

Abstract
Compared with normal cells, cancer cells show alterations in many cellular processes, including energy metabolism. Studies on cancer metabolism started with Otto Warburg's observation at the beginning of the last century. According to Warburg, cancer cells rely on glycolysis more than mitochondrial respiration for energy production. Considering that glycolysis yields much less energy compared with mitochondrial respiration, Warburg hypothesized that mitochondria must be dysfunctional and this is the initiating factor for cancer formation. However, this hypothesis did not convince every scientist in the field. Some believed the opposite: the reduction in mitochondrial activity is a result of increased glycolysis. This discrepancy of opinions is ongoing. In this review, we will discuss the alterations in glycolysis, pyruvate metabolism, and the Krebs cycle in cancer cells and focus on cause and consequence.
Introduction
Almost a century ago, Otto Warburg made a very significant observation that would start a long-lasting, heated discussion. He observed that cancer cells, unlike many other cells in the body, opt for glycolysis rather than mitochondrial respiration, even in the presence of oxygen (referred to here as the Warburg effect) [1]. Warburg proposed that the aerobic glycolysis phenotype that he observed stemmed from the fact that cancer cell mitochondria are irreversibly dysfunctional. He believed, in fact, that dysfunctional mitochondria are required and necessary to start all the biochemical events that eventually result in transformation to the cancerous state [2]. His findings went hand-in-hand with Pasteur's postulations. In 1861, Pasteur reported that yeast cells upregulate glycolysis under hypoxic conditions. Given that the inner regions of solid tumors are hypoxic because of anomalous vascularization, Pasteur's effect seemed to explain Warburg's observation. However, the biochemist Weinhouse was not convinced by Warburg's explanation of cancer initiation by damaged mitochondria [3,4]. As a pioneer of isotope tracer usage in biochemistry, he found that cancer cells are able to oxidize glucose and fatty acids to carbon dioxide at levels comparable to those of normal cells [5]. He argued that the reverse was true: cancer cells have reduced mitochondrial activity as a consequence of heightened glycolytic flux, which is known to inhibit mitochondria—the so-called Crabtree effect [6,7]. To this day, the field has not been able to reach a conclusive decision on this matter. To explore the relationship between these two views, we use the chicken-and-egg analogy: it is difficult to determine whether mitochondrial dysfunction emerges first, thereby forcing cells to rely on glycolysis, or whether the reverse occurs, whereby increased glycolytic flux takes place first, which in turn suppresses mitochondrial respiration. There are data supporting both of the models in different contexts. In this review, we will discuss the two different points of view in relation to glycolysis, pyruvate metabolism, and the Krebs cycle.
Changes in the glycolytic pathway during tumorigenesis
As Warburg noticed, cancer cells have elevated levels of glucose uptake compared with non-cancer cells. These findings have been confirmed by using recent technological developments that allow non-invasive monitoring of glucose uptake in vivo, called 2-fluoro-6-deoxyglucose positron emission tomography (FDG-PET). In this technique, a traceable glucose analog is used that is recognized and taken up by glucose transporters (GLUTs) but cannot be used for downstream glycolytic reactions. Hence, the glucose analog accumulates in cells and can be visualized and quantified by PET. This technique also allows diagnosis and localization of tumors. With FDG-PET, glucose uptake has been shown to increase in certain tumors [8], and higher FDG uptake has been correlated with poor cancer prognosis [9–11].
Multiple observations outlined below, however, suggest that glycolytic flux in cancer cells is upregulated as a result of multiple changes in signaling pathways and not necessarily as a result of impaired mitochondrial function. Glycolysis begins with cellular uptake of glucose via GLUTs on the cell surface (Figure 1). In 1974, Hatanaka proposed that cells upregulate GLUTs in order to meet increased glucose demand upon transformation [12]. Later studies showed that, indeed, levels of GLUTs, especially the high-affinity GLUTs 1 and 3, were upregulated in a plethora of tumor types (nicely reviewed in [13,14]). Moreover, GLUT1 transcription is upregulated in response to hypoxia [15,16] and inhibition of mitochondrial respiration [17], both conditions in which cells need to divert the metabolic flux from mitochondrial respiration to glycolysis. Furthermore, in tumors with high insulin signaling, GLUT4 is enriched at the cell membrane as a consequence of elevated PI3K/Akt signaling (reviewed in [18]) and GLUT1 transcription is upregulated via the serine/threonine kinase AKT [19]. The human genome actually has three families of GLUTs, namely SLC2A, SLC5A, and SLC50A, with a total of 27 members [20]. These members are differentially regulated in various tumor types. These findings could suggest that upregulation of glucose uptake and hence glycolytic flux is a primary alteration in cancer and not a consequence of impaired mitochondrial function. That said, activity of GLUTs is also strongly driven by activation of AMP-activated protein kinase (AMPK) [21], and AMPK can be activated by an adenosine triphosphate (ATP) decrease caused by mitochondrial dysfunction. Therefore, increased glucose uptake could also result from mitochondrial dysfunction. This nicely illustrates the fact that glycolytic flux and mitochondrial function are so intertwined that it is difficult to determine what is cause and what is consequence.
Figure 1. In cancer, glycolytic flux is increased through upstream parts of the glycolytic pathway up to pyruvate kinase and then decreased from pyruvate kinase downward, thereby generating a ‘bottleneck’.

