

Abstract
Background
Ninety percent of glucose filtered by the glomerulus is reabsorbed by a sodium-glucose cotransporter 2 (SGLT2), which is expressed mainly on the apical membrane of renal proximal tubules. Since SGLT-2-mediated glucose reabsorption is enhanced under diabetic conditions, selective inhibition of SGLT2 has been proposed as a potential therapeutic target for the treatment of patients with diabetes. However, it remains unclear which diabetes-associated factors are involved in overexpression of SGLT2.
Methods
Therefore, in this study, we examined whether insulin, high glucose, advanced glycation end products (AGEs), or H2O2 stimulated SGLT2 expression in human cultured proximal tubular cells, and then investigated the underlying molecular mechanisms.
Results
High glucose or AGEs did not affect SGLT2 expression in tubular cells. Insulin significantly increased tubular SGLT2 level in a dose-dependent manner, whereas bell-shaped dose-response curves were observed for H2O2-treated cells. An anti-oxidant, N-acetylcysteine completely blocked insulin-induced up-regulation of SGLT2 as well as increase in glucose absorption by tubular cells. Furthermore, insulin dose-dependently increased reactive oxygen species generation in tubular cells.
Conclusions
Our present study demonstrated that insulin could stimulate SGLT-2-mediated glucose entry into cultured proximal tubular cells via oxidative stress generation. Suppression of the insulin-induced overexpression of SGLT2 in tubular cells might be a novel therapeutic strategy for the treatment of diabetic nephropathy.
Keywords: SGLT2, Oxidative stress, Insulin, Diabetic nephropathy, AGEs
Introduction
Diabetic nephropathy is a leading cause of end-stage renal disease, which could account for disabilities and high mortality rates in patients with diabetes [1, 2]. Diabetic nephropathy is characterized by functional and structural changes in the glomerulus such as glomerular hyperfiltration, thickening of glomerular basement membrane, and an expansion of extracellular matrix in the mesangial areas, which could ultimately progress glomerular sclerosis associated with an increased urinary excretion rate of albumin and renal dysfunction [2, 3]. Indeed, characteristic histological changes of diabetic nephropathy are diffuse and nodular glomerulosclerosis [2, 3]. However, it is supposed that changes within the tubulointerstitium are more important than glomerulopathy in terms of renal dysfunction in diabetic nephropathy [4, 5].
Ninety percent of glucose filtered by the glomerulus is reabsorbed by a low-affinity/high capacity sodium-glucose cotransporter 2 (SGLT2), which is expressed mainly on S1 and S2 segment of renal proximal tubules [6–8]. Since blockade of SGLT2 promotes urinary glucose excretion and resultantly improves hyperglycemia in an insulin-independent manner, SGLT2 inhibitors are now one of the widely used agents for the treatment of diabetes [9–11]. Furthermore, we have previously shown that increased glucose uptake into cultured renal proximal tubular cells via SGLT2 stimulates oxidative stress generation and resultantly potentiates the pro-apoptotic effects of advanced glycation end products (AGEs), senescent macroprotein derivatives formed acceleratedly under diabetes, on tubular cells [12, 13]. Therefore, blockade of SGLT2 could also be a therapeutic target for preventing tubular apoptosis and atrophy in diabetic nephropathy. SGLT2 levels in tubular cells harvested from the urine of diabetic subjects are increased compared with non-diabetic subjects [14]. However, which diabetes-associated factors are involved in SGLT2 overexpression in diabetic kidney remains unclear. Therefore, in this study, we examined whether insulin, high glucose, AGEs, or H2O2 stimulated SGLT2 expression in human cultured proximal tubular cells, and then investigated the underlying molecular mechanisms.
