

Abstract
Hepatocellular carcinoma (HCC) is a major cause of cancer death worldwide. As in many other types of cancer, aberrant activation of the canonical Wnt/β-catenin signaling pathway is an important contributor to tumorigenesis. In HCC this frequently occurs through mutations in the N-terminal region of β-catenin that stabilize the protein and permit an elevated level of constitutive transcriptional activation by β-catenin/TCF complexes. In this article we review the abundant evidence that Wnt/β-catenin signaling contributes to liver carcinogenesis. We also discuss what is known about the roles of Wnt signaling in liver development, regeneration, and stem cell behavior, in an effort to understand the mechanisms by which activation of the canonical Wnt pathway promotes tumor formation in this organ. The Wnt/β-catenin pathway presents itself as an attractive target for developing novel rational therapies for HCC, a disease for which few successful treatment strategies are currently available.
Introduction
Hepatocellular carcinoma (HCC), the most common form of liver cancer, is one of the most frequent of all malignant cancers worldwide. The disease is most prevalent in parts of Africa and Asia, but incidence in the Western world is showing an alarming rise [1–3]. As a major public health problem, its high incidence is exacerbated by its lethality: overall 5-year survival rates in the U.S. are less than 9%, making it the second most lethal form of cancer after pancreatic carcinoma. Globally, HCC is responsible for over 500,000 deaths annually, and is thus the third most common cause of cancer death [1,4]. There is clearly an urgent need to develop improved prevention and treatment options for this disease. A promising approach will be to define novel targets for therapeutic strategies based on the identification of molecular pathways responsible for initiating and sustaining HCC. The canonical Wnt signaling pathway is one such signaling mechanism that is frequently activated in this disease and is clearly implicated in tumorigenesis. The purpose of this article is to review the evidence linking Wnt signaling to carcinogenesis in the liver, consider the possible functions of these signals at the cellular level, and to speculate on ways in which this knowledge could be translated into future therapeutic strategies.
Primary cancers of the liver
Cancers can arise from several of the different cellular components of liver but by far the most common (85–90%) is HCC, which originates from hepatocytes or their precursor cells [5]. The second most frequent form of liver cancer is cholangiocarcinoma, derived from the ductal epithelium of the intrahepatic bile duct. Although its global occurence is increasing, cholangiocarcinoma is still a rare cancer and the annual incidence in the U.S. is <1 per 100,000 people. Even more rare is hepatoblastoma, a tumor that arises from immature hepatic precursor cells and occurs mostly in children under the age of 4. Approximately 100 cases of hepatoblastoma per year are reported in the U.S. Benign tumors derived from hepatocytes are also observed, known as hepatocellular adenomas. These are also extremely rare (<1 in 106 in the U.S.). The low reported incidence may be in part because such lesions are generally asymptomatic and difficult to diagnose, but it also suggests that most HCCs arise directly from liver parenchyma or dysplastic nodules, without an identifiable adenomatous precursor stage.
HCC: risk factors, treatment, and outcomes
The age-standardized incidence of HCC ranges from approximately 4 in 100,000 per year in the United States to almost 10 times that frequency in parts of Central and Southeast Asia, sub-Saharan Africa, and Melanesia [1,2]. This wide variation in incidence is attributable to disparate environmental risk factors, especially viral hepatitis. Major risk factors for HCC include chronic infection with hepatitis viruses HBV and HCV, and other conditions that lead to cirrhosis of the liver such as alcoholism and non-alcoholic steatohepatitis [2]. HBV is currently the single most important underlying global cause of HCC, especially in undeveloped countries and some parts of Asia. Case-control studies have shown that HBV carriers have a 5- to 15-fold increased risk of HCC compared to the general population [6]. Effective vaccines that prevent HBV infection have been available since 1982. In contrast, HCV vaccines are still under development, the most advanced being in phase II clinical trials. HCV infection can increase risk of HCC by as much as 17-fold [7] and is a particularly important risk factor in the U.S., Europe, and Japan. HCV is now estimated to affect over 2 million Americans, and its increasing prevalence is largely responsible for the rising incidence of HCC in the U.S. recently [2,8]. There are also concerns that steatohepatitis, a form of chronic liver inflammation associated with obesity and diabetes, may assume increased importance as a risk factor for HCC in future as the prevalence of metabolic syndrome increases [3].
Cirrhosis is a degenerative condition resulting from chronic liver disease over a long period. It is characterized by the replacement of normal hepatocytes by fibrotic tissue and a consequent reduction in liver function. The vast majority of HCCs develop in cirrhotic liver, and hence cirrhosis is widely cited as a clinical risk factor for HCC. Since the various conditions that promote cirrhosis do so by causing chronic liver damage and inflammation, which themselves are associated with increased risk of carcinogenesis, this raises the question of whether cirrhosis itself is a direct mechanistic risk factor in terms of cause and effect. Indeed, since hepatocyte numbers are reduced in cirrhotic liver, the condition might have been expected to reduce the target size for development of HCC. Instead, the risk of HCC remains low during chronic liver disease but increases dramatically once cirrhosis is present [6]. This suggests either that cirrhosis is a consistent marker for tissue alterations that presage tumor formation, or that additional carcinogenic mechanisms are activated in cirrhotic tissue besides those imparted by the factors responsible for causing the cirrhosis. An excellent discussion of these issues has recently been presented by El-Serag and Rudolph [6].
Treatment choices for HCC are disappointingly limited, the most effective being surgical resection for patients with well-preserved liver function. However, advanced disease or extensive cirrhotic tissue frequently precludes this option since insufficient liver function would remain after resection [2]. Liver transplantation is the treatment of choice for some patients, especially those with early HCC in end stage cirrhosis. 5-year survival rates after transplant can be as high as 70%, but limited availability of donors severely restricts the application of this option [4]. For inoperable HCC, a variety of tumor ablation strategies may prolong life, and systemic chemotherapy is often used for patients who have metastatic disease. Novel kinase inhibitors are showing modest efficacy in clinical trials and may prolong overall survival [9–12], but standard chemotherapy drugs have generally not shown significant success against HCC in randomized trials [13]. Thus, HCC is frequently fatal. Indeed, a retrospective study that evaluated survival of North American patients with all stages of HCC found the median survival time to be 10 months [14].
Pathways of Wnt signaling
The biochemical mechanisms by which Wnt proteins elicit signaling responses in target cells have been described in detail by numerous excellent reviews [15–17]. Consequently, we will here confine our attention to the essential elements of Wnt signaling pathways as currently understood, and selected aspects that merit more emphasis.
The best characterized mechanism of Wnt signaling is the so-called ‘canonical’ or Wnt/β-catenin pathway that uses the multifunctional protein β-catenin as a signaling intermediate. This pathway is distinct from the one or more ‘non-canonical’ Wnt signaling mechanisms which are defined by being independent of β-catenin [16,18]. Unlike the canonical Wnt pathway, whose details have been elucidated from a concordance of genetic and direct biochemical evidence, the non-canonical Wnt pathways are much less well understood and their details more contentious. Non-canonical pathways include the ‘Wnt-PCP (Planar Cell Polarity) pathway, the ‘Wnt-Ca2+’ pathway, and a recently identified mechanism of Wnt signaling via the receptor tyrosine kinases Ror and Ryk [18]. It is likely that most Wnt proteins are capable of eliciting non-canonical signals, even if they also stimulate the β-catenin pathway [19]. However, we shall focus here on the Wnt β-catenin pathway since there is currently little evidence specifically linking non-canonical Wnt signaling with liver cancer.
A convenient way in which to think about the canonical Wnt/β-catenin signaling pathway is through the function of three distinct protein complexes: the ligand-receptor complex, the β-catenin destruction complex, and the β-catenin/TCF transcription complex (Fig. 1). Under normal circumstances, in the absence of Wnt receptor ligands, the free cytosolic pool of β-catenin is continuously degraded through the action of a multiprotein complex that includes the tumor suppressor proteins APC and Axin, and the Ser/Thr kinases CK1α and GSK3β [17,18]. Upon binding this complex, β-catenin becomes phosphorylated at a group of N-terminal phosphorylation sites and is consequently targeted for ubiquitination and proteolysis. Wnt receptor complexes at the cell surface are composed of a seven transmembrane domain Frizzled protein and a single-pass transmembrane protein of the LDL receptor-related protein family (specifically LRP5 or LRP6). Once Wnt ligands engage this complex, a signal is transduced that inactivates the β-catenin destruction complex and so permits unphosphorylated β-catenin to accumulate in both cytoplasm and nucleus. In the nucleus, β-catenin forms complexes with members of the TCF/LEF family of DNA-binding proteins and thereby modulates the transcription of numerous target genes with TCF-responsive promoters [17,18].