DHAP, dihydroxyacetone phosphate; F-1,6-bisP, fructose-1,6-bisphosphate; Fruc-6-P, fructose-6-phosphate; Gluc-6-P, glucose-6-phosphate; GLUT, glucose transporter; HIF-1, hypoxia-induced factor 1; HK, hexokinase; PEP, phosphoenolpyruvate; PFK, phosphofructokinase; PG, phosphoglycerate; PGAM1, phosphoglycerate mutase 1; PGI, phosphoglucose isomerase; PK, pyruvate kinase; pyr, pyruvate; PPP, pentose phosphate pathway; TIGAR, TP53-induced glycolysis and apoptosis regulator; VDAC, voltage-dependent anion channel; VEGF, vascular endothelial growth factor.
After uptake into the cell, the next step in glycolysis is phosphorylation of glucose to glucose-6-phosphate by hexokinase (HK) (Figure 1). There are four isoforms of HK and upon transformation, isoform II, the isoform with the highest enzymatic activity, becomes the prevalent isoform in the cell [22] and this is due in part to HIF1α (hypoxia-induced factor 1 α)-dependent transcriptional upregulation [23]. HKI and especially HKII are known to interact with voltage-dependent anion channels (VDACs) on the mitochondrial outer membrane of rapidly proliferating cells. This interaction is important for the inhibition of apoptosis by blocking cytochrome c release into the cytoplasm [24]. This helps cells evade apoptosis, one of the six hallmarks of cancer [25]. For these reasons, the VDAC-HKII interaction is a potential target for cancer therapy. There are data showing that the VDAC-HKII interaction favors glycolysis by inhibiting a negative feedback of HKII by its own product and by stabilizing HKII protein [26,27]. Thus, HKII offers a mechanistic explanation for the Warburg effect independently of mitochondrial dysfunction.
Glucose-6-phosphate is next converted to fructose-6-phosphate by phosphoglucose isomerase (PGI) (Figure 1). Expression of PGI is induced in response to HIF-1α and vascular endothelial growth factor (VEGF) signaling, both of which are often deregulated in tumors [28]. Interestingly, PGI acts as a cytokine outside of the cell. Secreted PGI is a tumor marker, as it can be detected in serum and urine of patients with cancer [29,30]. PGI is also called AMF (autocrine motility factor) because treatment of fibrosarcoma cells with purified PGI protein induces cell migration [31], implicating it in metastasis. Furthermore, mere PGI gain of function is sufficient to drive cell proliferation in 3T3 fibroblasts [32]. All in all, PGI can be considered an oncogene.
Next, fructose-6-phosphate is phosphorylated again to yield fructose-1,6-bisphosphate (Figure 1). This step is of particular importance for regulation of metabolism for several reasons. Firstly, it is the rate-limiting reaction. Secondly, the fact that ATP, its own substrate, allosterically inhibits phosphofructokinase 1 and 2 (PFK1 and 2) [33-35] offers an explanation for the Pasteur effect: inhibition of glycolysis by mitochondrial respiration. Thirdly, this is a decision point for glucose to enter further into glycolysis or to be diverted into the pentose phosphate pathway (PPP). The PPP has two branches: an irreversible, oxidative branch that starts with glucose-6-phosphate and generates reduced nicotinamide adenine dinucleotide phosphate (NADPH) and a reversible, non-oxidative branch that interconverts 5-carbon sugars and produces no NADPH (Figure 1). Hence, changes in PFK activity influence how much glucose enters the oxidative branch of PPP, yielding reducing equivalents for fatty acid biosynthesis and reactive oxygen species (ROS) defense (Figure 1). One critical factor determining PFK1 activity is the level of fructose-2-6-biphosphate, which is the product of the bifunctional enzyme phosphofructokinase 2/fructosebisphosphatase (PFKB). Fructose-2,6-bisphosphate allosterically activates PFK1, thereby counteracting ATP inhibition [36,37]. PFKB has four isozymes that are expressed in a tissue-specific manner; however, PFKBP3, which has the highest kinase activity, is upregulated in high-grade astrocytomas and malignant breast and colon tumors [38,39], tipping the scale of the bidirectional reaction in favor of fructose-2,6-bisphosphate production. This sustains increased glycolytic activity rather than PPP. Upregulation of PFKB expression and activity occurs in various ways. A recent publication showed that methylation stabilizes PFKB3 in U937 human leukemia cells [40]. All PFKB isoforms are upregulated in response to hypoxia in vivo, PFKBP3 being induced to the largest extent [41]. In addition to PFKB3, PFKB4 has been also found to be important for cancer cell survival via small interfering RNA (siRNA) screens in two different models: glioma stem-like cells [42] and prostate cancer cell lines [43]. Another critical factor controlling PFK activity is the p53 tumor suppressor-mediated induction of TIGAR (TP53-induced glycolysis and apoptosis regulator). TIGAR negatively regulates PFK2 activity and lowers fructose-2,6-bisphosphate levels, and thereby reduces PFK1 activity and diverts glucose into the PPP rather than glycolysis [44]. In tumors with p53 loss of function, TIGAR thereby contributes to the increase in glycolytic rate. p53 has additional regulatory roles in metabolism, such as downregulating GLUT1 and GLUT4 expression at the transcriptional level [45].
Phosphoglycerate mutase 1 (PGAM1) is another glycolytic enzyme whose activity is increased in several tumor types, including hepatocellular carcinoma [46]. Its protein level is negatively regulated by p53 [47]. PGAM1 converts 3-phosphoglycerate to 2-phosphoglycerate (Figure 1). Its activity is important in metabolic regulation because its substrate, 3-phosphoglycerate, inhibits flux through the oxidative branch of PPP [48], thereby diverting glucose into glycolysis. Therefore, upregulation of PGAM1 is advantageous for cell proliferation, although it renders tumor cells sensitive to oxidative stress due to reduction in NADPH production from the oxidative branch of PPP.
