

Abstract
FE65 and FE65L1 are cytoplasmic adaptor proteins that bind a variety of proteins, including the amyloid precursor protein, and that mediate the assembly of multimolecular complexes. We previously reported that FE65/FE65L1 double knockout (DKO) mice display disorganized laminin in meningeal fibroblasts and a cobblestone lissencephaly-like phenotype in the developing cortex. Here, we examined whether loss of FE65 and FE65L1 causes ocular and muscular deficits, 2 phenotypes that frequently accompany cobblestone lissencephaly. Eyes of FE65/FE65L1 DKO mice develop normally, but lens degeneration becomes apparent in young adult mice. Abnormal lens epithelial cell migration, widespread small vacuole formation, and increased laminin expression underneath lens capsules suggest impaired interaction between epithelial cells and capsular extracellular matrix in DKO lenses. Cortical cataracts develop in FE65L1 knockout (KO) mice aged 16 months or more but are absent in wild-type or FE65 KO mice. FE65 family KO mice show attenuated grip strength, and the nuclei of DKO muscle cells frequently locate in the middle of muscle fibers. These findings reveal that FE65 and FE65L1 are essential for the maintenance of lens transparency, and their loss produce phenotypes in brain, eye, and muscle that are comparable to the clinical features of congenital muscular dystrophies in humans.—Suh, J., Moncaster, J. A., Wang, L., Hafeez, I., Herz, J., Tanzi, R. E., Goldstein, L. E., Guénette, S. Y. FE65 and FE65L1 amyloid precursor protein–binding protein compound null mice display adult-onset cataract and muscle weakness.
Keywords: APP, congenital muscular dystrophy, extracellular matrix, laminin, lens
The FE65 protein family members FE65, FE65L1, and FE65L2 mediate the formation of multiprotein complexes via their consensus 3 protein–protein interaction domains: 1 N-terminal WW (tryptophan, tryptophan) domain and 2 C-terminal PTB (phosphotyrosine binding) domains. Among all the cellular proteins to which FE65 proteins bind, the best-studied interaction is that of FE65 with the amyloid precursor protein (APP). FE65 binds the cytoplasmic domain of APP and regulates its processing, including the generation of β-amyloid (Aβ), the main component of senile plaques in the brains of patients with Alzheimer disease (1–3). FE65 proteins are also implicated in a variety of other cellular mechanisms through their binding to receptors that interact with lipoproteins (4–6), molecules that regulate cytoskeletal remodeling and cell movement (7–9), and proteins that play key roles in nuclear signaling, DNA repair, and calcium homeostasis (10–12).
Insights into the in vivo functions of the FE65 protein family have come from knockout studies utilizing mice and worms (2, 13–16). These studies revealed a role for FE65 proteins in neuronal positioning (2), developmental neurogenesis (13, 17), neuromuscular transmission (14, 15), and learning and memory (16). Our previous studies reported marginal zone heterotopias resembling human cobblestone lissencephaly in the developing cortex of FE65/FE65L1 double knockout (DKO) mice (2). These cortical malformations, resulting from neuronal overmigration into the subarachnoid space, are also observed in the embryonic mouse brains lacking all 3 APP family proteins: APP, amyloid precursor-like protein (APLP) 1, and APLP2 (18). Disruptions of the meningeal basement membrane and altered laminin organization in the FE65/FE65L1 DKO mice suggest a pivotal role for FE65 proteins in basement membrane formation. Normal functions of APP include roles in cell adhesion and cell motility (19, 20), possibly through the binding of its ectodomain to cell adhesion molecules and extracellular matrix (ECM) proteins (21–24). In cultured cells, APP and FE65 colocalize at the edge of migrating filopodia, promote neurite outgrowth, and regulate actin-based motility (8, 9, 23, 25). The depletion of either APP or FE65 results in the disorganization of ECM proteins (2, 8, 18).
Cobblestone lissencephaly is a characteristic of autosomal recessive diseases with cerebral, ocular, and muscular deficits, such as Walker-Warburg syndrome, Fukuyama congenital muscular dystrophy, and muscle–eye–brain disease (26). These types of congenital muscular dystrophies are called dystroglycanopathies because they are caused by mutations that lead to the hypoglycosylation of α-dystroglycan, a linker protein at the cell surface (27, 28). In normal tissues, the heavily glycosylated region of α-dystroglycan binds to ECM proteins that possess laminin G domains (27, 29). Thus, the developmental abnormalities in muscle, eye, and brain in dystroglycanopathies are caused by defective interactions between α-dystroglycan at the cell surface and the ECM.
The similarities of brain phenotypes in FE65/FE65L1 DKO mice with cobblestone lissencephaly led us to ask whether FE65/FE65L1 DKO mice would display the other 2 phenotypes of dystroglycanopathies: ocular and muscular deficits. In addition to muscle weakness, microphthalmia, retinal dysplasia, and cataract are frequently observed in the eyes of dystroglycanopathy patients (27, 29). In this study, we examined the effects of FE65 and/or FE65L1 depletion on eye and muscle phenotypes with behavioral, optical, histologic, and biochemical approaches. We found that the deficiency of FE65 family proteins causes cataract and muscle weakness in a gene dose-dependent manner.
Materials and Methods
Mice and hanging wire test
FE65 knockout (KO), FE65L1 KO, and FE65/FE65L1 DKO mice of a C57BL/6 × 129SvEvBradley hybrid genetic background were generated and genotyped as previously described (2). Mice were tested for grip strength during the light phase of their light–dark cycle. The wire hang test was performed as follows. Each mouse was placed on the wire cage top and the cage top was inverted, so that the mouse was suspended above the cage. The latency to fall off the wire was measured up to 60 seconds. Tests were carried out 3 times for each mouse, with a rest period between tests. All animal generation, husbandry, and experimental procedures were approved by the Massachusetts General Hospital (MGH) Subcommittee on Research Animal Care and were performed in accordance with the MGH Institutional Animal Care and Use Committee.