Figure 1.

Key components of the Wnt/β-catenin signaling pathway. Proteins shaded in gray have an inhibitory effect on the stability of β-catenin and its accumulation in the nucleus. Unshaded components have a positive influence on Wnt/β-catenin signaling. See text for details.
The canonical Wnt pathway involves components that act as positive, and others as negative, modulators of downstream signaling (Figure 1). In the context of cancer, therefore, these can be thought of as the products of oncogenes and tumor suppressor genes, respectively. While most of the known cancer-associated mutations in this pathway thus far involve β-catenin, Axin, or APC [20], it is possible that additional gain- and/or loss-of-function mutations remain to be identified in other components, perhaps on a less frequent basis. In addition to the constitutive activation of signaling that can be achieved by specific types of mutation in Wnt pathway components, there is growing evidence that epigenetic changes in the expression of Wnt signaling modulators, including Wnt proteins themselves, may play an important role in the genesis of many cancers, including those of the liver. In this regard it is important to note that besides Wnt proteins, and alternative Frizzled or LRP5/6 ligands that can stimulate Wnt signaling [21,22], there are several species of soluble Wnt signaling antagonists whose loss of expression may result in excessive local signaling provided that Wnts or other agonists are also present. These inhibitors include members of the sFRP and Dkk families, and the Wnt-inhibitory factor WIF-1 [15].
While the canonical Wnt pathway is frequently represented diagrammatically in a way that implies specific regulation of TCF/β-catenin activity by signals transduced from Wnt receptors, a further complication is that β-catenin stability can also be regulated by cross-talk from other pathways. These include the PTEN/Akt pathway and several tyrosine kinases, of which the HGF receptor MET is of particular relevance to liver [23–25]. Thus, elevated levels of β-catenin in cancer may not be always be the result of increased Wnt receptor signaling. It is also important to recognize the functionally distinct intracellular pools of β-catenin protein which exist in most cells: a membrane-associated pool bound to cadherins and other transmembrane proteins, a ‘free’ cytosolic pool that can be isolated as a centrifugal supernatant or by ‘pull-down’ with an E-cadherin cytoplasmic domain, and a nuclear fraction that is engaged in transcriptional activation. The membrane pool of β-catenin is typically much more abundant than the other two, especially in cells of epithelial origin with plentiful adherens junctions. In most cases the membrane pool appears unaffected by Wnt signaling [26], perhaps because it is sequestered away from the β-catenin destruction complex. Thus, changes in β-catenin levels at the membrane will not necessarily correlate with altered levels of β-catenin signaling in the nucleus. This distinction is important in relation to IHC studies of human tissues that in some cases report increased membrane-associated β-catenin without evidence of increases in the cytoplasm or nucleus. It is the free cytosolic pool of β-catenin that is initially affected by Wnt signaling, and stabilization of this fraction is usually accompanied by nuclear translocation. The detection of nuclear β-catenin by IHC is generally taken as circumstantial evidence of β-catenin transcriptional activity, although in some cases there may be sufficient nuclear β-catenin to activate target genes without it being readily detectable by immunostaining in that compartment. Hence accumulation of β-catenin in the cytoplasm may itself be indicative of active signaling.
Wnt signaling in normal liver: hepatogenesis, homeostasis, regeneration
The Wnt/β-catenin signaling mechanism appears to be ubiquitous in animal tissues, playing key roles in the development of most organ systems. Liver is no exception and Wnt signaling components are thus found in hepatocytes and other cells of the liver. Of the 19 different Wnt ligands, systematic studies report that at least 8 are expressed in hepatocytes and as many as 12 in bile duct epithelium [27]. Similarly, 9 out of 10 members of the Frizzled family, and both LRP5 and LRP6, are expressed in normal liver [28,29]. Thus there is a broad repertoire of Wnt ligands and receptors upon which to base spatial and temporal signaling patterns, and these can be modulated further by soluble Wnt inhibitors such as sFRP and Dkk proteins.
Genetic studies in zebrafish have implicated Wnt signaling at the earliest stages of liver development. Mutations in the Wnt family member Wnt2bb (a relative of mouse Wnt2b/13) result in failure of specification of hepatic endoderm at the appropriate time [30]. Wnt2bb is expressed in the lateral plate mesoderm and exerts a paracrine signal to the endoderm. The ability of dominant negative Tcf to block liver specification at this stage implies that Wnt2bb acts via the canonical Wnt/β-catenin pathway [30]. The expression pattern of murine Wnt2b/13 is consistent with its involvement in liver specification in the mouse also [31,32].
Analysis of β-catenin levels during later hepatic development in the mouse embryo reveals dynamic changes, suggesting that canonical Wnt signaling is at a peak between E10–14, a period associated with cell proliferation [33]. Similarly, activated β-catenin is at peak levels in the mouse liver between 5 and 20 days after birth, another period coinciding with rapid cell proliferation [34]. In an elegant study using retroviruses for transduction of chick embryonic liver in ovo, activated β-catenin resulted in a 3-fold increase in liver size, while the LRP5/6 antagonist Dkk1 reduced the normal size of the organ [35]. Similarly, knock-down of β-catenin levels in mouse liver cultures or human hepatoma cell lines inhibited cell proliferation [36,37] and liver-specific β-catenin knockout mice have smaller livers than wild-type [34]. A particularly informative study was one that achieved hyperactivation of Wnt/β-catenin signaling in adult mouse liver by conditional inactivation of a floxed Apc allele in vivo with Adenovirus Cre [38]. This resulted in hepatocyte hyperplasia, hepatomegaly, and a dramatic incidence of HCC [38]. These data argue that Wnt/β-catenin signaling, at least when hyperactive, is sufficient to cause hyperproliferation in the liver, not only in the embryo but in the adult.
In other organ systems the Wnt/β-catenin pathway has attracted much attention recently because of its roles in stem cell self-renewal, precursor cell proliferation, and tissue regeneration [39–41]. This raises the idea that it is involved in similar functions in the liver. One possible expectation is that β-catenin signaling levels might be highest in actively proliferating cells. While data mentioned above support this possibility during hepatogenesis, studies of adult liver indicate that the story is more complicated. An association between β-catenin signaling and adult liver cell proliferation may be limited to specific types of liver injury and/or to specific cell types.
The parenchyma of adult liver is organized as a network of lobules, each of which is organized around a central, or centrilobular, vein (Figure 2). Hepatocytes within the lobule receive blood from the portal tracts, containing branches of the hepatic artery and portal vein, which are distributed around the periphery of each lobule. In terms of metabolic function, the hepatocytes in each lobule are organized in distinct zones along the portal to venous axis. The outer circumferential rows of hepatocytes, adjacent to the portal tracts, constitute the periportal zone, while those around the central venule constitute the perivenous zone. The significance of these zones is that there are distinct gradients of biochemical function along the portal to venous axis. The zones can thus be identified by differential expression of particular metabolic enzymes, such as those involved in pathways of ammonia detoxification [42,43]. For example, carbamoylphosphate synthase 1 (CPS 1) is expressed in a gradient whose peak is at the periportal zone, while glutamine sythase (GS) is a perivenous marker, localized to cells surrounding the central venule (Figure 2). An important study by Benhamouche et al. (45) showed that high levels of Wnt/β-catenin signaling coincide with the perivenous zone of hepatic lobules and, remarkably, that β-catenin signaling plays a major role in regulating the metabolic zonation of the liver. Immunohistochemical detection of β-catenin unphosphorylated by GSK3, the form associated with downstream signaling [44], showed staining throughout the perivenous zone and not in the periportal zone [45]. In contrast, the negative regulator of β-catenin, APC, showed a complementary expression pattern. Most importantly, experimental activation of β-catenin throughout the liver resulted in an expansion of the perivenous zone while antagonism of endogenous Wnt signaling converted perivenous hepatocytes to a periportal phenotype [45]. This result accounts for the previously unexplained observation that experimental activation of β-catenin in mouse liver results in the appearance throughout the entire lobule of markers such as GS that are normally confined to the proximal perivenous hepatocytes [46].