Materials and methods
Materials
Insulin, bovine serum albumin (BSA) (essentially fatty acid free and essentially globulin free, lyophilized powder), N-acetylcysteine (NAC), and poly-L-lysine were purchased from Sigma (St. Louis, MO, USA). D-Glucose and H2O2 from Wako Pure Chemical Industries Ltd. (Osaka, Japan). D-glyceraldehyde from Nakalai Tesque (Kyoto, Japan). Antibodies (Abs) directed against human SGLT2 and β-actin from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
Cells
Proximal tubular epithelial cells from human kidney were maintained in complete medium (basal medium supplemented with 5 % fetal bovine serum, 0.5 μg/ml hydrocortisone, 10 ng/ml human epidermal growth factor, 0.5 μg/ml epinephrine, 6.5 ng/ml triiodo-L-thyronine, 10 μg/ml transferrin, 5 μg/ml insulin, and GA-1000) according to the supplier’s instructions (Lonza Walkersville, Inc. Walkersville, MD, USA) [15]. Cells at 3-5 passages were used for the experiments. Insulin, H2O2, or other treatments were carried out in a serum-free basal medium containing 10 μg/ml transferrin and GA-1000.
Preparation of AGEs-BSA
AGEs-BSA was prepared as described previously [16]. In brief, BSA (25 mg/ml) was incubated under sterile conditions with 0.1 M D-glyceraldehyde in 0.2 M NaPO4 buffer (pH 7.4) for 7 days. Then unincorporated sugars were removed by PD-10 column chromatography and dialysis against phosphate-buffered saline. Control non-glycated BSA was incubated in the same conditions except for the absence of reducing sugars.
Western blot analysis
Tubular cells were treated with or without the indicated concentrations of insulin and H2O2, 11, 22 or 33 mM glucose, 100 μg/ml AGEs-BSA or non-glycated BSA in the presence or absence of 1 mM NAC. After 24 h, proteins were extracted from tubular cells with lysis buffer, and then separated by SDS-PAGE and transferred to nitrocellulose membranes as described previously [13]. Membranes were probed with Abs against SGLT2 or β-actin, and then immune complexes were visualized with an enhanced chemiluminescence detection system (Amersham Bioscience, Buckinghamshire, United Kingdom). Data were normalized by the intensity of β-actin-derived signals and related to the value of non-treated control cells.
Assay for sodium-dependent glucose uptake
Tubular cells were treated with or without 50 ng/ml insulin in the presence or absence of 1 mM NAC for 24 h. Tubular cells were incubated with complete medium containing 100 μM 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG, Peptide Institute Inc., Osaka, Japan), fluorescent derivative of glucose for 15 min. Then culture medium was removed and replaced with Hanks’ balanced salt solution, and fluorescence intensity in the cells was analyzed in an ARVO fluorescent plate reader (PerkinElmer, Inc., Winter Street Waltham, MA, USA) as described previously [13].
Measurement of reactive oxygen species (ROS) generation
Intracellular formation of ROS was detected using a fluorescent probe carboxy-H2DFFDA (Life Technologies Japan, Tokyo, Japan) as described previously [17]. In brief, 96-well plates (FALCON, New York, NY, USA) were coated with 0.01 % poly-L-lysin for 30 min at room temperature. Then tubular cells were seeded into the well, and incubated with 0.1 % dimethyl sulfoxide (DMSO) in the presence or absence of 10 μM carboxy-H2DFFDA for 1 h. Then the cells were washed with phosphate-buffered saline, and treated with or without the indicated concentrations of insulin. After 24 min, intracellular ROS generation was measured with an ARVO X3 fluorescent plate reader (PerkinElmer Japan, Yokohama, Japan). ROS production was calculated by subtracting the fluorescence for cells pre-incubated with DMSO only from that with carboxy-H2DFFDA. Under cell-free conditions, 10 μM carboxy-H2DFFDA was also incubated with the indicated concentrations of H2O2 for 24 min, and then the fluorescence was measured.
Statistical analysis
All values are presented as mean ± standard deviation. One-way analysis of variance followed Tukey’s test or student’s t-test was performed for statistical comparisons; p-values of less than 0.05 were considered significant.
Results
Effects of insulin, high glucose, AGEs, or H2O2 on SGLT2 expression
We first examined whether insulin, high glucose, AGEs, or H2O2 stimulated SGLT2 expression in cultured proximal tubular cells. High glucose up to 33 mM or 100 μg/ml AGEs, comparable levels with plasma concentrations under diabetic situations [18], did not affect SGLT2 expression in tubular cells (data not shown). However, as shown in Figs. 1a and b, insulin significantly increased tubular SGLT2 expression level in a dose-dependent manner, whereas bell-shaped dose-response curves were observed for H2O2-treated cells. Since maximum response was obtained in 50 ng/ml insulin-treated cells (Fig. 1a), we chose the condition of 50 ng/ml insulin for the following experiments.
