VSMC through dephosphorylation of IRβ, similar to other insulin sensitive tissues.
Interestingly, Dube et al. reported that PTP-1B –deficient cells displayed a decrease in Ras/MAPK activity and reduced proliferation, suggesting PTP-1B may be a positive regulator of Ras/MAPK (Dube et al., 2004). In addition, in vivo studies in an acute arterial injury model showed that PTP-1B mRNA expression was specifically up-regulated in proliferating and migrating SMC in response to vessel damage (Wright et al., 2000). Possibly, the up-regulation of PTP-1B in turn increases the mitogenic activation through MAPK pathway. Since PTP-1B appears to be a negative regulator for insulin-induced IRS/PI3K/Akt but positively regulates MAPK, our present findings are consistent with this contention.
Our data also showed that the activation of PTP-1B by Ang II in VSMC is via a PKA/JAK2 dependent mechanism. Consistent with this notion, PKA has been shown to be a positive regulator of PTP-1B through serine phosphorylation hence increase its activity (Brautigan and Pinault, 1993; Tao et al., 2001). However, PKA –induced activation of PTP-1B in fat and skeletal muscle is cAMP-dependent (Tao et al., 2001). Even though we cannot rule out the possibility of cAMP dependent PKA-induced PTP-1B activation in our system, the activation of PKA by Ang II in VSMC has been reported to occur via a cAMP-independent mechanism (Dulin et al., 2001). Since cAMP is known to be a second messenger for vasodilators (Rashid et al., 2006) and inhibits VSMC growth (Zhuplatov et al., 2006), thus antagonizing the Ang II actions in VSMC, the PKA-dependent activation of PTP-1B by Ang II in VSMC most likely occur through a non-cAMP-dependent. An alternative method of PKA activation is through IκB phosphorylation. It has been suggested that activation of PKA by vasoactive peptides involves IκB degradation rather than being cAMP-dependent (Dulin et al., 2001). Furthermore, JAK2 can phosphorylate IκB (Digicaylioglu and Lipton, 2001) which may subsequently increase PKA activity. Ang II-stimulated tyrosine phosphorylation and activation of JAK2 has been repeatedly reported by our group (Shaw et al., 2003a; Shaw et al., 2003b). Indeed, our results using AG-490 and H-89 suggest that Ang II-induced activation of PTP-1B can occur by this alternative mechanism. However, this hypothesis needs further investigation.
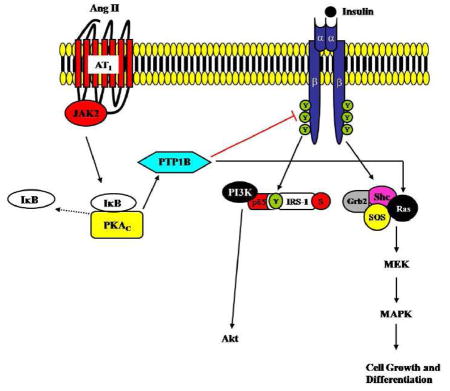
Previous studies have shown that the inhibitory effects of Ang II on insulin signaling occur through AT1R in various tissues such as skeletal muscle (Horiuchi et al., 2006), endothelium (Cooper et al., 2007) and VSMC (Cooper et al., 2007; Igarashi et al., 2007). Although we did not directly test that in these studies, our data here suggest that the induction of PTP-1B activity by Ang II in our system may depend upon the activation of AT1R. This notion is supported by our observations that Ang II-induced PKA activation was sensitive to JAK2 inhibition, suggesting JAK2 may be a crucial component of Ang II-induced activation of PTP-1B in VSMC. As our group repeatedly demonstrated that the activation of JAK2 by Ang II occurs via AT1R (Shaw et al., 2003a; Shaw et al., 2003b) in VSMC, we speculate that Ang II signaling through AT1R results in an increased PTP-1B activity and further causes insulin resistance at least at cellular levels. In addition to AT1R, other RAS (renin-angiotensin system) components have been shown to be involved with insulin signaling. For example, AT2R null mice exhibited a smaller insulin-induced glucose uptake in adipose tissues compared to control groups (Horiuchi et al., 2006), suggesting the function of AT2R in promoting activation of insulin signaling. In contrast, Igarachi et al. have demonstrated that inhibition of AT1R restores insulin resistance induced by Ang II in VSMC derived from both normal and diabetics rat while PD123319, an AT2R antagonist had no effect (Igarashi et al., 2007). Additionally, the expression of AT2R is extremely low in adult smooth muscle cells (Ichiki et al., 1996). Therefore, it is likely that the contribution of AT2R in our system is minimal. In conclusion (Figure 6), our findings led us to conclude that Ang II-modulated insulin signaling pathways in VSMC by activation of PTP-1B. PTP-1B impairs insulin-induced activation of IRS-1/PI3K/Akt pathway while enhances insulin-induced activation of MAPK pathway in VSMC. Imbalance of insulin signaling in VSMC could contribute to vascular complications in type 2 diabetes. More functional studies are needed to be done to test whether or not this mechanism can lead to vascular diseases in vivo. We also observed that Ang II-induced activation of PTP-1B in VSMC occurs via a PKA/JAK2-dependent mechanism. Our data emphasized the significant information of the crosstalk between cytokine and insulin signaling in development of vascular insulin resistance.