Figure 2.
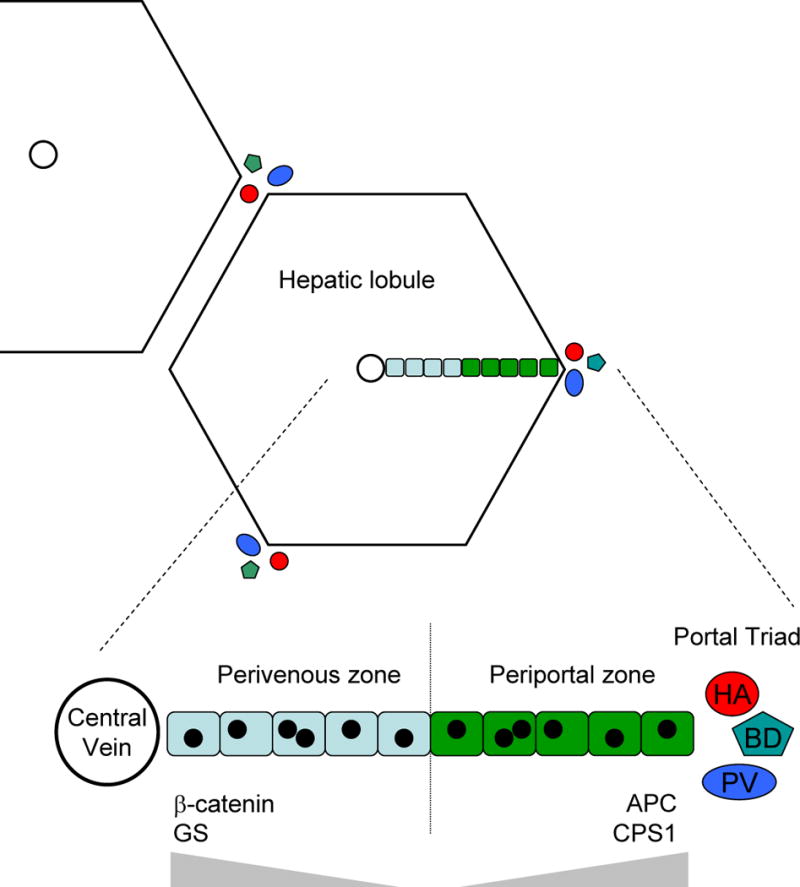
Zonation of hepatocytes within liver lobules reflects the level of Wnt/β-catenin signaling. Hepatocytes are organized in lobules around a central vein, and branches of the hepatic artery (HA), bile duct (BD), and hepatic portal vein (PV) are located at the periphery of each lobule. Expanded view of a single row of hepatocytes within a lobule showing the perivenous zone (pale blue), in which β-catenin is detected by IHC, and the periportal zone (green) in which APC is detected [45]. Enzymes associated with distinct metabolic functions, such as glutamine synthase (GS) and carbamoylphosphate synthase 1 (CPS1) are expressed in gradients (gray triangles).
Thus it appears that the level of Wnt/β-catenin signaling in adult hepatocytes determines the particular program of metabolic gene expression. Whether there is a continuous gradient of signaling, or two quantitatively distinct zones like a step gradient, is unclear. Immunohistochemical staining data disagree over the width of the zone positive for unphosphorylated β-catenin. One study reported such staining throughout the PV zone while another showed it confined to a single cell layer around the central vein [45,47]. Although that whole layer showed elevated cytoplasmic β-catenin by IHC, in reporter mice expressing either β-gal or EGFP from a β-catenin/Tcf-responsive promoter only a few isolated cells in the most proximal layer were reporter-positive [47]. One explanation for these discrepancies is that different assays of β-catenin signaling have different thresholds of sensitivity. However, a consistent conclusion is that the highest levels of β-catenin activity are found in cells most proximal to the central vein. This supports the notion that Wnt signaling regulates hepatic cell fate but raises the earlier question of whether high physiological β-catenin levels correlate with liver cell proliferation. In normal adult liver this may not be the case as the perivenous hepatocytes with high β-catenin levels are GS-positive differentiated cells that rarely proliferate [47,48]. In contrast, the immature precursors or stem-like cells are located near the periportal region where β-catenin levels are usually lowest. However, the rate of cell turnover in normal liver is very slow so it is difficult to draw firm conclusions about this issue. Instead, it is more informative to look in regenerating liver.
The liver is unique among mammalian organs in its capacity to regenerate following massive injury such as partial hepatectomy. Strictly speaking, the rapid regrowth that follows is compensatory growth of the liver that remains rather than regeneration of the resected tissue [48]. Surprisingly, this growth seems not to be dependent on stem cell recruitment but instead results from rapid induction of cell division in most of the remaining hepatocytes [48,49]. An examination of β-catenin distribution at early times after partial hepatectomy showed evidence of rapid nuclear translocation of β-catenin within 5 minutes, although this was followed by an overall decrease in cellular β-catenin levels [50]. Modulation of β-catenin may thus play a role in the initial response to liver injury. Consistent with this notion, experimental depletion of β-catenin levels in conjunction with partial hepatectomy resulted in reduced liver regeneration [51]. However, a recent study of β-catenin/TCF reporter mice found no change in distribution or intensity of the EGFP reporter signal following liver injury [47]. This suggests that hepatocyte proliferation can occur on a large scale in adult liver without an accompanying elevation in canonical Wnt signaling levels. However, given the potential sensitivity limits in transgenic reporter mice of this sort, signaling would need to be evaluated by independent methods in order to solidify this conclusion. In the reporter strain insTOP-EGFP, for example, the discrepancies between patterns of reporter expression and of β-catenin antibody staining suggest that the reporter may only be expressed in cells with a particularly high level of Wnt/β-catenin signaling [47].
Although hepatocytes have an unusual capacity to proliferate, in severe or prolonged cases of liver injury this property may be compromised as a result of the cells becoming damaged or senescent [48,52]. Under such circumstances a normally ‘covert’ population of facultative liver stem cells, known as oval cells, becomes activated. Oval cells appear to emanate from periportal areas and constitute a reserve compartment of progenitor cells. Once activated they proliferate, producing a transit-amplifying population which can then give rise to both hepatocytes and cholangiocytes [48]. Two important recent papers provide evidence that Wnt/β-catenin signaling is activated in the atypical ductal proliferations formed by oval cells, both in a mouse liver injury model using the hepatotoxin 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) and in a rat hepatectomy model [53,54]. This activation may occur through a paracrine mechanism since induced expression of several Wnt genes was observed in the neighboring hepatocytes and the oval cells were shown to respond to purified Wnt protein in culture [53]. Thus, as in other tissues, Wnt/β-catenin signaling is associated with regulating the behavior of stem-like progenitor cells. Since it is proposed that at least some HCCs arise from oval cells [52,55,56] these findings may have profound implications for understanding the role of canonical Wnt signaling in liver cancer.
In summary, while canonical Wnt signaling may regulate several aspects of liver specification, cell fate and proliferation during hepatogenesis, β-catenin signaling levels in normal adult liver correlate better with programs of metabolic cell fate than with hepatocyte proliferation. Experimental effects of β-catenin knock-down on hepatocyte growth must be interpreted with caution since basal levels of β-catenin may be needed for cell viability. However, it is clear that abnormally high levels of Wnt/β-catenin signaling are sufficient to cause hepatocyte proliferation and expansion of the liver, and that Wnt/β-catenin signaling is implicated in the control of stemlike precursor cells that are recruited into the parenchyma when hepatocyte proliferation is compromised.