Fig. 1.
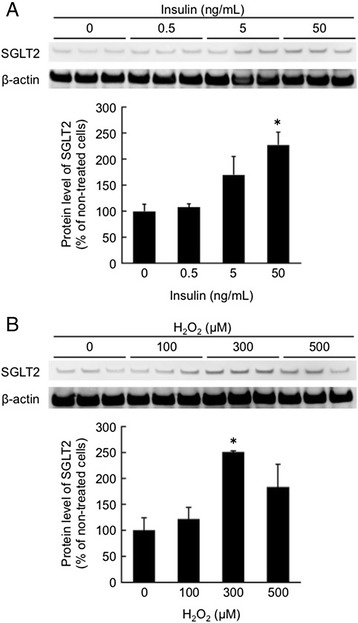
Effects of insulin (a) and H2O2 (b) on SGLT2 expression in tubular cells. Tubular cells were treated with or without the indicated concentrations of insulin and H2O2. After 24 h, proteins were extracted from tubular cells with lysis buffer, and then separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were probed with Abs against SGLT2 or β-actin, and then immune complexes were visualized with an enhanced chemiluminescence detection system. Each upper panel shows the representative bands of western blots. Each lower panel shows the quantitative data. Data were normalized by the intensity of β-actin-derived signals and related to the value of non-treated control cells. N = 3. *, p < 0.05 compared with the values of non-treated cells
Effects of NAC on insulin-induced SGLT2 expression and glucose uptake by tubular cells
We next studied the effects of an anti-oxidant NAC on insulin-induced SGLT2 expression and glucose uptake by tubular cells. As shown in Fig. 2a, 1 mM NAC completely blocked the insulin-induced up-regulation of SGLT2 level in tubular cells. Furthermore, 50 ng/ml insulin significantly increased glucose entry into tubular cells, which was also completely prevented by the treatment with 1 mM NAC (Fig. 2b).
Fig. 2.
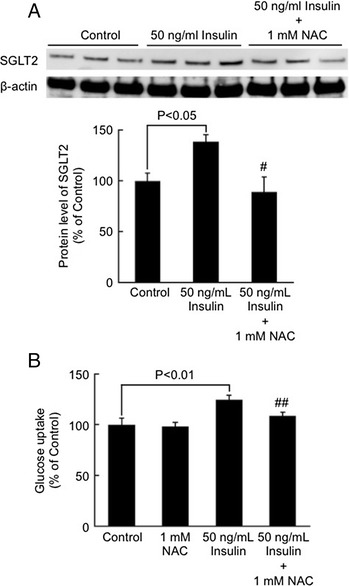
Effects of NAC on insulin-induced SGLT2 expression (a) and glucose uptake (b) by tubular cells. Tubular cells were treated with or without 50 ng/ml insulin in the presence or absence of 1 mM NAC for 24 h. a SGLT2 protein level was analyzed with western blot analysis. Upper panel shows the representative bands. Lower panel shows the quantitative data. N = 3. b Tubular cells were incubated with complete medium containing 100 μM 2-NBDG for 15 min. Then fluorescence intensity in the cells was analyzed. N = 6. Data of two independent experiments were combined. # and ##, p < 0.05 and p < 0.01, respectively compared with the values of 50 ng/ml insulin-treated cells
Effects of insulin on ROS generation in tubular cells
We investigated the effects of insulin on ROS generation in tubular cells. As shown in Fig. 3, insulin dose-dependently increased ROS generation in tubular cells. ROS production in tubular cells elicited by 50 ng/ml insulin was comparable with that of 300 μM H2O2.
Fig. 3.
