

Abstract
Cystic fibrosis is the most common autosomal recessive genetic disease in Caucasians and has been extensively studied for many decades. The cystic fibrosis transmembrane conductance regulator gene was identified in 1989. It encodes a complex protein which has numerous cellular functions. Our understanding of cystic fibrosis pathophysiology and genetics is constantly expanding and being refined, leading to improved management of the disease and increased life expectancy in affected individuals.
Introduction
Cystic fibrosis is a monogenic, predominantly Caucasian, autosomal recessive disease. Approximately 10,000 cystic fibrosis patients live in the UK today, of which more than 57% are adults. The median predicted survival in the UK is currently 37 years [1].
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene affect multiple organs, including the intestine, sweat glands, pancreas and the reproductive system, but cystic fibrosis lung disease causes most morbidity and leads to premature mortality in cystic fibrosis patients [2]. The CFTR protein generates a chloride channel present in the apical membrane of epithelial cells throughout the body. However, CFTR has many other functions and understanding the effects of CFTR mutations on disease pathophysiology is an important step towards improving the life expectancy of cystic fibrosis patients. The Cystic Fibrosis Mutation Database (CFTR1) currently lists 1,991 CFTR mutations (http://www.genet.sickkids.on.ca/cftr) and understanding the consequences of a particular mutation on protein function is central to the development of mutation-specific treatments.
Here, we will discuss how further improving our understanding of cystic fibrosis pathophysiology and genetics is continuously contributing to the improved management of cystic fibrosis lung disease.
Cystic fibrosis pathophysiology
It is undisputed that persistent cycles of lung infection and inflammation lead to mucous obstruction of the cystic fibrosis patient's airways, which ultimately causes organ failure in most cystic fibrosis patients. Although the role of CFTR in transepithelial ion transport and host defence is widely accepted, there is an ongoing debate about some of the key processes. The “low volume” hypothesis has long been the leading hypothesis [3]. This postulates that decreased transepithelial chloride transport, due to mutated CFTR, and increased transepithelial sodium absorption, due to lack of CFTR-dependent inhibition of epithelial sodium channels (ENaCs), leads to increased water absorptions into the tissue and, therefore, decreased airway surface liquid and reduced mucociliary clearance (Figure 1). This hypothesis is consistent with altered potential difference measurements in the nose and upper airways of cystic fibrosis patients [4,5] and has contributed to the development of nebulised hypertonic saline as an inexpensive and effective additional therapy for patients with cystic fibrosis [6]. Studies in the cystic fibrosis knockout pig confirmed the lack of chloride transport and sodium hyperabsorption in nasal epithelium [7], but highlighted that sodium hyperabsorption and depletion of airway surface liquid were not present in cystic fibrosis pig airways [8], thereby somewhat questioning the validity of the “low volume” hypothesis. However, consistent with cystic fibrosis patients, the cystic fibrosis pig also had reduced CFTR-dependent bicarbonate secretion in the airways [8]. In a subsequent study in cystic fibrosis pigs, the group suggests that reduced bicarbonate secretion leads to reduced airway surface pH which impairs innate bacterial defence mechanisms [9]. If these findings are further confirmed, strategies aimed at increasing airway surface pH may be of therapeutic benefit in cystic fibrosis. In addition to reducing airway surface pH, the impaired bicarbonate secretion has also been implicated in affecting mucous unfolding and rheology [10], thereby providing further opportunities for new treatment strategies.
Figure 1. Ion and water transport across airway epithelial cells.
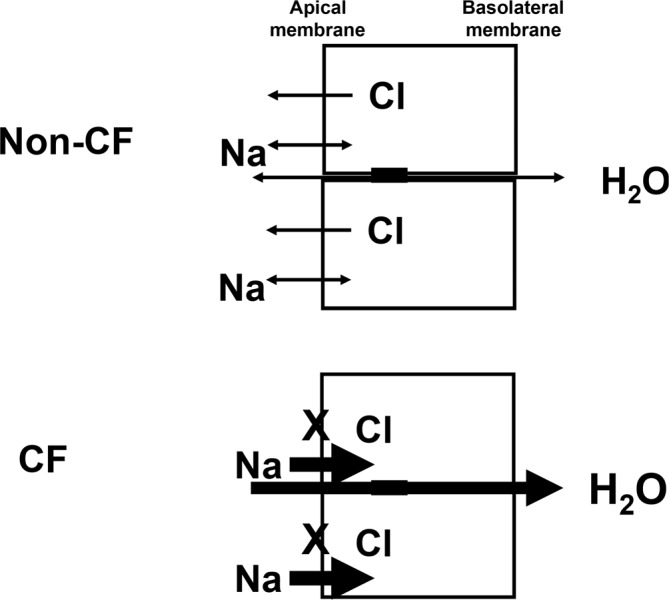
Chloride (Cl−) and sodium (Na+) transport across the apical membrane of airway epithelial cells regulate water transport. The “low volume” hypothesis postulates that, in cystic fibrosis patients, chloride transport is impaired and sodium absorption is upregulated, which leads to increased water absorption from the airways into the tissue and depletion of the airway surface liquid.