Aberrant Wnt signaling in liver carcinoma
Of several signaling pathways commonly activated in liver carcinoma, the canonical Wnt pathway is one of those most frequently reported [6,57,58]. For example, the incidence of activating mutations in β-catenin in HCC is as high or higher than p53 alterations [59]. As in other forms of human cancer, activation of the Wnt pathway can result from dominant-acting gain of function mutations in positive modulators of Wnt signaling such as β-catenin, loss-of-function mutations in negative modulators such as Axin and APC, or by epigenetic changes that result in altered gene expression without overt mutation. The known characteristics of the above mutations, and their parallels in other tumors, leave little doubt that they contribute to hepatic carcinogenesis. In comparison, the functional significance of the altered expression data is less definitive. Given the multiplicity of factors, both extracellular and intracellular, that can regulate Wnt signaling either positively or negatively, altered expression of a single individual component should be interpreted with caution. This is especially true of components such as Axin2, Dkk1, and Frizzled7, whose own expression can be directly regulated by Wnt/β-catenin signaling [60–62]. However, when correlated with alterations in signaling activity, such as revealed by immunostaining of cytoplasmic or nuclear β-catenin, the altered expression data gain greater significance. Indeed some authors have reported frequencies of β-catenin stabilization of over 65% in HCC, which is too high to be explained only by the incidence of known Wnt pathway mutations [3,63]. This implies that in several cases the pathway may be activated by epigenetic means.
Reports of altered expression of Wnt signaling components at the RNA level in liver cancer have thus far focused mostly on components of the ligand-receptor complexes and their inhibitors. While there have been no reports of individual Wnt ligands consistently overexpressed in cancerous tissues, a recent paper showed that several of the endogenously expressed Wnts are significantly overexpressed during chronic liver injury, especially in the portal tract regions [53]. Importantly, this is associated with activated nuclear β-catenin in oval cells, the proliferating progenitor cells in those regions of the liver [53]. Analysis of altered Frizzled expression has focused mostly on Fzd7. Quantitative RT-PCR data indicate that Fzd7 expression is elevated in up to 90% of HCCs sampled, in comparison to normal liver, although some increased expression was also observed in peritumoral normal tissue [64]. The same Fzd has been found consistently overexpressed in several mouse models of HCC and may be associated with elevated Wnt/β-catenin signaling [63].
Other Wnt signaling components whose altered expression has been examined in liver cancer include several of the secreted antagonists of Wnt signaling such as sFRP, WIF1, and Dkk. There is evidence of sFRP1 down-regulation in approximately 50% of HCCs, and this is attributable to hypermethylation of its promoter, which has also been shown for other sFRP family members [65–67]. The available data for Dkk1 expression are intriguing, although so far based on a single study that focused on hepatoblastoma (HB) [68]. Dkk1 is a secreted inhibitor of canonical Wnt signaling that works by binding specifically to LRP5/6 and preventing Wnt-mediated signal transduction. The gene is not detectably expressed in normal liver, but Dkk1 RNA was observed in 26 of 32 HB samples examined [68]. Since Dkk1 is a known transcriptional target for β-catenin/Tcf in other tissues [61], and HBs show a high frequency of activating mutations in β-catenin (see below), it is possible that the frequent induction of Dkk1 in HB is the result of a β-catenin-mediated feedback loop. If such a mechanism also pertains to HCC, overproduction of Wnt ligands might have only limited potential to achieve constitutive signaling because of the resulting attenuation of receptor function. However, activation of the pathway downstream of Wnt receptors, such as by activating mutations in β-catenin, would be resistant to this feedback limitation.
Wnt pathway mutations in HCC
By far the most common Wnt pathway mutations observed in HCC are activating mutations in β-catenin. As also found in many other types of cancer, these are dominant gain-of-function mutations that affect the amino-terminal phosphorylation sites. They result in a β-catenin protein that is intrinsically more stable, such that it accumulates in a manner that is largely ligand-independent. There are now more than 25 publications describing such mutations in HCC. The reported frequency of β-catenin mutations ranges from 8% to 44% (see Table 1) with a median of approximately 20%, although for technical reasons this is likely to be an underestimate. Several groups have investigated correlations between β-catenin mutations and specific sub-types of HCC, risk factors, or other genetic lesions. Such correlations are not absolute, but β-catenin mutations are found more commonly in tumors with little or no chromosomal instability or LOH [59,69], and more frequently in those associated with chronic infection with HCV, rather than with HBV [70–72]. For example, one study found a 31% incidence of β-catenin mutations in HCC associated with HCV, but only 19% and 13% for cancers associated with HBV and alcohol risk factors, respectively [72]. Another study put the frequency of β-catenin mutations in HCV-associated malignancy as high as 41% [73]. Since the rising incidence of HCC in the US is largely attributable to the increasing prevalence of HCV [2], it is likely that the overall observed frequency of β-catenin mutations in HCC will increase.
Table 1.
Frequency of mutations in components of the Wnt/β-catenin pathway in HCC.
| Component | Mutation Incidence (%) | Sample Size | Reference |
|---|---|---|---|
| APC | 0 | 22 | [73] |
| 0 | 30 | [69] | |
| GSK3β | 0 | 34 | [105] |
| Axin1 | 14.3 | 49 | [75] |
| 10.6 | 123 | [106] | |
| 6.2 | 81 | [107] | |
| 9.6 | 73 | [76] | |
| 8.8 | 137 | [59] | |
| 5.0 | 100 | [108] | |
| Axin2 | 2.7 | 73 | [76] |
| β-catenin | 24.1 | 29 | [109] |
| 39.5 | 152 | [110] | |
| 16.0 | 81 | [111] | |
| 27.6 | 123 | [106] | |
| 40.8 | 49 | [75] | |
| 16.0 | 81 | [107] | |
| 14.9 | 248 | [112] | |
| 13.5 | 325 | [113] | |
| 14.3 | 56 | [114] | |
| 42.1 | 38 | [115] | |
| 44.1 | 34 | [105] | |
| 19.2 | 73 | [76] | |
| 44.1 | 34 | [116] | |
| 11.7 | 60 | [70] | |
| 19.0 | 137 | [59] | |
| 18.8 | 32 | [117] | |
| 13.0 | 100 | [108] | |
| 13.1 | 434 | [71] | |
| 23.7 | 38 | [118] | |
| 40.9 | 22 | [73] | |
| 17.6 | 119 | [69] | |
| 19.2 | 73 | [119] | |
| 34.3 | 35 | [120] | |
| 23.1 | 26 | [79] | |
| 18.7 | 75 | [121] |
Typically, HCC-associated lesions in the β-catenin gene are missense mutations that affect an individual amino acid between residues 31 and 47, either a Ser/Thr or neighboring amino acid that forms part of the kinase recognition site. The mutations are found within a single exon (exon 3) and so are relatively easy to detect by PCR amplification and sequencing of genomic DNA. A chart showing the relative incidence of mutations in individual codons is shown in Figure 3. The clustering and distribution of target amino acids is remarkably similar to that observed in colorectal cancer. It is also notable that the most frequently mutated residue is Ser45. This has been shown to be the principal site for phosphorylation mediated by CK1α, a priming event necessary for efficient phosphorylation of the more upstream sites by GSK3β [74]. A mutation in codon 45, therefore, may the most effective way to ‘protect’ β-catenin from phosphorylation-mediated destruction.
Figure 3.
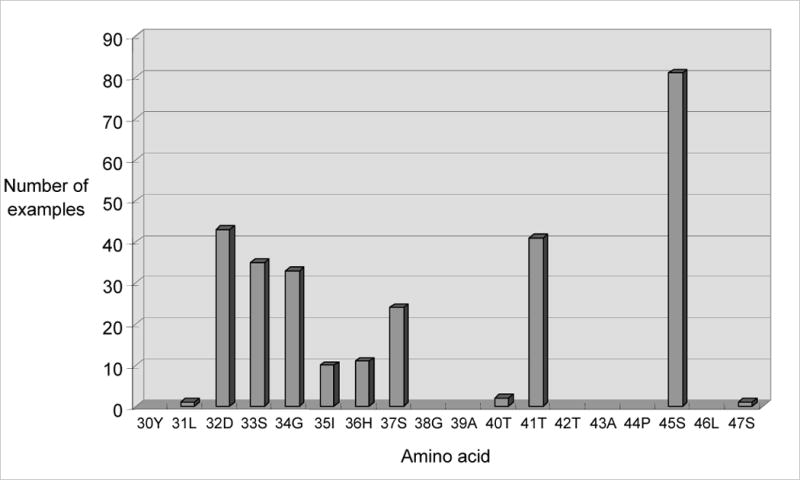
Distribution and frequency of β-catenin missense mutations leading to activation of canonical Wnt signaling in HCC. Specific amino acid residues of β-catenin are indicated by their position number and single letter code.
