

Abstract
Dietary methionine restriction (MR) by 80% increases energy expenditure (EE), reduces adiposity, and improves insulin sensitivity. We propose that the MR-induced increase in EE limits fat deposition by increasing sympathetic nervous system–dependent remodeling of white adipose tissue and increasing uncoupling protein 1 (UCP1) expression in both white and brown adipose tissue. In independent assessments of the role of UCP1 as a mediator of MR’s effects on EE and insulin sensitivity, EE did not differ between wild-type (WT) and Ucp1−/− mice on the control diet, but MR increased EE by 31% and reduced adiposity by 25% in WT mice. In contrast, MR failed to increase EE or reduce adiposity in Ucp1−/− mice. However, MR was able to increase overall insulin sensitivity by 2.2-fold in both genotypes. Housing temperatures used to minimize (28°C) or increase (23°C) sympathetic nervous system activity revealed temperature-independent effects of the diet on EE. Metabolomics analysis showed that genotypic and dietary effects on white adipose tissue remodeling resulted in profound increases in fatty acid metabolism within this tissue. These findings establish that UCP1 is required for the MR-induced increase in EE but not insulin sensitivity and suggest that diet-induced improvements in insulin sensitivity are not strictly derived from dietary effects on energy balance.—Wanders, D., Burk, D. H., Cortez, C. C., Van, N. T., Stone, K. P., Baker, M., Mendoza, T., Mynatt, R. L., Gettys, T. W. UCP1 is an essential mediator of the effects of methionine restriction on energy balance but not insulin sensitivity.
Keywords: adipose tissue, amino acid, FGF21, housing temperature, obesity
Dietary methionine restriction (MR) produces an integrated series of metabolic and physiologic responses that develop quickly after introduction of the diet and maintain the improvements in biomarkers of metabolic health for as long as the diet is consumed (1–3). The most prominent physiologic responses are coordinated increases in energy intake and expenditure (2), with the larger effect on energy expenditure (EE) limiting ongoing fat deposition by increasing the proportion of total energy intake required for maintenance of existing tissue. When the decrease in net energy available for growth is integrated over time, it significantly limits the normal age-associated expansion of adipose tissue (4, 5). The diet also increases in vivo insulin sensitivity through a combination of direct and indirect effects of MR on liver, adipose tissue, and muscle (6). These mechanisms notwithstanding, improvements in overall insulin sensitivity are predicted to accrue in part from diet-induced reductions in adiposity. However, the extent to which increased EE and reductions in adiposity are required for diet-induced improvements in insulin sensitivity are not known.
Dietary MR increases EE soon after its introduction by mimicking many of the responses observed during thermoregulatory thermogenesis. For example, dietary MR produces a rapid increase in Ucp1 (uncoupling protein 1) mRNA and protein expression in brown adipose tissue (BAT) while simultaneously remodeling the morphology of white adipose tissue (WAT) (1, 2). Although the magnitude of these changes is depot specific, their overall impact on thermogenic activity is most evident at night, when a 2-fold higher heat increment of feeding is observed in the MR group (2). This amplified increase in core temperature is temporally linked to an exaggerated increase in nocturnal EE, suggesting that induction and activation of UCP1 plays a key role in mediating the effects of MR on EE (2). In addition, the increase in EE and induction of Ucp1 expression by MR are dependent on β-adrenergic signaling (7), consistent with a mechanism involving diet-induced activation of the sympathetic nervous system (SNS). Together, these findings make a compelling case that dietary MR regulates the induction and activation of UCP1 by increasing SNS activity, but a key remaining question is whether these changes in UCP1 are essential to the diet-induced increase in EE.
Mice lacking Ucp1 are able to engage alternative thermogenic mechanisms when cold stressed (8–10) but are also differentially responsive to changes in housing temperature in the sense that they are more prone to developing obesity than wild-type (WT) mice when housed at thermoneutrality but not standard housing temperatures (22–23°C) (11). It is well established that rearing mice under standard housing temperatures produces significant activation of nonshivering thermogenesis through SNS-dependent norepinephrine turnover in BAT and WAT (12–15). The increased energy required to defend body temperature and weight at 23°C is provided by a commensurate increase in energy intake and EE (15–17). Given that dietary MR may also utilize the SNS as a motor arm to increase EE at 23°C, the strategy of the present work was to incorporate Ucp1 loss of function into an experimental design that also modulates SNS activity by varying housing temperature. Using Ucp1−/− mice housed at temperatures that either stimulate (23°C) or minimize (28°C) SNS activity, we report here that dietary MR increases EE and reduces fat deposition under both conditions and requires UCP1 to do so. In contrast, diet-induced enhancement of in vivo insulin sensitivity is fully intact in the absence of UCP1.
MATERIALS AND METHODS
Animals and diets
All vertebrate animal experiments were reviewed and approved by the Pennington Institutional Animal Care and Use Committee using guidelines established by the National Research Council, the Animal Welfare Act, and the PHS Policy on humane care and use of laboratory animals. The animals used in all experiments were male C57BL/6J mice obtained from Jackson Labs (Bar Harbor, ME, USA) at 4 weeks of age or age-matched male C57BL/6J Ucp1−/− from our breeding colony. All mice were adapted to the control diet for 7 days before assignment to dietary groups in each study. The feeding paradigm and diets were as described previously (7), with the control diet containing 0.86% methionine and the MR diet containing 0.17% methionine. The diets were formulated as extruded pellets and the energy content of both control and MR diets was 15.96 kJ/g, with 18.9% of energy coming from fat (corn oil), 64.9% carbohydrate, and 14.8% from a custom mixture of l-amino acids. Diets and water were provided ad libitum, and lights were on from 7 am to 7 pm. Housing temperatures were either 23°C or 28°C as described for specific experiments below.
Experiment 1
Age-matched wild-type (WT; e.g., Ucp1+/+) and Ucp1−/− mice adapted to the control diet were randomly assigned in equal numbers to housing temperatures of 23°C or 28°C. After adaption to the respective housing temperatures for 1 week, half the mice of each genotype were randomly assigned to receive the MR diet while the remaining mice continued to receive the control diet. The 2 housing temperatures were employed to evaluate the effects of each diet on each genotype under conditions designed to produce mild cold stress (23°C) or conditions near thermoneutrality (28°C) to minimize SNS activity (18). All mice were singly housed in shoebox cages with corncob bedding, and 12 mice per genotype were assigned to the 4 groups as follows: control diet at 23°C, control diet at 28°C, MR diet at 23°C, or MR diet at 28°C. The mice received their diets for 11 weeks, after which 4 mice from each genotype × diet × temperature combination were anesthetized and perfused with 10% neutral buffered formalin. Tissues from these mice were processed, embedded in paraffin, and used for histologic analysis. Tissues and serum were collected from the remaining mice (n = 7–8) in each genotype × diet × temperature combination.
Indirect calorimetry
EE was measured after mice (n = 7–8 from each genotype × diet × temperature combination) had been on the respective diets for 8 weeks using a Comprehensive Laboratory Animal Monitoring System (Columbus Instruments, Columbus, OH, USA). Power analyses suggested that 8 subjects would be required for these studies, as determined using the variance in our primary variables of interest at an effect size of 0.8 and an α level of 0.05. Power calculations were determined using SAS for Windows software (version 9.1; SAS Institute, Cary, NC, USA). The animal numbers suggested by the power analysis to be used in each group also coincides with our experience for the detection of differences in the majority of variables we would be interested in. It has been suggested that additional replication is required when using ANCOVA, particularly when comparing animals of similar size and composition (19). However, it was also noted that small sample size is not a valid reason to avoid ANCOVA, because if the study is insufficiently powered to detect treatment differences with ANCOVA, it will also be underpowered for analyses by other statistical approaches (20).