Figure 6.

Proposed signaling mechanisms of Ang II-induced activation of PTP-1B in VSMC leading to VSMC insulin resistance. (PKAc = catalytic subunit of PKA)
Acknowledgments
We gratefully acknowledge Dr. Amy K. L. Banes-Berceli for initial guidance and encouragement. We thank Dr. Henry L. Keen for helpful review of the manuscript.
Sources of Funding
This work was supported by National Institutes of Health Grants HL58139, DK50268 (MBM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Asante-Appiah E, Kennedy BP. Protein tyrosine phosphatases: the quest for negative regulators of insulin action. Am J Physiol Endocrinol Metab. 2003;284:E663–E670. doi: 10.1152/ajpendo.00462.2002. [DOI] [PubMed] [Google Scholar]
- Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- Brautigan DL, Pinault FM. Serine phosphorylation of protein tyrosine phosphatase (PTP1B) in HeLa cells in response to analogues of cAMP or diacylglycerol plus okadaic acid. Mol Cell Biochem. 1993;127–128:121–129. doi: 10.1007/BF01076763. [DOI] [PubMed] [Google Scholar]
- Byon JC, Kusari AB, Kusari J. Protein-tyrosine phosphatase-1B acts as a negative regulator of insulin signal transduction. Mol Cell Biochem. 1998;182:101–108. [PubMed] [Google Scholar]
- Carvalho CR, Thirone AC, Gontijo JA, Velloso LA, Saad MJ. Effect of captopril, losartan, and bradykinin on early steps of insulin action. Diabetes. 1997;46:1950–1957. doi: 10.2337/diab.46.12.1950. [DOI] [PubMed] [Google Scholar]
- Cooper SA, Whaley-Connell A, Habibi J, Wei Y, Lastra G, Manrique C, Stas S, Sowers JR. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol. 2007;293:H2009–H2023. doi: 10.1152/ajpheart.00522.2007. [DOI] [PubMed] [Google Scholar]
- Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- Dube N, Cheng A, Tremblay ML. The role of protein tyrosine phosphatase 1B in Ras signaling. Proc Natl Acad Sci U S A. 2004;101:1834–1839. doi: 10.1073/pnas.0304242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff JL, Monia BP, Berk BC. Mitogen-activated protein (MAP) kinase is regulated by the MAP kinase phosphatase (MKP-1) in vascular smooth muscle cells. Effect of actinomycin D and antisense oligonucleotides. J Biol Chem. 1995;270:7161–7166. doi: 10.1074/jbc.270.13.7161. [DOI] [PubMed] [Google Scholar]
- Dulin NO, Niu J, Browning DD, Ye RD, Voyno-Yasenetskaya T. Cyclic AMP-independent activation of protein kinase A by vasoactive peptides. J Biol Chem. 2001;276:20827–20830. doi: 10.1074/jbc.C100195200. [DOI] [PubMed] [Google Scholar]
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- Esposito DL, Li Y, Vanni C, Mammarella S, Veschi S, Della LF, Mariani-Costantini R, Battista P, Quon MJ, Cama A. A novel T608R missense mutation in insulin receptor substrate-1 identified in a subject with type 2 diabetes impairs metabolic insulin signaling. J Clin Endocrinol Metab. 2003;88:1468–1475. doi: 10.1210/jc.2002-020933. [DOI] [PubMed] [Google Scholar]
- Flint AJ, Gebbink MF, Franza BR, Jr, Hill DE, Tonks NK. Multi-site phosphorylation of the protein tyrosine phosphatase, PTP1B: identification of cell cycle regulated and phorbol ester stimulated sites of phosphorylation. EMBO J. 1993;12:1937–1946. doi: 10.1002/j.1460-2075.1993.tb05843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florian JA, Watts SW. Integration of mitogen-activated protein kinase kinase activation in vascular 5-hydroxytryptamine2A receptor signal transduction. J Pharmacol Exp Ther. 1998;284:346–355. [PubMed] [Google Scholar]