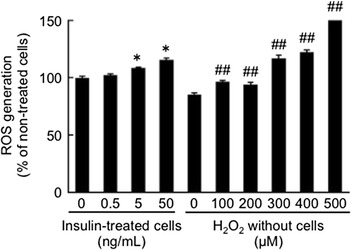
Effects of insulin on ROS generation in tubular cells. Tubular cells were incubated with 0.1 % DMSO in the presence or absence of 10 μM carboxy-H2DFFDA for 1 h. Then the cells were treated with or without the indicated concentrations of insulin. After 24 min, ROS generation was measured. Under cell-free conditions, 10 μM carboxy-H2DFFDA was incubated with the indicated concentrations of H2O2 for 24 min, and then the fluorescence was also measured. N = 6. *, p < 0.05 compared with the values of non-treated cells. ##, p < 0.01 compared with the values under cell-free conditions without H2O2
Discussion
In this study, we found that (1) 50 ng/ml insulin or 300 μM H2O2 significantly increased SGLT2 expression level in cultured proximal tubular cells, (2) an anti-oxidant NAC completely inhibited up-regulation of SGLT2 level in, and subsequently blocked the increase of glucose uptake by, 50 ng/ml insulin or exposed tubular cells, (3) 50 ng/ml insulin-evoked ROS generation in tubular cells was comparable with that of 300 μM H2O2, and (4) magnitude of increase in SGLT2 protein induced by 50 ng/ml insulin was similar to that by 300 μM H2O2. These observations suggest that insulin might stimulate SGLT-2-mediated glucose entry into cultured proximal tubular cells via oxidative stress generation.
There is some controversy about the expression level of SGLT2 in the diabetic kidney of type 1 diabetic animals; SGLT2 were decreased, unchanged or increased in streptozotocin-induced diabetic rats [19–22]. However, in contrast to type 1 diabetic animals, increased SGLT2 expression was consistently observed in the kidney of type 2 diabetic animals [23–25] and in tubular cells harvested from the urine of type 2 diabetic subjects [17], the latter of which was correlated with glucose reabsorption capacity in these patients. Therefore, insulin resistance and resultant hyperinsulinemia may contribute to SGLT2 overexpression in the kidney of type 2 diabetes. The ability of insulin to stimulate sodium reabsorption in proximal tubules is preserved in insulin-resistant subjects despite resistance to insulin metabolic effects [26]. So, the selective insulin resistance might be involved in SGLT2 induction under hyperinsulinemic conditions.
In the present study, although high glucose or AGEs have been reported to stimulate ROS generation in tubular cells [12, 13, 17], neither of them increased tubular SGLT2 expression (data not shown). High glucose or AGEs has been reported to induce apoptotic cell death of cultured proximal tubular cells [12, 13, 17], whereas insulin has anti-apoptotic properties in tubular cells and stimulates proliferation of this cell type [27, 28]. Furthermore, while high level of ROS is toxic to various types of cells, including proximal tubular cells [29–31], relatively low level of intracellular ROS could function as a second messenger in signaling cascades involved in gene expression [30–32]. Indeed, insulin-induced ROS generation has been coupled with its action in insulin-sensitive cells [30]. Therefore, the action of ROS on SGLT2 expression in tubular cells might also depend on its concentration. ROS generation evoked by high glucose or AGEs may be higher than 300 μM H2O2 and toxic to cells, which might partly explain the reason why these two agents cannot induce tubular SGLT2 expression. In this study, (1) SGLT2 level induced by 500 μM H2O2 was less than that by 300 μM H2O2 (Fig. 1b), and (2) total protein amounts obtained from 500 μM H2O2-exposed tubular cells were decreased to about 70 % of those of non-treated controls or 300 μM H2O2-exposed cells, thus supporting the concept that toxic level of ROS could affect SGLT2 expression in tubular cells.
We have previously shown that SGLT2-mediated glucose overload in tubular cells could augment the cells’ susceptibility toward pro-apoptotic effects of AGEs via overexpression of receptor for AGEs (RAGE) [13]. Furthermore, we, along with others, have recently found that empagliflozin, an inhibitor of SGLT2, suppresses oxidative, inflammatory and fibrotic reactions in the kidney and aorta of diabetic rats partly via suppression of the AGEs-RAGE axis [21, 33]. Apoptosis of proximal tubular cells plays a central role in tubular atrophy and atubular glomeruli of diabetic nephropathy [34, 35], which are most closely correlated with declining creatinine clearance in patients with diabetes [4, 5]. Given the pathological role of the AGEs-RAGE axis in tubular cell apoptosis [12, 13, 17], blockade of insulin-induced SGLT2 overexpression may not just improve hyperglycemia by promoting urinary glucose excretion, but could also directly inhibit glucotoxicity to proximal tubular cells, thus protecting against tubulointerstitial damage in diabetic nephropathy.