Body composition of each mouse was measured by NMR before entry and upon exit from the metabolic chambers. Mice were acclimated in the metabolic chambers for 24 hours before continuous measurement of O2 consumption (Vo2), CO2 production (Vco2), energy intake, and voluntary activity at 16 minute intervals for 72 hours. EE was calculated as {Vo2 × [3.815 + (1.232 × respiratory exchange ratio)] × 4.019}, while respiratory exchange ratios were calculated as the ratio of Vco2 produced to Vo2 consumed. Group differences in EE (kJ/mouse/h) were compared by ANCOVA (JMP Statistical Software, version 11; SAS Institute), calculating least squares means that account for variation in EE attributable to differences in lean mass, fat mass, activity, and energy intake among the mice. The least squares means ± sem for each genotype, diet, and temperature were compared by 3-way analysis of variance, and the significance of the model effects were compared by residual variance within ANCOVA.
Experiment 2
The second experiment used hyperinsulinemic–euglycemic clamps conducted on cohorts of WT and Ucp1−/− mice (n = 6–9 from each genotype × diet combination) by the Vanderbilt Mouse Metabolic Phenotyping Center (MMPC, Nashville, TN, USA). WT and Ucp1−/− mice were shipped to the Vanderbilt facility at 4 weeks of age. The animals were adapted to the control diet for 1 wk, then randomly assigned to receive either the control or MR diet for a period of 9 weeks before the clamp procedure. Animals were provided the diets and water ad libitum and housed at standard room temperature of (23°C) on a 12 h light cycle. One week before the clamp procedure, catheters were surgically placed in the carotid artery and jugular vein for sampling and infusions, respectively. Mice were withheld food for 5 hours before conducting the clamp procedure, as described previously (21). Erythrocytes were replaced to prevent a decline in hematocrit that occurs with repeated blood sampling. A primed (1.5 μCi) continuous (0.075 μCi/min) [3-3H]glucose infusion was started at −20 min. The clamp was initiated at 0 min with a continuous insulin infusion (2.5 mU/kg/min) that was maintained for 145 min. Arterial glucose was measured at 10 min intervals to provide feedback to adjust the glucose infusion rate (GIR) containing [3-3H]glucose as needed to clamp glucose concentration and specific activity. [3-3H]Glucose kinetics were determined at −10 min and at 10 min intervals between 80 and 120 min because insulin action is in a steady state by this interval. Plasma insulin was measured at −10 min, 100 min, and 120 min during the procedure. A 13 µCi intravenous bolus of 2[14C]deoxyglucose was administered at 120 min and used to determine the glucose metabolic index (Rg), an indication of tissue-specific glucose uptake. Blood samples were collected at 2, 5, 10, and 25 min after injection to measure disappearance of 2[14C]deoxyglucose from plasma. Tissues were collected to assess tissue specific Rg. Whole-body glucose appearance and endogenous glucose production (endo Ra), a measure of hepatic glucose production, were calculated as previously described (21, 22).
Immunoblotting of UCP1
Mitochondrial extracts were prepared from BAT homogenates, and tissue lysates were prepared from inguinal white adipose tissue (IWAT). UCP1 expression was determined by Western blot analysis using an affinity-purified antibody raised against the peptide sequence corresponding to amino acids 145 to 159 in mouse UCP1 (23). The respective blots for UCP1 in BAT and IWAT were reprobed for PDC-E2 (BAT) and β-actin (IWAT) as loading controls for the respective tissues. Detected proteins were quantitated using scanning laser densitometry and the relative expression of UCP1 versus the corresponding loading control for each tissue was calculated to test for diet-induced differences in expression at each rearing temperature. The β-actin antibody was from Sigma-Aldrich (St. Louis, MO, USA), and the PDC-E2 antibody was from Santa Cruz Biotechnology (Dallas, TX, USA).
RNA isolation and quantitative real-time PCR
Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA). One microgram of total RNA was reverse transcribed to produce complementary DNA. Gene expression was measured by RT-PCR (Applied Biosystems, Foster City, CA, USA) by measurement of SYBR Green. mRNA concentrations were normalized to cyclophilin expression.
Histology
Paraffin-embedded tissues from experiment 1 were sectioned at 5 µm and stained with hematoxylin and eosin (H&E). For adipocyte size measurements, H&E-stained slides were scanned on a Hamamatsu NanoZoomer, and the resulting image files (NDPI) were converted to equivalent ×5 magnification JPEG files using a conversion algorithm (http://www.matthias-baldauf.at/software/ndpi-converter/). The JPEG images were then batch processed within Fiji, an open-source platform for biologic image analysis (24), using minimal image processing before thresholding and subsequent identification/measurement of adipocytes using the “Analyze Particles” plug-in with fixed size/shape cutoffs. The software counted and measured all spherical cells on each slide from size 200 to 50,000 µm2. On average, over 32,000 adipocytes were counted and measured for each slide, with 2 slides per animal analyzed and 4 animals per treatment group.
To assess the extent of “browning” within IWAT, paraffin-embedded sections were dewaxed before heat-induced epitope retrieval in a pressure cooker for 20 min at 100°C in sodium citrate buffer (pH 6.0). Slides were blocked with normal goat serum (5%) in Tris-buffered saline and Tween-20 (TBST) buffer for 1 hour before incubation overnight with rabbit anti-perilipin (Cell Signaling Technology, Danvers, MA, USA). After washing in TBST and incubation with an Alexa Fluor–conjugated goat anti-rabbit secondary (Invitrogen, Carlsbad, CA, USA) for 1 hour, the slides were incubated with WGA-Oregon Green (20 µg/ml) and DAPI (500 nM) for 15 min. After washing briefly in TBST, the slides were mounted with aquamount (Thermo Fisher Scientific, Waltham, MA, USA) and subsequently scanned on a Hamamatsu NanoZoomer in fluorescence mode (DAPI/FITC/TxRed triple cube). The resulting images were cropped and exported as JPEGs at ×5 or ×10 resolution for analysis in Cell Profiler (http://www.cellprofiler.org/citations.shtml). Briefly, a pipeline was generated to identify cells containing both a green (cell membrane) and red component (perilipin), as nonadipocytes would lack an obvious perilipin signal. “Brownlike” cells were identified as those adipocytes that, in addition to the peripheral green ring of WGA in membranes, also had a critical level of perilipin staining within the cells that is characteristic of multilocular adipocytes.
Metabolomics analysis
A portion of inguinal WAT from control and Ucp1−/− mice housed at 23°C and fed the respective control and MR diets was sent to Metabolon (Durham, NC, USA) for metabolomics analysis using gas chromatography–mass spectrometry and ultra-performance liquid chromatography–tandem mass spectrometry analytical platforms. Nonesterified fatty acids were extracted from the adipose tissue without saponification using a methanol extraction solution. The quality control analysis included several technical replicate samples that were created from a homogeneous pool containing a small amount of all study samples. Instrument and process variability met Metabolon’s acceptance criteria, and a total of 257 metabolites were detected and quantified in the inguinal WAT samples. Metabolites associated with medium chain, long chain, branched chain, and polyunsaturated fatty acids were analyzed by a 2-way ANOVA to test for effects of genotype, diet, and genotype by diet interaction. Group means were compared using residual variance as the error term.
Serum analysis
Blood glucose concentrations were measured via tail nick following a 6-hour food withdrawal through use of a OneTouch UltraSmart glucometer. All other serum measurements were determined after a 4-hour food withdrawal upon completion of the study. Serum insulin and fibroblast growth factor 21 (FGF21) concentrations were measured with ELISA (Millipore, Billerica, MA, USA, and R&D Systems, Minneapolis, MN, USA, respectively).