Numerous reports suggest additional defects in innate and adaptive immune responses in cystic fibrosis patients. For example, Kelly et al. [11] reported that the Toll-like receptor 4, which is responsible for lipopolysaccharide (LPS)-mediated activation of nuclear factor-κB (NFκB) is not processed appropriately, which may account for the prolonged cycle of chronic inflammation associated with cystic fibrosis. Production of hypothiocyanite, an important host defence molecule, also appears to be altered in the cystic fibrosis lung [12]. The role of CFTR expression in inflammatory cells — such as neutrophils, macrophages and, more recently, T cells — has been widely, and so far inconclusively, debated [13–17] but studies overall appear to suggest a potential defect in adaptive immune responses in cystic fibrosis patients, which may explain the exaggerated pulmonary inflammatory responses which have been generally observed, an area that requires further studies.
Our understanding of cystic fibrosis pathophysiology has recently been complicated by studies showing that external factors, such as cigarette smoke, can alter CFTR function in the lungs as well as in extrapulmonary tissues [18,19]. These studies further highlight the complex interaction between the CFTR genotype and the environment (see below) [20].
The gender gap
The issue of whether female cystic fibrosis patients have worse outcomes compared to men has been widely debated and generated conflicting results. The “gender gap” has largely been studied retrospectively using databases of various sizes. The most recent and currently largest retrospective study assessed a cohort of almost 33,000 cystic fibrosis patients and showed that female cystic fibrosis patients die earlier and have a higher risk of bacterial infection [21]. However, a recent, but comparatively small (~130 subjects) prospective study did not confirm these data [22].
A gender gap has also been documented for asthma and chronic obstructive pulmonary disease (COPD) [23,24]. The reasons are still unclear, but sex hormones are obvious candidates. In the context of cystic fibrosis, studies have shown that oestrogens affect epithelial ion transport and reduce airway surface liquid, as well as reduce cilia beat frequency in airway epithelial cells [25–27]. In addition, it has recently been shown that oestrogen exposure leads to the conversion of non-mucoid to mucoid Pseudomonas aeroginosa, which are more difficult to eradicate [28]. However, a retrospective study using UK registry data did not show positive or negative effects in cystic fibrosis females using oral contraception [29]. Numerous other factors, including physical activity, nutrition, adherence to treatment, as well as socioeconomic and environmental factors, may play a role. Prospective longitudinal studies aimed at untangling and identifying key contributors will be complex, but are warranted and will further improve understanding and management of cystic fibrosis.
Atypical cystic fibrosis and CFTR-related metabolic syndrome (CRMS)
Although most individuals with cystic fibrosis have respiratory and gastrointestinal symptoms (classical cystic fibrosis), some mutations, generally milder genetic variants which retain residual CFTR function (see below), can lead to isolated, milder symptoms, such as chronic sinusitis, idiopathic pancreatitis, non-cystic fibrosis bronchiectasis, and congenital bilateral absence of the vas deferens [30]. Based on symptoms, these patients have been classified as atypical cystic fibrosis, non-classical cystic fibrosis, CFTR-related disorders, low-risk genotypes, or mild variant cystic fibrosis. Individuals with atypical cystic fibrosis presentations usually do not have pancreatic insufficiency and they are typically diagnosed at an older age.
The introduction of newborn screening programmes (see below) led to a further refinement of disease categorisation. In contrast to patients classified as having atypical cystic fibrosis, babies identified through newborn screening programmes are generally symptom free. This leads to the identification of cases in whom cystic fibrosis cannot clearly be diagnosed or ruled out because, although up to two CFTR mutations may have been identified, at least one of these can currently not be clearly categorised as disease-causing. The term CFTR-related metabolic syndrome (CRMS, US nomenclature) or CF-SPID (cystic fibrosis screen positive, inconclusive diagnosis, EU nomenclature) are now used to describe this category of subjects, and guidelines for clinical management have been published [31].
CFTR mutations
As mentioned above, the cystic fibrosis mutation database (CFTR1) currently lists 1,991 mutations, but not all can be conclusively categorized as disease-causing. By far the most common mutation worldwide is a deletion of phenylalanine (F508del, previously called deltaF508). In the UK, 90% of cystic fibrosis patients are homozygote or compound heterozygotes for F508del, although the absolute frequency varies amongst different populations. The missense mutations G551D and R117H are present in 5.8% and 4.5% of UK cystic fibrosis patients, followed by the nonsense mutation G542X (3.6%). The vast majority of other mutations are very rare, which makes them difficult to study. CFTR mutations are currently grouped into six classes depending on the effects on the protein [32] (Table 1). F508del is a class II mutation leading to the defective processing of the CFTR protein, G551D is a class III mutation leading to defective chloride channel gating, R117H is a class IV mutation leading to defective channel conductance, and G542X is an example of a class I mutation where a premature stop codon results in a complete lack of protein expression.