Histologic and immunohistochemical staining of mouse eyes
Mice were deeply anesthetized with isoflurane, then euthanized by cervical dislocation. Eyes were dissected and fixed overnight in 4% paraformaldehyde. For embryonic eyes, whole heads of embryos were fixed and processed. The fixed eyes were processed and embedded in paraffin at the MGH Pathology Core. Seven-micron-thick sagittal sections were used for hematoxylin and eosin (H&E) staining and immunohistochemistry. For each mouse, at least 10 sections for adult eyes and 5 sections for embryonic eyes were stained with H&E for the examination of overall morphology. For laminin immunohistochemistry, paraffin sections were deparaffinized, rehydrated, and incubated with 0.25% Triton X-100 and 0.3% H2O2 for 15 minutes at room temperature. Sections were then blocked in 10% horse serum for 1 hour at room temperature, followed by incubation overnight at 4°C with anti-laminin antibody (Sigma-Aldrich, St. Louis, MO, USA) in 10% horse serum. The bound primary antibodies were detected by biotin-conjugated anti-rabbit antibody for 2 hours, followed by incubation in avidin–biotin–peroxidase complex and peroxidase activity detection with DAB (3,3′-diaminobenzidine; Vector Laboratories, Burlingame, CA, USA). Stained sections were dehydrated with increasing concentration of ethanol and mounted with Cytoseal (Thermo Fisher Scientific, Waltham, MA, USA). Staining was visualized with a microscope (Nikon TE300), and photomicrographs were taken with an attached digital camera and associated software (Spot InSight). Areas underneath the anterior lens capsule stained with anti-laminin antibody were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
TUNEL assay
Paraffin was removed from the paraffin-embedded sections, and a peroxidase in situ apoptosis kit (ApopTag Plus; Chemicon, Atlanta, GA, USA) was used to detect apoptotic nuclei in mouse eyes. Rodent mammary gland tissue was included as a positive control, and methyl green was used for counterstaining of cell nuclei. The TUNEL assay procedure was performed in accordance with the manufacturer’s instructions.
Western blot analysis
Mouse tissues were dissected and homogenized on ice in Tris-buffered saline (muscle) or in RIPA buffer (lens and retina; 50 mM Tris HCl pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% SDS) containing protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN, USA). Muscle homogenates were further diluted in RIPA buffer. After incubation on ice for 10 minutes, tissue debris was removed by centrifugation at 17,000 g for 10 minutes at 4°C. Protein samples for SDS-PAGE were prepared in NuPAGE sample buffer containing β-mercaptoethanol (2.5%) and separated on 4% to 12% NuPAGE gels (Invitrogen, Carlsbad, CA, USA). Proteins were transferred to PVDF membranes (EMD Millipore, Billerica, MA, USA) for Western blot analysis. Antibodies used were as follows: pan-FE65 antibody detecting both FE65 and FE65L1 (30), APP (A8717; Sigma-Aldrich), laminin (Sigma-Aldrich), α-dystroglycan (VIA4-1, Millipore; IIH6, Upstate Biotechnology, Lake Placid, NY, USA) and pan-actin (Sigma-Aldrich). Immunoreactive bands on Western blots were detected with ECL reagents (Pierce, Rockford, IL, USA) on X-ray film. Band intensities were quantified by densitometric analysis using Image Gauge software v 3.12 (Fuji Photo Film) and ImageJ software.
In vivo and ex vivo slit lamp microscopy
Mouse pupils were dilated with 1% tropicamide ophthalmic solution for in vivo slit lamp (IVSL) microscopy. Both eyes were examined without anesthesia with the aid of a zoom photograph slit-lamp biomicroscope at Boston University School of Medicine (31). After the examination, mice were euthanized, and eyes were dissected for ex vivo lens analysis. Dissected lenses were bathed in artificial aqueous humor (295 mOsm, pH 7.2) and visualized with a custom-adapted surgical slit lamp stereophotomicroscope (Carl Zeiss GmbH, Jena, Germany) equipped with a stereo beamsplitter (Urban Engineering, Burbank, CA, USA) and side-arm digital camera assembly (Nikon, Tokyo, Japan) (32). This system permits high resolution stereophotomicroscopic imaging of living ex vivo lens specimens for phenotype analysis (33).
Neuron culture
Primary neurons derived from wild-type (WT) and FE65/FE65L1 DKO mouse embryos (E15) were prepared and cultured as previously described (3). Neuronal RIPA lysates were analyzed for α-dystroglycan expression by Western blot analysis as described above.
Histologic analysis of mouse muscles
Mouse quadriceps muscles were prepared as previously described (34). Briefly, excised muscles were frozen in liquid nitrogen-cooled isopentane and the frozen tissues were kept at −80°C. The frozen muscle tissues were sectioned in transverse direction at 12 μm thickness and mounted on glass slides. For each mouse, at least 10 sections of muscle tissue were stained with H&E for examination of their overall morphology. Immunohistochemistry for laminin was performed as described above.
RESULTS
Loss of FE65 and FE65L1 causes lens degeneration in young adult mice
During the phenotypic characterization of mice lacking the FE65 and FE65L1 genes, we noticed opacity in the lenses of 10-month-old FE65/FE65L1 DKO mice that was not evident in age-matched WT mice (Fig. 1A, B). To investigate further the cause of lens opacification, we dissected and examined the eyes from WT, FE65 KO, FE65L1 KO, and FE65/FE65L1 DKO mice. H&E staining of ocular sections from WT and FE65/FE65L1 DKO mice revealed severe lens disruption and degeneration in the DKO mice (Fig. 1C, D). In contrast to WT lenses, massive vacuolization, lens capsule rupture, and the disruption of lens fiber cell organization were consistently observed in the DKO lenses (Fig. 1C–F). However, no notable morphologic changes were observed in any other parts of the eye, including laminar organization of the retina (Fig. 1G, H). To determine the age at onset, DKO eyes at different ages were dissected for histologic analysis. Lenses from FE65/FE65L1 DKO embryos (E15) and newborn pups (P0) did not show any gross morphologic differences compared to age-matched WT controls (Fig. 1I, J). This suggests that FE65 and FE65L1 proteins are not essential for the initial stages of lens development. Lens disruption was detected in FE65/FE65L1 DKO mice as early as 1 month of age with a low (10%) incidence (Table 1). The prevalence rose to 72% in mice aged between 2 and 10 months, and was 91% in mice older than 12 months. Some mice had only one affected lens. The overall size and morphology of lenses from the FE65 and FE65L1 single KO mice were indistinguishable from those of WT mice (data not shown).
Figure 1.
