

Abstract
Phosphatidylserine (PS) and monosialotetrahexosylganglioside (GM1) are examples of two host-derived lipids in the membrane of enveloped virus particles that are known to contribute to virus attachment, uptake, and ultimately dissemination. A quantitative characterization of their contribution to the functionality of the virus requires information about their relative concentrations in the viral membrane. Here, a gold nanoparticle (NP) binding assay for probing relative PS and GM1 lipid concentrations in the outer leaflet of different HIV-1 and Ebola virus-like particles (VLPs) using sample sizes of less than 3×106 particles is introduced. The assay evaluates both scattering intensity and resonance wavelength and determines relative NP densities through plasmon coupling as a measure for the target lipid concentrations in the NP-labeled VLP membrane. A correlation of the optical observables with absolute lipid contents is achieved by calibration of the plasmon coupling-based methodology with unilamellar liposomes of known PS or GM1 concentration. The performed studies reveal significant differences in the membrane of VLPs that assemble at different intracellular sites and pave the way to an optical quantification of lipid concentration in virus particles at physiological titers.
Keywords: Plasmon Coupling, Bioplasmonics, Viral Lipidomics, Lipid Quantification
1. Introduction
It is becoming increasingly clear that the host-derived membrane of an enveloped virus contributes more to the infection mechanism than simply forming a molecular scaffold for the presentation of virus encoded membrane glycoproteins. Different lipids have been shown to play a role in virion capture and uptake through their interactions with corresponding receptors on the host cell surface. These multivalent interactions stabilize virus – cell interactions and, thus, induce subsequent virus-induced cellular processes. Phosphatidylserine (PS), for instance, has been shown to facilitate apoptotic mimicry and enhance glycoprotein-independent uptake of Vaccinia,[1] Ebola[2] and Dengue viruses.[3] PS is also a cofactor in infectivity of human immunodeficiency virus (HIV) in monocytic cells.[4] Glycosphingolipids (GSLs) are another important class of lipids that mediate interactions between virus particles and host cells. GSLs enable the glycoprotein-independent binding of HIV-1 particles to mature dendritic cells (mDCs),[5] and they have been indicated to trigger the segregation of HIV-1 particles in non-lysosomal plasma membrane invaginations.[6] The functionality of GSLs is, however, not limited to HIV-1, instead GSLs act as attachment and entry factors for a diverse group of viruses.[7] All of the examples above corroborate the hypothesis that lipids contribute significantly to mediating virus – host-cell interactions, and this realization has motivated great interest in a quantitative analysis of viral lipidome to identify potential diagnostic and therapeutic targets.[8]
Electrospray ionization (ESI) mass spectroscopy (MS) is a powerful analytical tool that has become the method of choice in quantitative lipidomics.[9] However, even for ESI-MS the quantification of viral lipids can pose significant challenges due to the need for adequate mass standards[10] and the relatively low physiological concentrations of most virus particles. In the case of HIV-1, for instance, the virus concentration is only ~106 particles/mL of blood at the time of seroconversion, which provides at best ~1.3×10−12 moles of total lipid per mL.[11] Considering additional losses during virus isolation and lipid extraction, sample quantities in the picomole range or above, as is required for state-of-the-art MS-based approaches,[9c] are difficult to obtain especially for non-abundant lipids. Sample quantities are less of a concern for virus samples propagated in vitro in cell cultures where arbitrary amounts of virus can be generated. However, the clarification of important human health related questions, such as the role of specific lipids in in vivo virulence, require the ability to quantify relative concentrations of specific lipid species from patient-isolated samples. In response to this need, we introduce here an alternative gold nanoparticle (NP) based optical approach for the quantification of selected lipids in the viral membrane that is compatible with small sample quantities.
The binding affinity of NP labels for a specific lipid depends on target concentration in the viral membrane. A NP binding assay is, consequently, a viable approach for characterizing the targeted lipid concentration, provided adequate assays for the quantification of the bound NPs are available. Gold NPs have unique optical properties[12] that greatly aid the quantification of NP binding. The optical properties of noble metal NPs are determined by coherent conduction band electron density oscillations, so-called localized surface plasmon resonances (LSPRs)[13] that give rise to large scattering cross-sections at resonant excitation.[12a,14] The peak scattering intensity, Iscat, of individual virus particles is – in first approximation – proportional to the number of bound NP labels, and the LSPR wavelength, λres, of the bound NPs encodes information about the NP density, ρ, on the viral membrane. With growing ρ, the average separation between the NP labels decreases, which results in measurable changes in the scattering spectra (Figure 1). The plasmons in close-by nanoparticles couple,[15] and, as a consequence, λres red-shifts with decreasing interparticle separation.[16] Plasmon coupling has been utilized before as analytical tool to probe the spatial clustering of nanoparticle labeled cellular surface receptors,[17] to monitor nanoparticle uptake,[18] and to study the enzymatic cleavage of DNA or proteins tethered between nanoparticles.[19] In this manuscript, we demonstrate that the combination of Iscat and λres into one metric facilitates the quantification of NP-labeled target lipids in viral membranes. Similar as in a conventional quantitative immunoassay, the proposed assay determines binding affinities by evaluating the binding of specific labels. Unlike in a conventional immunoassay our assay uses the brightness of plasmonic NPs and near-field interactions between them as a transducer to quantify the binding with very high sensitivity. We apply this technique to characterize the content of PS and the model GSL, GM1, in the membrane of HIV-1 and Ebola virus-like-particles (VLPs). The compositions of these VLPs are believed to closely mimic those of the corresponding infectious virus particles due to identical assembly and budding mechanisms.[20] The extraordinary brightness of NPs facilitates the monitoring of lipid labeling for many individual VLPs in parallel in a darkfield microscope. Characterizing lipid contents in a massively parallel single virus particle assay has the advantage that the necessary sample quantity is no longer determined by the sensitivity of the detector, losses during lipid extraction, or other experimental considerations, but only by the number of virus particles required to adequately sample the ensemble.
Figure 1.

Simulated scattering spectra of gold NP labeled VLPs. a) Schematics of three random configurations of gold NP binding to VLPs. b) Simulated peak intensity and wavelength as function of the number of bound NPs, m. Solid lines show the average spectra calculated from 25 configurations. The numbers, 1-3, correspond to the structures shown in (a). c) Average peak wavelength as a function of bound NP density, ρ. d) Average peak scattering intensity as a function of ρ. Standard deviations are included as error bars.