Limitations
We did experiments of Figs. 1a, b and 2a separately. So the 50 ng/ml column in Figs. 1a and 2a was not the same data. Since exposure time of an enhanced chemiluminescence detection system in each experiment differed, we were not able to show the data with the same units. This was a reason why we showed the data of non-treated cells in each experiment as a control. The physiological concentration of insulin in humans is 0.5-5 ng/ml. Oral administration of NAC for the treatment of acetaminophen poisoning obtained a plasma level of NAC at 10 μM [36]. Therefore, the concentration of insulin (50 ng/ml) and NAC (1 mM) having biological effects on tubular cells used in the present experiments may be in the superphysiologic range. This study was only analyzed in cell culture, not investigated about animal models and human. Therefore, further study is needed to clarify whether hyperinsulinemia may contribute to SGLT2 overexpression in animal model or human diabetic kidneys.
Conclusions
Our present study demonstrated that insulin could stimulate SGLT-2-mediated glucose entry into cultured proximal tubular cells via oxidative stress generation. Suppression of the insulin-induced overexpression of SGLT2 in tubular cells might be a novel therapeutic strategy for the treatment of diabetic nephropathy.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research (B) 22390111 (SY) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Abbreviations
- Abs
Antibodies
- AGEs
Advanced glycation end products
- BSA
Bovine serum albumin
- DMSO
Dimethyl sulfoxide
- NAC
N-acetylcysteine
- 2-NBDG
2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose
- ROS
Reactive oxygen species
- SGLT2
Sodium-glucose cotransporter 2
- RAGE
Receptor for AGEs
Footnotes
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
NN, TM, and YI acquired, analyzed, and interpreted data. SY mainly contributed to the present study, conceptualized and designed the study, acquired, analyzed, and interpreted data, and drafted the manuscript, and took responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Contributor Information
Nobutaka Nakamura, Email: nakamura_nobutaka@med.kurume-u.ac.jp.
Takanori Matsui, Email: matsui_takanori@med.kurume-u.ac.jp.
Yuji Ishibashi, Email: ishibashi_yuuji@med.kurume-u.ac.jp.
Sho-ichi Yamagishi, Phone: +81-942-31-7873, Email: shoichi@med.kurume-u.ac.jp.
References
- 1.Yamagishi S, Imaizumi T. Diabetic vascular complications: pathophysiology, biochemical basis and potential therapeutic strategy. Curr Pharm Des. 2005;11:2279–99. doi: 10.2174/1381612054367300. [DOI] [PubMed] [Google Scholar]
- 2.Yamagishi S, Matsui T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev. 2010;3:101–8. doi: 10.4161/oxim.3.2.11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukami K, Taguchi K, Yamagishi S, Okuda S. Receptor for advanced glycation endproducts and progressive kidney disease. Curr Opin Nephrol Hypertens. 2015;24:54–60. doi: 10.1097/MNH.0000000000000091. [DOI] [PubMed] [Google Scholar]
- 4.Taft JL, Nolan CJ, Yeung SP, Hewitson TD, Martin FI. Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes. 1994;43:1046–51. doi: 10.2337/diab.43.8.1046. [DOI] [PubMed] [Google Scholar]
- 5.Ziyadeh FN, Goldfarb S. The renal tubulointerstitium in diabetes mellitus. Kidney Int. 1991;39:464–75. doi: 10.1038/ki.1991.57. [DOI] [PubMed] [Google Scholar]
- 6.Lee YJ, Lee YJ, Han HJ. Regulatory mechanims of Na+/glucose cotransporters in renal proximal tubule cells. Kidney Int. 2007;72:527–35. doi: 10.1038/sj.ki.5002490. [DOI] [PubMed] [Google Scholar]
- 7.Santer R, Calado J. Familial renal glucosuria and SGLT2: from a mendelian trait to a therapeutic target. Clin J Am Soc Nephrol. 2010;5:133–41. doi: 10.2215/CJN.04010609. [DOI] [PubMed] [Google Scholar]