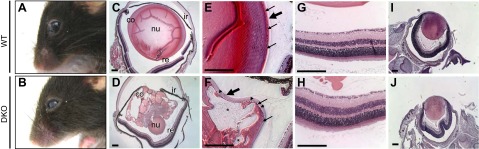
Cataract and lens degeneration in FE65/FE65L1 DKO mice. Gross appearance of 10-month-old WT (A) and FE65/FE65L1 DKO (B) mouse eyes showing opacity in DKO lens. H&E staining of paraffin-embedded sections of 4-month-old WT (C, E) and DKO (D, F) eyes revealed severe lens disruption and degeneration in DKO mice. Breaks in retina are artifacts from tissue processing for paraffin embedding. nu, lens nucleus; co, lens cortex; re, retina; ir, iris. High magnification (E, F) of lens equatorial region reveals a thickened anterior lens capsule (large arrow) and small vacuolization subjacent to capsule (arrowheads) in DKO lens. Small arrows indicate nuclei of lens epithelial cells; asterisk indicates extensive vacuolization in DKO lens cortex. (G, H) Retina lamination of WT and DKO mice. Compared to WT (I), no overt morphologic difference was detected in lens of a DKO pup at P0 (J). Scale bar, 200 μm.
TABLE 1.
Lens disorganization in FE65/FE65L1 mice
| Genotype | Average age | No. of mice (eyes) with lens disruption |
|---|---|---|
| WT | >12 mo (13.7 ± 0.2) | 0/7 (0/14) |
| FE65 KO | >12 mo (12.3 ± 0.0) | 0/4 (0/8) |
| FE65L1 KO | >12 mo (12.3 ± 0.0) | 0/3 (0/6) |
| FE65/FE65L1 DKO | <1 d | 0/18 (0/36) |
| 1 mo | 1/5 (1/10) | |
| 2–10 mo (5.7 ± 0.7) | 7/9 (13/18) | |
| >12 mo (15.8 ± 0.4) | 13/14 (21/23) |
FE65/FE65L1 DKO lenses display abnormal epithelial cell positioning and increased expression of laminin and APP
In addition to the disorganized fiber cells and extensive vacuolization, the lenses of FE65/FE65L1 DKO mice showed several defects with regard to the lens capsule. The lens capsule is a specialized basement membrane, and the components of this ECM, including laminin and collagen, are generated by lens epithelial cells subjacent to the anterior capsule (Fig. 1E) (35). In the degenerating DKO lens, the anterior capsule was thicker than in control mice, and the posterior region of the capsule was frequently ruptured, resulting in subluxation of the lens nucleus (Fig. 1D–F). Under higher magnification, small vacuoles were apparent subjacent to the anterior capsule, and some epithelial cells exhibited ectopic positioning in the lens (Fig. 2A, B). We next examined laminin expression in DKO lenses, as abnormal distribution and poor organization of laminin were previously found in the cortex and meningeal fibroblasts of these mice (2). Compared to the expression in lenses from WT mice, excessive laminin accumulation was detected subjacent to the anterior capsule in the degenerating DKO lenses (Fig. 2C–G). Quantitation of laminin immunostaining underneath the lens capsule shows a significant increase in laminin staining in DKO lenses (Fig. 2I). Interestingly, the high level of laminin expression underneath the capsule was detected even in 1-month-old nondisrupted DKO lenses (Fig. 2F). This indicates that laminin up-regulation precedes lens disorganization. Increased levels of laminin in the DKO lens was further confirmed by Western blot analysis of the lens lysates (Fig. 2H, J). The abnormal laminin accumulation may interfere with proper interaction between epithelial cells and capsular ECM. In support of this notion, severely disorganized migration and proliferation of lens epithelial cells, which are highly dependent on proper cell–ECM interaction (35, 36), were found in the degenerating DKO lenses (Fig. 2A). TUNEL staining showed no evidence of cell death in either lens fiber or epithelial cells (data not shown), indicating that cell death is not causative for the observed lens disruption and degeneration.
Figure 2.

Disordered lens epithelial cells and increased laminin in FE65/FE65L1 DKO lens. (A, B) Photomicrographs of degenerating lens of H&E-stained FE65/FE65L1 DKO mice. Arrows indicate abnormal distribution of lens epithelial cell nuclei in lens. Higher magnification (B) reveals widespread small vacuoles (arrowheads) in epithelial cell layer subjacent to lens capsule. (C–G) Laminin immunohistochemistry of adult WT (C, 13 months) and DKO (D, 9 months), and young WT (E, 1 month) and DKO (F, G, 1 month) eyes reveals increased laminin (small arrow) underneath the anterior capsule (large arrow) of DKO lenses. Gray-black color in (E–G) is due to addition of nickel chloride in DAB substrate development (brown, C, D). ir, iris; cn, cornea. Asterisk indicates high level of laminin expression in corneal epithelium. Scale bar, 50 μm. H) Western blot of laminin expression in lenses from 1- and 4-month-old WT and DKO mice. Brain lysates were used as control. I) Laminin immunopositive areas underneath lens capsule in adult WT (13.6 ± 0.2-month-old) and DKO (13.4 ± 2.0-month-old) mice; n = 3. J) Densitometric quantification of laminin in (H). Values are mean ± sem, Student’s t test. **P < 0.01.
We next examined whether loss of FE65 and FE65L1 in the lens affects APP expression. Western blot analysis showed that both APP and FE65/FE65L1 proteins are expressed in the lens, and the expression levels of both proteins are much lower than in the brain (Fig. 3A). While APP levels in the intact 1-month-old DKO lens are similar to that of WT (Fig. 3B, C), in the degenerating 4-month-old DKO lens, APP expression is significantly elevated compared to WT controls (Fig. 3B, C). In the retina of DKO mice, APP levels remained similar to those of WT (Fig. 3B, C). Given the severe lens fiber degeneration in DKO mice, the increase of APP is in part due to the massive loss of crystallins, which represent over 90% of lens proteins. However, densitometric analysis showed that the increase in APP is much more robust than that of actin (Fig. 3B, C) and other housekeeping proteins in the lens (data not shown). The increase of APP in the FE65- and FE65L1-depleted lens may be a compensatory mechanism for the loss of FE65/FE65L1-mediated cell–ECM interaction.
Figure 3.

APP and FE65/FE65L1 expression in mouse lens. A) APP and FE65/FE65L1 expression in 1-month-old WT and FE65/FE65L1 DKO mice. Brain lysates were used as control for detection. B) Lens APP levels in 1- and 4-month-old WT and DKO mice. Both APP and actin levels in WT lenses decreased from 1 to 4 months of age. Compared to APP immunoblots for lens lysates, lighter exposure is shown for retinal lysates. C) Densitometric quantification of APP levels over actin; n = 4. Statistical analyses were performed by Student-Newman-Keuls test. *P < 0.05.