Statistical analysis
Response variables in experiment 1 were analyzed by a 3-way ANOVA, with genotype, diet, and housing temperature as main effects. The analysis tested for genotype × diet, genotype × temperature, diet × temperature, and genotype × diet × temperature interactions using residual variance as the error term. Body weight, adiposity, energy intake, cell sizing data, cellular “browning” data, gene expression data, and the insulin clamp data were compared within each housing temperature using a 2-way ANOVA, and least squares means of each variable were compared using residual variance as the error term and making the Bonferroni correction. For metabolomics analysis, the data for each identified metabolite was analyzed using a 2-way ANOVA to test for effects of genotype, diet, and any genotype × diet interaction. Group differences in metabolite concentrations were then assessed relative to all other groups following log transformation and imputation of missing values, if any, with the minimum observed value for each compound. Protection against type I errors was set at 5%. GraphPad Prism software (La Jolla, CA, USA) was used to generate figures.
RESULTS
Effects of housing temperature and dietary MR on energy balance
To assess the role of UCP1 as a mediator of the effects of dietary MR on energy balance, EE was measured by indirect calorimetry in cohorts of WT and Ucp1−/− mice housed under standard conditions (e.g., 23°C) or within the thermoneutral zone at 28°C (17, 18). This approach was taken to deconflate the effects of dietary MR on EE from the effects of rearing temperature on EE and test whether induction and activation of UCP1 is essential to the ability of dietary MR to increase EE. ANCOVA was used to assess the impact of genotype, diet, and temperature on EE after adjusting for variation in total EE associated with variation in activity, lean mass, fat mass, and food intake of mice within each group. The relative contributions of model components in accounting for variation in total EE is shown as the t ratio of each variable’s impact on EE (Fig. 1). Although all model effects were significant, Fig. 1 shows that activity was a significant contributor to variation in EE among the mice. However, activity did not differ between the genotypes at either housing temperature or on either diet (Table 1), indicating that it is not the basis for either genotypic or diet-induced differences in EE. Temperature had a significant independent effect on EE such that irrespective of genotype, EE in mice on the control diet at 23°C was 30% to 40% higher than EE in mice at 28°C. Genotype and diet also had significant independent effects on EE (Fig. 1), and a significant genotype × diet interaction was detected based on the differential effects of dietary MR on EE in each genotype. For example, dietary MR produced a comparable 30% increase in EE in WT mice at both housing temperatures, while the diet had no effect on EE in Ucp1−/− mice at either temperature (Table 1).
Figure 1.

Assessment of components contributing to variation in EE. The relative contributions of model components in accounting for variation in total EE is shown as the t ratio of each variable’s impact on EE. EE was measured in mice (n = 7–8) by indirect calorimetry after consuming control (CON) or MR diets for 8 wk at 23°C or 28°C.
TABLE 1.
Energy balance parameters for WT and Ucp1−/− mice housed at 23°C or 28°C and fed control or methionine-restricted diets for 11 weeks
| Characteristic | Housing temperature |
|||||||
|---|---|---|---|---|---|---|---|---|
| 23°C |
28°C |
|||||||
| WT |
Ucp1−/− |
WT |
Ucp1−/− |
|||||
| Control | MR | Control | MR | Control | MR | Control | MR | |
| Initial body weight (g) | 16.0 ± 0.5a | 15.9 ± 0.5a | 13.7 ± 0.8a | 13.5 ± 0.7a | 15.9 ± 0.5a | 15.9 ± 0.5a | 13.6 ± 0.6a | 13.7 ± 0.7a |
| Final body weight (g) | 26.1 ± 1.2a | 22.8 ± 0.5b | 21.3 ± 0.5a | 15.5 ± 0.8b | 25.9 ± 0.4a | 21.5 ± 0.3b | 24.0 ± 0.5a | 13.6 ± 0.6b |
| Initial % adiposity | 15.6 ± 0.5a | 16.0 ± 0.6a | 20.8 ± 0.9b | 20.1 ± 0.7b | 15.6 ± 0.5a | 16.1 ± 0.4a | 20.1 ± 0.9b | 19.4 ± 1.0b |
| Final % adiposity | 18.4 ± 1.3a | 13.9 ± 0.7b | 18.0 ± 0.5a | 23.3 ± 0.9b | 16.0 ± 0.4a | 13.6 ± 0.7a | 19.6 ± 0.6b | 20.4 ± 1.0b |
| Energy intake (kJ/d/mouse) | 41.0 ± 0.5a | 47.7 ± 0.6b | 36.9 ± 0.6c | 27.1 ± 0.8d | 31.0 ± 0.3c | 36.6 ± 1.1c | 29.8 ± 0.3c | 19.0 ± 0.5d |
| Energy intake (kJ/d/g) | 1.88 ± 0.03a | 2.27 ± 0.03b | 1.87 ± 0.02a | 1.89 ± 0.03a | 1.46 ± 0.02c | 1.90 ± 0.05a | 1.55 ± 0.02c | 1.47 ± 0.02c |
| Energy expenditure (kJ/h/mouse) | 1.66 ± 0.02a | 2.17 ± 0.01b | 1.68 ± 0.01a | 1.67 ± 0.01a | 1.02 ± 0.01c | 1.31 ± 0.01d | 1.16 ± 0.01e | 1.21 ± 0.02e |
| Total activity | 621 ± 50a | 621 ± 33a | 536 ± 49a | 440 ± 68a | 629 ± 46a | 661 ± 34a | 577 ± 34a | 448 ± 62a |
Energy expenditure was measured 8 weeks after initiating diets using indirect calorimetry and analyzed by analysis of covariance as described in the Materials and Methods using genotype, housing temperature, and diet as main effects and calculating least squares means using lean mass, fat mass, and activity as covariates. Body weight, adiposity, energy intake, and total activity were compared using a 3-way ANOVAs using genotype, housing temperature, and diet as main effects and testing for interactions. Least squares means were compared using residual variance as the error term and presented as means ± sem (n = 8–9/group). Means denoted with different superscripts (a–d) within housing temperatures differ at P < 0.05. Body composition was measured by NMR as described in the Materials and Methods, and percentage adiposity was calculated as [(fat mass/body weight) × 100]. The average food intake less spillage was determined over a 24-hour period each week in mice of each genotype on each diet and at each housing temperature, converted to kilojoules, and expressed per mouse or per unit of body weight. Means represent average daily energy intake over the entire study and were compared as described above.
The effect of temperature on energy intake in mice on the control diet was comparable between WT and Ucp1−/− mice because irrespective of genotype, mice housed at 23°C consumed 25% to 30% more food than mice housed at 28°C (Table 1). In contrast, the effect of diet on energy intake differed between the genotypes, creating a significant genotype × diet interaction. For example, WT mice on the MR diet increased energy intake by 16% and 18% at 23°C and 28°C, respectively, while Ucp1−/− mice on the MR diet reduced their energy intake by 36% at 23°C and 57% at 28°C (Table 1).
The combined effects of dietary MR on energy intake and expenditure at the 2 housing temperatures affected energy balance in WT mice in the predicted manner. For example, the diet-induced increase in EE in WT mice at both temperatures was larger than the corresponding diet-induced increases in energy intake. This translated into significant diet-induced reductions in accumulation of body weight and adiposity at both temperatures (Table 1). In contrast, the predominant effect of dietary MR in Ucp1−/− mice was to reduce energy intake without affecting EE. Despite the resulting negative energy balance, the Ucp1−/− mice on the MR diet had a surprising expansion (23°C) or retention (28°C) of adiposity at the 2 temperatures (Table 1). The Ucp1−/− mice consuming the MR diet gained little to no weight at either temperature over the course of the study, but when energy intake is expressed per unit body weight, weight-adjusted energy intake was the same as Ucp1−/− mice on the control diet (Table 1). This further supports the conclusion that dietary MR had no effect on EE in Ucp1−/− mice and affected energy balance solely through effects on energy intake. Collectively, these findings show that UCP1 is essential to the ability of dietary MR to increase EE, and rather than increase energy intake as expected, the absence of Ucp1 results in a paradoxical reduction in consumption of the diet.