2. Results and Discussion
2.1. Density-Dependent Spectral Response of Gold Nanoparticle Labels
The quantification of NP binding in an all-optical fashion requires a correlation of the NP signal, given by the peak scattering intensity, Iscat, and the plasmon resonance wavelength, λres, with the concentration of membrane-bound NPs. In the first step of our analysis we set out to verify this relationship through rigorous electromagnetic finite-difference time-domain (FDTD) simulations (for details see Methods). We simulated the spectral response of gold NPs bound to a 150 nm diameter dielectric sphere with a refractive index of nr = 1.500, which resembles the VLP – or complete virus particle for that manner – as a function of NP surface density. The latter is defined as ρ = m/A, where m is the number of membrane-bound NPs and A is the surface area of the virus particle. Up to m = 20 NPs were distributed across the surface in a random fashion (see Methods) with at least l = 25 different configurations for each m. Figure 1a illustrates schematically three of the investigated structures. The simulations assumed plane wave excitation at an angle of incidence of φ = 60° and were averaged over two orthogonal polarizations as indicated to emulate unpolarized white light excitation through a darkfield condenser. The peak positions of the scattering spectra for different m,l configurations are summarized in Figure 1b. The average spectra for each m are included as solid lines. In Figure 1c we plot the resulting average peak plasmon resonance wavelength ± std as function of m (and ρ). The fitted resonance wavelengths for the different m,l configurations have some spread since random morphological differences impact the electromagnetic coupling between the NPs, but shows a clear continuous red-shift as function of m. The simulations indicate an average increase in of ~24 nm going from m = 1 to m = 20. This red-shift is consistent with an increasing plasmon coupling due to decreasing average interparticle separations as m grows. The slope of the graph is steepest for small m (or ρ) and then levels off with increasing m. The electromagnetic coupling does not continue to intensify in larger NP clusters of random geometry as plasmon coupling is a nearest neighbor effect.[21] Figure 1d contains a plot of the average peak scattering intensity, ± std as function of m and ρ increases approximately 26-fold between m = 1 and 20. Interestingly, the increase in is weaker for the m-range in which increases the most. This behavior indicates that the fraction of the incident power localized in the near-field is larger for smaller m. For larger m the scattering into the far-field dominates.
2.2. HIV-1 VLP Panel with Different Membrane Origins
We used a test panel of four different enhanced green fluorescent protein (eGFP) tagged HIV-1 Gag-derived VLPs in this study. All VLPs were produced in vitro upon transient transfection of HEK 293T cells and had an average hydrodynamic diameter of ~160 nm (Figure S1). Our VLP panel included Wild Type (WT) VLPs, VLPs generated in 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP)-treated host cells (PDMP VLPs), and VLPs derived from two Gag mutants: 29/31KE and ΔMA. PDMP inhibits GSL synthesis[22] in VLP-producing cells, so VLPs derived from PDMP-treated cells contain lower amounts of GSLs.[5a] Note that 29/31KE Gag-eGFP and ΔMA Gag-eGFP contain mutations in the matrix (MA) protein component of Gag and have been shown by us and others to result in an alteration of VLP and infectious virus assembly sites.[23] In 29/31KE Gag-eGFP, two positively charged lysines at positions 29 and 31 have been replaced with acidic glutamates, while ΔMA Gag-eGFP contains an in-frame deletion between amino acids 15 through 99 that alters plasma membrane targeting specificity of Gag. Mutations in the matrix protein interfere with the ability of Gag to properly assemble at the cytoplasmic side of the plasma membrane, thus resulting in assembly of these mutant VLPs at intracellular membranes.[23b,23c,24] This is illustrated in Figure 2a-c which shows the intracellular distribution of WT, ΔMA, and 29/31KE Gag-eGFP proteins in HEK 293T cells, similar to the results that we have shown before.[23c] While WT Gag-eGFP is preferentially associated with the plasma membrane (Figure 2a), ΔMA Gag-eGFP and 29/31KE Gag-eGFP are associated with diverse intracellular endocytic compartments (Figure 2a and c) that are invariably CD63 and lamp1 positive.[23b,25] The differences in intracellular spatial distribution of wild-type and mutant Gag-eGFP proteins are consistent with a differential budding phenotype of mutant Gags (ΔMA and 29/31KE) from different intracellular membranes. The different budding sites have direct influence on the capture of the VLPs by mDCs. The capture efficiencies of ΔMA and 29/31KE VLPs by mDCs are significantly lower than that observed for WT VLPs (Figure 2d). VLP binding to mDCs has been shown to be mediated by interactions between GSLs (in particular GM1 and GM3) on VLPs and CD169 on mDCs.[5b,5c] The observed decrease in capture for the Gag mutants, ΔMA and 29/31KE, is consistent with a decrease of these lipid species in the VLP membranes.[23c] Similarly, previous studies have shown that VLPs derived from PDMP-treated host cells also show reduced capture by mDCs due to decreased GSL content in VLPs.[26] Given these ostensible differences in their membrane composition, WT, ΔMA, 29/31KE, and PDMP VLPs form a pertinent test panel for the development and validation of the proposed plasmonic NP based assay.
Figure 2.
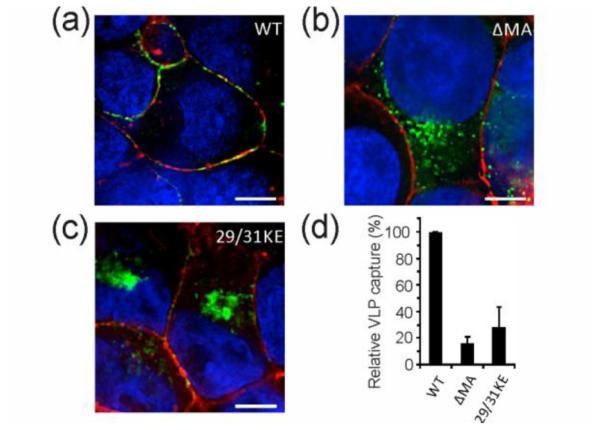
Mutations in the matrix domain of the Gag redirect the formation of VLPs from plasma membrane to intracellular compartments. Fluorescence images of HEK293T cells 1 day post transfection with a) WT Gag-eGFP, b) ΔMA Gag-eGFP and c) 29/31KE Gag-eGFP expression plasmids. Plasma membrane (GM1) and nucleus were stained in red and blue, respectively. In (a) WT Gag-eGFP colocalizes with the plasma membrane where WT VLPs assemble. In contrast, ΔMA and 29/31KE Gag-eGFP in (b) and (c) are associated with intracellular compartments. d) Relative capture of WT, ΔMA and 29/31KE VLPs by mDCs.
2.3. PS and GM1 Labeling with Gold NPs
The Gag VLPs were labeled using annexin V (AnxV) and cholera toxin B (CTB) as highly specific recognition elements for PS and GM1, respectively,[27] in combination with established biotin-Neutravidin binding chemistries (Figure 3a).[19c] After tethering biotins to the VLP surface using AnxV or CTB, biotinylated gold NPs were bound using the tetrameric Neutravidin protein as linker. Under otherwise identical experimental conditions (ligand type, ligand concentration and incubation time), the average number of bound gold NPs in this labeling approach is determined by the surface concentration of the targeted lipid as this quantity determines the total number of biotins on the VLP surface. Figure 3b shows a transmission electron microscopy (TEM) image of OsO4-fixed and negatively-stained WT VLPs after labeling with gold NPs. The NPs are clearly discernible due to their large contrast. Additional TEM images as well as fluorescence-correlated scanning electron microscopic (SEM) images are provided in Figure S2. We characterized the average number of bound NPs for selected lipid-VLP combinations (GM1-WT; PS-WT; PS-29/31KE) as well as for a control (missing required lipid-binding ligand, AnxV and CTB) in the SEM. The resulting relative labeling efficiencies are plotted in Figure 3c. The SEM data reveal a lower concentration of GM1 than PS in WT VLPs and a lower PS content in 29/31KE VLPs than in WT VLPs.
Figure 3.