In several mutational studies of β-catenin in HCC, samples were analyzed for elevated β-catenin protein levels by IHC. Some of these data are difficult to assimilate because of variable staining protocols and the differential significance of nuclear/cytoplasmic versus membranous β-catenin. However, elevated levels of staining have been reported in 17–66% of the samples examined, with most authors agreeing that immunohistochemical evidence of Wnt pathway activation is more common than the observed incidence of Wnt pathway mutations could account for [3]. This suggests either that there are mutations in other Wnt signaling components not yet rigorously examined, that β-catenin is stabilized by cross-talk from other signaling pathways, or that epigenetic changes account for altered Wnt signaling in some cases, as discussed above.
The only other Wnt pathway components in which mutations have been observed at a significant level in HCC are Axin1 and its paralog Axin2, each of which functions in the β-catenin destruction complex and normally acts as an antagonist of Wnt/β-catenin signaling. The observed mutations result in loss of Axin function and can be more challenging to catalog because of the multiple ways in which this loss can be achieved. For Axin1 these mutations have been observed in 6–14% of cases of HCC (see Table 1). As in colon cancer, Axin1 mutations are usually mutually exclusive of β-catenin mutations. However, a recent report suggests that their functional consequences may be distinct [75]. Only two examples of Axin2 mutation have thus far been reported in HCC [76]. The lack of observed mutations in other Wnt pathway components is in some cases based on solid negative data. For example, two studies examined HCC for the type of APC mutations commonly found in colorectal cancer but found no examples in a total of 52 cases examined [69,73]. This is consistent with a lack of genetic predisposition to HCC among FAP patients, even though targeted mutation of Apc in the mouse liver can lead to HCC [38].
Mouse models of HCC
Genetic manipulation of the canonical Wnt pathway in mouse liver is a powerful means of modeling the activating mutations observed in human HCC. Widespread activation of Wnt/β-catenin signaling, such as by liver-specific expression of an activated β-catenin transgene, by Cre-mediated deletion of β-catenin exon 3, or by homozygous inactivation of Apc, leads to rapid hepatomegaly and early death of the mice [38,77,78]. This is consistent with a positive effect on hepatocyte growth, at least when the signal is active in large numbers of cells. In contrast, when β-catenin was stabilized in a small proportion of cells in the liver, the mice survived for more than 6 months without signs of abnormal growth or neoplasia [78]. In another study, the β-catenin pathway was activated by Cre-mediated deletion of Apc in a small proportion of liver cells [38]. These mice survived much longer and 67% developed HCC, suggesting that this is in many ways an appropriate model of β-catenin-mediated HCC, albeit one driven by a mutation that rarely if ever occurs in the human disease [38,79]. Collectively the above results indicate that in the liver, as in other tissues, activation of β-catenin is not sufficient for tumorigenesis and that additional oncogenic changes are required [38,80]. However, while the phenotype of an individual β-catenin mutant cell in resting liver may be relatively subtle, its effects might be more dramatic under conditions of chronic liver damage and/or if the mutation occurs in a stem cell.
A number of other mouse models of HCC have been generated by expression in the liver of oncogenes not specifically related to the Wnt pathway. These include c-myc, H-ras, and N-myc2 [79,81]. Importantly, the frequency of activating mutations in β-catenin in these tumors is even higher than in human HCC, ranging from 25% of the N-myc mediated tumors, to as many as 55% of those driven by a c-myc transgene [79,81]. Although the proportion of small intragenic deletions relative to missense mutations in β-catenin is somewhat higher in the mouse than it is in humans, the mutations occur within the same cluster of N-terminal amino acids as in human HCC. As with human tumors, an even higher proportion of HCCs in mouse transgenic models display nuclear accumulation of β-catenin, and in some cases these are associated with changes in expression of Wnt pathway modulators [82,83].
Murine HCCs induced by chronic liver damage with chemicals also display a remarkably high frequency of β-catenin activating mutations. These models include induction of lesions with diethynitrosamine (54% β-catenin mutations), oxazepam (43%) and methyleugenol (69%) [84,85]. From the perspective of evaluating drug targets, these transgenic and chemically induced mouse models of HCC provide a valuable resource in which to evaluate novel therapeutic strategies against autochthonous liver tumors in which mutational activated β-catenin contributes to tumorigenesis.
Wnt pathway alterations in hepatoblastoma
As mentioned earlier, hepatoblastoma is a childhood malignancy that appears to originate from embryonic liver precursor cells. While the disease can sometimes be detected in utero, affected children are usually diagnosed between 6 months and 3 years of age. Hepatoblastoma is a rare disease but is the most frequent liver cancer of childhood and is a malignancy with one of the highest known frequencies of β-catenin mutation. The incidence of such mutations in 8 independent studies ranges from 46% to 89% and in aggregate is approximately 60% [76,86–91](Table 2). The data from mouse models are even more dramatic, since hepatoblastomas induced by anthraquinone, oxazapam, or diethanolamine all have shown a 100% incidence of β-catenin mutations [92,93]. These numbers imply that β-catenin activation is effectively a ‘gatekeeper’ mutation in hepatoblastoma, at least in the mouse, and may reflect an essential role of the canonical Wnt pathway in regulating proliferation of hepatic precursor cells. One curious difference between the mutation data for hepatoblastoma versus HCC is that more than half the β-catenin mutations in the former are interstitial deletions affecting exon 3 (or exon 2 in the mouse) while in HCC missense mutations are much more common [76,86,89,92,93]. However, this might reflect differences in prevailing mutational mechanisms in different tumor precursor cells rather than distinct phenotypic consequences of the different mutations.
Table 2.
Incidence of β-catenin mutations in all forms of liver cancer
Stem Cells in Liver Cancer
In the past few years there has been a resurgence of evidence that the growth of many types of human carcinoma is driven by a small sub-population of stem-like cells, known as ‘cancer stem cells’, which are distinguished by their robust tumor-initiating capacity in experimental models. One form of the cancer stem cell hypothesis incorporates the concept that tissue stem cells, in an aberrant or mutant state, are the cells of origin of the tumor. For liver cancer, one possibility is that HCC may frequently be derived from oval cells [52,55,56]. This notion would tie together the frequent mutational activation of β-catenin observed in HCC with recent reports linking Wnt/β-catenin signaling to oval cell proliferation [53,54]. However, the extent to which the cancer stem cell concept applies to liver tumors remains to be determined. Initial studies showed that hepatoma cell lines contain subsets of highly tumorigenic cells that can be identified either as a ‘side population’ in Hoechst dye exclusion flow cytometry [94] or by expression of the putative stem cell marker CD133 [95–97]. The latter has been reported as a marker of rare cancer-initiating cells in several other human cancers but in some hepatoma lines can be detected in as many as 90% of the cell population [97,98]. More recently it has been reported that the surface marker CD90, in conjunction with CD44, is associated with highly tumorigenic subsets of both HCC cell lines and primary liver carcinomas [99,100]. The analysis of stem-like cells in liver cancer will surely be an experimental priority for many laboratories in the immediate future.
Therapeutic targets
Although much remains to be understood about the role of canonical Wnt/β-catenin signaling in normal and diseased liver, the pathway presents itself as a prime target for rational therapies. As well as antagonism of the harmful effects of activated β-catenin signaling in HCC and other liver cancers, one might envisage harnessing the growth-stimulating functions in a controlled manner for the purpose of stimulating the regeneration of healthy liver in conditions of cirrhosis or partial hepatectomy. An ability to activate oval cells and transiently stimulate hepatocyte precursors with Wnt agonists might be a future goal of regenerative medicine.