Table 1. CFTR Gene Mutation Classes.
| Class | Effect on Protein | Key examples of disease-causing mutations |
|---|---|---|
| I | Defective protein production | G542X |
| II | Defective protein processing | F508del |
| III | Defective protein regulation | G551D |
| IV | Defective protein conductance | R117H |
| V | Reduced protein synthesis | A455E |
| VI | Reduced protein surface retention | c.120del23 |
Our understanding of how DNA sequence alterations affect CFTR protein function is constantly increasing and helped by the Clinical and Functional Translation of CFTR (CFTR2) Project (www.cftr2.org), which aims to further characterise the effects of CFTR mutation on protein function and clinical presentation [33]. CFTR2 will contribute to improving diagnosis, newborn screening, and carrier testing, as well as understanding the complex relationship between genotype and phenotype (see below) and the further development of mutation-specific treatments.
Cystic fibrosis newborn screening
Newborn screening for cystic fibrosis has now been implemented in many European countries, North America and Australia. In London and the South East of England the cystic fibrosis screening programme was rolled out in 2007 [34]. Immunoreactive trypsinogen (IRT) is first quantified in a dried blood sample. If IRT is elevated, a screen for the four most common CFTR mutations is performed. If no or only one CFTR mutation is identified, IRT is quantified in a second blood sample to confirm the results. In babies in whom one CFTR mutation was identified, a further genetic test assessing an extended panel of 30 or 32 CFTR mutations (95% pick-up rate in Caucasians) is carried out (Figure 2). This protocol was developed to minimise false positive detection rates, reduce the identification of carriers and increase the chances of detecting cystic fibrosis in ethnic minorities, in which the extended mutation panel only picks up 65% of cases. (Of note: the described protocol is based on UK guidelines. Protocols in other countries vary.)
Figure 2. Schematic of the UK Newborn Screening Programme.
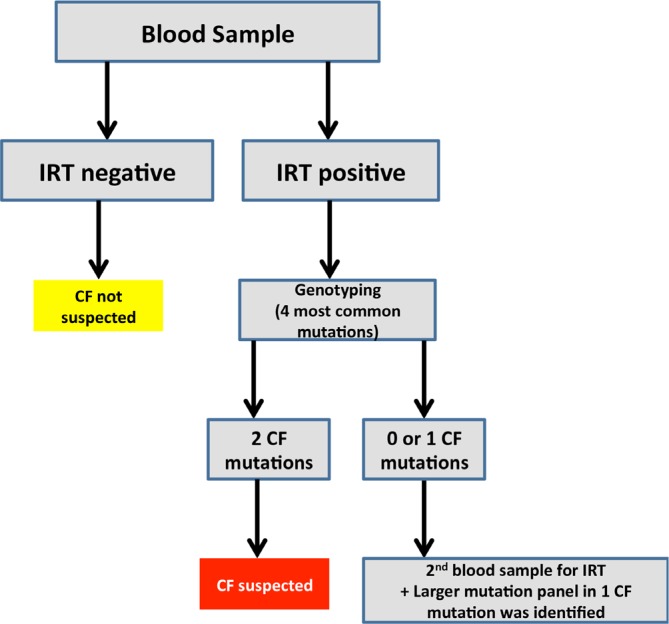
CF, cystic fibrosis; IRT, immunoreactive trypsinogen.
The benefits of newborn screening and the inherent earlier diagnosis are now well recognised, but ethical questions related to the increased detection of cystic fibrosis carriers and uncertainties related to the CFTR-related metabolic disorders status (see above) have to be carefully managed.
Genotype/phenotype correlations and modifier genes
CFTR mutation classes I-III are generally considered to be more severe than classes IV-VI, which retain varying degrees of residual CFTR function. However, the genotype/phenotype correlation varies significantly between affected organs. The correlation is, for example, strong for pancreatic disease, showing that severe mutations (classes I-III) lead to pancreatic insufficiency whereas milder mutations (classes IV-VI) are associated with pancreatic sufficiency [35]. In contrast, lung disease severity is very variable and difficult to predict, even in siblings with the same CFTR mutations, highlighting the significant contribution of external factors (for example, the environment) and endogenous factors (modifier genes) to disease severity [36].