- Folli F, Kahn CR, Hansen H, Bouchie JL, Feener EP. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J Clin Invest. 1997;100:2158–2169. doi: 10.1172/JCI119752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folli F, Saad MJ, Velloso L, Hansen H, Carandente O, Feener EP, Kahn CR. Crosstalk between insulin and angiotensin II signalling systems. Exp Clin Endocrinol Diabetes. 1999;107:133–139. doi: 10.1055/s-0029-1212088. [DOI] [PubMed] [Google Scholar]
- Gual P, Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Jacob S, Kinnick TR, Teachey MK, Krekler M. Selective angiotensin II receptor receptor antagonism reduces insulin resistance in obese Zucker rats. Hypertension. 2001;38:884–890. doi: 10.1161/hy1101.092970. [DOI] [PubMed] [Google Scholar]
- Horiuchi M, Mogi M, Iwai M. Signaling crosstalk angiotensin II receptor subtypes and insulin. Endocr J. 2006;53:1–5. doi: 10.1507/endocrj.53.1. [DOI] [PubMed] [Google Scholar]
- Hsueh WA, Lyon CJ, Quinones MJ. Insulin resistance and the endothelium. Am J Med. 2004;117:109–117. doi: 10.1016/j.amjmed.2004.02.042. [DOI] [PubMed] [Google Scholar]
- Ichiki T, Kambayashi Y, Inagami T. Differential inducibility of angiotensin II AT2 receptor between SHR and WKY vascular smooth muscle cells. Kidney Int Suppl. 1996;55:S14–S17. [PubMed] [Google Scholar]
- Igarashi M, Hirata A, Nozaki H, Kadomoto-Antsuki Y, Tominaga M. Role of angiotensin II type-1 and type-2 receptors on vascular smooth muscle cell growth and glucose metabolism in diabetic rats. Diabetes Res Clin Pract. 2007;75:267–277. doi: 10.1016/j.diabres.2006.06.032. [DOI] [PubMed] [Google Scholar]
- Izawa Y, Yoshizumi M, Fujita Y, Ali N, Kanematsu Y, Ishizawa K, Tsuchiya K, Obata T, Ebina Y, Tomita S, Tamaki T. ERK1/2 activation by angiotensin II inhibits insulin-induced glucose uptake in vascular smooth muscle cells. Exp Cell Res. 2005;308:291–299. doi: 10.1016/j.yexcr.2005.04.028. [DOI] [PubMed] [Google Scholar]
- Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–457. doi: 10.1172/JCI5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook A, Kawano Y, Song XM, Efendic S, Roth RA, Wallberg-Henriksson H, Zierath JR. Improved glucose tolerance restores insulin-stimulated Akt kinase activity and glucose transport in skeletal muscle from diabetic Goto-Kakizaki rats. Diabetes. 1997;46:2110–2114. doi: 10.2337/diab.46.12.2110. [DOI] [PubMed] [Google Scholar]
- Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, Feener EP, Herbert TP, Rhodes CJ, King GL. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin. Circulation. 2000;101:676–681. doi: 10.1161/01.cir.101.6.676. [DOI] [PubMed] [Google Scholar]
- Marrero MB, Fulton D, Stepp D, Stern DM. Angiotensin II-induced insulin resistance and protein tyrosine phosphatases. Arterioscler Thromb Vasc Biol. 2004;24:2009–2013. doi: 10.1161/01.ATV.0000140059.04717.f3. [DOI] [PubMed] [Google Scholar]
- Marrero MB, Schieffer B, Li B, Sun J, Harp JB, Ling BN. Role of Janus kinase/signal transducer and activator of transcription and mitogen-activated protein kinase cascades in angiotensin II- and platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem. 1997;272:24684–24690. doi: 10.1074/jbc.272.39.24684. [DOI] [PubMed] [Google Scholar]
- Marrero MB, Venema VJ, Ju H, Eaton DC, Venema RC. Regulation of angiotensin II-induced JAK2 tyrosine phosphorylation: roles of SHP-1 and SHP-2. Am J Physiol. 1998;275:C1216–C1223. doi: 10.1152/ajpcell.1998.275.5.C1216. [DOI] [PubMed] [Google Scholar]
- Navarro-Cid J, Maeso R, Perez-Vizcaino F, Cachofeiro V, Ruilope LM, Tamargo J, Lahera V. Effects of losartan on blood pressure, metabolic alterations, and vascular reactivity in the fructose-induced hypertensive rat. Hypertension. 1995;26:1074–1078. doi: 10.1161/01.hyp.26.6.1074. [DOI] [PubMed] [Google Scholar]