As discussed above, there is abundant evidence that hyperactive Wnt/β-catenin signaling contributes to tumorigenesis and that signal attenuation can inhibit cancer cell growth. As in other tissues, however, it is important to recognize that physiological levels of canonical Wnt signaling may provide important or essential functions for normal cells of the liver. Individual components of the pathway also participate in other processes besides signal transduction. β-catenin, for example, plays a critical role in cell-cell adhesive junctions [101]. Unless Wnt signaling antagonists can be delivered selectively to tumor cells, an appealing strategy would be one that preferentially targets the high levels of β-catenin signaling associated with cancer and so minimizes collateral damage to normal tissue. Alternatively, it is possible that Wnt antagonists could be used at doses that provide a safe therapeutic window.
What would be the likely molecular targets of such therapies? Several small molecule antagonists of canonical Wnt signaling have already been described, some of which interfere with the interaction of β-catenin with TCF/LEF proteins or the transcriptional coactivator CEBP [102–104]. This is a promising approach. Intervention at the level of the Wnt ligand-receptor complex is another possibility, especially in those cases of HCC in which there is evidence for β-catenin activation without known mutations in the pathway. Such tumors may be partially dependent on Wnt signals that could be antagonized at the cell surface. For those tumors with oncogenic β-catenin mutations, we suggest that the respective mutant β-catenin RNA may be an attractive target for RNAi therapeutics, and one that has the advantage of being tumor-specific.
Acknowledgments
We thank Louise R. Howe for critical comments on the manuscript. The authors’ research on Wnt signaling in cancer has been supported by the National Institutes of Health (CA47207, CA123238), the New York State Department of Health (NYS C021339), and by charitable donations to Strang Cancer Prevention Center.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Bruix J, Boix L, Sala M, Llovet JM. Cancer Cell. 2004;5(3):215–219. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 3.Branda M, Wands JR. Hepatology. 2006;43(5):891–902. doi: 10.1002/hep.21196. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Burroughs A, Bruix J. Lancet. 2003;362(9399):1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 5.Suriawinata AA, Thung SN. Clin Liver Dis. 2002;6(2):527–554. ix. doi: 10.1016/s1089-3261(02)00005-3. [DOI] [PubMed] [Google Scholar]
- 6.El-Serag HB, Rudolph KL. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 7.Donato F, Tagger A, Gelatti U, Parrinello G, Boffetta P, Albertini A, Decarli A, Trevisi P, Ribero ML, Martelli C, Porru S, Nardi G. Am J Epidemiol. 2002;155(4):323–331. doi: 10.1093/aje/155.4.323. [DOI] [PubMed] [Google Scholar]
- 8.Parkin DM, Bray F, Ferlay J, Pisani P. Int J Cancer. 2001;94(2):153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 9.Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz B, Taylor I, Moscovici M, Saltz LB. J Clin Oncol. 2006;24(26):4293–4300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- 10.Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, Donehower RC, Fitch T, Picus J, Erlichman C. J Clin Oncol. 2005;23(27):6657–6663. doi: 10.1200/JCO.2005.14.696. [DOI] [PubMed] [Google Scholar]
- 11.Thomas MB, Chadha R, Glover K, Wang X, Morris J, Brown T, Rashid A, Dancey J, Abbruzzese JL. Cancer. 2007 doi: 10.1002/cncr.22886. [DOI] [PubMed] [Google Scholar]
- 12.Zhu AX. Cancer 2007 [Google Scholar]
- 13.Llovet JM, Bruix J. Hepatology. 2003;37(2):429–442. doi: 10.1053/jhep.2003.50047. [DOI] [PubMed] [Google Scholar]
- 14.Stuart KE, Anand AJ, Jenkins RL. Cancer. 1996;77(11):2217–2222. doi: 10.1002/(SICI)1097-0142(19960601)77:11<2217::AID-CNCR6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Logan CY, Nusse R. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 16.Veeman MT, Axelrod JD, Moon RT. Dev Cell. 2003;5(3):367–377. doi: 10.1016/s1534-5807(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 17.Clevers H. Cell. 2006;127(3):469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Gordon MD, Nusse R. J Biol Chem. 2006;281(32):22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez-Sancho JM, Brennan KR, Castelo-Soccio LA, Brown AMC. Mol Cell Biol. 2004;24(11):4757–4768. doi: 10.1128/MCB.24.11.4757-4768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giles RH, van Es JH, Clevers H. Biochim Biophys Acta. 2003;1653(1):1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 21.Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Cell. 2004;116(6):883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 22.Nam JS, Turcotte TJ, Smith PF, Choi S, Yoon JK. J Biol Chem. 2006;281(19):13247–13257. doi: 10.1074/jbc.M508324200. [DOI] [PubMed] [Google Scholar]
- 23.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. J Biol Chem. 2007;282(15):11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lilien J, Balsamo J. Curr Opin Cell Biol. 2005;17(5):459–465. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, Wang X, Zarnegar R, Michalopoulos GK. Cancer Res. 2002;62(7):2064–2071. [PubMed] [Google Scholar]
- 26.Giarre M, Semenov MV, Brown AMC. Ann N Y Acad Sci. 1998;857:43–55. doi: 10.1111/j.1749-6632.1998.tb10106.x. [DOI] [PubMed] [Google Scholar]
- 27.Thompson MD, Monga SP. Hepatology. 2007;45(5):1298–1305. doi: 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]
- 28.Fujino T, Asaba H, Kang MJ, Ikeda Y, Sone H, Takada S, Kim DH, Ioka RX, Ono M, Tomoyori H, Okubo M, Murase T, Kamataki A, Yamamoto J, Magoori K, Takahashi S, Miyamoto Y, Oishi H, Nose M, Okazaki M, Usui S, Imaizumi K, Yanagisawa M, Sakai J, Yamamoto TT. Proc Natl Acad Sci U S A. 2003;100(1):229–234. doi: 10.1073/pnas.0133792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng G, Awan F, Otruba W, Muller P, Apte U, Tan X, Gandhi C, Demetris AJ, Monga SP. Hepatology. 2007;45(1):195–204. doi: 10.1002/hep.21473. [DOI] [PubMed] [Google Scholar]
- 30.Ober EA, Verkade H, Field HA, Stainier DY. Nature. 2006;442(7103):688–691. doi: 10.1038/nature04888. [DOI] [PubMed] [Google Scholar]
- 31.Zakin LD, Mazan S, Maury M, Martin N, Guenet JL, Brulet P. Mech Dev. 1998;73(1):107–116. doi: 10.1016/s0925-4773(98)00040-9. [DOI] [PubMed] [Google Scholar]
- 32.Burke ZD, Thowfeequ S, Tosh D. Curr Biol. 2006;16(17):R688–R690. doi: 10.1016/j.cub.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 33.Micsenyi A, Tan X, Sneddon T, Luo JH, Michalopoulos GK, Monga SP. Gastroenterology. 2004;126(4):1134–1146. doi: 10.1053/j.gastro.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 34.Apte U, Zeng G, Thompson MD, Muller P, Micsenyi A, Cieply B, Kaestner KH, Monga SP. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1578–G1585. doi: 10.1152/ajpgi.00359.2006. [DOI] [PubMed] [Google Scholar]