- 8.Sabolic I, Vrhovac I, Eror DB, Gerasimova M, Rose M, Breljak D, Ljubojevic M, Brzica H, Sebastiani A, Thal SC, Sauvant C, Kipp H, Vallon V, Koepsell H. Expression of Na + -D-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am J Physiol Cell Physiol. 2012;302:C1174–88. doi: 10.1152/ajpcell.00450.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–33. doi: 10.1016/S0140-6736(10)60407-2. [DOI] [PubMed] [Google Scholar]
- 10.Musso G, Gambino R, Cassader M, Pagano G. A novel approach to control hyperglycemia in type 2 diabetes: sodium glucose co-transport (SGLT) inhibitors: systematic review and meta-analysis of randomized trials. Ann Med. 2012;44:375–93. doi: 10.3109/07853890.2011.560181. [DOI] [PubMed] [Google Scholar]
- 11.Mikhail N. Place of sodium-glucose co-transporter type 2 inhibitors for treatment of type 2 diabetes. World J Diabetes. 2014;5:854–9. doi: 10.4239/wjd.v5.i6.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Beneficial effects of metformin and irbesartan on advanced glycation end products (AGEs)-RAGE-induced proximal tubular cell injury. Pharmacol Res. 2012;65:297–302. doi: 10.1016/j.phrs.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Maeda S, Matsui T, Takeuchi M, Yamagishi S. Sodium-glucose cotransporter 2-mediated oxidative stress augments advanced glycation end products-induced tubular cell apoptosis. Diabetes Metab Res Rev. 2013;29:406–12. doi: 10.1002/dmrr.2407. [DOI] [PubMed] [Google Scholar]
- 14.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54:3427–34. doi: 10.2337/diabetes.54.12.3427. [DOI] [PubMed] [Google Scholar]
- 15.Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M. Advanced glycation end products inhibit de novo protein synthesis and induce TGF-beta overexpression in proximal tubular cells. Kidney Int. 2003;63:464–73. doi: 10.1046/j.1523-1755.2003.00752.x. [DOI] [PubMed] [Google Scholar]
- 16.Yamagishi S, Nakamura K, Matsui T, Inagaki Y, Takenaka K, Jinnouchi Y, Yoshida Y, Matsuura T, Narama I, Motomiya Y, Takeuchi M, Inoue H, Yoshimura A, Bucala R, Imaizumi T. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J Biol Chem. 2006;281:20213–20. doi: 10.1074/jbc.M602110200. [DOI] [PubMed] [Google Scholar]
- 17.Matsui T, Yamagishi S, Takeuchi M, Ueda S, Fukami K, Okuda S. Irbesartan inhibits advanced glycation end product (AGE)-induced proximal tubular cell injury in vitro by suppressing receptor for AGEs (RAGE) expression. Pharmacol Res. 2010;61:34–9. doi: 10.1016/j.phrs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Miura J, Yamagishi S, Uchigata Y, Takeuchi M, Yamamoto H, Makita Z, Iwamoto Y. Serum levels of non-carboxymethyllysine advanced glycation endproducts are correlated to severity of microvascular complications in patients with Type 1 diabetes. J Diabetes Compl. 2003;17:16–21. doi: 10.1016/S1056-8727(02)00183-6. [DOI] [PubMed] [Google Scholar]
- 19.Albertoni Borghese MF, Majowicz MP, Ortiz MC, Passalacqua Mdel R, Sterin Speziale NB, Vidal NA. Expression and activity of SGLT2 in diabetes induced by streptozotocin: relationship with the lipid environment. Nephron Physiol. 2009;112:45–52. doi: 10.1159/000214214. [DOI] [PubMed] [Google Scholar]
- 20.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol. 2013;304:F156–67. doi: 10.1152/ajprenal.00409.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ojima A, Matsui T, Nishino Y, Nakamura N, Yamagishi S. Empagliflozin, an inhibitor of sodium-glucose cotransporter 2 exerts anti-inflammatory and anti-fibrotic effects on experimental diabetic nephropathy partly by suppressing AGEs-receptor axis. Horm Metab Res. 2015; doi:10.1055/s-0034-1395609. [DOI] [PubMed]