Even though cystic fibrosis is defined as a single gene disorder, it is clear that other genes (modifiers) will affect disease progression. Identification of modifier genes may improve prognosis and identify new drug targets. A large number of genome-wide and candidate gene approaches have been conducted to identify cystic fibrosis modifier genes and have recently been reviewed [36]. Results are not always consistent and it has become clear that large sample sizes and well-defined homogeneous patient cohorts, as well as independent confirmation of results in separate cohorts of patients, are essential in order to generate reliable data. It has also become apparent that the number of putative modifier genes may be large and organ dependent. In addition, it is obvious that, particularly for cystic fibrosis lung disease, environmental factors will play a significant role.
Our understanding of cystic fibrosis is further complicated by studies implying that gain-of-function mutations/polymorphisms causing hyperactivity of the ENaCs can, in the absence of CFTR mutations, lead to cystic fibrosis-like disease [37–39]. These findings strengthen the search for ENaC inhibitors in the context of new treatments for cystic fibrosis.
New treatments
Over the last two decades, several therapies — including, amongst others, novel antibiotics, such as inhaled tobramycin and macrolides, and novel mucolytics, such as dornase alpha (a recombinant DNase) and hypertonic saline — have progressed from clinical trials into mainstream treatment. Here, we will briefly describe key mutation-specific and mutation-independent approaches that are currently assessed in clinical trials or have recently been licensed.
Mutation-specific drugs
CFTR potentiators to treat class III mutations
CFTR potentiators are defined as agents that potentiate mutated but apically localised CFTR and may therefore be of therapeutic value for class III and IV mutations. The recently licensed drug ivacaftor (also known as Kalydeco or Vx-770) is a CFTR potentiator that was developed for class III mutations and first assessed in patients with at least one G551D mutation. Oral administration led to rapid improvement in lung function, weight gain, reduced pulmonary exacerbations, and improved quality of life ([40] and references therein). More recently, ivacaftor was also assessed in patients with other class III gating mutations and demonstrated comparable efficacy for eight other class III gating mutations [41]. However, ivacaftor was ineffective in patients homozygous for F508del [42]. The identification and licensing of ivacaftor was an important milestone because this provided, for the first time, proof-of-concept that the molecular defect in CFTR is targetable and correctable. However, it is important to note that only ~5% of cystic fibrosis patients carry mutations that respond to ivacaftor and a number of important points have been raised, including the high costs of the treatment (although exact costs are not publically available), clinical trial design when targeting very rare mutations, appropriate endpoints for small and comparatively short trials, and lower age and severity cut-off for trial participants (all recently reviewed in [43]). Importantly, the successful development of ivacaftor has renewed interest and investment into other therapies aimed at targeting the molecular defect, which may be more applicable for larger numbers of cystic fibrosis patients (see below).
CFTR correctors to treat class II mutations
CFTR correctors are defined as agents that correct the processing defect of class II mutations, such as F508del, and therefore promise to be suitable for the vast majority of cystic fibrosis patients. The CFTR corrector lumacaftor (also called Vx-809) reduced sweat chloride, but did not improve lung function in F508del patients when given alone [44]. Although, as mentioned above, ivacaftor monotherapy was also ineffective in F508del patients, the combination therapy of lumacaftor plus ivacaftor was assessed in homozygote and compound heterozygote F508del patients [45]. This is based on the rationale that lumacaftor may progress F508del protein to the apical membrane, but that the mutant protein requires the potentiator to function fully. The co-administration of lumacaftor with ivacaftor led to improvements in lung function (albeit lower than achieved with ivacaftor monotherapy in class III patients) and appeared to be less effective in F508del compound heterozygotes (recently reviewed in [43]). It is currently widely debated whether the magnitude of the therapeutic benefit observed warrants licensing of the combination therapy, or whether more potent correctors will be required.
Readthrough drugs to treat class I mutations
Proof-of-concept for drug-induced readthrough of premature nonsense mutations has been established in vitro and in vivo more than 10 years ago. This early research led to the development of ataluren (also known as PTC124) which was recently assessed in a placebo-controlled phase 3 trial [46]. Although the trial did not reach its primary endpoint (FEV1% predicted) a pre-defined subgroup analysis determined that inhaled antibiotics which were taken as part of routine clinical care negatively affected ataluren-mediated readthrough of the nonsense mutation. These findings may warrant further assessment of ataluren in patients not taking concomitant antibiotics.
Mutation-independent drugs
Gene therapy
Gene therapy, defined as the addition of a functional CFTR gene into airway epithelial cells, is the classical example of a mutation-independent treatment strategy and has been developed for the last 20 years. A phrase similar to “cystic fibrosis gene therapy has failed to demonstrate clinical benefit” which is frequently stated in the literature requires some clarification. It is undoubtedly true that the development of pulmonary gene therapy has been slower than initially predicted and hoped for. This is in part due to powerful external and internal barriers (including mucus, cilia beating and the nuclear membrane) which affect the efficiency of gene transfer [47]. In the past, 26 clinical trials involving approximately 450 patients have been carried out [48]. The vast majority of these trials were short, included small numbers of patients, and were not designed to assess clinical benefit, but to establish safety and proof-of-concept for gene transfer using molecular endpoints, such as the detection of recombinant mRNA or correction of the ion transport defect. Early trials focussed on delivery of gene transfer agents to the nasal epithelium, used as a surrogate for lung delivery. In the majority of trials adenoviral and adeno-associated viral (AAV) vectors were used.