Effects of housing temperature on UCP1 expression by dietary MR
It is well established that mice reared at 22°C to 23°C experience significant cold stress and increase SNS-dependent browning of WAT and expression of UCP1 in BAT to defend body temperature by adaptive thermogenesis (15, 17, 18, 25). Given that dietary MR is thought to use the SNS to increase UCP1 and EE through similar mechanisms (7), and that mice lacking Ucp1 may be more sensitive to thermal stress than WT mice (18, 25), the effects of genotype, diet, and housing temperature on thermogenic capacity were evaluated in BAT and WAT from the respective animals. In WT mice housed at 23°C, dietary MR produced a significant increase in Ucp1 mRNA and protein expression in both BAT (Figs. 2A, B) and IWAT (Figs. 2C, D). Compared to mice on the control diet at 23°C, housing at 28°C produced a significant reduction in Ucp1 mRNA in both BAT (Fig. 2A) and IWAT (Fig. 2C). A corresponding temperature-dependent decrease in UCP1 protein in the 2 tissues was not detected. However, in both BAT and IWAT from mice housed at 28°C, dietary MR produced a significant increase in Ucp1 mRNA and protein compared to controls (Fig. 2A–D). Together, these findings indicate that the effects of housing temperature and dietary MR on UCP1 expression are independent. Thus, although temperature plays an important role in matching UCP1 expression to environmental conditions, dietary MR retains the ability to independently increase UCP1 expression under both conditions.
Figure 2.

Effect of diet and housing temperature on UCP1 expression. Ucp1 gene and protein expression in BAT (A and B, respectively) and IWAT (C and D, respectively) from WT mice fed the control (CON) or MR diets housed at either 23°C or 28°C for 11 weeks. mRNA expression was normalized to cyclophilin. Relative band densities of UCP1 protein expression were normalized to PDC-E2 (BAT) or β-actin (IWAT). Means ± sem are presented. Different letters denote statistically significant differences.
Effects of UCP1 and housing temperature on remodeling of WAT
Mice lacking Ucp1 are able to engage alternative thermogenic mechanisms to defend body temperature when gradually adapted to cold (9, 25), but the extent to which standard housing conditions, relative to thermoneutrality, provoke adaptive changes in SNS-dependent activity and adipose tissue remodeling is unclear. Given the potential of SNS-dependent remodeling of adipose tissue to affect EE and glucose utilization, even in the absence of UCP1 (26), remodeling of adipose tissue by dietary MR was evaluated in WT and Ucp1−/− mice housed at 23°C and 28°C. Representative H&E stained sections of IWAT from each genotype, diet, and housing temperature are shown in Fig. 3A. Unbiased analysis of cell size illustrates a genotype × diet interaction at both temperatures (Fig. 3B). For example, dietary MR increased the number of small adipocytes in WT but not Ucp1−/− mice housed at 23°C, but at 28°C, dietary MR increased the number of small adipocytes in Ucp1−/− mice but not WT mice (Fig. 3B). In MR-fed Ucp1−/− mice at 28°C, this was complemented by a corresponding decrease in intermediate size adipocytes (Fig. 3B). Perhaps most interesting was the genotype × diet interaction at 23°C, where the number of small adipocytes in Ucp1−/− mice on the control diet was much higher than the number in WT mice (Fig. 3). Dietary MR produced no additional effects on cell size in Ucp1−/− mice housed at 23°C. These data suggest that IWAT has undergone significant remodeling in Ucp1−/− mice on control diet housed at 23°C compared to WT mice. In addition, the change in cell morphology between 28°C and 23°C is much more evident in Ucp1−/− mice compared to WT mice (Fig. 3A).
Figure 3.
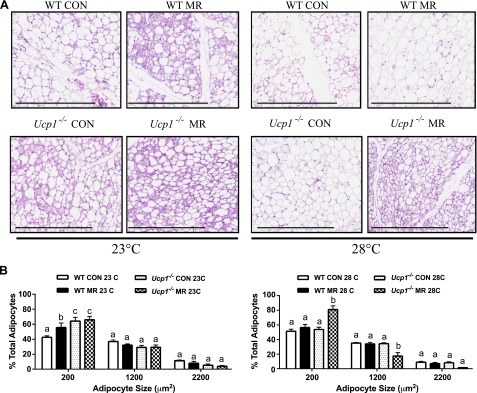
Effect of genotype, diet, and housing temperature on adipose tissue morphology. A) Representative H&E stains of IWAT from WT and Ucp1−/− mice fed the control (CON) or MR diets housed at either 23°C or 28°C for 11 weeks. Scale bars, 400 µm. B) Adipocyte sizes (area per adipocyte, µm2) in control (CON) or MR-fed WT and Ucp1−/− mice graphed as percent of total adipocytes. X axis indicates bin center with bin sizes of 1000. Means ± sem are presented for each variable and are based on n = 4 per group. Different letters denote statistically significant differences within each adipocyte size bin.
Remodeling of IWAT was evaluated using a second imaging-based approach to detect changes in the number of multilocular cells within the depot. Figure 4 shows that dietary MR produced a significant 3-fold increase in the percentage of multilocular adipocytes within IWAT of WT mice at 23°C, but less than a 2-fold increase in WT mice housed at 28°C. Ucp1−/− mice on the control diet at 23°C had a 4-fold greater percentage of multilocular adipocytes in IWAT compared to WT mice, and not surprisingly, dietary MR produced only a modest further increase in remodeling at this temperature (Fig. 4). Housing Ucp1−/− mice on the control diet at 28°C resulted in a reciprocal decrease in the percentage of multilocular adipocytes to levels similar to WT mice housed at 23°C (Fig. 4). Dietary MR produced a 3-fold increase in percentage of multilocular adipocytes in IWAT of Ucp1−/− mice at 28°C (Fig. 4).
Figure 4.
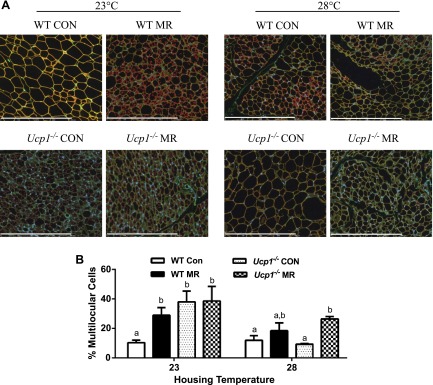
Effect of genotype, diet, and housing temperature on browning of WAT. A) Representative images of IWAT from WT and Ucp1−/− mice fed the control (CON) or MR diets housed at either 23°C or 28°C for 11 weeks. Scale bars, 400 µm. Cell membranes were stained with wheat germ agglutinin (green), lipid droplet membranes were stained with perilipin (red), and nuclei were stained with DAPI (blue). B) Relative number of multilocular, brownlike adipocytes graphed as percentage of total adipocytes. Means ± sem are presented for each variable and are based on n = 4 per group. Different letters denote statistically significant differences within each housing temperature.
Together, these data illustrate the involvement of a complex but unexpected genotype × temperature interaction in IWAT morphology. On the basis of the involvement of FGF21 in remodeling of WAT (27–29) and the demonstration of its regulation by dietary MR (6), Fgf21 expression was evaluated to test for a potential role in this interaction. Figure 5 shows that in mice on both the control and MR diets, hepatic Fgf21mRNA and serum FGF21 levels were significantly higher in Ucp1−/− than WT mice at both housing temperatures. Dietary MR also produced a greater absolute increase in serum FGF21 levels in Ucp1−/− mice compared to WT mice, but the fold increase in serum FGF21 produced by MR was comparable between the genotypes at both temperatures (Fig. 5B). Although not significant, there was a strong suggestion of a temperature-dependent increase in both hepatic mRNA and serum FGF21 in WT mice between 23°C and 28°C, irrespective of diet (Fig. 5). In contrast, temperature had no effect on serum FGF21 levels of Ucp1−/− mice on either diet. These data are suggestive of a potential role for FGF21 in the exaggerated remodeling of IWAT in Ucp1−/− mice housed at 23°C, although the reversion of adipocyte morphology to a more unilocular phenotype when these mice were housed at 28°C, despite their elevated FGF21, may be indicative of a requirement for coincident SNS input. The intense remodeling of IWAT by MR in Ucp1−/− mice at 28°C is consistent with this suggestion, although future studies will be required to address this possibility in detail.