NP labeling strategy for PS and GM1. a) PS or GM1 are labeled using AnxV or CTB in combination with Biotin-Neutravidin binding chemistries. b) TEM images of fixed and negatively-stained VLPs after NP binding to PS. Scale bars are 40 nm. c) Relative Labeling efficiencies for different lipid-VLP combinations: PS-WT; PS-29/31KE, GM1-WT and control (lacking AnxV and CTB).
Our SEM and TEM studies validate that our NP labeling strategy was successful. A detailed quantitative analysis of NP binding is, however, challenging via electron microscopy for multiple reasons. First, it requires the analysis of hydrated VLPs to retain the structural integrity and labeling of the VLPs. Meniscus forces acting on the NP labeled VLPs during de-hydration can perturb the structural distribution of the NP labels on the VLP surface. Furthermore, due to the large magnification required to count NP binding in the electron microscope, it is very time-consuming to gather information from statistically significant sample sizes in both SEM and TEM. Our aim was to avoid these complications entirely by developing an uncomplicated optical approach for monitoring NP labeling of hydrated VLPs based on multispectral imaging.
2.4. Multispectral Characterization of VLP Labeling
The optical set-up used in this work (Figure 4a) was designed to facilitate the acquisition of both fluorescence and scattering images (Figure 4b). We found that the scattering spectra of individual NP labeled VLPs obtained through multispectral imaging (see Methods for details) reproduce full spectra acquired with a conventional imaging spectrometer very well (Figure 4c). At the same time, the multispectral imaging provides the ability to acquire spectral information from all of the VLPs in the field of view in a short time without the need of raster-scanning the field of view. We used a 60x oil objective in our studies and routinely acquired spectral data of ~100 VLPs in a field of view of 75 by 75 μm2 within 100 seconds. By translating the stage and recording data from different areas of the substrate, we collected the scattering spectra of hundreds of VLPs per sample within a few minutes.
Figure 4.
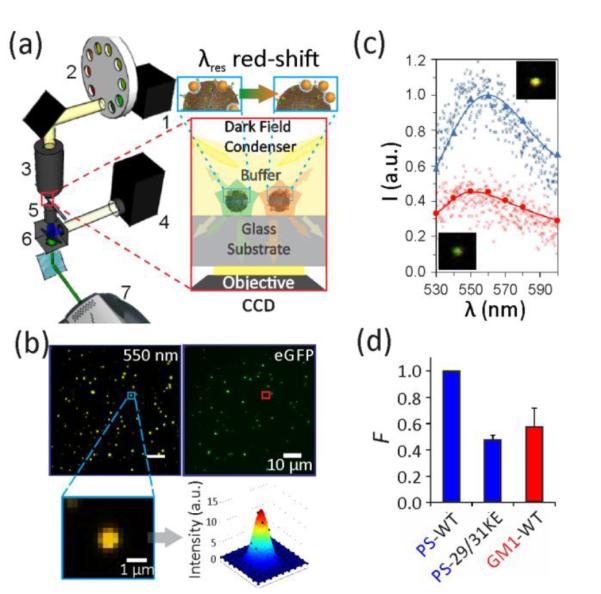
Optical set-up for correlated fluorescence / multispectral darkfield imaging. a) Scheme of the optical set-up to characterize NP labeled VLPs (inset). 1-Tungsten lamp, 2-Filter wheel, 3-Darkfield condenser, 4-Mercury lamp, 5-60x oil objective, 6-Fluorescence filter set, 7-EMCCD. b) Scattering (left) and fluorescence (right) image for WT VLP labeled for PS with NPs. A set of 9 scattering images in the range between 530 to 600 nm were recorded. Inset shows fitted point-spread-function for one scatterer. c) Comparison of spectra obtained through multispectral analysis (continuous lines) and imaging spectrometer (small markers) for an individual VLP with low (red, bottom) and high (blue, top) NP coverage. Insets show the corresponding scattering images. d) The binding probability, F, obtained through three or more optical measurements of three different samples. Error bars indicate standard deviations.
The second derivative of the single VLP spectra provided a quantifiable measure to determine whether a VLP was labeled. We only included spectra that showed a clear plasmon resonance peak, defined by a threshold of the second derivative of ≤ −2 (see Methods) and determined the fraction, F, of labeled VLPs in a sample. In Figure 4d, we plot the F values for the following lipid-VLP combinations: PS-WT; PS-29/31KE; GM1-WT. The F values from the multispectral analysis reproduce the relative NP labeling efficiencies obtained through SEM in Figure 3c for the same lipid-VLP combinations. The good agreement confirms that the optical F value increases with growing number of gold NPs per VLP.
2.5. Calibration of Multispectral Imaging with Liposomes
The multispectral imaging approach measures the peak scattering intensity, Iscat, and the resonance wavelength of NP labels, λres. These values are defined through the maxima in the scattering spectra of individual VLPs obtained via multispectral imaging. Both observables are functions of the NP surface density on the VLPs, ρ. The conversion of Iscat and λres measurements into a concentration of the targeted lipid requires a calibration with a membrane system of known composition. To that end, we fabricated unilamellar liposomes of similar size as the VLPs with a varying PS or GM1 content between 0% and 20%. The liposomes were labeled using the experimental strategy described above for VLPs before they were immobilized on the surface of a rectangular glass capillary to acquire multispectral data. These spectra were then fitted to obtain Iscat and λres (see Methods). Figure 5a shows scatter plots of Iscat versus λres for different liposome samples, each containing the data of >500 individual liposomes. The figure shows the distribution of single liposome measurements in the (Iscat, λres) plane as black markers and a fitted distribution function, P(Iscat, λres), as a heat map (see Methods). P(Iscat, λres) describes the statistical weight of a specific Iscat, λres combination. Figure 5b contains the binding probability, F, for the same samples of Figure 5a. Figure 5a shows that Iscat increases and λres red-shifts with increasing target lipid composition, which is consistent with a growing number of bound NP labels. Electromagnetic simulations (Figure 1) confirm that binding of a few NPs onto a VLP induces sufficient electromagnetic coupling to shift the resonance wavelength. In a random binding process, short and long interparticle distances will be formed simultaneously on the VLPs to generate a broad range of different NP scattering responses. The experimentally observed spread in Figure 5a is again consistent with the prediction from electromagnetic simulations (Figure 1). Both the scattering intensity and the resonance wavelength distribution of NP labeled VLPs contain information about the number of bound labels and these parameters are not independent. Pearson correlation coefficients of Iscat and λres in the range between 0.4-0.6 (Figure S3) indicate correlated intensity and spectral changes. Instead of characterizing NP labeling in terms of two individual observables, we therefore chose to describe the labeling through the fitted P(Iscat, λres) distribution as a metric that depends on both Iscat and λres. As a reference value for comparing the width of different P(Iscat, λres) distributions, we calculated the area, A90%, in the Iscat, λres plane containing ~90% of all measurements around the P(Iscat, λres) maximum. This value was normalized by dividing through the corresponding area of the Au NP monomer ensemble (F=1 and A90%=1, Figure S4). We found good reproducibility in obtaining F and A90% values, and independent experiments using the same VLP batch showed deviations in the B values of less than 1% (Figure S5).
Figure 5.