The only trial currently designed and powered to assess clinical efficacy (defined as improvement in lung function) administered AAV2-CFTR to the lungs of cystic fibrosis patients on three occasions (monthly dosing intervals) and failed to demonstrate an improvement in lung function. There are several reasons for these results: (a) it has been shown that AAV2 is very inefficient in transducing airway epithelial cells via the apical membrane and more potent serotypes have since been identified; (b) the long terminal repeat (LTR) promoter, which was used to drive expression of the 4.7 kb CFTR cDNA due to the limited packaging capacity (about 5 kb) of the virus, is too weak; and (c) repeat administration of AAV2 to the lung was not possible due to the development of an anti-viral immune response. Induction of immune responses to most viral vectors, including adenovirus and AAV, severely restrict their use and these vectors are now deemed not to be suitable for repeated administration which is required to treat a life-long disease, such as cystic fibrosis.
In addition to viral vectors, numerous non-viral vectors have been studied in cystic fibrosis clinical trials but, as mentioned above, none of these was designed to assess clinical efficacy [48]. Importantly, the UK Cystic Fibrosis Gene Therapy Consortium has recently (Autumn 2014) completed the first non-viral gene therapy trial designed to answer whether repeated non-viral gene transfer (12 doses over 12 months) can lead to clinical benefit. The trial was placebo-controlled, included 130 patients and was powered to detect an improvement in lung function. Until the results of this trial are available, the statement “cystic fibrosis gene therapy has failed to demonstrate clinical benefit” may be premature. Importantly, in addition to being mutation-independent, gene therapy has the potential to help reduce the significant treatment burden which causes treatment fatigue and poor compliance in the cystic fibrosis community. We are, for example, currently developing a lentiviral vector which efficiently transduces the airways and leads to stable gene expression in mouse airways for >20 months after a single dose [49,50].
Pharmacological inhibitors and activators of other ion channels
Several ion channels have been targeted to date. One of the leading strategies is the down-regulation of the hyperactive ENaC. Amiloride, an ENaC inhibitor used for the treatment of hypertension and congestive heart failure, was the first example assessed in the context of cystic fibrosis. However, the drug was ineffective, possibly due to its short half-life. Unfortunately, longer acting and more potent ENaC inhibitors such as benzamil and PS552 have, to date, also not shown convincing efficacy in cystic fibrosis trials [51,52].
An alternative approach is the activation of alternative calcium-activated chloride channels, via activation of purinergic receptors using ATP, UTP or denufosol. However, the clinical efficacy of this approach has not yet been convincingly demonstrated [43].
Conclusions
Our understanding of cystic fibrosis pathophysiology and genetics is constantly expanding and this continuously leads to tangible improvements for patients; most notably the recent licensing of the first drug, ivacaftor, targeting the molecular defect in the CFTR protein in approximately 5% of cystic fibrosis patients. The next few years will show whether other classes of small molecule drugs or gene therapy (applicable to most cystic fibrosis patients) will be equally successful. However, significant challenges remain. Firstly, the costs of many new drugs will likely be high and may place significant burdens on available health budgets. Secondly, treatment fatigue and poor compliance are real problems in the cystic fibrosis community and need to be tackled. Attention has to be paid to reducing the overall treatment burden. Thirdly, although novels insights into pathophysiology and genetics have led to significant improvements in disease management in the developed world, this has not translated into less well-developed countries (including Eastern Europe). Strategies to introduce state-of-the-art care into these countries need to be developed. Lastly, although cystic fibrosis is largely known as a Caucasian disease, increasing numbers of patients with cystic fibrosis are being identified in other large populations, including China and India. Raising awareness of cystic fibrosis and improving diagnostic methods in these countries is essential.
Acknowledgments
We thank Samia Soussi for help with preparing the manuscript. The project was supported by the National Institute for Health Research (NIHR) Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. The views expressed in this publication are those of the authors and not necessarily those of the NHS, The National Institute for Health Research or the Department of Health.