Pyruvate: the intersection
So far, we have discussed the steps of glycolysis through which flux is increased in cancer cells. Interestingly, cancer cells do not increase flux in all steps of glycolysis. Although flux through the upstream steps is increased, they often exhibit a bottleneck in the following steps of glycolysis leading from phosphoenolpyruvate (PEP) to the Krebs cycle and this is for the reasons discussed below.
Pyruvate kinase (PK) is an important node of control in cancer cell metabolism. It catalyzes the conversion of PEP to pyruvate (Figure 1). PK has two isoforms produced by alternative splicing of PKM, namely M1, and M2, which are spatially and temporally differentially expressed. PKM1 is expressed mainly during adulthood, and PKM2 is expressed during embryogenesis. The alternatively spliced transcripts of PKLR, the L and R isoforms, on the other hand, are expressed specifically in liver [49]. In 2008, it was shown that expressing PKM2 confers a proliferative advantage to tumor cells [50]. In fact, depletion of PKM2 expression in cancer cell lines and reconstitution with PKM1 led to inhibition of growth and reversal of the Warburg phenotype [50]. This finding was unexpected because at first glance it does not fit into the model that cancer cells have increased glycolytic flux. PKM2 is the isoform with lower enzymatic activity and therefore it slows down this glycolytic reaction. However, generating a bottleneck at the end of the glycolytic pathway appears to be necessary for giving intermediates the time to flux through alternative pathways, such as PPP, that are of crucial importance for replenishing building blocks required for growth and proliferation [49]. PPP flux is especially needed in cancer cells for the production of pentose phosphates, which are used for nucleotide biosynthesis, and for the production of NADPH, which is a reducing equivalent needed both for fatty acid and sterol biosynthesis [51] and for mounting an anti-oxidant response to ROS by re-oxidizing glutathione [52]. PKM2 is also subject to negative regulation by phosphotyrosine peptides. Since tyrosine kinase signaling is often deregulated in a cancer setting, this regulation could be another way of pushing glycolytic intermediates into anabolic pathways such as the PPP [53]. Another advantage that selective PKM2 expression confers to cancer cells is the accumulation of its substrate, PEP. PEP acts as a phosphate donor to phosphorylate PGAM1 at the catalytic histidine, thereby increasing its activity [54]. Interestingly, the functional relevance of PKM2 in tumor development was analyzed in a recent study in which mice specifically lacking the PKM2 isoform were found to develop breast tumors and liver metastases induced by BRCA1 loss of function, indicating that PKM2 per se is not required for tumor development [55]. Tumor cells had compensatory PKM1 expression, although PKM1 levels were heterogenous throughout the tumor. Whereas non-proliferating tumor cells exhibited higher PKM1 expression, proliferating cells did not [55]. The same study also analyzed human tumor samples and reported the presence of tumor samples with no detectable PK expression. Even though further studies are required, these findings hint that proliferating cells can lack PK activity altogether. It appears that PKM2 expression per se is not required. Rather, the outcome of reduced PK activity is what favors cell proliferation.
Pyruvate is the critical node where the flux of glucose-derived carbons is determined, either toward lactate which is usually secreted or into mitochondria (Figure 2). As Warburg observed, cancer cells metabolize pyruvate by aerobic glycolysis and produce lactate. Here, we will discuss the mechanisms of aerobic glycolysis induction which do not necessarily stem from dysfunctional mitochondria, which would have pleased Weinhouse.
Figure 2. In cancer cells, reduction of pyruvate to lactate and its secretion is favored rather than pyruvate entry into mitochondria and the Krebs cycle.
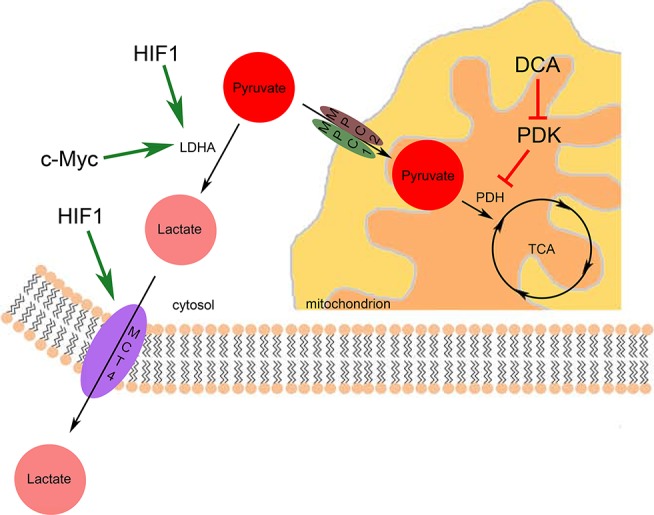
DCA, dichloroacetate; HIF1, hypoxia-induced factor 1; LDHA, lactate dehydrogenase A; MCT4, monocarboxylate transporter 4; MPC, mitochondrial pyruvate carrier; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; TCA, tricarboxylic acid.
The first possibility for pyruvate is to be reduced to lactate by lactate dehydrogenase (LDH). The prevalent isoform of LDH is LDHA. Like many other glycolytic genes, LDHA is induced by both HIF-1α [56] and c-myc [57]. Another means of inducing LDHA activity in the context of cancer is via phosphorylation at the tyrosine 10 residue [58]. Reflecting the importance of the Warburg effect for cancer cell survival, loss of function of LDHA by means of either siRNA depletion or pharmacological inhibition forces mitochondria to respire and slows down proliferation [59,60]. To maintain intracellular pH homeostasis, cells need to evacuate the resulting lactate to the extracellular space. Indeed, loss of function of the hypoxia-responsive lactate/H+ symporter MCT4 (monocarboxylate transporter 4) [61] impairs tumor growth [62], thereby rendering MCT4 a good therapeutic target.