- Nigro J, Osman N, Dart AM, Little PJ. Insulin resistance and atherosclerosis. Endocr Rev. 2006;27:242–259. doi: 10.1210/er.2005-0007. [DOI] [PubMed] [Google Scholar]
- Ogihara T, Asano T, Ando K, Chiba Y, Sakoda H, Anai M, Shojima N, Ono H, Onishi Y, Fujishiro M, Katagiri H, Fukushima Y, Kikuchi M, Noguchi N, Aburatani H, Komuro I, Fujita T. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension. 2002;40:872–879. doi: 10.1161/01.hyp.0000040262.48405.a8. [DOI] [PubMed] [Google Scholar]
- Rashid N, Morin FC, III, Swartz DD, Ryan RM, Wynn KA, Wang H, Lakshminrusimha S, Kumar VH. Effects of prostacyclin and milrinone on pulmonary hemodynamics in newborn lambs with persistent pulmonary hypertension induced by ductal ligation. Pediatr Res. 2006;60:624–629. doi: 10.1203/01.pdr.0000242343.84510.81. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Shao J, Yamashita H, Qiao L, Friedman JE. Decreased Akt kinase activity and insulin resistance in C57BL/KsJ-Leprdb/db mice. J Endocrinol. 2000;167:107–115. doi: 10.1677/joe.0.1670107. [DOI] [PubMed] [Google Scholar]
- Shaw S, Wang X, Redd H, Alexander GD, Isales CM, Marrero MB. High glucose augments the angiotensin II-induced activation of JAK2 in vascular smooth muscle cells via the polyol pathway. J Biol Chem. 2003a;278:30634–30641. doi: 10.1074/jbc.M305008200. [DOI] [PubMed] [Google Scholar]
- Shaw SS, Schmidt AM, Banes AK, Wang X, Stern DM, Marrero MB. S100B-RAGE-mediated augmentation of angiotensin II-induced activation of JAK2 in vascular smooth muscle cells is dependent on PLD2. Diabetes. 2003b;52:2381–2388. doi: 10.2337/diabetes.52.9.2381. [DOI] [PubMed] [Google Scholar]
- Tao J, Malbon CC, Wang HY. Insulin stimulates tyrosine phosphorylation and inactivation of protein-tyrosine phosphatase 1B in vivo. J Biol Chem. 2001;276:29520–29525. doi: 10.1074/jbc.M103721200. [DOI] [PubMed] [Google Scholar]
- Wang CC, Gurevich I, Draznin B. Insulin affects vascular smooth muscle cell phenotype and migration via distinct signaling pathways. Diabetes. 2003;52:2562–2569. doi: 10.2337/diabetes.52.10.2562. [DOI] [PubMed] [Google Scholar]
- Wright MB, Seifert RA, Bowen-Pope DF. Protein-tyrosine phosphatases in the vessel wall: differential expression after acute arterial injury. Arterioscler Thromb Vasc Biol. 2000;20:1189–1198. doi: 10.1161/01.atv.20.5.1189. [DOI] [PubMed] [Google Scholar]
- Wu Y, Ouyang JP, Wu K, Wang SS, Wen CY, Xia ZY. Rosiglitazone ameliorates abnormal expression and activity of protein tyrosine phosphatase 1B in the skeletal muscle of fat-fed, streptozotocin-treated diabetic rats. Br J Pharmacol. 2005;146:234–243. doi: 10.1038/sj.bjp.0706306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–1545. doi: 10.1161/01.cir.101.13.1539. [DOI] [PubMed] [Google Scholar]
- Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. 1996;98:894–898. doi: 10.1172/JCI118871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuplatov SB, Masaki T, Blumenthal DK, Cheung AK. Mechanism of dipyridamole’s action in inhibition of venous and arterial smooth muscle cell proliferation. Basic Clin Pharmacol Toxicol. 2006;99:431–439. doi: 10.1111/j.1742-7843.2006.pto_516.x. [DOI] [PubMed] [Google Scholar]
- Zinker BA, Rondinone CM, Trevillyan JM, Gum RJ, Clampit JE, Waring JF, Xie N, Wilcox D, Jacobson P, Frost L, Kroeger PE, Reilly RM, Koterski S, Opgenorth TJ, Ulrich RG, Crosby S, Butler M, Murray SF, McKay RA, Bhanot S, Monia BP, Jirousek MR. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc Natl Acad Sci U S A. 2002;99:11357–11362. doi: 10.1073/pnas.142298199. [DOI] [PMC free article] [PubMed] [Google Scholar]