Development of cortical cataract in the aged FE65L1 KO mice
The majority of lenses (34 of 41) from adult FE65/FE65L1 DKO mice (>2 months old, Table 1) showed severe disruption and degeneration in histologic staining. However, the remaining DKO lenses (7 of 41) were normal and indistinguishable from WT. Lenses with subtle morphologic defects, such as mild cataract, were not found in any of the DKO mice examined. This clear difference in lens morphology among DKO mice suggests that once the lens deterioration starts, the collapse of lens integrity and the subsequent lens degeneration progress very rapidly in the adult DKO eyes. Because FE65 and FE65L1 have high sequence homology and DKO lenses degenerate in young mice, we hypothesized that loss of either FE65 or FE65L1 alone might result in a milder phenotype in aged mouse lenses. To explore this hypothesis, and to increase the sensitivity of cataract detection in live mice, we used IVSL micrography, a technique routinely used to detect human cataract (31, 32) (Fig. 4A–C). As expected, IVSL examination of aged FE65/FE65L1 DKO mice (16 months old) revealed highly opaque lenses that produced light scattering in IVSL (Fig. 4C, Table 2). In contrast, none of the age-matched WT (n = 4) or FE65 KO (n = 5) mice showed signs of cataract in the IVSL examination (Table 2). Interestingly, 1 of 5 of the 16-month-old FE65L1 KO mice exhibited light scattering indicative of cataract (Table 2). Moreover, 2 more FE65L1 KO mice, aged between 19 to 20 months, demonstrated cataract in the IVSL test.
Figure 4.

Cortical cataract in aged FE65L1 KO mice. A–C) IVSL examination of 16-month-old WT (A), FE65L1 KO (B), and FE65/FE65L1 DKO (C) mouse eyes. No cataract was detected in either WT or FE65 KO mice. C, cornea; LC, lens capsule; L, lens; PR, Purkinje reflex. D–F) Stereophotomicrograph images of ex vivo lenses dissected from 16-month-old WT, FE65L1 KO, and DKO mice. Yellow dots indicate extent of slit beam. Arrowheads (E) indicate opacification in lens cortex. G) Stereo image showing cortical cataract in lens from 20-month-old FE65L1 KO mouse. White dots are for superimposing. H–K) Compared to transparent WT lens (H), lenses from 19- to 20-month-old FE65L1 KO mice exhibit opacity, ranging from partial cortical cataract (I) to circumferential cortical cataract (J) and entire lens opacification (K).
TABLE 2.
Cataract and corneal defect in aged FE65 protein family KO mice
| Genotype | Age (mo) | Mouse no. | Cataract detectiona |
No. of corneal defectsc | Lens weight (mg) | |
|---|---|---|---|---|---|---|
| In vivo | Ex vivo | |||||
| WT | 13–19 | 4 | 0/4 | 0/4 (0)b | 0 | 9.6 ± 0.6 |
| FE65 KO | 16 | 5 | 0/5 | 0/5 (0) | 0 | 9.5 ± 0.2 |
| FE65L1 KO | 16 | 6 | 1/5 | 2/6 (2) | 1 | 9.6 ± 0.5 |
| 19–20a | 5 | 3/4 | 4/5 (3) | 3 | 9.6 ± 0.5 | |
| FE65/FE65L1 DKO | 16 | 5 | 3/3 | 5/5 (0) | 2 | 3.0 ± 1.2* |
Data for both in vivo and ex vivo slit lamp lens microscopy were obtained from same eye (left) of each mouse, except for 19 to 20-month-old FE65L1 KO mice. In these mice, both eyes were examined in vivo, but only left eyes were dissected for ex vivo examination.
Number in parentheses is the number of lenses that have cortical cataract.
Number of mice with corneal ulceration; these mice were excluded from IVSL examination.
Significantly different from WT (P < 0.05).
In addition to cataract, some of the aged FE65L1 KO (4 of 11) and FE65/FE65L1 DKO (2 of 5) mice displayed ulcerlike defects in the cornea (Table 2). This corneal abnormality precluded IVSL examination of those affected mice. To examine further the characteristics of the observed cataract, we dissected lenses from the FE65 protein family KO mice (Fig. 4D–F). All lenses from 16-month-old DKO mice were degenerated, and in all cases, the lens nucleus was dissociated from the cortex (Fig. 4F). Notably, some lenses (2 of 6) from 16-month-old FE65L1 KO mice exhibited a narrow opacity in the deep cortex (Fig. 4E, G), and cataract incidence (4 of 5) increased in 19- to 20-month-old FE65L1 KO mice. In some mice at this age, opacity was limited to the lens cortex (Fig. 4I, J), while in others opacity was evident throughout the lens (Fig. 4K). While all lenses from the DKO mice were degenerated and small (Fig. 4F), the opaque FE65L1 KO lenses remained similar in size and weight compared to WT lenses (Fig. 4H–K, Table 2). Taken together, these findings suggest that FE65L1 plays an important role in the maintenance of lens transparency and that deficiency of FE65 family proteins lead to age-related cataract in mice.
FE65 family protein KO mice show muscular deficits
We next asked whether the depletion of FE65 family genes would cause muscle weakness, another phenotype frequently associated with cobblestone lissencephaly(26–28). Previous studies of APP gene family KO mice showed limb muscle weakness (37, 38) and neuromuscular junction defects (39, 40). Phenotypic characterization of the FE65/FE65L1 DKO mice revealed abnormal hind limb clasping (Fig. 5A), indicative of neurologic or motor deficits (41). Mice lacking either FE65 or FE65L1 did not show the abnormal clasping behavior and were indistinguishable from WT mice (data not shown). Instead, the hanging wire grip test showed that the latency to fall is shorter for both FE65 and FE65L1 single KO than WT mice (Fig. 5B). An even shorter latency was observed for FE65/FE65L1 DKO mice, suggesting cumulative effects of FE65 and FE65L1 proteins on muscle strength.
Figure 5.

Behavioral abnormalities in FE65/FE65L1 DKO mice. A) Hind limb clasping tests with 10-month-old WT and FE65/FE65L1 DKO mice reveal altered plantar reaction in DKO mice. B) Hanging wire grip test, measured as latency to fall, was examined in WT (12.5 ± 1.5 months old), FE65 (12.8 ± 0.2), and FE65L1 (12.8 ± 0.1) single KO and FE65/FE65L1 DKO (12.2 ± 0.6) mice. Number of mice used per genotype is shown in parentheses. Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate further the cause of muscle weakness, we examined quadriceps from FE65/FE65L1 DKO mice. H&E staining of the muscle sections did not reveal any sign of severe deterioration in DKO mice aged over 12 months (Fig. 6A, B). Instead, compared to the peripheral location of nuclei in the WT muscle cells, 14% of DKO cells harbor nuclei in the middle of muscle fibers (Fig. 6C). In muscle cells, centralized nuclei are characteristic of pathologic conditions, such as myotonic dystrophies (42, 43) and denervation (44). The molecular mechanism for central nuclei in the DKO muscle cells is unclear, but this phenotype is a manifestation of an abnormal muscle condition, which may contribute to the observed skeletal muscle strength deficits.