The second possibility of pyruvate utilization is to send it to mitochondria for further oxidation in the Krebs cycle. The limiting step here is the entry of pyruvate into mitochondria by mitochondrial pyruvate carriers (MPCs) (Figure 2). The activity and chemical inhibition of a MPC were shown in 1974 [63], but the molecular identities of these carriers remained a secret until 2012, when they were identified as MPC1 and MPC2 [64,65]. The cancer relevance of these carriers was thoroughly investigated very recently [66–68]. In MPC1 loss-of-function tumor models, forced expression of the protein leads to activation of mitochondrial pyruvate oxidation, which inhibits anchorage-independent growth of colon cancer cells [68]. These studies indicate that MPC loss of function could be one means of rewiring cancer metabolism toward aerobic glycolysis, the Warburg effect. In tumor types with reduced MPC activity, it could be used as a therapeutic target. The fact that forced activation of MPC can activate mitochondrial respiration suggests that mitochondria are not necessarily irreversibly damaged, as Warburg claimed. Upon entry into mitochondria, pyruvate is converted to acetyl coenzyme A (CoA) and joins the Krebs cycle, during which reduced nicotinamide adenine dinucleotide (NADH) and precursors for anabolic pathways are generated (Figure 3A).
Figure 3. Krebs cycle reactions are altered but not completely abrogated in cancer cells.


(A) Full Krebs cycle in the presence of functional mitochondria and oxygen. Cancer cells use glutamine more than pyruvate for anaplerosis. (B) A truncated form of the Krebs cycle is favored in cancer cells with defective mitochondria or under hypoxic conditions (or both) to generate metabolic precursors with reductive carboxylation. 2-HG, 2-hydroxyglutarate; AcCoA, acetyl coenzyme A; ACL, ATP citrate lyase; ASCT2, sodium-dependent neutral amino acid transporter type 2; CS, citrate synthase; DCA, dichloroacetate; FA, fatty acid; FAD, flavin adenine dinucleotide; FH, fumarate hydratase; GLS, glutaminase; HIF1, hypoxia-induced factor 1; IDH, isocitrate dehydrogenase; ME, malic enzyme; NAD, nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, reduced nicotinamide adenine dinucleotide phosphate; OAA, oxaloacetate; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; pyr, pyruvate; SDH, succinate dehydrogenase.
At this stage, there is one more regulation point, pyruvate dehydrogenase (PDH). PDH is negatively regulated by NADH and acetyl CoA and through phosphorylation by pyruvate dehydrogenase kinase (PDK) [69,70]. PDK is a direct target of HIF1 [71,72]. HIF1 upregulates PDK transcription in response to hypoxia to reduce mitochondrial utilization of glucose. Moreover, oncogenic tyrosine kinases contribute to the reduction of PDH activity in two ways: they phosphorylate PDK to increase its activity [73] and they phosphorylate and inhibit a phosphatase that acts on PDH [74]. Dicholoroacetate (DCA) is a pharmacological inhibitor of PDK [75–78], which is used in treatment of lactic acidosis and cancer. The fact that DCA treatment is able to reverse Warburg phenotypes [76] is another indication that mitochondria do not have to be irreversibly damaged.
‘Hacking’ the Krebs cycle
Even though cancer cells tend to use glucose in aerobic glycolysis rather than oxidizing it in the Krebs cycle, the Krebs cycle is not dispensable altogether. The Krebs cycle constitutes a chain of reactions that are essential for the production of building blocks for growth and proliferation. As anabolic reactions use substrates from the Krebs cycle, thereby depleting the Krebs cycle, the cell needs to replenish these intermediates via a set of so-called anaplerotic reactions [79]. For example, Vacanti and colleagues [66] made the observation that, upon inhibition of pyruvate entry into mitochondria in C2C12 myotubes, cells were able to maintain Krebs cycle metabolism without any drastic changes in the levels of intermediates, even though they exhibit a reduction in the mitochondrial oxidation of pyruvate. Cancer cells that divert glucose metabolism away from mitochondria face a situation similar to pyruvate entry inhibition and therefore employ strategies to keep the cycle going, discussed below. By this means, they use mitochondria for biosynthesis rather than degradation.
One of the anaplerosis strategies is glutaminolysis (Figure 3A). Cells use glutamine as a carbon source to feed the Krebs cycle [80]. Glutamine is first converted to glutamate by glutaminase (GLS). Next, glutamate is converted to α-ketoglutarate (α-KG) either by a transamination reaction of the amine group onto another ketoacid or by deamination [81]. Myc, which is activated in most cancer types, increases glutaminolytic activity by increasing expression of GLS and the glutamine transporter ASCT2 (ASC amino acid transporter 2) [82]. If a particular tumor uses this strategy to keep the Krebs cycle filled, it renders the cancer cells addicted to glutamine. The filling of the Krebs cycle via glutaminolysis is then used for several cataplerotic reactions important for cancer cell lipid biosynthesis: (1) malate is converted to pyruvate by malic enzyme (ME) in the cytosol, thereby yielding NADPH (Figure 3A), which is a reducing equivalent used for fatty acid biosynthesis; (2) oxaloacetate (OAA) is converted back to malate and also exported to yield NADPH; and (3) citrate is used as a source of acetyl CoA for fatty acid synthesis [83] (Figure 3A). The relative contribution of these reactions via ME to the NADPH pool is quite significant and is roughly equivalent to the NADPH production by the PPP [84].