Figure 5.

Effect of genotype, diet, and housing temperature on FGF21. Hepatic Fgf21 mRNA expression (A) and serum concentrations (B) from WT and Ucp1−/− mice fed the control (CON) or MR diets housed at either 23°C or 28°C for 11 weeks. mRNA expression was normalized to cyclophilin. Serum samples were collected after a 4-hour food withdrawal. Means ± sem are presented for each variable and based on n = 7–8 per group. Different letters denote statistically significant differences.
Effects of genotype and diet-dependent remodeling of WAT on fatty acid composition
To assess the impact of genotype- and diet-dependent remodeling of IWAT on lipid composition at 23°C, targeted analysis of medium chain, long chain, branched chain, and polyunsaturated nonesterifed fatty acids was conducted in this depot. Figure 6 shows a heat map of the genotype- and diet-dependent effects on each metabolite, along with tests for main effect interactions. The highly significant degree of remodeling of IWAT noted in Ucp1−/− mice on the control diet compared to WT mice (Figs. 3 and 4) is mirrored by significant increases in 2 of 4 medium chain fatty acids, 12 of 15 long chain fatty acids, 6 of 11 polyunsaturated fatty acids, and 1 of 2 branched chain fatty acids in this group. In contrast, dietary MR produced essentially no further increase in remodeling of IWAT in Ucp1−/− mice (Fig. 4B) or in fatty acid metabolites (Fig. 6). The most profound impact on fatty acid metabolism was seen in WT mice on the MR diet compared to the control diet, where only 6 of 32 metabolites were unaffected by the diet (Fig. 6). In this comparison, it is particularly noteworthy that every long chain fatty acid in our profile was significantly increased by dietary MR (Fig. 6). The increase in free fatty acid content of the IWAT may be indicative of increased lipolysis. We have previously shown that MR increases fatty acid oxidation in IWAT (1). We would also expect increased de novo lipogenesis, which occurs at a higher rate in beige adipocytes compared to white (30), but given that lipid synthesis and metabolism are constantly cycling, these data provide a snapshot of the alteration in lipid metabolism induced by the MR diet and genetic deletion of Ucp1. Overall, these data provide strong evidence that the genotypic and dietary effects on remodeling of IWAT resulted in profound changes in fatty acid metabolism within this depot.
Figure 6.

Group contrasts are represented as mean ratios, with red- and green-shaded cells indicating significant (P ≤ 0.05) increases (red) or decreases (green) in the mean ratios. Cells shaded with light red and light green indicate that the mean ratios were trending (e.g., 0.05 < P < 0.10) higher (light red) or lower (light green) for that comparison. For ANOVA of main effects, blue-shaded cells indicate P ≤ 0.05, while light blue-shaded cells indicate 0.05 < P < 0.10.
Effects of temperature, genotype, and diet on fasting insulin and glucose
In mice housed at 23°C, there were no meaningful differences in fasting glucose among the groups, although a clear suggestion of a glucose lowering effect of MR in both genotypes was evident (Table 2). Both glucose and fasting insulin were lower in Ucp1−/− mice on the control diet compared to WT mice on the same diet (Table 2). Dietary MR reduced fasting insulin in both genotypes, but only the decrease in WT mice reached statistical significance. Housing at 28°C versus 23°C had essentially no effect on fasting insulin in WT mice on the control diet and no effect on the ability of MR to reduce insulin levels in this genotype (Table 2). In contrast, housing Ucp1−/− mice on the control diet at 28°C increased fasting insulin to levels observed in WT mice at 23°C (Table 3). Despite this increase, MR reduced fasting insulin by 4-fold in Ucp1−/− mice housed at 28°C (Table 2).
TABLE 2.
Fasting glucose and insulin concentrations in WT and Ucp1−/− mice housed at 23°C or 28°C and fed control or methionine-restricted diets for 11 weeks
| Characteristic | Housing temperature |
|||||||
|---|---|---|---|---|---|---|---|---|
| 23°C |
28°C |
|||||||
| WT |
Ucp1−/− |
WT |
Ucp1−/− |
|||||
| Control | MR | Control | MR | Control | MR | Control | MR | |
| Glucose (mg/dl) | 209 ± 14b | 189 ± 7b,c | 185 ± 5b,c | 160 ± 11c | 177 ± 9b,c | 166 ± 14c | 148 ± 5c | 90 ± 10d |
| Insulin (ng/ml) | 1.3 ± 0.2b | 0.37 ± 0.1c | 0.47 ± 0.1c | 0.26 ± 0.1c | 1.3 ± 0.2b | 0.42 ± 0.1c | 1.4 ± 0.3b | 0.31 ± 0.1c |
Blood glucose was measured after a 6-hour food withdrawal after 10 weeks on diet; serum insulin was measured after 11 weeks on diet after a 4-hour food withdrawal. The serum parameters were compared by a 3-way analysis of variances using genotype, housing temperature, and diet as main effects and testing for interactions. Least squares means were compared using residual variance as the error term and presented as means ± sem (n = 8/group). Means denoted with different superscripts (a–d) differ at P < 0.05.
Effects of genotype and diet on in vivo insulin sensitivity
To assess the effects of genotype and dietary MR on insulin sensitivity in mice housed at 23°C, hyperinsulinemic–euglycemic clamps were employed after mice received the control or MR diets for 8 weeks. The GIRs needed to establish euglycemia after initiation of insulin infusion at time 0 were achieved within 40 minutes in all 4 treatment groups, and steady-state glucose levels were maintained during the final hour of the clamp across the groups (Fig. 7A). The GIRs needed to establish steady-state glucose levels were essentially identical in WT and Ucp1−/− mice on the control diet (Fig. 7A), despite the significantly lower fasting insulin levels detected in Ucp1−/− mice on the control diet at 23°C (Table 2). The steady-state GIR needed to maintain euglycemia was 2.2-fold higher in mice on the MR diet, regardless of genotype (Fig. 7A). Thus, the absence of Ucp1 affected neither overall insulin sensitivity nor the ability of the mice to increase sensitivity in response to dietary MR. However, further evaluation of tissue-specific responses to insulin revealed several significant differences between genotypes. Liver glucose metabolism was assessed using 3-[3H]-glucose infusion to calculate basal endo Ra and compare insulin-dependent suppression of endo Ra during the hyperinsulinemic–euglycemic clamp. Suppression of hepatic glucose production by insulin was comparable in WT and Ucp1−/− mice on the control diet, and MR enhanced the efficacy of insulin to suppress hepatic glucose production in both genotypes (Fig. 7B). The Rg provides a direct measure of insulin sensitivity in each tissue, and it showed that MR produced significant increases in uptake of [14C]2-deoxyglucose (2-DG) in gastrocnemius muscle (2.6-fold), vastus lateralis muscle (3.1-fold), epididymal white adipose tissue (3.6-fold), IWAT (8.3-fold), BAT (1.5-fold), and heart (2-fold) of WT mice (Fig. 7C, D). The responses of Ucp1−/− mice to MR were similar in many respects but fundamentally different in others. For example, MR increased Rg in gastrocnemius muscle and epididymal white adipose tissue to the same degree in Ucp1−/− and WT mice, while in vastus lateralis, IWAT, BAT, and heart, the effect of MR in Ucp1−/− mice did not reach significance (Fig. 7C, D). Particularly noteworthy was the diet-independent effect of genotype on Rg in IWAT, where insulin-dependent [14C]2-DG uptake in Ucp1−/− mice on the control diet was 19-fold higher than uptake in WT controls (Fig. 1C). Therefore, it is not surprising that dietary MR produced no further increase in insulin sensitivity here. In other tissues in Ucp1−/− mice where MR failed to increase Rg, there was a noticeable effect of genotype on [14C]2-DG uptake that was not attributable to diet (Fig. 7C, D). These tissue-specific differences notwithstanding, overall insulin sensitivity was comparable between genotypes, and dietary MR produced an almost identical increase in insulin-dependent GIR between the genotypes.