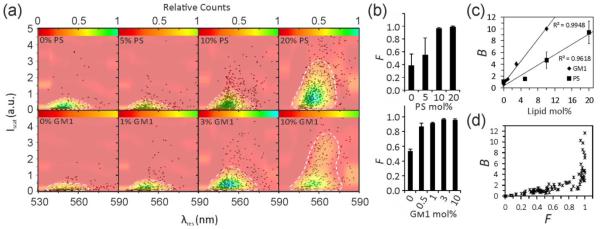
Calibration of concentration-dependent optical NP response with liposomes. a) (Iscat, λres) scatter plots for liposomes with varying PS (top row) or GM1 (bottom row) concentrations as specified after labeling with gold NPs. Data are plotted as black markers, and fitted distribution functions, P(Iscat, λres), are overlaid as color maps. The white dashed lines comprise the Iscat × λres, area, A90%, that contains 90% of the data points. Each plot contains 500-1500 liposomes in total. b) Fraction, F, of labeled liposomes as a function of PS (top) and GM1 (bottom) concentration. c) Plot of the spectral function B = F × A90% for GM1and PS. d) B versus F scatterplot for various liposome and VLP measurements. The data plotted in (a) – (c) were collected in three independent experiments.
F and A90% values are synergistic. At low NP labeling levels, when the contribution from plasmon coupling to the optical signal is negligible, F is more useful to distinguish different samples than A90%. This changes, however, for higher NP labeling levels where A90% can facilitate the differentiation between samples in which all VLPs are labeled. In this case, the major difference lies in the number of bound NPs per VLP and, thus, ρ. To describe the NP labeling of VLPs over a broad range of target lipid concentration with a single quantity, we introduced the overall binding parameter, B = F × A90%. Figure 5c plots the B values obtained for the investigated PS and GM1 liposomes as a function of lipid composition. The plotted data are averages of 3 independent experiments performed on specific batches of liposomes. The B values exhibit a linear increase with growing lipid concentration in the investigated concentration range as described by the following fit functions:
| (1) |
| (2) |
where mPS = 2.18, mGM1 = 1.07, bPS = −0.37, bGM1 = −0.83 and [PS] and [GM1] are in mo1%. Figure 5d contains a plot of B vs. F for ~100 measurements of different types of liposomes and VLPs. This plot shows that B increases linearly as function of F due to negligible changes in A90%. However, for F > 0.8, the slope of B increases abruptly, indicative of the onset of plasmon coupling due to multiple NP binding events to one object. For F > 0.95, the B value becomes almost exclusively a function of A90%, as plasmon coupling dominates the B value in this range.
2.6. Characterizing GM1 and PS Contents in the HIV-1 VLP Test Panel
Figure 6a shows (Iscat, λres) scatter plots as well as fitted P(Iscat, λres) surfaces as color maps for the PS and GM1 labeled VLPs of our HIV-1 test panel. The F values for the HIV-1 VLPs as well as for a WT VLP negative control that lacked any AnxV or CTB treatment are plotted in Figure 6b. The GM1 labeled VLPs in Figure 6a have lower intensities and more blue-shifted resonance wavelengths than their PS counterparts. This finding, together with the systematically lower F values in Figure 6b, confirm that the HIV-1 VLP membrane contains significantly less GM1 than PS. To substantiate this observation and to quantify the PS and GM1 lipid concentrations, we determined B values based on the experimentally obtained F values and the width of the (Iscat, λres) distributions, quantified as A90%. Equations (1) and (2) were then applied to convert the B values into PS and GM1 concentrations. These values are summarized in Figure 6c. Interestingly, our results indicate differences not only between PS and GM1 but also between PS in different HIV-1 VLPs. A two sample t-test showed significant (p<0.05) differences between all the samples except for PS levels in PDMP and 29/31KE VLPs and GM1 levels in WT and ΔMA VLPs.
Figure 6.
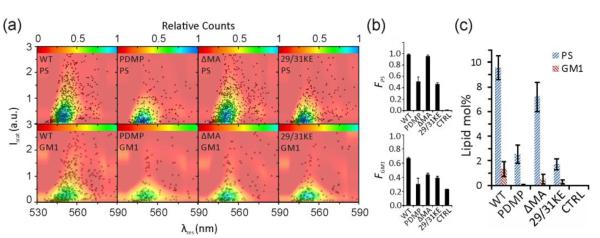
Optical quantification of PS and GM1 contents in 4 different HIV-1 VLPs. a) Iscat versus λres, scatter plot after labeling PS (top row) and GM1 (bottom row) in (from left to right) WT, PDMP, ΔMA and 29/31KE VLPs. Each plot contains the data of 500-1500 VLPs obtained in 3 or more independent experiments with one batch of VLPs. Data are plotted as black markers and fitted distribution functions, P(Iscat, λres), are overlaid as color maps. b) F values for lipid-VLP combinations shown in (a) and a WT VLP negative control that lacked any AnxV or CTB treatment. c) PS and GM1 content determined with Eq. 1 and 2 from the data in (a) and (b).
We observed that the PS content in different batches of WT VLPs varied between 5.5-10.1%. For the batch of VLPs used for obtaining the results displayed in Figure 6, the PS content was determined as 9.5±1.0 mol%. These values are in good agreement with mass spectroscopic studies of in vitro propagated HIV-1 virus particles that reported PS contents in the range between 8.4% - 13.4%, depending on host cell and conditions[10,28]. Interestingly, the PS content in 29/31KE VLPs (1.7±0.4%) and PDMP VLPs (2.5±0.7%) are both significantly lower. The PS content in ΔMA VLPs with 7.2±1.2% is comparable to that in WT VLPs. In the case of GM1, we determined lipid contents of 1.3±0.6%, 0.03±0.04%, 0.5±0.4% and 0.3±0.1% for WT, PDMP, ΔMA and 29/31KE HIV-1 VLPs, respectively. These concentrations are close to the detection limit of our assay and we conclude that the GM1 content in HIV-1 is low. The obtained data imply that the GM1 level is highest in WT VLPs and that it is decreased to less than 1 mol% in other mutants.
2.7. GM1 and PS Contents in WT and PDMP EBOV VP40-Derived VLPs
We also applied the optical lipid quantification assay to Ebola virus (EBOV) matrix protein VP40-derived VLPs, produced in untreated HEK-293T cells (WT VLPs) or PDMP-treated cells (PDMP VLPs). The (Iscat, λres) scatter plot and F histograms are shown in Figure 7a and b. Negative controls without AnxV and CTB confirmed that the binding was PS or GM1 specific (Figure S6). In analogy to our analysis of the HIV-1 VLPs, we converted these raw data into PS and GM1 contents (Figure 7c).
Figure 7.

Optical quantification of PS and GM1 contents WT and PDMP EBOV VP40-derived VLPs. a) Iscat versus λres, scatter plot after labeling PS (top row) and GM1 (bottom row). Each plot contains the data of 500-700 VLPs obtained in 3 or more independent experiments. Data are plotted as black markers and fitted distribution functions, P(Iscat, λres), are overlaid as color maps. b) F values associated with the same measurements. c) PS and GM1 concentrations (in mol%) resulting from the optical measurements.