Although glutaminolysis can replenish Krebs cycle intermediates, it still requires functional mitochondria and oxygen, which are not always available in the tumor context. Cancer cells use reductive carboxylation in cases of defective mitochondria [85] and hypoxia [86,87] (Figure 3B). This pathway involves NADPH-dependent reductive carboxylation of α-KG (that derives from glutaminolysis, see above paragraph) by isocitrate dehydrogenase (IDH) into isocitrate, which is the reverse reaction of the conventional Krebs cycle. Isocitrate is subsequently isomerized to citrate, which is essential for lipid biosynthesis. Substrate availability also influences the direction of the reaction: that is, reductive carboxylation or oxidative decarboxylation of α-KG. A high NADPH/NADP+ (nicotinamide adenine dinucleotide phosphate) ratio, low citrate levels, and high α-KG abundance increase the reductive carboxylation activity of cells [88–91].
Cancer cells need to upregulate their fatty acid biosynthesis upon neoplastic transformation [92,93] in order to meet the increased demand of membrane production due to increased proliferation. Citrate production is of importance for fatty acid biosynthesis because the citrate shuttle transports mitochondrial acetyl CoA across the membranes to the cytoplasm, where it is converted back to OAA and acetyl CoA by ATP citrate lyase [94] (Figure 3A and B). This is one of the reasons why anaplerotic reactions are crucial for supporting growth and proliferation of the tumor cells.
Warburg was not totally wrong
So far, we have discussed mechanisms that promote aerobic glycolysis independently of possible mitochondrial defects. However, there are cases where Warburg was right, whereby mutation of genes whose products act in mitochondria are enough to induce transformation. Discussed below are examples of such cases.
Various cancer types have been found to harbor neomorphic mutations in IDH1 and IDH2 enzymes, causing them to lose their activity of decarboxylation of isocitrate to α-KG, and to gain the activity of converting α-KG to the oncometabolite 2-hydroxyglutarate (2-HG) [95,96] (Figure 3A). The prevalent view on the mechanism of 2-HG-induced tumor formation is that it alters cellular methylation of both DNA and proteins. There is evidence that 2-HG competitively inhibits α-KG-dependent dioxygenases, such as methylcysteine hydroxylases and the Jumonji C (JmjC) family of histone demethylases [97–99]. Data also show that 2-HG stabilizes HIF-1α by inhibiting prolyl hydroxylases (PHDs) (also α-KG-dependent dioxygenases) [98].
Some enzymes in the Krebs cycle function as tumor suppressors. For example, mutations in succinate dehydrogenase (SDH) subunits have been associated with hereditary paraganglioma [100,101], and fumarate hydratase (FH) mutations were implicated in renal cell cancers and smooth muscle tumors [102]. In both cases, substrates of the enzymes, succinate and fumarate, respectively, have been shown to accumulate. The mechanism of SDH and FH stemmed carcinogenesis is induction of a ‘pseudohypoxia’ state. Hypoxia-responsive genes are regulated by the transcription factor HIF1α. Under normoxic conditions, HIF1α is constantly hydroxylated at proline residues by PHDs. Upon hydroxylation, HIF1α is readily recognized and ubiquitinated by Von Hippel-Lindau complex that has E3 ubiquitin ligase activity [103,104]. PHDs use α-KG as cofactors and generate succinate at the end of the reaction [105]. In the context of SHD and FH mutations, cytosolic succinate [106,107] and fumarate [108] concentrations increase and they inhibit the activity of PHDs, leading to stabilization of HIF1α (Figure 3A) even in the presence of oxygen and induction of an aberrant hypoxic response [109].
Conclusions
There has been discussion for almost a century about the reasons for the metabolic phenotypes that Warburg initially observed, i.e. that cells use glucose via glycolytic reactions instead of respiration, even in the presence of oxygen. It is a phenomenon that appears counterintuitive and wasteful, given how little ATP glycolysis yields compared with oxidative phosphorylation. The main disagreement revolves around Warburg's proposal that irreversible damage of mitochondria is the main reason for carcinogenesis. In some cases, Warburg's proposal holds true. For example, the fact that mutations in some Krebs cycle enzymes, such as SDH and FH, are able to induce renal tumors indicates that mitochondrial dysfunction can be sufficient for tumor formation. However, there are reports that in some cases cancer cell mitochondrial function is intact. When they are forced to respire, they are able to. In sum, it seems that mitochondrial dysfunction is usually the consequence of tumor formation, yet there are cases in which mitochondrial dysfunction is the cause of the tumor formation. Indeed, one recent study suggests that glycolysis is needed for sustaining stemness and proliferation, whereas forced activation of mitochondrial respiration induces differentiation and reduces proliferation [110]. There seems to be a very strong, yet not very well-understood, crosstalk between the regulation of proliferation and a preference for glycolysis.