- 22.Osorio H, Coronel I, Arellano A, Franco M, Escalante B, Bautista R. Ursodeoxycholic acid decreases sodium-glucose cotransporter (SGLT2) expression and oxidative stress in the kidney of diabetic rats. Diabetes Res Clin Pract. 2012;97:276–82. doi: 10.1016/j.diabres.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 23.Freitas HS, Anhê GF, Melo KF, Okamoto MM, Oliveira-Souza M, Bordin S, Machado UF. Na+-glucose transporter-2 messenger ribonucleic acid expression in kidney of diabetic rats correlates with glycemic levels: involvement of hepatocyte nuclear factor-1alpha expression and activity. Endocrinology. 2008;149:717–24. doi: 10.1210/en.2007-1088. [DOI] [PubMed] [Google Scholar]
- 24.Osorio H, Bautista R, Rios A, Franco M, Santamaría J, Escalante B. Effect of treatment with losartan on salt sensitivity and SGLT2 expression in hypertensive diabetic rats. Diabetes Res Clin Pract. 2009;86:e46–9. doi: 10.1016/j.diabres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Renal Physiol. 2011;300:R1009–22. doi: 10.1152/ajpregu.00809.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horita S, Seki G, Yamada H, Suzuki M, Koike K, Fujita T. Insulin resistance, obesity, hypertension, and renal sodium transport. Int J Hypertens. 2011;2011:391762. doi: 10.4061/2011/391762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gansler T, Hsu WC, Gramling TS, Robinson KA, Buse MG, Blocker N, Roy L, Green S, Garvin AJ, Sens DA. Growth factor binding and bioactivity in human kidney epithelial cell cultures. In Vitro Cell Dev Biol. 1990;26:285–90. doi: 10.1007/BF02624459. [DOI] [PubMed] [Google Scholar]
- 28.Meier M, Nitschke M, Hocke C, Kramer J, Jabs W, Steinhoff J, Schutt M. Insulin inhibits caspase-3 activity in human renal tubular epithelial cells via the PI3-kinase/Akt pathway. Cell Physiol Biochem. 2008;21:279–86. doi: 10.1159/000129386. [DOI] [PubMed] [Google Scholar]
- 29.Schaaf GJ, Nijmeijer SM, Maas RF, Roestenberg P, de Groene EM, Fink-Gremmels J. The role of oxidative stress in the ochratoxin A-mediated toxicity in proximal tubular cells. Biochim Biophys Acta. 2002;1588:149–58. doi: 10.1016/S0925-4439(02)00159-X. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–21. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oelze M, Kröller-Schön S, Welschof P, Jansen T, Hausding M, Mikhed Y, Stamm P, Mader M, Zinßius E, Agdauletova S, Gottschlich A, Steven S, Schulz E, Bottari SP, Mayoux E, Münzel T, Daiber A. The sodium-glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS One. 2014;9 doi: 10.1371/journal.pone.0112394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugden PH, Clerk A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal. 2006;8:2111–24. doi: 10.1089/ars.2006.8.2111. [DOI] [PubMed] [Google Scholar]
- 33.Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11:173–86. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 34.Brezniceanu ML, Lau CJ, Godin N, Chénier I, Duclos A, Ethier J, Filep JG, Ingelfinger JR, Zhang SL, Chan JS. Reactive oxygen species promote caspase-12 expression and tubular apoptosis in diabetic nephropathy. J Am Soc Nephrol. 2010;2:943–54. doi: 10.1681/ASN.2009030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White KE, Marshall SM, Bilous RW. Prevalence of atubular glomeruli in type 2 diabetic patients with nephropathy. Nephrol Dial Transplant. 2008;23:3539–45. doi: 10.1093/ndt/gfn351. [DOI] [PubMed] [Google Scholar]
- 36.Olsson B, Johansson M, Gabrielsson J, Bolme P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur J Clin Pharmacol. 1988;34:77–82. doi: 10.1007/BF01061422. [DOI] [PubMed] [Google Scholar]