Abbreviations
- AAV
adeno-associated virus
- CFTR
cystic fibrosis transmembrane conductance regulator
- CFTR1
cystic fibrosis mutation database
- CRMS
CFTR-related metabolic syndrome
- ENaC
epithelial sodium channel
- IRT
immunoreactive trypsinogen
Disclosures
The authors declare that they have no disclosures but acknowledge that they receive funding for cystic fibrosis gene therapy-based research.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/7/64
References
- 1.UK Cystic Fibrosis Registry: Annual Data report. [ www.cysticfibrosis.org.uk/media/598466/annual-data-report-2013-jul14.pdf]
- 2.Boyle MP, Boeck K de. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. The Lancet. Respiratory medicine. 2013;1:158–63. doi: 10.1016/S2213-2600(12)70057-7. [DOI] [PubMed] [Google Scholar]
- 3.Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annual review of medicine. 2007;58:157–70. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- 4.Naehrlich L, Ballmann M, Davies J, Derichs N, Gonska T, Hjelte L, van Konigsbruggen-Rietschel S, Leal T, Melotti P, Middleton P, Tümmler B, Vermeulen F, Wilschanski M. Nasal potential difference measurements in diagnosis of cystic fibrosis: an international survey. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2014;13:24–8. doi: 10.1016/j.jcf.2013.08.006. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718232878
- 5.Davies JC, Davies M, McShane D, Smith S, Chadwick S, Jaffe A, Farley R, Collins L, Bush A, Scallon M, Pepper J, Geddes DM, Alton Eric WFW. Potential difference measurements in the lower airway of children with and without cystic fibrosis. American journal of respiratory and critical care medicine. 2005;171:1015–9. doi: 10.1164/rccm.200408-1116OC. [DOI] [PubMed] [Google Scholar]
- 6.Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, Belousova EG, Xuan W, Bye Peter TP. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. The New England journal of medicine. 2006;354:229–40. doi: 10.1056/NEJMoa043900. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/11399
- 7.Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB, Zabner J, Prather RS, Welsh MJ. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science (New York, NY) 2008;321:1837–41. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1123954
- 8.Chen J, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, Zabner J, Welsh MJ. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–23. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/7064957
- 9.Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa Mahmoud H Abou, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Bánfi B, Horswill AR, Stoltz DA, McCray PB, Welsh MJ, Zabner J. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–13. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717948506
- 10.Quinton PM. Role of epithelial HCO3− transport in mucin secretion: lessons from cystic fibrosis. American journal of physiology. Cell physiology. 2010;299:C1222–33. doi: 10.1152/ajpcell.00362.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/722351688
- 11.Kelly C, Canning P, Buchanan PJ, Williams MT, Brown V, Gruenert DC, Elborn JS, Ennis M, Schock BC. Toll-like receptor 4 is not targeted to the lysosome in cystic fibrosis airway epithelial cells. American journal of physiology. Lung cellular and molecular physiology. 2013;304:L371–82. doi: 10.1152/ajplung.00372.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717986343
- 12.Smith DJ, Lamont IL, Anderson GJ, Reid DW. Targeting iron uptake to control Pseudomonas aeruginosa infections in cystic fibrosis. The European respiratory journal. 2013;42:1723–36. doi: 10.1183/09031936.00124012. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718194673
- 13.Goldstein W, Döring G. Lysosomal enzymes from polymorphonuclear leukocytes and proteinase inhibitors in patients with cystic fibrosis. The American review of respiratory disease. 1986;134:49–56. doi: 10.1164/arrd.1986.134.1.49. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura H, Yoshimura K, McElvaney NG, Crystal RG. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. The Journal of clinical investigation. 1992;89:1478–84. doi: 10.1172/JCI115738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haggie PM, Verkman AS. Cystic fibrosis transmembrane conductance regulator-independent phagosomal acidification in macrophages. The Journal of biological chemistry. 2007;282:31422–8. doi: 10.1074/jbc.M705296200. [DOI] [PubMed] [Google Scholar]
- 16.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, Nelson DJ. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nature cell biology. 2006;8:933–44. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1040460
- 17.Mueller C, Braag SA, Keeler A, Hodges C, Drumm M, Flotte TR. Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. American journal of respiratory cell and molecular biology. 2011;44:922–9. doi: 10.1165/rcmb.2010-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/722979439