Abbreviations
- 2-HG
2-hydroxyglutarate
- α-KG
α-ketoglutarate
- AMPK
AMP-activated protein kinase
- ATP
adenosine triphosphate
- CoA
coenzyme A
- DCA
dicholoroacetate
- FDG
2-fluoro-6-deoxyglucose
- FH
fumarate hydratase
- GLS
glutaminase
- GLUT
glucose transporter
- HIF
hypoxia-induced factor
- HK
hexokinase
- IDH
isocitrate dehydrogenase
- LDH
lactate dehydrogenase
- MCT4
monocarboxylate transporter 4
- ME
malic enzyme
- MPC
mitochondrial pyruvate carrier
- NADH
reduced nicotinamide adenine dinucleotide
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- OAA
oxaloacetate
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- PEP
phosphoenolpyruvate
- PET
positron emission tomography
- PFK
phosphofructokinase
- PFKBP
phosphofructokinase/fructosebisphosphatase
- PGAM
phosphoglycerate mutase
- PGI
phosphoglucose isomerase
- PHD
prolyl hydroxylase
- PK
pyruvate kinase
- PPP
pentose phosphate pathway
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- siRNA
small interfering RNA
- TIGAR
TP53-induced glycolysis and apoptosis regulator
- VDAC
voltage-dependent anion channel
Disclosures
The authors declare that they have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/b/7/41
References
- 1.Warburg O. The Metabolism of Carcinoma Cells. J Cancer Res. 1925;9:148–63. doi: 10.1158/jcr.1925.148. [DOI] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Weinhouse S. The Warburg hypothesis fifty years later. Cancer Res Clin Oncol. 1976;87:115–26. doi: 10.1007/BF00284370. [DOI] [PubMed] [Google Scholar]
- 4.Weinhouse S. On respiratory impairment in cancer cells. Science. 1956;124:267–9. doi: 10.1126/science.124.3215.267. [DOI] [PubMed] [Google Scholar]
- 5.Weinhouse S, Millington RH, Wenner CE. Metabolism of neoplastic tissue. I. The oxidation of carbohydrate and fatty acids in transplanted tumors. Cancer Res. 1951;11:845–50. [PubMed] [Google Scholar]
- 6.Sussman I, Erecinska M, Wilson DF. Regulation of cellular energy metabolism: the Crabtree effect. Biochim Biophys Acta. 1980;591:209–23. doi: 10.1016/0005-2728(80)90153-X. [DOI] [PubMed] [Google Scholar]
- 7.Crabtree H.G. Observations on the carbohydrate metabolism of tumours. Biochem J. 1929;23:536–45. doi: 10.1042/bj0230536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endo K, Oriuchi N, Higuchi T, Iida Y, Hanaoka H, Miyakubo M, Ishikita T, Koyama K. PET and PET/CT using 18F-FDG in the diagnosis and management of cancer patients. Int J Clin Oncol. 2006;11:286–96. doi: 10.1007/s10147-006-0595-0. [DOI] [PubMed] [Google Scholar]
- 9.Kitajima K, Suenaga Y, Kanda T, Miyawaki D, Yoshida K, Ejima Y, Sasaki R, Komatsu H, Saito M, Otsuki N, Nibu K, Kiyota N, Minamikawa T, Sugimura K. Prognostic value of FDG PET imaging in patients with laryngeal cancer. PLoS One. 2014;9:e96999. doi: 10.1371/journal.pone.0096999. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718384686
- 10.Dooms C, van Baardwijk A, Verbeken E, van Suylen RJ, Stroobants S, De Ruysscher D, Vansteenkiste J. Association between 18F-fluoro-2-deoxy-D-glucose uptake values and tumor vitality: prognostic value of positron emission tomography in early-stage non-small cell lung cancer. J Thorac Oncol. 2009;4:822–8. doi: 10.1097/JTO.0b013e3181a97df7. [DOI] [PubMed] [Google Scholar]
- 11.Halfpenny W, et al. FDG-PET. A possible prognostic factor in head and neck cancer. Br J Cancer. 2002;86:512–6. doi: 10.1038/sj.bjc.6600114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatanaka M, Hain SF, Biassoni L, Maisey MN, Sherman JA, McGurk M. Transport of sugars in tumor cell membranes. Biochim Biophys Acta. 1974;355(1):77–104. doi: 10.1016/0304-419X(74)90008-0. [DOI] [PubMed] [Google Scholar]
- 13.Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta. 2013;1835:164–9. doi: 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Medina RA, Owen GI. Glucose transporters: expression, regulation and cancer. Biol Res. 2002;35:9–26. doi: 10.4067/S0716-97602002000100004. [DOI] [PubMed] [Google Scholar]
- 15.Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–25. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- 16.Murakami T, Nishiyama T, Shirotani T, Shinohara Y, Kan M, Ishii K, Kanai F, Nakazuru S, Ebina Y. Identification of two enhancer elements in the gene encoding the type 1 glucose transporter from the mouse which are responsive to serum, growth factor, and oncogenes. J Biol Chem. 1992;26:9300–6. [PubMed] [Google Scholar]
- 17.Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995;270:29083–9. doi: 10.1074/jbc.270.49.29083. [DOI] [PubMed] [Google Scholar]
- 18.Holman GD, Cushman SW. Subcellular localization and trafficking of the GLUT4 glucose transporter isoform in insulin-responsive cells. Bioessays. 1994;16:753–9. doi: 10.1002/bies.950161010. [DOI] [PubMed] [Google Scholar]
- 19.Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–6. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- 20.Wright EM. Glucose transport families SLC5 and SLC50. Mol Aspects Med. 2013;34:183–96. doi: 10.1016/j.mam.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–86. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–93. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377:347–55. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 26.Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci U S A. 1977;74:3735–9. doi: 10.1073/pnas.74.9.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. 2002;1555:14–20. doi: 10.1016/S0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 28.Funasaka T, Yanagawa T, Hogan V, Raz A. Regulation of phosphoglucose isomerase/autocrine motility factor expression by hypoxia. FASEB J. 2005;19:1422–30. doi: 10.1096/fj.05-3699com. [DOI] [PubMed] [Google Scholar]
- 29.Filella X, Molina R, Jo J, Mas E, Ballesta AM. Serum phosphohexose isomerase activities in patients with colorectal cancer. Tumour Biol. 1991;1:360–7. doi: 10.1159/000217737. [DOI] [PubMed] [Google Scholar]
- 30.Baumann M, Kappl A, Lang T, Brand K, Siegfried W, Paterok E. The diagnostic validity of the serum tumor marker phosphohexose isomerase (PHI) in patients with gastrointestinal, kidney, and breast cancer. Cancer Invest. 1990;8:351–6. doi: 10.3109/07357909009012053. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe H, Carmi P, Hogan V, Raz T, Silletti S, Nabi IR, Raz A. Purification of human tumor cell autocrine motility factor and molecular cloning of its receptor. J Biol Chem. 1991;266:13442–8. [PubMed] [Google Scholar]