Even though we anticipate that the derived lipid concentrations have a larger error for EBOV than HIV-1, as the calibration was derived for spherical particles whereas EBOV VP40 VLPs have a filamentous shape, they still provide valuable information about relative differences. We find that the gap between PS and GM1 content has significantly dropped in EBOV VP40 WT VLPs when compared to HIV-1 WT VLPs. The PS concentration is decreased, whereas the GM1 concentration is increased. We conclude that the lipid composition of WT VLPs of EBOV and HIV-1 show significant differences. The most important one (vide infra) is the increased GM1 concentration in EBOV VP40-derived VLPs. The GM1 content is – as expected – decreased in EBOV PDMP VLPs. As in the case of HIV-1 VLPs, we find that PDMP treatment also decreases the PS concentration, albeit to a lesser degree.
2.8. Discussion of PS and GM1 Quantification in HIV-1 and Ebola VLPs
Our NP-based lipid quantification reveals distinct PS contents in the membrane of HIV-1 VLPs budding from different cellular membranes. This experimental observation confirms measurable differences in the composition of cellular membranes. Consistent with previous studies that demonstrated an enrichment of PS in the plasma membrane,[29] the PS concentration was at the highest 9.5±1.0% in membranes of WT HIV-1 Gag-eGFP VLPs that assemble and bud from the plasma membrane. Interestingly, ΔMA VLPs, that bud from intracellular membranes (Figure 2), exhibited a PS content of 7.2±1.2%. This concentration is only slightly lower than that of the WT VLPs. In contrast, 29/31KE VLP membranes had a substantially lower PS content of 1.7±0.4%. The striking differences in membrane composition between ΔMA and 29/31KE VLPs suggests different intracellular budding sites. The measured PS level of ΔMA VLPs is close to that of the endoplasmic reticulum (ER).[24] Given the observed striking difference in PS content, it is unlikely that 29/31KE VLPs also bud from the ER, instead, other budding sites, such as late endosomal membrane, seem more plausible.[23b]
Our measurements show clear differences in the GM1 content of WT EBOV and HIV-1 VLPs. While both EBOV and HIV-1 are known to bud from GSL-enriched lipid rafts in the plasma membrane,[10,30] there is experimental evidence for the existence of different sub-sets of lipid rafts that are selectively enriched in GM1 or GM3.[31] The differences in GM1 content between EBOV and HIV-1 VLPs observed in this work is consistent with the previous findings that EBOV and HIV-1 bud from separate lipid raft microdomains.[32] While EBOV VP40-derived VLPs preferentially bud from the GM1 enriched domains,[30] HIV-1 Gag-eGFP VLPs assemble and bud from GM3 enriched plasma membrane sites.[5c] The low GM1 content in HIV-1 VLPs may be the reason why GM1 is not as effective as GM3 in mediating the capture of HIV-1 particles by DCs,[5c] although CD169 is capable of binding to both GSLs equally well,[5b] Another surprising finding of our studies is that the PDMP treatment of host cells not only reduces the GM1 content in the VLP membranes derived from these cells, but also the PS content. Since PDMP VLPs still bud from the plasma membrane,[26] this finding indicates that PDMP treatment of the host cell not only inhibits the GSL synthesis pathway, but also affects other cellular processes that lead to a concomitant reduction of PS in the VLP membrane.
3. Conclusion
We have introduced a new optical assay for measuring the lipid contents in VLP and viral envelope membranes based on the spectral analysis of gold nano-label binding and plasmon coupling. In this work we used 3×106 VLPs per experiment, although data collection from ~103 VLPs was sufficient to determine the membrane content of the targeted lipids. The large excess of more than 3 orders of magnitude of VLPs was necessary to achieve a sufficient binding of VLPs to the glass surface of our optical instrument in an adequate amount of time. We envision that the required sample amount can be dramatically reduced by improving the handling of the sample using microfluidics.
The NP-based approach only provides information about labeled lipid species and, thus, does not provide the same breadth of information as label-free techniques, such as MS. Nevertheless, the ability to quantify selected lipid species in an optical approach with very small virus samples will facilitate new applications. A monitoring of surface features of patient-isolated virus particles is thus now within reach, which will eventually result in a more complete molecular characterization of viral species in different individuals and at different stages of an infection.
4. Experimental Section
Electromagnetic Simulations
FDTD simulations were performed with the Lumerical Solutions 8.7.1 software package. The mesh size of these simulations was always 5 nm. The substrate was chosen as glass with a refractive index of 1.517. Virus-like particles (VLPs) and liposome were emulated as dielectric beads with a diameter of 150 nm and a refractive index of 1.500. The refractive index of the medium was set to 1.335. Gold nanoparticles (NPs) were assumed to be 40 nm in diameter and we used the gold dielectric function given by Johnson and Christy.[33] The light source in these simulations was always a planar wave with a single wavelength. The angle of incidence was 60° and we averaged over two orthogonal polarizations. The simulations were performed for nine discrete wavelengths (corresponding to the center wavelength of the filters used in the multispectral imaging: 530 nm, 540 nm, 546 nm, 550 nm, 555 nm, 560 nm, 570 nm, 580 nm, 600 nm). The detector in this simulation was a single planar far-field detector placed in the glass beneath the VLP/NP complex. The maximum possible number of NPs is 38 in the case of close packing. We distributed m NPs randomly over these coordinates. We considered m = 1 – 10, 15 and 20. For each m, 25 different unique random configurations were created. We kept a minimum spacing of 10 nm between the NPs. The NPs were placed 5 nm away from the VLP surface, assuming a 5 nm thickness for the ssDNA shell on the NPs under experimental conditions.
Liposomes Preparation
In the first step, lipids in a defined ratio were dissolved in chloroform. The lipid mix consisted of 1,2-dipalmitoleoyl-sn-glycero-3-phosphocholine (DPPC), Cholesterol, 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) and ganglioside GM1, all purchased from Avanti Polar Lipids, Inc. The cholesterol concentration was kept constant at 45 mol%, 0.1% of which was fluorescently labeled with TopFluor. In the case of PS liposomes, the concentration of GM1 was fixed at 5%, while the concentration of DOPS was either 0, 5, 10 or 20%. The DPPC concentration of these liposomes was 50% 45, 40 and 30%. Similarly, in the case of GM1 liposomes, the concentration of DOPS was kept constant at 10%, and the concentration of GM1 was varied between 0, 0.5, 1, 3 and 10%. The DPPC concentration in the liposomes was accordingly 45, 44.5, 44, 42 and 35%. The total concentration of lipids in 150 μl of chloroform was 7 mM.
The chloroform in the lipid solution was removed with a rotary evaporator connected to vacuum at room temperature. The flasks containing the lipids were left in vacuum overnight. Then, 1 mL of 20 mM HEPES buffer (pH=7) was added to each flask and it was sonicated in a bath sonicator for 60 min at room temperature. This colloid of synthesized liposomes was extruded through a 100 nm polycarbonate membrane 9 times while maintaining a temperature of 40°C on a hot plate. The liposomes were subsequently stored at 4°C. Average size and concentration of the liposomes were measured through dynamic light scattering (DLS) with a Malvern ZetaSizer. Concentration was measured based on the count rates in dynamic light scattering tests on serial dilutions of the sample and using Concentration Utilities of ZetaSizer software.