Figure 6.

Histologic and biochemical analysis of muscles in FE65/FE65L1 DKO mice. A, B) Representative H&E-stained images of transverse sections of quadriceps from adult WT (A) and DKO (B) mice. Arrows indicate nuclei in muscle cells. Scale bar, 50 μm. C) Percentile of muscle cells harboring central nuclei in WT (14.1 ± 1.7 months old, n = 4) and DKO (13.9 ± 2.5, n = 3) quadriceps. Student’s t test. **P < 0.01. D, E) Laminin staining of quadriceps from adult WT and DKO mice. F, G) Western blot analysis of FE65/FE65L1, APP, laminin, and α-dystroglycan (α-DG) in muscles (F) and α-dystroglycan in primary neurons (G). DIV, days in vitro.
Western blot analysis of the muscle lysates revealed that FE65/FE65L1 proteins are present in muscle and that the expression level is comparable to that of lens (Figs. 3A and 6F). However, APP expression levels were very low and not changed in the DKO muscle, which implies a minimal role for APP in cell–ECM interaction in muscle. In contrast to the brain and lens (Fig. 2H), laminin is highly expressed in the endomysium, the extracellular proteoglycan matrix between muscle cells (Fig. 6D–F). However, no notable change in laminin expression was detected in DKO muscle. Hypoglycosylation of α-dystroglycan, which leads to muscle deterioration in congenital muscular dystrophies (27, 28), was not detected in the DKO muscles (Fig. 6F). While α-dystroglycan was barely detectable in the lens (data not shown), Western blot analysis of primary neurons derived from DKO mice (Fig. 6G) suggests that lens degeneration and muscle weakness in the DKO mice is not due to a decrease in α-dystroglycan glycosylation. Collectively, these findings indicate that the extent and the underlying mechanism for the muscular deficits in the FE65 protein family KO mice are different from those of dystroglycanopathies.
DISCUSSION
In this study, we showed the targeted deletion of FE65 family genes causes cataract in adult mouse eyes. In FE65L1 KO mice, cataracts were found in the lens cortex of 16-month-old mice. In FE65/FE65L1 DKO mice, lenses become disrupted and degenerated at a much earlier age (average, ∼6 months). We also report that depletion of FE65 and FE65L1 produces limb muscle weakness in a gene dose-dependent manner. These ocular and muscular deficits, coupled with the brain malformations in FE65/FE65L1 DKO mice (2), are comparable to the 3 main clinical features of dystroglycanopathies, a subset of congenital muscular dystrophies (26, 28).
The lens maintains transparency by the ordered packing of crystallin proteins and the regular arrangement of lens fiber cells (45–47). In addition to aging, genetic determinants or environmental factors that disturb lens homeostasis can cause cataract (48–50). In this study, we found that the absence of both FE65 and FE65L1 leads to severe lens fiber cell disorganization and degeneration. In addition, the cortical cataract observed in aged FE65L1 single KO mice suggests that the etiology underlying lens degeneration in the DKO is similar to that of cataract. Several additional morphologic and biochemical abnormalities provide clues indicating a potential underlying pathogenic mechanism in the FE65/FE65L1 DKO eyes. First, in the degenerating lenses of the DKO mice, the anterior lens capsule is thicker than in controls, and the posterior lens is ruptured. Second, numerous small vacuoles are present underneath the anterior capsule. Third, the proliferation and migration of lens epithelial cells are severely deregulated. Finally, the expression of laminin, one of the major components of the lens capsule, is abnormally increased underneath the anterior lens capsule. This accumulation of laminin occurs prior to disruption of the DKO lens. While further studies are needed to elucidate the precise molecular mechanism, our findings suggest that a lack of proper interaction between lens epithelial cells and the anterior capsule is responsible, at least in part, for the lens disruption and degeneration in the FE65/FE65L1 DKO mice.
The lens capsule is an uninterrupted basement membrane that surrounds the lens. This special ECM is composed of interacting networks of laminin, collagen, and several other proteoglycans, all of which are secreted by lens cells (35). Binding to these ECM proteins is a fundamental requirement for lens epithelial cells to properly migrate and differentiate (35, 36). Deficiency of ECM components, such as perlecan and SPARC (secreted protein acidic and rich in cysteine), can lead to cataract formation in mice (36, 51). Given the essential role of cell–ECM interactions for cell differentiation and migration, the abnormalities in FE65/FE65L1 DKO lenses shown herein and those previously published in brain (2) suggest a mechanistic similarity. First, the pial basement membrane (ECM) of FE65/FE65L1 DKO mice is disrupted and neurons are ectopically localized in the marginal zone of the developing cortex. Second, laminin is severely disorganized in meningeal fibroblasts derived from DKO mouse brains. Third, eye abnormalities, including cataract, are frequently observed in patients with cobblestone lissencephaly (27, 28). Therefore, it is feasible to speculate that the observed phenotypes in the adult lenses and the developing brains of FE65/FE65L1 DKO mice share a common pathogenic mechanism: a disrupted cell–ECM interaction.
In addition to the lens defects, we report that loss of FE65 protein family members decreases peripheral muscle strength. This muscular deficit is concordant with the previous findings in APP and APLP2 KO animals. APP KO mice showed a decrease in forelimb grip strength (37), and APP/APLP2 KO mice displayed defects in neuromuscular synapses (39). In Caenorhabditis elegans, depletion of the APP or FE65 orthologs reduced acetylcholinesterase activity at the neuromuscular junction and altered the rate of pharyngeal contraction (14, 15). Interestingly, in the histologic analysis of the FE65/FE65L1 DKO mouse quadriceps muscle, 14% of muscle cells harbor their nuclei in the middle of muscle fibers. Centralized nuclei are found in muscles only in transitional states (development, regeneration) and in pathologic conditions (42, 43, 52, 53). While severe muscle cell degeneration was not detected, the centralized nuclei are indicative of an abnormal muscle condition in the DKO mice. Given that central nucleation occurs in denervated muscles (44), it warrants a further study to examine whether FE65/FE65L1 DKO mice has neuromuscular junction defects, similar to those found in APP/APLP2 DKO mice.