Figure 7.

Hyperinsulinemic-euglycemic clamps in WT and Ucp1−/− mice housed at 23°C after 9 weeks of dietary MR. The clamp procedures were conducted as described in the Materials and Methods. A) GIR required to maintain euglycemia during the insulin clamps. B) The ability of insulin to suppress hepatic glucose production (% suppression of endo Ra) during the clamp procedure. Different letters denote statistically significant differences. C) Rg, an indication of tissue-specific insulin-dependent glucose uptake. Means ± sem are presented for each variable and are based on n = 6–9 per group. *Means differ from mice of the same genotype fed the control (CON) diet at P < 0.05.
DISCUSSION
Previous studies establish that dietary MR produces a coordinated series of biochemical and physiologic responses that develop soon after introduction of the diet and persist for as long as the diet is consumed (3, 7, 31–34). The biologic significance of these effects in aggregate is substantial, resulting in animals that are leaner, that are more insulin sensitive, and that live longer (1, 2, 35–37). An important unresolved question is whether the effects of dietary MR on overall insulin sensitivity are the result of and dependent on the effects of the diet on energy balance and adiposity. The present work sought to address this question while also assessing the importance of UCP1 in the mechanistic effects of the MR diet on EE. Using an experimental approach designed to separate environmental and dietary inputs to transcriptional regulation of Ucp1, the most significant findings of this work are that UCP1 is essential to the ability of dietary MR to increase EE but not to its ability to increase insulin sensitivity. A potential caveat is that energy intake was unexpectedly decreased by dietary MR in Ucp1−/− mice opposite from the hyperphagic response normally produced by the diet (7). Thus, even in the absence of a dietary effect on EE in Ucp1−/− mice, the reduction in energy intake was sufficient to limit growth. More surprisingly, despite near cessation of growth in Ucp1−/− mice on the MR diet, fat deposition was preserved at the expense of lean tissue. By study’s end, this unexpected pattern of nutrient partitioning resulted in Ucp1−/− mice that were significantly smaller but had greater adiposity than WT mice on the MR diet. Despite these differences in body composition, we found no evidence that dietary MR affected EE in Ucp1−/− mice at either housing temperature. This is fully consistent with our data showing that decreased body weight in Ucp1−/−mice on the MR diet was fully accounted for by the observed reductions in energy intake. These findings point to an unexpected role for UCP1 in the hyperphagic response to dietary MR. One possibility is that the hyperphagic response is compensatory and the result of the UCP1-dependent increase in EE. In such a scenario, the absence of a diet-induced increase in EE in Ucp1−/− mice would remove the signal for a compensatory increase in energy intake. However, there is no direct evidence to support such a mechanism, and it remains unclear how the absence of a diet-induced increase in EE could trigger the observed negative effect on energy intake. Alternatively, recent work suggests that UCP1 is involved in the anorexigenic effects of leptin (38) based on findings that exogenous leptin was less able to reduce food intake in Ucp1−/− compared to WT mice. Previous work has shown that dietary MR significantly lowers circulating leptin by reducing its expression in adipose tissue (2), but dietary MR did not lower leptin mRNA in BAT or IWAT of Ucp1−/− mice here (data not shown). This finding notwithstanding, the observed negative effect of the diet on energy intake suggests the involvement of an additional mechanism beyond the absence of the normal hyperphagic leptin signal generated by MR in WT mice.
Mice perceive mild cold stress at normal housing temperatures (e.g., 23°C) and respond by increasing UCP1-dependent thermogenesis in BAT and WAT (15, 17). The associated expansion of thermogenic capacity in each site is regulated by norepinephrine from sympathetic nerves innervating the respective fat depots. The intensity of SNS-dependent recruitment and activation of UCP1 in BAT and WAT is regulated along a thermal continuum, with mice housed in the thermoneutral zone (28°C to 36°C) having little need to activate thermogenesis, while housing at 4°C requires maximal recruitment and mobilization of thermogenic capacity for survival (18, 25). As recently noted by Nedergaard and Cannon (15), changes in the integrity of an animal’s insulation (e.g., hair, skin) can enhance heat loss at temperatures below thermoneutrality, amplifying the perception of cold and necessitating a greater than normal thermogenic response to defend body temperature. This raises the question of whether the absence of the critical thermoregulatory molecule, UCP1, intensifies the perception of cold in Ucp1−/− mice such that normal housing temperatures produce a compensatory increase in SNS stimulation of adipose tissue. Such a view is supported by the extensive remodeling observed in inguinal WAT of Ucp1−/− mice housed at 23°C compared to WT controls ((10) and this study). Additionally, the higher FGF21 levels in Ucp1−/− mice at 23°C could be acting as an endocrine enhancer of browning in WAT (28, 29). Recent work proposes that FGF21 also acts centrally to induce SNS activity (39), providing a potential mechanism for FGF21 to act through a combination of direct effects on WAT and indirectly through centrally mediated SNS-dependent recruitment of thermogenic activity. We recently proposed that the MR-induced increase in hepatic Fgf21 expression serves as an endocrine signal linking the direct effects of MR in the liver to increased insulin sensitivity and glucose uptake in WAT (6). Thus, it seems possible that FGF21 could be a critical link between MR’s effects on adipose tissue remodeling and enhanced insulin sensitivity.
The present findings make a compelling case that housing temperature and dietary MR interact to produce genotype-specific patterns of remodeling within the IWAT depot. Housing Ucp1−/− mice at 23°C alone is sufficient to produce extensive remodeling of the depot, leaving little additional effect of dietary MR under these conditions. Housing Ucp1−/− mice at 28°C causes a reversion of adipocyte morphology to large unilocular cells, while providing them the MR diet under these conditions restores its efficacy to increase the number of small, multilocular, beige/brown adipocytes within the depot. An important question is the extent to which the remodeling impacts the observed metabolic phenotype within the context of each genotype. Ex vivo and in vivo data from IWAT in mice housed at 23°C imply that both genotype and diet had significant effects on fatty acid metabolism and glucose uptake under these conditions. For example, in the highly remodeled IWAT from Ucp1−/− mice on the control diet, both insulin-dependent glucose uptake and fatty acid metabolism were significantly enhanced compared to WT controls. This is consistent with recent reports that browned WAT displays enhanced basal glucose uptake and has altered lipid metabolism (30, 40). Interestingly, no additional effect of dietary MR on adipocyte morphology, glucose uptake, or fatty acid metabolism was discernable in Ucp1−/− mice housed at 23°C. The extensive remodeling of IWAT in Ucp1−/− mice at 23°C may be the result of a compensatory intensification of SNS activity, but the unexpectedly higher FGF21 levels in these mice could also be a contributing factor. However, the failure of the elevated FGF21 in Ucp1−/− mice housed at 28°C to effect IWAT remodeling, coupled with the highly effective remodeling produced by dietary MR in these mice suggests that SNS activity is the primary regulator of browning in this experimental context. Compared to Ucp1−/− mice, WT mice at 23°C on the control diet showed little evidence of remodeling of IWAT, with adipocytes retaining a unilocular morphology. Dietary MR was highly effective in these mice, increasing the number of small multilocular adipocytes with increased thermogenic capacity, increased capacity to metabolize fatty acids, and enhanced capacity to take up glucose. Considered together, the data provide compelling evidence that the remodeling of IWAT by dietary MR has a significant physiologic impact in each genotype that contributes to the resulting metabolic phenotype of the mice.