- 32.Tsutsumi S, Yanagawa T, Shimura T, Fukumori T, Hogan V, Kuwano H, Raz A. Regulation of cell proliferation by autocrine motility factor/phosphoglucose isomerase signaling. J Biol Chem. 2003;278:32165–72. doi: 10.1074/jbc.M304537200. [DOI] [PubMed] [Google Scholar]
- 33.Cabrera R, Baez M, Pereira HM, Caniuguir A, Garratt RC, Babul J. The crystal complex of phosphofructokinase-2 of Escherichia coli with fructose-6-phosphate: kinetic and structural analysis of the allosteric ATP inhibition. J Biol Chem. 2011;286:5774–83. doi: 10.1074/jbc.M110.163162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kotlarz D, Buc H. Regulatory properties of phosphofructokinase 2 from Escherichia coli. Eur J Biochem. 1981;117:569–74. doi: 10.1111/j.1432-1033.1981.tb06375.x. [DOI] [PubMed] [Google Scholar]
- 35.Atkinson DE, Walton GM. Kinetics of Regulatory Enzymes. Escherichia Coli Phosphofructokinase. J Biol Chem. 1965;240:757–63. [PubMed] [Google Scholar]
- 36.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86:174–9. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Wu C, Khan SA, Peng LJ, Lange AJ. Roles for fructose-2,6-bisphosphate in the control of fuel metabolism: beyond its allosteric effects on glycolytic and gluconeogenic enzymes. Adv Enzyme Regul. 2006;46:72–88. doi: 10.1016/j.advenzreg.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 38.Kessler R, Bleichert F, Warnke JP, Eschrich K. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) is up-regulated in high-grade astrocytomas. J Neurooncol. 2008;86:257–64. doi: 10.1007/s11060-007-9471-7. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/721742682
- 39.Minchenko OH, Ochiai A, Opentanova IL, Ogura T, Minchenko DO, Caro J, Komisarenko SV, Esumi H. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie. 2005;87:1005–10. doi: 10.1016/j.biochi.2005.04.007. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/721601695
- 40.Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T, Kamata A, Sakamoto K, Nakanishi T, Kubo A, Hishiki T, Suematsu M. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. 2014;5:3480. doi: 10.1038/ncomms4480. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718312104
- 41.Minchenko OI, Opentanova I, Caro J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003;554:264–70. doi: 10.1016/S0014-5793(03)01179-7. [DOI] [PubMed] [Google Scholar]
- 42.Goidts V, Bageritz J, Puccio L, Nakata S, Zapatka M, Barbus S, Toedt G, Campos B, Korshunov A, Momma S, Van Schaftingen E, Reifenberger G, Herold-Mende C, Lichter P, Radlwimmer B. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene. 2012;31:3235–43. doi: 10.1038/onc.2011.490. [DOI] [PubMed] [Google Scholar]
- 43.Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, Zamboni N, Schulze A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012;2:328–43. doi: 10.1158/2159-8290.CD-11-0234. [DOI] [PubMed] [Google Scholar]
- 44.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 45.Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013;18:617–33. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren F, Wu H, Lei Y, Zhang H, Liu R, Zhao Y, Chen X, Zeng D, Tong A, Chen L, Wei Y, Huang C. Quantitative proteomics identification of phosphoglycerate mutase 1 as a novel therapeutic target in hepatocellular carcinoma. Mol Cancer. 2010;9:81. doi: 10.1186/1476-4598-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–85. [PubMed] [Google Scholar]
- 48.Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, Gu TL, Jin P, Alečković M, LeRoy G, Kang Y, Sudderth JA, DeBerardinis RJ, Luan CH, Chen GZ, Muller S, Shin DM, Owonikoko TK, Lonial S, Arellano ML, Khoury HJ, Khuri FR, Lee BH, Ye K, Boggon TJ, Kang S, He C, Chen J. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718384368
- 49.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 50.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1104536
- 51.Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med. 2012;53:421–36. doi: 10.1016/j.freeradbiomed.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Deneke SM, Fanburg BL. Regulation of cellular glutathione. Am J Physiol. 1989;257:163–73. doi: 10.1152/ajplung.1989.257.4.L163. [DOI] [PubMed] [Google Scholar]
- 53.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–6. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1104388
- 54.Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–9. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/5752959
- 55.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, Burga LN, Xie J, Jurczak MJ, DePinho RA, Clish CB, Jacks T, Kibbey RG, Wulf GM, Di Vizio D, Mills GB, Cantley LC, Vander Heiden MG. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409. doi: 10.1016/j.cell.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718140955
- 56.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol (1985) 2000;88:1474–80. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 57.Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–63. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan J, Hitosugi T, Chung TW, Xie J, Ge Q, Gu TL, Polakiewicz RD, Chen GZ, Boggon TJ, Lonial S, Khuri FR, Kang S, Chen J. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol Cell Biol. 2011;31:4938–50. doi: 10.1128/MCB.06120-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/719780298
- 59.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037–42. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/723912270
- 60.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–34. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1032849
- 61.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281:9030–7. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 62.Le Floch R, Chiche J, Marchiq I, Naiken T, Ilc K, Murray CM, Critchlow SE, Roux D, Simon MP, Pouysségur J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc Natl Acad Sci U S A. 2011;108:16663–8. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/723902386
- 63.Halestrap AP, Brand MD, Denton RM. Inhibition of mitochondrial pyruvate transport by phenylpyruvate and alpha-ketoisocaproate. Biochim Biophys Acta. 1974;367:102–8. doi: 10.1016/0005-2736(74)90140-0. [DOI] [PubMed] [Google Scholar]
- 64.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717952788
- 65.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–6. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717952983
- 66.Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, Metallo CM. Regulation of Substrate Utilization by the Mitochondrial Pyruvate Carrier. Mol Cell. 2014;56:425–35. doi: 10.1016/j.molcel.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/725385148