- 35.Suksaweang S, Lin CM, Jiang TX, Hughes MW, Widelitz RB, Chuong CM. Dev Biol. 2004;266(1):109–122. doi: 10.1016/j.ydbio.2003.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monga SP, Monga HK, Tan X, Mule K, Pediaditakis P, Michalopoulos GK. Gastroenterology. 2003;124(1):202–216. doi: 10.1053/gast.2003.50000. [DOI] [PubMed] [Google Scholar]
- 37.Sangkhathat S, Kusafuka T, Miao J, Yoneda A, Nara K, Yamamoto S, Kaneda Y, Fukuzawa M. Int J Oncol. 2006;28(3):715–722. [PubMed] [Google Scholar]
- 38.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C. Proc Natl Acad Sci U S A. 2004;101(49):17216–17221. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. Nature. 2003;423(6938):409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 40.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Nat Rev Genet. 2004;5(9):691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 41.Lowry WE, Blanpain C, Nowak JA, Guasch G, Lewis L, Fuchs E. Genes Dev. 2005;19(13):1596–1611. doi: 10.1101/gad.1324905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burke ZD, Tosh D. Bioessays. 2006;28(11):1072–1077. doi: 10.1002/bies.20485. [DOI] [PubMed] [Google Scholar]
- 43.Jungermann K, Kietzmann T. Annu Rev Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- 44.Staal FJ, Noort Mv M, Strous GJ, Clevers HC. EMBO Rep. 2002;3(1):63–68. doi: 10.1093/embo-reports/kvf002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, Kuo CJ, Kahn A, Perret C, Colnot S. Dev Cell. 2006;10(6):759–770. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, Kitajewski J, Kahn A, Perret C. Oncogene. 2002;21(54):8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 47.Moriyama A, Kii I, Sunabori T, Kurihara S, Takayama I, Shimazaki M, Tanabe H, Oginuma M, Fukayama M, Matsuzaki Y, Saga Y, Kudo A. Genesis. 2007;45(2):90–100. doi: 10.1002/dvg.20268. [DOI] [PubMed] [Google Scholar]
- 48.Fausto N, Campbell JS. Mech Dev. 2003;120(1):117–130. doi: 10.1016/s0925-4773(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 49.Michalopoulos GK, DeFrances MC. Science. 1997;276(5309):60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 50.Monga SP, Pediaditakis P, Mule K, Stolz DB, Michalopoulos GK. Hepatology. 2001;33(5):1098–1109. doi: 10.1053/jhep.2001.23786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sodhi D, Micsenyi A, Bowen WC, Monga DK, Talavera JC, Monga SP. J Hepatol. 2005;43(1):132–141. doi: 10.1016/j.jhep.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 52.Roskams T. Oncogene. 2006;25(27):3818–3822. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 53.Hu M, Kurobe M, Jeong YJ, Fuerer C, Ghole S, Nusse R, Sylvester KG. Gastroenterology. 2007;133(5):1579–1591. doi: 10.1053/j.gastro.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 54.Apte U, Thompson MD, Cui S, Liu B, Cieply B, Monga SP. Hepatology. 2007 doi: 10.1002/hep.21973. [DOI] [PubMed] [Google Scholar]
- 55.Kuhlmann WD, Peschke P. Int J Exp Pathol. 2006;87(5):343–359. doi: 10.1111/j.1365-2613.2006.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alison MR. Stem Cell Rev. 2005;1(3):253–260. doi: 10.1385/SCR:1:3:253. [DOI] [PubMed] [Google Scholar]
- 57.Lee HC, Kim M, Wands JR. Front Biosci. 2006;11:1901–1915. doi: 10.2741/1933. [DOI] [PubMed] [Google Scholar]
- 58.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Semin Liver Dis. 2007;27(1):55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 59.Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, Monges G, Thomas G, Bioulac-Sage P, Zucman-Rossi J. Gastroenterology. 2001;120(7):1763–1773. doi: 10.1053/gast.2001.24798. [DOI] [PubMed] [Google Scholar]
- 60.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Mol Cell Biol. 2002;22(4):1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, Sugano S, Akiyama T. Oncogene. 2004;23(52):8520–8526. doi: 10.1038/sj.onc.1207892. [DOI] [PubMed] [Google Scholar]
- 62.Willert J, Epping M, Pollack JR, Brown PO, Nusse R. BMC Dev Biol. 2002;2(1):8. doi: 10.1186/1471-213x-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Merle P, Kim M, Herrmann M, Gupte A, Lefrancois L, Califano S, Trepo C, Tanaka S, Vitvitski L, de la Monte S, Wands JR. J Hepatol. 2005;43(5):854–862. doi: 10.1016/j.jhep.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 64.Merle P, de la Monte S, Kim M, Herrmann M, Tanaka S, Von Dem Bussche A, Kew MC, Trepo C, Wands JR. Gastroenterology. 2004;127(4):1110–1122. doi: 10.1053/j.gastro.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 65.Shih YL, Hsieh CB, Lai HC, Yan MD, Hsieh TY, Chao YC, Lin YW. Int J Cancer. 2007;121(5):1028–1035. doi: 10.1002/ijc.22750. [DOI] [PubMed] [Google Scholar]
- 66.Shih YL, Shyu RY, Hsieh CB, Lai HC, Liu KY, Chu TY, Lin YW. Cancer. 2006;107(3):579–590. doi: 10.1002/cncr.22023. [DOI] [PubMed] [Google Scholar]
- 67.Huang J, Zhang YL, Teng XM, Lin Y, Zheng DL, Yang PY, Han ZG. BMC Cancer. 2007;7(126) doi: 10.1186/1471-2407-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wirths O, Waha A, Weggen S, Schirmacher P, Kuhne T, Goodyer CG, Albrecht S, Von Schweinitz D, Pietsch T. Lab Invest. 2003;83(3):429–434. doi: 10.1097/01.lab.0000059926.66359.bd. [DOI] [PubMed] [Google Scholar]
- 69.Legoix P, Bluteau O, Bayer J, Perret C, Balabaud C, Belghiti J, Franco D, Thomas G, Laurent-Puig P, Zucman-Rossi J. Oncogene. 1999;18(27):4044–4046. doi: 10.1038/sj.onc.1202800. [DOI] [PubMed] [Google Scholar]
- 70.Wong CM, Fan ST, Ng IO. Cancer. 2001;92(1):136–145. doi: 10.1002/1097-0142(20010701)92:1<136::aid-cncr1301>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 71.Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, Peng SY. Am J Pathol. 2000;157(3):763–770. doi: 10.1016/s0002-9440(10)64590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle HM, Matsuda M, Fujii H, Scoazec JY, Ohgaki H. Int J Cancer. 2003;106(3):334–341. doi: 10.1002/ijc.11254. [DOI] [PubMed] [Google Scholar]
- 73.Huang H, Fujii H, Sankila A, Mahler-Araujo BM, Matsuda M, Cathomas G, Ohgaki H. Am J Pathol. 1999;155(6):1795–1801. doi: 10.1016/s0002-9440(10)65496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Cell. 2002;108(6):837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 75.Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, Cunha AS, Bioulac-Sage P, Perret C. Oncogene. 2007;26(5):774–780. doi: 10.1038/sj.onc.1209824. [DOI] [PubMed] [Google Scholar]
- 76.Taniguchi K, Roberts LR, Aderca IN, Dong X, Qian C, Murphy LM, Nagorney DM, Burgart LJ, Roche PC, Smith DI, Ross JA, Liu W. Oncogene. 2002;21(31):4863–4871. doi: 10.1038/sj.onc.1205591. [DOI] [PubMed] [Google Scholar]
- 77.Cadoret A, Ovejero C, Saadi-Kheddouci S, Souil E, Fabre M, Romagnolo B, Kahn A, Perret C. Cancer Res. 2001;61(8):3245–3249. [PubMed] [Google Scholar]
- 78.Harada N, Miyoshi H, Murai N, Oshima H, Tamai Y, Oshima M, Taketo MM. Cancer Res. 2002;62(7):1971–1977. [PubMed] [Google Scholar]
- 79.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Proc Natl Acad Sci U S A. 1998;95(15):8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Cancer Res. 2004;64(1):48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- 81.Renard CA, Fourel G, Bralet MP, Degott C, De La Coste A, Perret C, Tiollais P, Buendia MA. Oncogene. 2000;19(22):2678–2686. doi: 10.1038/sj.onc.1203617. [DOI] [PubMed] [Google Scholar]