A key objective of the present studies was to determine whether the ability of dietary MR to influence insulin sensitivity was dependent on its ability to increase EE and limit fat deposition. A loss of function approach showed that UCP1 was essential to the ability of dietary MR to increase EE but not to its ability to enhance insulin sensitivity. That conclusion is supported by the identical improvements in overall insulin sensitivity that were produced by dietary MR in WT and Ucp1−/− mice. In recent work, we proposed that dietary MR enhances insulin sensitivity through a combination of direct effects in the liver to amplify insulin signaling and indirect effects in adipose tissue through MR-dependent increases in hepatic transcription and release of FGF21 (6). The diet-induced increases in both basal and insulin-dependent glucose uptake in isolated brown and white adipocytes (6) are consistent with established effects of FGF21 on glucose transport in human primary adipocytes (27). On the basis of recent studies showing that FGF21 is also involved in browning of WAT (28) while acting centrally to regulate SNS activity (39), the higher levels of FGF21 (basal and MR induced) may be important to retention of the insulin sensitizing effects of MR in Ucp1−/− mice. The greater remodeling of IWAT in Ucp1−/− mice and the higher Rg in this tissue are consistent with this suggestion. Although the complex effects of Ucp1’s absence on temperature sensitivity, SNS-dependent remodeling among fat depots, and hepatic FGF21 production are poorly understood, the current results provide compelling evidence that the ability of dietary MR to enhance in vivo insulin sensitivity are not compromised by its absence.
The potential to translate the reported benefits of dietary MR from preclinical studies to the clinic was assessed in an initial proof of concept study in subjects with metabolic syndrome (41). The approach involved elimination of meat, poultry, dairy, and grains from the diet and replacement of 100% of the daily protein requirement with the commercial food, Hominex-2, a semisynthetic mixture of l-amino acids low in cystine and lacking methionine. Hominex-2 was developed for children with pyridoxine-insensitive homocystinemia or hypermethionemia. The poor palatability of Hominex-2 may have led to poor compliance, because the reduction in plasma methionine in the MR group was only 14% (41). This limitation notwithstanding, dietary MR increased 24-hour fat oxidation and reduced hepatic lipid content. Thus, preexisting obesity and insulin resistance did not prevent subjects from responding to the MR diet, although their responses were clearly not as robust as observed in preclinical studies. We believe that the presence of cysteine in Hominex-2 may provide the key to the small reductions in plasma methionine and modest physiologic responses to the diet. The methionine-sparing effect of cysteine is well documented (42), so the rodent MR diets are formulated without cysteine. Recent work has shown that adding back relatively high amounts of cysteine (e.g., 0.5%) to the MR diet completely reversed its beneficial metabolic effects (43). Moreover, recent unpublished studies in our lab found that adding back as little as 0.2% cysteine to the MR diet reversed all but the direct transcriptional effects of MR on specific hepatic genes associated with de novo lipogenesis (e.g., stearoyl-CoA desaturase-1). This could explain why the presence of cysteine in Hominex-2 limited the effects of the diet to those on hepatic lipid metabolism (41). Viewed together, these findings argue that successful implementation of dietary MR in a clinical setting would require both restricting methionine to a specific range and eliminating cysteine. Thus, the keys to using MR as a dietary approach to treat metabolic disease will involve developing palatable foods that eliminate cysteine and provide methionine within the defined range shown to be biologically effective.
Acknowledgments
The authors thank C. Tramonte for administrative support. This work was supported in part by the American Diabetes Association 1-12-BS-58 (T.W.G.), U.S. National Institutes of Health (NIH) Grants DK096311 (to T.W.G.), DK098687 (to R.L.M.), DK089641 (to R.L.M.), and the Mouse Metabolic Phenotyping Center Consortium (NIH U24 DK059637). This work also made use of the Genomics and Cell Biology and Bioimaging core facilities supported by NIH P20 GM103528 (to T.W.G.) and NIH 2P30 DK072476. This research project used the Transgenic and Animal Phenotyping core facilities that are supported in part by the NIH Nutrition Obesity Research Centers Grant 2P30 DK072476) (to R.L.M.). D.W. is supported by NIH National Research Service Award 1 F32 DK098918. The authors thank M. Orgeron, K. Dille, and A. Pierse for their technical support.
Glossary
- 2-DG
2-deoxyglucose
- BAT
brown adipose tissue
- EE
energy expenditure
- endo Ra
endogenous glucose production
- FGF21
fibroblast growth factor 21
- GIR
glucose infusion rate
- H&E
hematoxylin and eosin
- IWAT
inguinal white adipose tissue
- MR
methionine restriction
- Rg
glucose metabolic index
- SNS
sympathetic nervous system
- TBST
Tris-buffered saline and Tween-20
- UCP1
uncoupling protein 1
- WAT
white adipose tissue
- WT
wild-type
REFERENCES
- 1.Hasek B. E., Boudreau A., Shin J., Feng D., Hulver M., Van N. T., Laque A., Stewart L. K., Stone K. P., Wanders D., Ghosh S., Pessin J. E., Gettys T. W. (2013) Remodeling the integration of lipid metabolism between liver and adipose tissue by dietary methionine restriction in rats. Diabetes 62, 3362–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hasek B. E., Stewart L. K., Henagan T. M., Boudreau A., Lenard N. R., Black C., Shin J., Huypens P., Malloy V. L., Plaisance E. P., Krajcik R. A., Orentreich N., Gettys T. W. (2010) Dietary methionine restriction enhances metabolic flexibility and increases uncoupled respiration in both fed and fasted states. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R728–R739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malloy V. L., Krajcik R. A., Bailey S. J., Hristopoulos G., Plummer J. D., Orentreich N. (2006) Methionine restriction decreases visceral fat mass and preserves insulin action in aging male Fischer 344 rats independent of energy restriction. Aging Cell 5, 305–314 [DOI] [PubMed] [Google Scholar]
- 4.Munzberg, H., Henagan, T. M., and Gettys, T. W. (2013) Animal models of obesity: perspectives on evolution of strategies for the development and analysis of their phenotypes. In Handbook of Obesity, (Bray, G.A. and Bouchard, C., eds.), pp. 137–148, Informa Books, London [Google Scholar]
- 5.Blaxter K. L. (1986) Bioenergetics and growth: the whole and the parts. J. Anim. Sci. 63 (Suppl. 2), 1–103733583 [Google Scholar]
- 6.Stone K. P., Wanders D., Orgeron M., Cortez C. C., Gettys T. W. (2014) Mechanisms of increased in vivo insulin sensitivity by dietary methionine restriction in mice. Diabetes 63, 3721–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plaisance E. P., Henagan T. M., Echlin H., Boudreau A., Hill K. L., Lenard N. R., Hasek B. E., Orentreich N., Gettys T. W. (2010) Role of β-adrenergic receptors in the hyperphagic and hypermetabolic responses to dietary methionine restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R740–R750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enerbäck S., Jacobsson A., Simpson E. M., Guerra C., Yamashita H., Harper M. E., Kozak L. P. (1997) Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94 [DOI] [PubMed] [Google Scholar]
- 9.Ukropec J., Anunciado R. P., Ravussin Y., Hulver M. W., Kozak L. P. (2006) UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1−/− mice. J. Biol. Chem. 281, 31894–31908 [DOI] [PubMed] [Google Scholar]