- 18.Cantin AM, Hanrahan JW, Bilodeau G, Ellis L, Dupuis A, Liao J, Zielenski J, Durie P. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. American journal of respiratory and critical care medicine. 2006;173:1139–44. doi: 10.1164/rccm.200508-1330OC. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725183253
- 19.Raju SV, Jackson PL, Courville CA, McNicholas CM, Sloane PA, Sabbatini G, Tidwell S, Tang LP, Liu B, Fortenberry JA, Jones CW, Boydston JA, Clancy JP, Bowen LE, Accurso FJ, Blalock JE, Dransfield MT, Rowe SM. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. American journal of respiratory and critical care medicine. 2013;188:1321–30. doi: 10.1164/rccm.201304-0733OC. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718113095
- 20.Crystal RG. Are the smoking-induced diseases an acquired form of cystic fibrosis? American journal of respiratory and critical care medicine. 2013;188:1277–8. doi: 10.1164/rccm.201311-1947ED. [DOI] [PubMed] [Google Scholar]
- 21.Harness-Brumley CL, Elliott AC, Rosenbluth DB, Raghavan D, Jain R. Gender differences in outcomes of patients with cystic fibrosis. Journal of women's health (2002) 2014;23:1012–20. doi: 10.1089/jwh.2014.4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders DB, Li Z, Laxova A, Rock MJ, Levy H, Collins J, Ferec C, Farrell PM. Risk factors for the progression of cystic fibrosis lung disease throughout childhood. Annals of the American Thoracic Society. 2014;11:63–72. doi: 10.1513/AnnalsATS.201309-303OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ford ES. Trends in mortality from chronic obstructive pulmonary disease among adults in the United States. Chest. 2014 doi: 10.1378/chest.14-2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin RY, Lee GB. The gender disparity in adult asthma hospitalizations dynamically relates to age. The Journal of asthma: official journal of the Association for the Care of Asthma. 2008;45:931–5. doi: 10.1080/02770900802395504. [DOI] [PubMed] [Google Scholar]
- 25.Singh AK, Schultz BD, Katzenellenbogen JA, Price EM, Bridges RJ, Bradbury NA. Estrogen inhibition of cystic fibrosis transmembrane conductance regulator-mediated chloride secretion. The Journal of pharmacology and experimental therapeutics. 2000;295:195–204. [PubMed] [Google Scholar]
- 26.Coakley RD, Sun H, Clunes LA, Rasmussen JE, Stackhouse JR, Okada SF, Fricks I, Young SL, Tarran R. 17beta-Estradiol inhibits Ca2+-dependent homeostasis of airway surface liquid volume in human cystic fibrosis airway epithelia. The Journal of clinical investigation. 2008;118:4025–35. doi: 10.1172/JCI33893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain R, Ray JM, Pan J, Brody SL. Sex hormone-dependent regulation of cilia beat frequency in airway epithelium. American journal of respiratory cell and molecular biology. 2012;46:446–53. doi: 10.1165/rcmb.2011-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chotirmall SH, Smith SG, Gunaratnam C, Cosgrove S, Dimitrov BD, O'Neill SJ, Harvey BJ, Greene CM, McElvaney NG. Effect of estrogen on pseudomonas mucoidy and exacerbations in cystic fibrosis. The New England journal of medicine. 2012;366:1978–86. doi: 10.1056/NEJMoa1106126. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/716447805
- 29.Kernan NG, Alton Eric WFW, Cullinan P, Griesenbach U, Bilton D. Oral contraceptives do not appear to affect cystic fibrosis disease severity. The European respiratory journal. 2013;41:67–73. doi: 10.1183/09031936.00018712. [DOI] [PubMed] [Google Scholar]
- 30.Jain M, Goss CH. Update in cystic fibrosis 2013. American journal of respiratory and critical care medicine. 2014;189:1181–6. doi: 10.1164/rccm.201402-0203UP. [DOI] [PubMed] [Google Scholar]
- 31.Borowitz D, Parad RB, Sharp JK, Sabadosa KA, Robinson KA, Rock MJ, Farrell PM, Sontag MK, Rosenfeld M, Davis SD, Marshall BC, Accurso FJ. Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. The Journal of pediatrics. 2009;155:S106–16. doi: 10.1016/j.jpeds.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/724904901
- 32.Wang Y, Wrennall JA, Cai Z, Li H, Sheppard DN. Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. The international journal of biochemistry & cell biology. 2014;52:47–57. doi: 10.1016/j.biocel.2014.04.001. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718349329
- 33.Castellani C. CFTR2: How will it help care? Paediatric respiratory reviews. 2013;14(Suppl 1):2–5. doi: 10.1016/j.prrv.2013.01.006. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725446622
- 34.Lim MTC, Wallis C, Price JF, Carr SB, Chavasse RJ, Shankar A, Seddon P, Balfour-Lynn IM. Diagnosis of cystic fibrosis in London and South East England before and after the introduction of newborn screening. Archives of disease in childhood. 2014;99:197–202. doi: 10.1136/archdischild-2013-304766. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718178886
- 35.Ferrari M, Cremonesi L. Genotype-phenotype correlation in cystic fibrosis patients. Annales de biologie clinique. 1996;54:235–41. [PubMed] [Google Scholar]