Collectively, our present and previous characterization of the FE65/FE65L1 DKO mice identified 3 phenotypes that occur in brain, eye, and muscle. The presence of abnormalities in these 3 organs/tissues of FE65/FE65L1 DKO mice led us to compare the defects observed in our mice to the main clinical features of dystroglycanopathies. In these congenital disorders, poor glycosylation of α-dystroglycan, a protein that links the ECM protein laminin to the cytoskeleton, is responsible for the disorganization of basement membrane in affected tissues/organs (29). The cobblestone lissencephaly–like phenotype of FE65/FE65L1 DKO mice, which results from overmigration of neuronal cells beyond the pial basement membrane during brain development, is quite similar to that observed in dystroglycanopathies. However, phenotypes detected in the DKO mouse eyes and muscles have notable differences from those of dystroglycanopathies. In the DKO eyes, no ocular phenotype other than the lens and cornea deficits was detected. However, retinal dysplasia is also frequently observed in dystroglycanopathies. Moreover, lens defects in the DKO and FE65L1 KO occur in adult mice, whereas ocular deficits in dystroglycanopathy are notable from a very young age (27, 28). In the DKO mice, no severe muscle cell degeneration was noted, however, this is apparent in dystroglycanopathies. The mild muscle phenotype in the DKO mice is concordant with the daily motor behaviors of these mice (2). Beyond infrequent bilateral circling after cage disturbance, no other motor behavioral defects were visually detected in mice up to 16 months old.
Despite these differences, our findings of disturbed basement membranes in the cortex and in the lens suggest that the 2 similar pathologies in the DKO mice and in dystroglycanopathies are due to a similar underlying molecular mechanism, a disruption of cell–ECM interaction. Thus, we speculate that FE65 family proteins play a key role in linking the ECM to the cytoskeleton. As illustrated in Fig. 7, in normal tissue, the C-terminal PTB and WW domains of FE65/FE65L1 bind APP and Mena/Vasp, respectively (54). At the cell surface, APP can bind either laminin (22, 55) or other ECM proteins (24, 56–58). Lack of FE65 family proteins would destabilize this transmembrane multiprotein complex and produce disorganized basement membranes: the pial basal membrane in the cortex and the capsule in the lens. Altered laminin distribution has been found both in meningeal fibroblasts (2) and in the lens capsule of DKO mice. Furthermore, similar cortical phenotypes and muscle strength deficits between APP (18, 37, 39) and FE65 family protein KO mice support the hypothesis that FE65/FE65L1 stabilize the cell–ECM connection through their binding to APP family proteins. In this regard, it would be interesting to examine whether APP family KO mice develop cataract or other ocular deficits. The functional redundancy of the APP protein family members and the early lethality of some compound KO mice (e.g., APP/APLP2 DKO and APP/APLP1/APLP2 triple KO mice) may have thus far interfered with this assessment (18, 39).
Figure 7.

Schematic model for disrupted cell–ECM interaction in FE65/FE65 DKO mouse tissues. In WT mice, cytosolic FE65/FE65L1 proteins ensure linkage of cell–ECM interaction through their binding to the cytoplasmic domain of transmembrane APP. Absence of these adaptor proteins can produce altered laminin distribution and disorganized ECM (broken lines), which lead to brain malformation and lens cataract in DKO mice.
Notably, other lines of evidence indicate that changes in APP expression and its processing are causative for cataract development. Transgenic mice expressing an extra copy of the human APP gene produced lens fiber degeneration in the lens cortex (59), and Down syndrome patients carrying an additional copy of the APP-containing chromosome 21 develop cataract deep in the lens cortex (supranuclear cataract) with high frequency (32, 60). Moreover, an increase of Aβ, an APP metabolite, which is associated with cortical cataract, has been reported in lenses from Down syndrome and Alzheimer disease patients (32, 33, 61, 62). In this regard, the findings of cortical cataract in aged FE65L1 KO mice are concordant with the notion that improper regulation of APP can induce cataract in the lens cortex (32, 59). However, as the endogenous mouse Aβ levels in the brains and neurons of FE65/FE65L1 DKO mice were lower than those of WT mice (2, 3), it is unlikely that the cortical cataract or lens disruption phenotypes of the FE65 family protein KO mice were initiated by Aβ accumulation in the lens (63).
Taken together, our findings in the present study revealed essential roles of FE65 and FE65L1 in lens transparency and muscle strength. We propose that the lack of interactions between FE65 and APP family proteins is responsible for the impaired cell–ECM interaction, especially in the migration of lens epithelial and neuronal cells, which leads to lens disruption and brain malformations in FE65/FE65L1 DKO mice. While the DKO mice show pathologic condition in muscles (e.g., central nucleation), the molecular mechanism underlying the muscle weakness remains to be determined. Thus far, many studies addressing FE65 protein family function have focused on understanding the biological significance of FE65/APP interactions and its impact on APP processing and Aβ generation for their relevance to Alzheimer disease. Our characterization of defects in the FE65 family protein KO mice, associated with altered laminin regulation in brain and lens, suggest that FE65 and FE65L1 play key roles in the maintenance of basement membranes and in the interaction between cells and the ECM.
Acknowledgments
The authors thank D. Romano for her technical assistance in muscle pathology experiments. The authors also thank M. Lawlor (Boston Children Hospital) for his advice on muscle tissue preparation and C. Vanderburg (Massachusetts General Hospital) for his scientific expertise and advice on the muscle study. This work was supported by grants from the U.S. National Institutes of Health (to S.Y.G. and R.E.T.) and by a fellowship from the Korea Research Foundation (to J.S.).