- 10.Liu X., Rossmeisl M., McClaine J., Riachi M., Harper M. E., Kozak L. P. (2003) Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J. Clin. Invest. 111, 399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feldmann H. M., Golozoubova V., Cannon B., Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 [DOI] [PubMed] [Google Scholar]
- 12.Landsberg L., Saville M. E., Young J. B. (1984) Sympathoadrenal system and regulation of thermogenesis. Am. J. Physiol. 247, E181–E189 [DOI] [PubMed] [Google Scholar]
- 13.Ashwell M., Dunnett S. B. (1985) Fluorescent histochemical demonstration of catecholamines in brown adipose tissue from obese (ob/ob) and lean mice acclimated at different temperatures. J. Auton. Nerv. Syst. 14, 377–386 [DOI] [PubMed] [Google Scholar]
- 14.Ashwell M., Jennings G., Richard D., Stirling D. M., Trayhurn P. (1983) Effect of acclimation temperature on the concentration of the mitochondrial “uncoupling” protein measured by radioimmunoassay in mouse brown adipose tissue. FEBS Lett. 161, 108–112 [DOI] [PubMed] [Google Scholar]
- 15.Nedergaard J., Cannon B. (2014) The browning of white adipose tissue: some burning issues. Cell Metab. 20, 396–407 [DOI] [PubMed] [Google Scholar]
- 16.Zhao Z. J., Chi Q. S., Cao J., Han Y. D. (2010) The energy budget, thermogenic capacity and behavior in Swiss mice exposed to a consecutive decrease in temperatures. J. Exp. Biol. 213, 3988–3997 [DOI] [PubMed] [Google Scholar]
- 17.Cannon B., Nedergaard J. (2011) Nonshivering thermogenesis and its adequate measurement in metabolic studies. J. Exp. Biol. 214, 242–253 [DOI] [PubMed] [Google Scholar]
- 18.Gordon C. J. (2012) Thermal physiology of laboratory mice: defining thermoneutrality. J. Therm. Biol. 37, 654–685 [Google Scholar]
- 19.Butler A. A., Kozak L. P. (2010) A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes 59, 323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tschöp M. H., Speakman J. R., Arch J. R., Auwerx J., Brüning J. C., Chan L., Eckel R. H., Farese R. V. Jr., Galgani J. E., Hambly C., Herman M. A., Horvath T. L., Kahn B. B., Kozma S. C., Maratos-Flier E., Müller T. D., Münzberg H., Pfluger P. T., Plum L., Reitman M. L., Rahmouni K., Shulman G. I., Thomas G., Kahn C. R., Ravussin E. (2011) A guide to analysis of mouse energy metabolism. Nat. Methods 9, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayala J. E., Bracy D. P., McGuinness O. P., Wasserman D. H. (2006) Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55, 390–397 [DOI] [PubMed] [Google Scholar]
- 22.Berglund E. D., Li C. Y., Poffenberger G., Ayala J. E., Fueger P. T., Willis S. E., Jewell M. M., Powers A. C., Wasserman D. H. (2008) Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes 57, 1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Commins S. P., Watson P. M., Padgett M. A., Dudley A., Argyropoulos G., Gettys T. W. (1999) Induction of uncoupling protein expression in brown and white adipose tissue by leptin. Endocrinology 140, 292–300 [DOI] [PubMed] [Google Scholar]
- 24.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer C. W., Willershäuser M., Jastroch M., Rourke B. C., Fromme T., Oelkrug R., Heldmaier G., Klingenspor M. (2010) Adaptive thermogenesis and thermal conductance in wild-type and UCP1-KO mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R1396–R1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Granneman J. G., Burnazi M., Zhu Z., Schwamb L. A. (2003) White adipose tissue contributes to UCP1-independent thermogenesis. Am. J. Physiol. Endocrinol. Metab. 285, E1230–E1236 [DOI] [PubMed] [Google Scholar]
- 27.Kharitonenkov A., Shiyanova T. L., Koester A., Ford A. M., Micanovic R., Galbreath E. J., Sandusky G. E., Hammond L. J., Moyers J. S., Owens R. A., Gromada J., Brozinick J. T., Hawkins E. D., Wroblewski V. J., Li D. S., Mehrbod F., Jaskunas S. R., Shanafelt A. B. (2005) FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher F. M., Kleiner S., Douris N., Fox E. C., Mepani R. J., Verdeguer F., Wu J., Kharitonenkov A., Flier J. S., Maratos-Flier E., Spiegelman B. M. (2012) FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muise E. S., Souza S., Chi A., Tan Y., Zhao X., Liu F., Dallas-Yang Q., Wu M., Sarr T., Zhu L., Guo H., Li Z., Li W., Hu W., Jiang G., Paweletz C. P., Hendrickson R. C., Thompson J. R., Mu J., Berger J. P., Mehmet H. (2013) Downstream signaling pathways in mouse adipose tissues following acute in vivo administration of fibroblast growth factor 21. PLoS ONE 8, e73011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mottillo E. P., Balasubramanian P., Lee Y. H., Weng C., Kershaw E. E., Granneman J. G. (2014) Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic β3-adrenergic receptor activation. J. Lipid Res. 55, 2276–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orentreich N., Matias J. R., DeFelice A., Zimmerman J. A. (1993) Low methionine ingestion by rats extends life span. J. Nutr. 123, 269–274 [DOI] [PubMed] [Google Scholar]
- 32.Zimmerman J. A., Malloy V., Krajcik R., Orentreich N. (2003) Nutritional control of aging. Exp. Gerontol. 38, 47–52 [DOI] [PubMed] [Google Scholar]
- 33.Miller R. A., Buehner G., Chang Y., Harper J. M., Sigler R., Smith-Wheelock M. (2005) Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 4, 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun L., Sadighi Akha A. A., Miller R. A., Harper J. M. (2009) Life-span extension in mice by preweaning food restriction and by methionine restriction in middle age. J. Gerontol. A Biol. Sci. Med. Sci. 64, 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh S., Wanders D., Stone K. P., Van N. T., Cortez C. C., Gettys T. W. (2014) A systems biology analysis of the unique and overlapping transcriptional responses to caloric restriction and dietary methionine restriction in rats. FASEB J. 28, 2577–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richie J. P. Jr., Leutzinger Y., Parthasarathy S., Malloy V., Orentreich N., Zimmerman J. A. (1994) Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB J. 8, 1302–1307 [DOI] [PubMed] [Google Scholar]
- 37.Lees E. K., Król E., Grant L., Shearer K., Wyse C., Moncur E., Bykowska A. S., Mody N., Gettys T. W., Delibegovic M. (2014) Methionine restriction restores a younger metabolic phenotype in adult mice with alterations in fibroblast growth factor 21. Aging Cell 13, 817–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okamatsu-Ogura Y., Nio-Kobayashi J., Iwanaga T., Terao A., Kimura K., Saito M. (2011) Possible involvement of uncoupling protein 1 in appetite control by leptin. Exp. Biol. Med. (Maywood) 236, 1274–1281 [DOI] [PubMed] [Google Scholar]
- 39.Owen B. M., Ding X., Morgan D. A., Coate K. C., Bookout A. L., Rahmouni K., Kliewer S. A., Mangelsdorf D. J. (2014) FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. 20, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mössenböck K., Vegiopoulos A., Rose A. J., Sijmonsma T. P., Herzig S., Schafmeier T. (2014) Browning of white adipose tissue uncouples glucose uptake from insulin signaling. PLoS ONE 9, e110428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plaisance E. P., Greenway F. L., Boudreau A., Hill K. L., Johnson W. D., Krajcik R. A., Perrone C. E., Orentreich N., Cefalu W. T., Gettys T. W. (2011) Dietary methionine restriction increases fat oxidation in obese adults with metabolic syndrome. J. Clin. Endocrinol. Metab. 96, E836–E840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Buono M., Wykes L. J., Ball R. O., Pencharz P. B. (2001) Dietary cysteine reduces the methionine requirement in men. Am. J. Clin. Nutr. 74, 761–766 [DOI] [PubMed] [Google Scholar]
- 43.Elshorbagy A. K., Valdivia-Garcia M., Mattocks D. A., Plummer J. D., Smith A. D., Drevon C. A., Refsum H., Perrone C. E. (2011) Cysteine supplementation reverses methionine restriction effects on rat adiposity: significance of stearoyl-coenzyme A desaturase. J. Lipid Res. 52, 104–112 [DOI] [PMC free article] [PubMed] [Google Scholar]