- 82.Calvisi DF, Conner EA, Ladu S, Lemmer ER, Factor VM, Thorgeirsson SS. J Hepatol. 2005;42(6):842–849. doi: 10.1016/j.jhep.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 83.Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS. Gastroenterology. 2004;126(5):1374–1386. doi: 10.1053/j.gastro.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 84.Ogawa K, Yamada Y, Kishibe K, Ishizaki K, Tokusashi Y. Cancer Res. 1999;59(8):1830–1833. [PubMed] [Google Scholar]
- 85.Devereux TR, Anna CH, Foley JF, White CM, Sills RC, Barrett JC. Oncogene. 1999;18(33):4726–4733. doi: 10.1038/sj.onc.1202858. [DOI] [PubMed] [Google Scholar]
- 86.Jeng YM, Wu MZ, Mao TL, Chang MH, Hsu HC. Cancer Lett. 2000;152(1):45–51. doi: 10.1016/s0304-3835(99)00433-4. [DOI] [PubMed] [Google Scholar]
- 87.Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T. Cancer Res. 1999;59(2):269–273. [PubMed] [Google Scholar]
- 88.Takayasu H, Horie H, Hiyama E, Matsunaga T, Hayashi Y, Watanabe Y, Suita S, Kaneko M, Sasaki F, Hashizume K, Ozaki T, Furuuchi K, Tada M, Ohnuma N, Nakagawara A. Clin Cancer Res. 2001;7(4):901–908. [PubMed] [Google Scholar]
- 89.Udatsu Y, Kusafuka T, Kuroda S, Miao J, Okada A. Pediatr Surg Int. 2001;17(7):508–512. doi: 10.1007/s003830000576. [DOI] [PubMed] [Google Scholar]
- 90.von Schweinitz D, Kraus JA, Albrecht S, Koch A, Fuchs J, Pietsch T. Med Pediatr Oncol. 2002;38(2):104–108. doi: 10.1002/mpo.1280. [DOI] [PubMed] [Google Scholar]
- 91.Wei Y, Fabre M, Branchereau S, Gauthier F, Perilongo G, Buendia MA. Oncogene. 2000;19(4):498–504. doi: 10.1038/sj.onc.1203356. [DOI] [PubMed] [Google Scholar]
- 92.Anna CH, Sills RC, Foley JF, Stockton PS, Ton TV, Devereux TR. Cancer Res. 2000;60(11):2864–2868. [PubMed] [Google Scholar]
- 93.Hayashi SM, Ton TV, Hong HH, Irwin RD, Haseman JK, Devereux TR, Sills RC. Chem Biol Interact. 2003;146(3):251–261. doi: 10.1016/j.cbi.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 94.Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, Nakauchi H, Taniguchi H. Hepatology. 2006;44(1):240–251. doi: 10.1002/hep.21227. [DOI] [PubMed] [Google Scholar]
- 95.Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Biochem Biophys Res Commun. 2006;351(4):820–824. doi: 10.1016/j.bbrc.2006.10.128. [DOI] [PubMed] [Google Scholar]
- 96.Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, Yang S, Zheng S, Gu J. Int J Cancer. 2007;120(7):1444–1450. doi: 10.1002/ijc.22476. [DOI] [PubMed] [Google Scholar]
- 97.Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Gastroenterology. 2007;132(7):2542–2556. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 98.Mizrak D, Brittan M, Alison M. J Pathol. 2008;214(1):3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 99.Yang ZF, Ngai P, Ho DW, Yu WC, Ng MN, Lau CK, Li ML, Tam KH, Lam CT, Poon RT, Fan ST. Hepatology. 2008;47(3):919–928. doi: 10.1002/hep.22082. [DOI] [PubMed] [Google Scholar]
- 100.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST. Cancer Cell. 2008;13(2):153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 101.Nelson WJ, Nusse R. Science. 2004;303(5663):1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Cancer Cell. 2004;5(1):91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 103.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, Ha JR, Kahn M. Proc Natl Acad Sci U S A. 2004;101(34):12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barker N, Clevers H. Nat Rev Drug Discov. 2006;5(12):997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 105.Cui J, Zhou X, Liu Y, Tang Z, Romeih M. J Gastroenterol Hepatol. 2003;18(3):280–287. doi: 10.1046/j.1440-1746.2003.02973.x. [DOI] [PubMed] [Google Scholar]
- 106.Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, Herault A, Saric J, Belghiti J, Franco D, Bioulac-Sage P, Laurent-Puig P, Zucman-Rossi J. Hepatology. 2007;45(1):42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 107.Park JY, Park WS, Nam SW, Kim SY, Lee SH, Yoo NJ, Lee JY, Park CK. Liver Int. 2005;25(1):70–76. doi: 10.1111/j.1478-3231.2004.0995.x. [DOI] [PubMed] [Google Scholar]
- 108.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T, Yamaoka Y, Nakamura Y. Nat Genet. 2000;24(3):245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 109.Wang H, Zhang J, Feng W, Zhang S, Liang H, Wang Y, Zheng Q, Li Z. J Huazhong Univ Sci Technolog Med Sci. 2007;27(1):54–57. doi: 10.1007/s11596-007-0116-z. [DOI] [PubMed] [Google Scholar]
- 110.Audard V, Grimber G, Elie C, Radenen B, Audebourg A, Letourneur F, Soubrane O, Vacher-Lavenu MC, Perret C, Cavard C, Terris B. J Pathol. 2007;212(3):345–352. doi: 10.1002/path.2169. [DOI] [PubMed] [Google Scholar]
- 111.Nishida N, Nishimura T, Nagasaka T, Ikai I, Goel A, Boland CR. Cancer Res. 2007;67(10):4586–4594. doi: 10.1158/0008-5472.CAN-06-3464. [DOI] [PubMed] [Google Scholar]
- 112.Yuan RH, Jeng YM, Chen HL, Hsieh FJ, Yang CY, Lee PH, Hsu HC. Clin Cancer Res. 2005;11(7):2568–2575. doi: 10.1158/1078-0432.CCR-04-2039. [DOI] [PubMed] [Google Scholar]
- 113.Peng SY, Chen WJ, Lai PL, Jeng YM, Sheu JC, Hsu HC. Int J Cancer. 2004;112(1):44–50. doi: 10.1002/ijc.20279. [DOI] [PubMed] [Google Scholar]
- 114.Prange W, Breuhahn K, Fischer F, Zilkens C, Pietsch T, Petmecky K, Eilers R, Dienes HP, Schirmacher P. J Pathol. 2003;201(2):250–259. doi: 10.1002/path.1448. [DOI] [PubMed] [Google Scholar]
- 115.Yamamoto Y, Sakamoto M, Fujii G, Tsuiji H, Kenetaka K, Asaka M, Hirohashi S. Hepatology. 2003;37(3):528–533. doi: 10.1053/jhep.2003.50029. [DOI] [PubMed] [Google Scholar]
- 116.Cui J, Zhou X, Liu Y, Tang Z. J Cancer Res Clin Oncol. 2001;127(9):577–581. doi: 10.1007/s004320100259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fujie H, Moriya K, Shintani Y, Tsutsumi T, Takayama T, Makuuchi M, Kimura S, Koike K. Hepatol Res. 2001;20(1):39–51. doi: 10.1016/s1386-6346(00)00116-9. [DOI] [PubMed] [Google Scholar]
- 118.Kondo Y, Kanai Y, Sakamoto M, Genda T, Mizokami M, Ueda R, Hirohashi S. Jpn J Cancer Res. 1999;90(12):1301–1309. doi: 10.1111/j.1349-7006.1999.tb00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Terris B, Pineau P, Bregeaud L, Valla D, Belghiti J, Tiollais P, Degott C, Dejean A. Oncogene. 1999;18(47):6583–6588. doi: 10.1038/sj.onc.1203051. [DOI] [PubMed] [Google Scholar]
- 120.Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Am J Pathol. 1999;155(3):703–710. doi: 10.1016/s0002-9440(10)65168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y. Cancer Res. 1998;58(12):2524–2527. [PubMed] [Google Scholar]
- 122.Rebouissou S, Bioulac-Sage P, Zucman-Rossi J. J Hepatol. 2008;48(1):163–170. doi: 10.1016/j.jhep.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 123.Chen YW, Jeng YM, Yeh SH, Chen PJ. Hepatology. 2002;36(4 Pt 1):927–935. doi: 10.1053/jhep.2002.36126. [DOI] [PubMed] [Google Scholar]
- 124.Tokumoto N, Ikeda S, Ishizaki Y, Kurihara T, Ozaki S, Iseki M, Shimizu Y, Itamoto T, Arihiro K, Okajima M, Asahara T. Int J Oncol. 2005;27(4):973–980. [PubMed] [Google Scholar]
- 125.Sugimachi K, Aishima S, Taguchi K, Tanaka S, Shimada M, Kajiyama K, Sugimachi K, Tsuneyoshi M. J Hepatol. 2001;35(1):74–79. doi: 10.1016/s0168-8278(01)00079-4. [DOI] [PubMed] [Google Scholar]