- 36.Gallati S. Disease-modifying genes and monogenic disorders: experience in cystic fibrosis. The application of clinical genetics. 2014;7:133–46. doi: 10.2147/TACG.S18675. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/725446623
- 37.Sheridan MB, Fong P, Groman JD, Conrad C, Flume P, Diaz R, Harris C, Knowles M, Cutting GR. Mutations in the beta-subunit of the epithelial Na+ channel in patients with a cystic fibrosis-like syndrome. Human molecular genetics. 2005;14:3493–8. doi: 10.1093/hmg/ddi374. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718866463
- 38.Azad AK, Rauh R, Vermeulen F, Jaspers M, Korbmacher J, Boissier B, Bassinet L, Fichou Y, Des Georges M, Stanke F, Boeck K de, Dupont L, Balascáková M, Hjelte L, Lebecque P, Radojkovic D, Castellani C, Schwartz M, Stuhrmann M, Schwarz M, Skalicka V, Monestrol I de, Girodon E, Férec C, Claustres M, Tümmler B, Cassiman J, Korbmacher C, Cuppens H. Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Human mutation. 2009;30:1093–103. doi: 10.1002/humu.21011. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718839657
- 39.Ramos MD, Trujillano D, Olivar R, Sotillo F, Ossowski S, Manzanares J, Costa J, Gartner S, Oliva C, Quintana E, Gonzalez MI, Vazquez C, Estivill X, Casals T. Extensive sequence analysis of CFTR, SCNN1A, SCNN1B, SCNN1G and SERPINA1 suggests an oligogenic basis for cystic fibrosis-like phenotypes. Clinical genetics. 2014;86:91–5. doi: 10.1111/cge.12234. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718029167
- 40.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C, Elborn JS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365:1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/13960984
- 41.Boeck K de, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, Higgins M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2014;13:674–80. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718895188
- 42.Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordoñez CL, Geller DE. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142:718–24. doi: 10.1378/chest.11-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/725446624
- 43.Bell SC, Boeck K de, Amaral MD. New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacology & therapeutics. 2015;145:19–34. doi: 10.1016/j.pharmthera.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, Boeck K de, Donaldson SH, Dorkin HL, Dunitz JM, Durie PR, Jain M, Leonard A, McCoy KS, Moss RB, Pilewski JM, Rosenbluth DB, Rubenstein RC, Schechter MS, Botfield M, Ordoñez CL, Spencer-Green GT, Vernillet L, Wisseh S, Yen K, Konstan MW. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–8. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/714647852
- 45.Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. The Lancet. Respiratory medicine. 2014;2:527–38. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725446625
- 46.Kerem E, Konstan MW, Boeck K de, Accurso FJ, Sermet-Gaudelus I, Wilschanski M, Elborn JS, Melotti P, Bronsveld I, Fajac I, Malfroot A, Rosenbluth DB, Walker PA, McColley SA, Knoop C, Quattrucci S, Rietschel E, Zeitlin PL, Barth J, Elfring GL, Welch EM, Branstrom A, Spiegel RJ, Peltz SW, Ajayi T, Rowe SM. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet. Respiratory medicine. 2014;2:539–47. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718393259
- 47.Oakland M, Sinn PL, McCray PB. Advances in cell and gene-based therapies for cystic fibrosis lung disease. Molecular therapy: the journal of the American Society of Gene Therapy. 2012;20:1108–15. doi: 10.1038/mt.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Griesenbach U, Alton Eric WFW. Moving forward: cystic fibrosis gene therapy. Human molecular genetics. 2013;22:R52–8. doi: 10.1093/hmg/ddt372. [DOI] [PubMed] [Google Scholar]
- 49.Mitomo K, Griesenbach U, Inoue M, Somerton L, Meng C, Akiba E, Tabata T, Ueda Y, Frankel GM, Farley R, Singh C, Chan M, Munkonge F, Brum A, Xenariou S, Escudero-Garcia S, Hasegawa M, Alton Eric WFW. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Molecular therapy: the journal of the American Society of Gene Therapy. 2010;18:1173–82. doi: 10.1038/mt.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griesenbach U, Inoue M, Meng C, Farley R, Chan M, Newman NK, Brum A, You J, Kerton A, Shoemark A, Boyd AC, Davies JC, Higgins TE, Gill DR, Hyde SC, Innes JA, Porteous DJ, Hasegawa M, Alton Eric WFW. Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. American journal of respiratory and critical care medicine. 2012;186:846–56. doi: 10.1164/rccm.201206-1056OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirsh AJ, Molino BF, Zhang J, Astakhova N, Geiss WB, Sargent BJ, Swenson BD, Usyatinsky A, Wyle MJ, Boucher RC, Smith RT, Zamurs A, Johnson MR. Design, synthesis, and structure-activity relationships of novel 2-substituted pyrazinoylguanidine epithelial sodium channel blockers: drugs for cystic fibrosis and chronic bronchitis. Journal of medicinal chemistry. 2006;49:4098–115. doi: 10.1021/jm051134w. [DOI] [PubMed] [Google Scholar]
- 52.Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631–6. doi: 10.1378/chest.07-0288. [DOI] [PubMed] [Google Scholar]