Glossary
- APLP1
amyloid precursor-like protein 1
- APP
amyloid precursor protein
- Aβ
β-amyloid
- DAB
3,3′-diaminobenzidine
- DKO
double knockout
- ECM
extracellular matrix
- H&E
hematoxylin and eosin
- IVSL
in vivo slit lamp
- KO
knockout
- SPARC
secreted protein acidic and rich in cysteine
- WT
wild-type
REFERENCES
- 1.McLoughlin D. M., Miller C. C. (2008) The FE65 proteins and Alzheimer’s disease. J. Neurosci. Res. 86, 744–754 [DOI] [PubMed] [Google Scholar]
- 2.Guénette S., Chang Y., Hiesberger T., Richardson J. A., Eckman C. B., Eckman E. A., Hammer R. E., Herz J. (2006) Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 25, 420–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suh J., Lyckman A., Wang L., Eckman E. A., Guénette S. Y. (2011) FE65 proteins regulate NMDA receptor activation-induced amyloid precursor protein processing. J. Neurochem. 119, 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dumanis S. B., Chamberlain K. A., Jin Sohn Y., Jin Lee Y., Guénette S. Y., Suzuki T., Mathews P. M., Pak D. Ts., Rebeck G. W., Suh Y. H., Park H. S., Hoe H. S. (2012) FE65 as a link between VLDLR and APP to regulate their trafficking and processing. Mol. Neurodegener. 7, 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoe H. S., Magill L. A., Guenette S., Fu Z., Vicini S., Rebeck G. W. (2006) FE65 interaction with the ApoE receptor ApoEr2. J. Biol. Chem. 281, 24521–24530 [DOI] [PubMed] [Google Scholar]
- 6.Trommsdorff M., Borg J. P., Margolis B., Herz J. (1998) Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J. Biol. Chem. 273, 33556–33560 [DOI] [PubMed] [Google Scholar]
- 7.Ermekova K. S., Zambrano N., Linn H., Minopoli G., Gertler F., Russo T., Sudol M. (1997) The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J. Biol. Chem. 272, 32869–32877 [DOI] [PubMed] [Google Scholar]
- 8.Sabo S. L., Ikin A. F., Buxbaum J. D., Greengard P. (2001) The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 153, 1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabo S. L., Ikin A. F., Buxbaum J. D., Greengard P. (2003) The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 23, 5407–5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minopoli G., Gargiulo A., Parisi S., Russo T. (2012) Fe65 matters: new light on an old molecule. IUBMB Life 64, 936–942 [DOI] [PubMed] [Google Scholar]
- 11.Cao X., Südhof T. C. (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120 [DOI] [PubMed] [Google Scholar]
- 12.Nensa F. M., Neumann M. H., Schrötter A., Przyborski A., Mastalski T., Susdalzew S., Looße C., Helling S., El Magraoui F., Erdmann R., Meyer H. E., Uszkoreit J., Eisenacher M., Suh J., Guenette S. Y., Rohner N., Kogel D., Theiss C., Marcus K., Müller T. (2014) Amyloid beta a4 precursor protein-binding family B member 1 (FE65) interactomics revealed synaptic vesicle glycoprotein 2A (SV2A) and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) as new binding proteins in the human brain. Mol. Cell. Proteomics 13, 475–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Q. H., Futagawa T., Yang W. L., Jiang X. D., Zeng L., Takeda Y., Xu R. X., Bagnard D., Schachner M., Furley A. J., Karagogeos D., Watanabe K., Dawe G. S., Xiao Z. C. (2008) A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat. Cell Biol. 10, 283–294 [DOI] [PubMed] [Google Scholar]
- 14.Zambrano N., Bimonte M., Arbucci S., Gianni D., Russo T., Bazzicalupo P. (2002) feh-1 and apl-1, the Caenorhabditis elegans orthologues of mammalian Fe65 and beta-amyloid precursor protein genes, are involved in the same pathway that controls nematode pharyngeal pumping. J. Cell Sci. 115, 1411–1422 [DOI] [PubMed] [Google Scholar]
- 15.Bimonte M., Gianni D., Allegra D., Russo T., Zambrano N. (2004) Mutation of the feh-1 gene, the Caenorhabditis elegans orthologue of mammalian Fe65, decreases the expression of two acetylcholinesterase genes. Eur. J. Neurosci. 20, 1483–1488 [DOI] [PubMed] [Google Scholar]
- 16.Wang Y., Zhang M., Moon C., Hu Q., Wang B., Martin G., Sun Z., Wang H. (2009) The APP-interacting protein FE65 is required for hippocampus-dependent learning and long-term potentiation. Learn. Mem. 16, 537–544 [DOI] [PubMed] [Google Scholar]
- 17.Forni P. E., Fornaro M., Guénette S., Wray S. (2011) A role for FE65 in controlling GnRH-1 neurogenesis. J. Neurosci. 31, 480–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herms J., Anliker B., Heber S., Ring S., Fuhrmann M., Kretzschmar H., Sisodia S., Müller U. (2004) Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23, 4106–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soba P., Eggert S., Wagner K., Zentgraf H., Siehl K., Kreger S., Löwer A., Langer A., Merdes G., Paro R., Masters C. L., Müller U., Kins S., Beyreuther K. (2005) Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 24, 3624–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young-Pearse T. L., Bai J., Chang R., Zheng J. B., LoTurco J. J., Selkoe D. J. (2007) A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 27, 14459–14469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beher D., Hesse L., Masters C. L., Multhaup G. (1996) Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J. Biol. Chem. 271, 1613–1620 [DOI] [PubMed] [Google Scholar]
- 22.Kibbey M. C., Jucker M., Weeks B. S., Neve R. L., Van Nostrand W. E., Kleinman H. K. (1993) beta-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc. Natl. Acad. Sci. USA 90, 10150–10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young-Pearse T. L., Chen A. C., Chang R., Marquez C., Selkoe D. J. (2008) Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 3, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perreau V. M., Orchard S., Adlard P. A., Bellingham S. A., Cappai R., Ciccotosto G. D., Cowie T. F., Crouch P. J., Duce J. A., Evin G., Faux N. G., Hill A. F., Hung Y. H., James S. A., Li Q. X., Mok S. S., Tew D. J., White A. R., Bush A. I., Hermjakob H., Masters C. L. (2010) A domain level interaction network of amyloid precursor protein and Abeta of Alzheimer’s disease. Proteomics 10, 2377–2395 [DOI] [PubMed] [Google Scholar]
- 25.Cheung H. N., Dunbar C., Mórotz G. M., Cheng W. H., Chan H. Y., Miller C. C., Lau K. F. (2014) FE65 interacts with ADP-ribosylation factor 6 to promote neurite outgrowth. FASEB J. 28, 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waite A., Brown S. C., Blake D. J. (2012) The dystrophin–glycoprotein complex in brain development and disease. Trends Neurosci. 35, 487–496 [DOI] [PubMed] [Google Scholar]