Preparation of Biotinylated Gold NPs
40 nm gold NPs (from TedPella Inc.) were functionalized with single stranded DNA oligonucleotides using the method by Liu et al.[34] The oligonucleotides HS-AAAAAAAAAACTCACGCTAC-GACTGACACC and HS-AAAAAAAAAAGACCTACTAAGACTACTACACAACCAGAGA-Biotin oligonucleotide were mixed in a 4:1 ratio. This mix was added to gold nanoparticle colloid in DDI water to yield a final concentration of 25 μM DNA and 1.5 nM nanoparticles in a total volume of 10 μl and incubated for 30 min at room temperature. The pH was then lowered to pH = 3 by adding 10 mM sodium citrate buffer in a negligible volume. The mix was incubated for another 30 min. After that, the gold nanoparticles were washed by repeated centrifugation (2,400 ×g, 10 min) and resuspension in 1.5 mL in DDI water. After the third washing step, the NPs were passed through a 0.22 μm syringe filter to remove any agglomerates or impurities. The particles were then collected by centrifugation at 2,200×g. The resulting DNA-conjugated gold NP pellet was diluted with DDI water before 10x PBS was added to reach a final concentration of 0.15 nM NPs in 1x PBS. The volume of added PBS was adjusted in experiments that required higher/lower concentrations. The concentration, size and zeta potential of the NPs were measured in 1x PBS buffer by UV-Vis spectrometer and Malvern ZetaSizer right before use.
VLP/Liposome Functionalization
GM1 and PS in the VLP/Liposome membrane were functionalized with biotinylated Annexin V (Abcam) or cholera toxin subunit B (Life Sciences Inc.), respectively, using a 10000:1 ligand:VLP ratio. For this purpose, 107 VLPs/liposomes in 0.5-2 μL of stock were added to 10 μL of 1x Annexin Binding Buffer (ABB: 10 mM HEPES pH = 7.4, 137 mM NaCl, 2.5 mM CaCl2) and 0.5 μg of AnxV or CTB was added and incubated for 30 min at room temperature. Then, an 8-fold excess of neutravidin (NTV, Thermoscientific Inc.) in PBS and 1% Bovine Serum Albumin (BSA) was added and incubated for 30 min at room temperature. Subsequently, the functionalized VLPs were purified from excess proteins in solution through ultracentrifugation (Beckman-Coulter Optima TLX) on 60% sucrose cushion (100k ×g, 30 min, 4°C), followed by 24-48 h of dialysis against 1x ABB using a 100 nm pore size polycarbonate (PC) membrane in a micro-dialyzer (Harvard Apparatus Inc.) in an ice bath. Liposomes were purified by dialysis as described above. The functionalized VLPs/liposomes were diluted to 150 μL in ABB, and stored at 4°C for experiments during the following week. This amount of functionalized VLPs/liposomes was enough for at least 3 optical measurements or 5-10 electron microscopy samples. The Ebola VLPs were functionalized for PS and GM1 lipids in a similar procedure, with the exception that a 200 nm PC membrane was used.
Sample Preparation for Optical Measurements
The functionalized VLPs, in 50 μL ABB, were flushed into rectangular borosilicate capillaries (100 × 2 × 0.1 mm3, from Vitrocom), where they stuck non-specifically to the fused silica surface. Under typical experimental conditions, the VLPs were incubated until approx. 100 VLPs were bound in the field of view (75 μm×75 μm area of the image) to avoid interference between the individual VLPs (this step took normally around 30 min at 4°C). We used VLP samples with a concentration of 6x104 VLPs/μL and consumed a total of ~3x106 VLPs per experiment. After that, excess VLPs were removed by flushing the chamber with 1x ABB. The surface was then blocked by 10% BSA in 1x ABB for 2 hr at 4°C. After washing the samples with 1%BSA in 1xPBS the samples were incubated with 0.1 mg/mL polyinosinic acid (PI) in 1%BSA/1xPBS for 30 min at room temperature. PI was subsequently removed by flushing the flow chamber with 1%BSA in 1xPBS. In the next step, the immobilized VLPs were incubated with a 1.5 nM solution of biotin-functionalized gold nanoparticle labels in 1x PBS for 15 min at room temperature. Unbound gold nanoparticles were flushed out with 1x PBS. In case of Ebola VLPs, anti-EBOV GP goat IgG (antibody against EBOV glycoprotein) in PBS was incubated after this step in the capillary for 15 min at room temperature, followed by 15 min incubation of anti-goat IgG-Alexa488 (from Life Sciences Inc.). This procedure labeled the glycoproteins on the surface of the VLPs for VLP localization. The samples were then washed with PBS. After that, the samples were ready for optical characterization.
EM Sample Preparation
Electron Microscopy samples were prepared similar to optical measurement samples, except the substrate was a 5 mm × 5 mm quartz chip for SEM samples and a 9-window SiN grid for TEM samples (from Ted Pella Inc.). Also, the EM samples went through fixation or negative staining or both after gold nanoparticles binding. For SEM samples, 20 μl of the functionalized VLP stock was initially incubated on a quartz chip for 1 hr at 4°C. Then, the surface of the chip was flushed by pipetting 50 μL of ABB on the chip 3 times. Then, 10%BSA in ABB was incubated on the chip for 2 hr at 4°C. The chip was subsequently washed with PBS 3 times before 0.1 mg/mL PI in PBS was incubated on the sample for 30 min at room temperature. Then, the sample was washed with 1%BSA in 1x PBS and conjugated gold NPs in PBS were incubated on the substrate for 15 min at room temperature. Then the sample was washed again with PBS. NP-labeled VLP samples were incubated with 2.5% glutaraldehyde (Sigma-Aldrich) in 1x PBS for 30 min and subsequently with 1% OsO4 in PBS (50 mM salt) for 30 min at room temperature. The SEM samples were then washed with water and air dried. TEM samples went through a similar process on a SiN grid. The samples were stained with 1% sodium phosphotungstate in water (pH=7.4) for 30 seconds, blotted and then dried.
SEM and TEM Characterization
SEM samples were coated by a thin layer of Au/Pd using a sputter coater to provide conductivity. SEM imaging was performed on a Zeiss Supra 55 VP scanning electron microscope with 10 kV EHT. TEM imaging was performed by a JEOL JEM 2010 scope with 200 kV acceleration voltage.
Multispectral Imaging (MSI) and Spectrometry
All imaging experiments were performed with an inverted microscope (Olympus IX71) equipped with an oil immersion darkfield condenser and 60x objective. For each field of view, nine monochromatic scattering images were recorded under darkfield illumination using 10 nm bandpass filters with center wavelengths of 530, 540, 546, 550, 555, 560, 570, 580, and 600 nm. In addition to the 9 scattering images, one fluorescence image (400/530 nm excitation/emission filters and a 470 nm dichroic) of the same field of view was recorded. Images were all recorded by an Andor iXon EMCCD camera with a 512×512 pixels detector. For scattering images we accumulated 10 acquisitions with an exposure time of 1 sec and electron multiplier setting of 10. For fluorescence measurements we used an exposure time of 0.1 sec and an electron multiplier setting of 100. The 60x objective had a variable numerical aperture (NA), so the scattering images were recorded with NA = 0.65 and the fluorescence images were recorded with NA = 1.25. Full spectra were recorded using a Shamrock spectrometer with a CCD detector connected to the same microscope setup. To account for the spectral profile of the Tungsten lamp used as excitation source MSI and spectral data were corrected by dividing through the corresponding average scattering data obtained from unlabeled VLPs.