- 67.Yang C, Ko B1, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, Rutter J, Merritt ME, DeBerardinis RJ. Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during Impaired Mitochondrial Pyruvate Transport. Mol Cell. 2014;56:414–24. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/725391828
- 68.Schell JC, Olson KA, Jiang L, Hawkins AJ, Van Vranken JG, Xie J, Egnatchik RA, Earl EG, DeBerardinis RJ, Rutter J. A Role for the Mitochondrial Pyruvate Carrier as a Repressor of the Warburg Effect and Colon Cancer Cell Growth. Mol Cell. 2014;56:400–413. doi: 10.1016/j.molcel.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/725254231
- 69.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143–51. doi: 10.1042/BST0311143. [DOI] [PubMed] [Google Scholar]
- 70.Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003;284:E855–62. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- 71.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1031718
- 72.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1031760
- 73.Hitosugi T, Fan J, Chung TW, Lythgoe K, Wang X, Xie J, Ge Q, Gu TL, Polakiewicz RD, Roesel JL, Chen GZ, Boggon TJ, Lonial S, Fu H, Khuri FR, Kang S, Chen J. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell. 2011;44:864–77. doi: 10.1016/j.molcel.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/722241314
- 74.Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, Gu TL, Aguiar M, Lonning S, Chen H3, Mohammadi M, Britton LM, Garcia BA, Alečković M, Kang Y, Kaluz S, Devi N, Van Meir EG, Hitosugi T, Seo JH, Lonial S, Gaddh M, Arellano M, Khoury HJ, Khuri FR, Boggon TJ, Kang S, Chen J. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell. 2014;53:534–48. doi: 10.1016/j.molcel.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718262176
- 75.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329:191–6. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 77.Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry. 2006;45:402–15. doi: 10.1021/bi051402s. [DOI] [PubMed] [Google Scholar]
- 78.Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38:1124–44. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- 79.Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49:388–98. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–24. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1383974
- 81.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–84. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–7. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/13411029
- 83.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718379390
- 85.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718559290
- 86.Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–6. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/723901372
- 87.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–4. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/716647808
- 88.Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, Chandel NS, DeBerardinis RJ. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7:1679–90. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gameiro PA, Laviolette LA, Kelleher JK, Iliopoulos O, Stephanopoulos G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J Biol Chem. 2013;288:12967–77. doi: 10.1074/jbc.M112.396796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gameiro PA, Yang J, Metelo AM, Pérez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, López-Larrubia P, Stephanopoulos G, Iliopoulos O. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17:372–85. doi: 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem. 2012;287:14615–20. doi: 10.1074/jbc.C112.353946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. 2009;100:1369–72. doi: 10.1038/sj.bjc.6605007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang YA, Han WF, Morin PJ, Chrest FJ, Pizer ES. Activation of fatty acid synthesis during neoplastic transformation: role of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Exp Cell Res. 2002;279:80–90. doi: 10.1006/excr.2002.5600. [DOI] [PubMed] [Google Scholar]
- 94.Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–22. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- 95.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/720539740
- 96.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1168419
- 97.Ye D, Xiong Y, Guan KL. The mechanisms of IDH mutations in tumorigenesis. Cell Res. 2012;22:1102–4. doi: 10.1038/cr.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/8445959
- 99.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/6799956
- 100.Baysal BE. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab. 2003;14:453–9. doi: 10.1016/j.tem.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 101.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, 3rd, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–51. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 102.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomäki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA Multiple Leiomyoma Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–10. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 103.Kaelin WG, Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 104.Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/S0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 105.Schofield CJ, Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr Opin Struct Biol. 1999;9:722–31. doi: 10.1016/S0959-440X(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 106.Smith EH, Janknecht R, Maher LJ., 3rd Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16:3136–48. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- 107.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1022674
- 108.Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–9. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 109.Morin A, Letouzé E, Gimenez-Roqueplo AP, Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int J Cancer. 2014;135:2237–48. doi: 10.1002/ijc.29080. [DOI] [PubMed] [Google Scholar]
- 110.Vega-Naredo I, Loureiro R, Mesquita KA, Barbosa IA, Tavares LC, Branco AF, Erickson JR, Holy J, Perkins EL, Carvalho RA, Oliveira PJ. Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells. Cell Death Differ. 2014;21:1560–74. doi: 10.1038/cdd.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718389545