Image Processing and Spectral Analysis
All image processing and data analysis was performed using a home-written Matlab code. The point-spread-function (PSF) of every VLP/liposome in monochromatic scattering images was fitted with a Gaussian function of the form:
where xo and yo are the Gaussian peak position, σx and σy are the full width at half maximum of the Gaussian along x and y directions, A is the Gaussian amplitude, and BG is the background. After subtracting the background (BG), the integrated intensity of the PSF was taken as the scattering intensity of the VLP/liposome at that specific wavelength. Then, a white light correction was performed by dividing the intensities by the intensities of unlabeled VLPs (which we assumed to be whitelight scatterers) at the corresponding wavelengths. While the VLP spectrum was flat, binding of NPs led to an increase in intensity on one or more of the monitored wavelength channels.
We only included VLPs in our analysis of (Iscat, λres) distribution plots and A90% calculations whose intensity was in the third quartile (or above) of the intensity distribution of the unlabeled VLPs in all of the monitored wavelength channels. For the VLPs/liposomes that passed the intensity threshold, a second order Gaussian curve was fitted around the maximum-intensity wavelength and the second derivatives of the spectra at peak position were calculated. We chose a second order Gaussian fit function as the spectra showed some slight asymmetries. We included only VLPs with a second derivative at peak wavelength of −2 or lower (95% of the population of monomer NP labels passed this criterion) to further separate labeled from unlabeled VLPs. The fraction of labeled VLPs/liposomes defined the F value.
Peak Scattering Intensity versus Resonance Wavelength (Iscat, λres) Plots and B Value Calculations
We plotted the peak scattering intensity (Iscat) versus resonance wavelength (λres) for all VLPs and then histogrammed the data. The bin size of the histogram was 5 on the wavelength axis from 530 nm to 600 nm, and 0.2 on the intensity axis from 0 to 5 in arbitrary units. A 3D surface was then fitted to this histogram. For this purpose, the data of the 3D histogram were imported to TableCurve 3D v4.0 software and a Chebyshev bivariate polynomial order 7 was fitted on the data.As a measure of the width of the peak intensity versus wavelength distribution, we calculated the area enclosed by the fit at a height corresponding to 20% of the fitted surface peak. We found that this area enclosed 90%±7% of the measurements in all the samples, and we consequently refer to it as A90% throughout the text. The binding value, B, of each sample was calculated as B=F×A90%. The A90% value was set to 0 for samples with F ≤ 0.05.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Health through grants R01CA138509 (B.M.R.), RO1AI064099 (S.G.) and 1R56Al104393 (B.M.R. and S.G.).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Mercer J, Helenius A. Ann. N. Y. Acad. Sci. 2010;1209:49. doi: 10.1111/j.1749-6632.2010.05772.x. [DOI] [PubMed] [Google Scholar]
- [2].Moller-Tank S, Kondratowicz AS, Davey RA, Rennert PD, Maury W. J. Virol. 2013;87:8327. doi: 10.1128/JVI.01025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, Monahan S, Bu X, Farzan M, Freeman GJ, Umetsu DT, Dekruyff RH, Choe H. PLoS Pathog. 2013;9:e1003232. doi: 10.1371/journal.ppat.1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ. J. Immunol. 2003;170:4840. doi: 10.4049/jimmunol.170.9.4840. [DOI] [PubMed] [Google Scholar]
- [5].a) Puryear WB, Akiyama H, Geer SD, Ramirez NP, Yu X, Reinhard BM, Gummuluru S. PLoS Pathog. 2013;9:e1003291. doi: 10.1371/journal.ppat.1003291. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Izquierdo-Useros N, Lorizate M, Contreras FX, Rodriguez-Plata MT, Glass B, Erkizia I, Prado JG, Casas J, Fabrias G, Krausslich HG, Martinez-Picado J. PLoS Biol. 2012;10:e1001315. doi: 10.1371/journal.pbio.1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Puryear WB, Yu X, Ramirez NP, Reinhard BM, Gummuluru S. Proc. Natl. Acad. Sci. U. S. A. 2012;109:7475. doi: 10.1073/pnas.1201104109. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Puryear WB, Gummuluru S. Adv. Exp. Med. Biol. 2013;762:131. doi: 10.1007/978-1-4614-4433-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Yu HJ, Reuter MA, McDonald D. PLoS Pathog. 2008;4:e1000134. doi: 10.1371/journal.ppat.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yu X, Feizpour A, Ramirez NG, Wu L, Akiyama H, Xu F, Gummuluru S, Reinhard BM. Nat. Commun. 2014;5:4136. doi: 10.1038/ncomms5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Suzuki Y, Matsunaga M, Matsumoto M. J. Biol. Chem. 1985;260:1362. [PubMed] [Google Scholar]; b) Grassme H, Riehle A, Wilker B, Gulbins E. J. Biol. Chem. 2005;280:26256. doi: 10.1074/jbc.M500835200. [DOI] [PubMed] [Google Scholar]; c) Epand RM, Nir S, Parolin M, Flanagan TD. Biochemistry. 1995;34:1084. doi: 10.1021/bi00003a045. [DOI] [PubMed] [Google Scholar]; d) Cooling LLW, Koerner TAW, Naides SJ. J. Infect. Dis. 1995;172:1198. doi: 10.1093/infdis/172.5.1198. [DOI] [PubMed] [Google Scholar]; e) Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. EMBO J. 2003;22:4346. doi: 10.1093/emboj/cdg439. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Haslam SM, Julien S, Burchell JM, Monk CR, Ceroni A, Garden OA, Dell A. Immunol. Cell Biol. 2008;86:564. doi: 10.1038/icb.2008.54. [DOI] [PubMed] [Google Scholar]
- [8].a) Soares MM, King SW, Thorpe PE. Nat. Med. 2008;14:1357. doi: 10.1038/nm.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kawasaki N, Vela JL, Nycholat CM, Rademacher C, Khurana A, van Rooijen N, Crocker PR, Kronenberg M, Paulson JC. Proc. Natl. Acad. Sci. U. S. A. 2013;110:7826. doi: 10.1073/pnas.1219888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Koivusalo M, Haimi P, Heikinheimo L, Kostiainen R, Somerharju P. J. Lipid Res. 2001;42:663. [PubMed] [Google Scholar]; b) Han X, Gross RW. Mass Spectrom. Rev. 2005;24:367. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]; c) Brugger B. Annu. Rev. Biochem. 2014;83:79. doi: 10.1146/annurev-biochem-060713-035324. [DOI] [PubMed] [Google Scholar]
- [10].Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. J. Virol. 2008;82:11228. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fiebig EW, Wright DJ, Rawal BD, Garrett PE, Schumacher RT, Peddada L, Heldebrant C, Smith R, Conrad A, Kleinman SH, Busch MP. AIDS. 2003;17:1871. doi: 10.1097/00002030-200309050-00005. [DOI] [PubMed] [Google Scholar]
- [12].a) Yguerabide J, Yguerabide EE. Anal. Biochem. 1998;262:137. doi: 10.1006/abio.1998.2759. [DOI] [PubMed] [Google Scholar]; b) Yguerabide J, Yguerabide EE. Anal. Biochem. 1998;262:157. doi: 10.1006/abio.1998.2760. [DOI] [PubMed] [Google Scholar]; c) Jain PK, Huang X, El-Sayed IH, El-Sayad MA. Plasmonics. 2007;2:107. [Google Scholar]; d) Halas NJ. MRS Bull. 2005;30:362. [Google Scholar]; e) Nehl CL, Liao HW, Hafner JH. Nano Lett. 2006;6:683. doi: 10.1021/nl052409y. [DOI] [PubMed] [Google Scholar]
- [13].a) Kelly KL, Coronado E, Zhao LL, Schatz GC. J. Phys. Chem. B. 2003;107:668. [Google Scholar]; b) Hu M, Novo C, Funston A, Wang HN, Staleva H, Zou SL, Mulvaney P, Xia YN, Hartland GV. J. Mater. Chem. 2008;18:1949. doi: 10.1039/b714759g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA. Chem. Soc. Rev. 2012;41:2740. doi: 10.1039/c1cs15237h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Willets KA, Van Duyne RP. Annu. Rev. Phys. Chem. 2007;58:267. doi: 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- [15].a) Nordlander P, Oubre C, Prodan E, Li K, Stockman MI. Nano Lett. 2004;4:899. [Google Scholar]; b) Rechberger W, Hohenau A, Leitner A, Krenn JR, Lamprecht B, Aussenegg FR. Opt. Commun. 2003;220:137. [Google Scholar]; c) Wu L, Reinhard BM. Chem. Soc. Rev. 2014;43:3884. doi: 10.1039/c3cs60340g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Encina ER, Coronado EA. J. Phys. Chem. C. 2010;114:3918. [Google Scholar]
- [16].a) H. Su K, Wei QH, Zhang X, Mock JJ, Smith DR, Schultz S. Nano Lett. 2003;3:1087. [Google Scholar]; b) Reinhard BM, Siu M, Agarwal H, Alivisatos AP. J. Liphardt, Nano Lett. 2005;5:2246. doi: 10.1021/nl051592s. [DOI] [PubMed] [Google Scholar]; c) Jain PK, Huang WY, El-Sayed MA. Nano Lett. 2007;7:2080. [Google Scholar]; d) Yang L, Wang H, Yan B, Reinhard BM. J. Phys. Chem. C. 2010;114:4901. doi: 10.1021/jp911858v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Crow MJ, Grant G, Provenzale JM, Wax A. Am. J. Roentgenol. 2009;192:1021. doi: 10.2214/AJR.07.3535. [DOI] [PubMed] [Google Scholar]; b) Wang J, Yu X, Boriskina SV, Reinhard BM. Nano Lett. 2012;12:3231. doi: 10.1021/nl3012227. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang J, Boriskina SV, Wang H, Reinhard BM. ACS Nano. 2011;5:6619. doi: 10.1021/nn202055b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hu Q, Tay LL, Noestheden M, Pezacki JP. J. Am. Chem. Soc. 2007;129:14. doi: 10.1021/ja0670005. [DOI] [PubMed] [Google Scholar]
- [18].a) Aaron J, Travis K, Harrison N, Sokolov K. Nano Lett. 2009;9:3612. doi: 10.1021/nl9018275. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang H, Wu L, Reinhard BM. ACS Nano. 2012;6:7122. doi: 10.1021/nn302186n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Elghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Science. 1997;277:1078. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]; b) Jun YW, Sheikholeslami S, Hostetter DR, Tajon C, Craik CS, Alivisatos AP. Proc. Natl. Acad. Sci. U. S. A. 2009;106:17735. doi: 10.1073/pnas.0907367106. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sonnichsen C, Reinhard BM, Liphardt J, Alivisatos AP. Nat. Biotechnol. 2005;23:741. doi: 10.1038/nbt1100. [DOI] [PubMed] [Google Scholar]; d) Reinhard BM, Sheikholeslami S, Mastroianni A, Alivisatos AP, Liphardt J. Proc. Natl. Acad. Sci. U. S. A. 2007;104:2667. doi: 10.1073/pnas.0607826104. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Skewis LR, Reinhard BM. Nano Lett. 2008;8:214. doi: 10.1021/nl0725042. [DOI] [PubMed] [Google Scholar]
- [20].Grgacic EV, Anderson DA. Methods. 2006;40:60. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Yan B, Boriskina SV, Reinhard BM. J. Phys. Chem. C. 2011;115:24437. doi: 10.1021/jp207821t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yan B, Boriskina SV, Reinhard BM. J. Phys. Chem. C. 2011;115:4578. doi: 10.1021/jp112146d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yan B, Thubagere A, Premasiri R, Ziegler L, Dal Negro L, Reinhard BM. ACS Nano. 2009;3:1190. doi: 10.1021/nn800836f. [DOI] [PubMed] [Google Scholar]
- [22].Makino A, Ishii K, Murate M, Hayakawa T, Suzuki Y, Suzuki M, Ito K, Fujisawa T, Matsuo H, Ishitsuka R, Kobayashi T. Biochemistry. 2006;45:4530. doi: 10.1021/bi052104y. [DOI] [PubMed] [Google Scholar]
- [23].a) Ono A, Freed EO. J. Virol. 2004;78:1552. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Joshi A, Ablan SD, Soheilian F, Nagashima K, Freed EO. J. Virol. 2009;83:5375. doi: 10.1128/JVI.00109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Akiyama H, Miller C, Patel HV, Hatch SC, Archer J, Ramirez NG, Gummuluru S. J. Virol. 2014;88:8813. doi: 10.1128/JVI.00992-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Facke M, Janetzko A, Shoeman RL, Krausslich HG. J. Virol. 1993;67:4972. doi: 10.1128/jvi.67.8.4972-4980.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schiralli Lester GM, Akiyama H, Evans E, Singh J, Gummuluru S, Henderson AJ. Virology. 2013;436:235. doi: 10.1016/j.virol.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hatch SC, Archer J, Gummuluru S. J. Virol. 2009;83:3496. doi: 10.1128/JVI.02249-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. Cytometry. 1998;31:1. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]; b) Kuziemko GM, Stroh M, Stevens RC. Biochemistry. 1996;35:6375. doi: 10.1021/bi952314i. [DOI] [PubMed] [Google Scholar]
- [28].Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2641. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Leventis PA, Grinstein S. Annu. Rev. Biophys. 2010;39:407. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- [30].Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmaljohn C, Schmaljohn A, Aman MJ. J. Exp. Med. 2002;195:593. doi: 10.1084/jem.20011500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Lehmann M, Rocha S, Mangeat B, Blanchet F, Uji IH, Hofkens J, Piguet V. PLoS Pathog. 2011;7:e1002456. doi: 10.1371/journal.ppat.1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fujita A, Cheng J, Hirakawa M, Furukawa K, Kusunoki S, Fujimoto T. Mol. Biol. Cell. 2007;18:2112. doi: 10.1091/mbc.E07-01-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Leung K, Kim JO, Ganesh L, Kabat J, Schwartz O, Nabel GJ. Cell Host Microbe. 2008;3:285. doi: 10.1016/j.chom.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Johnson PB, Christy RW. Phys. Rev. B. 1972;6:4370. [Google Scholar]
- [34].a) Zhang X, Servos MR, Liu J. J. Am. Chem. Soc. 2012;134:7266. doi: 10.1021/ja3014055. [DOI] [PubMed] [Google Scholar]; b) Zhang X, Gouriye T, Goeken K, Servos MR, Gill R, Liu JW. J. Phys. Chem. C. 2013;117:15677. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.