- 27.Mendell J. R., Boué D. R., Martin P. T. (2006) The congenital muscular dystrophies: recent advances and molecular insights. Pediatr. Dev. Pathol. 9, 427–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devisme L., Bouchet C., Gonzalès M., Alanio E., Bazin A., Bessières B., Bigi N., Blanchet P., Bonneau D., Bonnières M., Bucourt M., Carles D., Clarisse B., Delahaye S., Fallet-Bianco C., Figarella-Branger D., Gaillard D., Gasser B., Delezoide A. L., Guimiot F., Joubert M., Laurent N., Laquerrière A., Liprandi A., Loget P., Marcorelles P., Martinovic J., Menez F., Patrier S., Pelluard F., Perez M. J., Rouleau C., Triau S., Attié-Bitach T., Vuillaumier-Barrot S., Seta N., Encha-Razavi F. (2012) Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain 135, 469–482 [DOI] [PubMed] [Google Scholar]
- 29.Sciandra F., Gawlik K. I., Brancaccio A., Durbeej M. (2007) Dystroglycan: a possible mediator for reducing congenital muscular dystrophy? Trends Biotechnol. 25, 262–268 [DOI] [PubMed] [Google Scholar]
- 30.Chang Y., Tesco G., Jeong W. J., Lindsley L., Eckman E. A., Eckman C. B., Tanzi R. E., Guénette S. Y. (2003) Generation of the beta-amyloid peptide and the amyloid precursor protein C-terminal fragment gamma are potentiated by FE65L1. J. Biol. Chem. 278, 51100–51107 [DOI] [PubMed] [Google Scholar]
- 31.Kasaikina M. V., Fomenko D. E., Labunskyy V. M., Lachke S. A., Qiu W., Moncaster J. A., Zhang J., Wojnarowicz M. W. Jr., Natarajan S. K., Malinouski M., Schweizer U., Tsuji P. A., Carlson B. A., Maas R. L., Lou M. F., Goldstein L. E., Hatfield D. L., Gladyshev V. N. (2011) Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J. Biol. Chem. 286, 33203–33212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moncaster J. A., Pineda R., Moir R. D., Lu S., Burton M. A., Ghosh J. G., Ericsson M., Soscia S. J., Mocofanescu A., Folkerth R. D., Robb R. M., Kuszak J. R., Clark J. I., Tanzi R. E., Hunter D. G., Goldstein L. E. (2010) Alzheimer’s disease amyloid-beta links lens and brain pathology in Down syndrome. PLoS ONE 5, e10659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldstein L. E., Muffat J. A., Cherny R. A., Moir R. D., Ericsson M. H., Huang X., Mavros C., Coccia J. A., Faget K. Y., Fitch K. A., Masters C. L., Tanzi R. E., Chylack L. T. Jr., Bush A. I. (2003) Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet 361, 1258–1265 [DOI] [PubMed] [Google Scholar]
- 34.Meng H., Janssen P. M., Grange R. W., Yang L., Beggs A. H., Swanson L. C., Cossette S. A., Frase A., Childers M. K., Granzier H., Gussoni E., Lawlor M. W. (2014) Tissue triage and freezing for models of skeletal muscle disease. J. Vis. Exp. 89, 51586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danysh B. P., Duncan M. K. (2009) The lens capsule. Exp. Eye Res. 88, 151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossi M., Morita H., Sormunen R., Airenne S., Kreivi M., Wang L., Fukai N., Olsen B. R., Tryggvason K., Soininen R. (2003) Heparan sulfate chains of perlecan are indispensable in the lens capsule but not in the kidney. EMBO J. 22, 236–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng H., Jiang M., Trumbauer M. E., Sirinathsinghji D. J., Hopkins R., Smith D. W., Heavens R. P., Dawson G. R., Boyce S., Conner M. W., Stevens K. A., Slunt H. H., Sisoda S. S., Chen H. Y., Van der Ploeg L. H. (1995) beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81, 525–531 [DOI] [PubMed] [Google Scholar]
- 38.Ring S., Weyer S. W., Kilian S. B., Waldron E., Pietrzik C. U., Filippov M. A., Herms J., Buchholz C., Eckman C. B., Korte M., Wolfer D. P., Müller U. C. (2007) The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 27, 7817–7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang P., Yang G., Mosier D. R., Chang P., Zaidi T., Gong Y. D., Zhao N. M., Dominguez B., Lee K. F., Gan W. B., Zheng H. (2005) Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-like protein 2. J. Neurosci. 25, 1219–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weyer S. W., Klevanski M., Delekate A., Voikar V., Aydin D., Hick M., Filippov M., Drost N., Schaller K. L., Saar M., Vogt M. A., Gass P., Samanta A., Jäschke A., Korte M., Wolfer D. P., Caldwell J. H., Müller U. C. (2011) APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 30, 2266–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeda S., Kondo M., Sasaki J., Kurahashi H., Kano H., Arai K., Misaki K., Fukui T., Kobayashi K., Tachikawa M., Imamura M., Nakamura Y., Shimizu T., Murakami T., Sunada Y., Fujikado T., Matsumura K., Terashima T., Toda T. (2003) Fukutin is required for maintenance of muscle integrity, cortical histiogenesis and normal eye development. Hum. Mol. Genet. 12, 1449–1459 [DOI] [PubMed] [Google Scholar]
- 42.Bassez G., Chapoy E., Bastuji-Garin S., Radvanyi-Hoffman H., Authier F. J., Pellissier J. F., Eymard B., Gherardi R. K. (2008) Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fiber central nucleation and scattered atrophy. J. Neuropathol. Exp. Neurol. 67, 319–325 [DOI] [PubMed] [Google Scholar]
- 43.Schoser B. G., Schneider-Gold C., Kress W., Goebel H. H., Reilich P., Koch M. C., Pongratz D. E., Toyka K. V., Lochmüller H., Ricker K. (2004) Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve 29, 275–281 [DOI] [PubMed] [Google Scholar]
- 44.De Castro Rodrigues A., Andreo J. C., Rosa G. M. Jr., dos Santos N. B., Moraes L. H., Lauris J. R. (2007) Fat cell invasion in long-term denervated skeletal muscle. Microsurgery 27, 664–667 [DOI] [PubMed] [Google Scholar]
- 45.Greiling T. M., Clark J. I. (2008) The transparent lens and cornea in the mouse and zebra fish eye. Semin. Cell Dev. Biol. 19, 94–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beebe D. C. (2008) Maintaining transparency: a review of the developmental physiology and pathophysiology of two avascular tissues. Semin. Cell Dev. Biol. 19, 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Francis P. J., Berry V., Moore A. T., Bhattacharya S. (1999) Lens biology: development and human cataractogenesis. Trends Genet. 15, 191–196 [DOI] [PubMed] [Google Scholar]
- 48.Truscott R. J. (2003) Human cataract: the mechanisms responsible; light and butterfly eyes. Int. J. Biochem. Cell Biol. 35, 1500–1504 [DOI] [PubMed] [Google Scholar]
- 49.Hejtmancik J. F. (2008) Congenital cataracts and their molecular genetics. Semin. Cell Dev. Biol. 19, 134–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benedek G. B. (1997) Cataract as a protein condensation disease: the Proctor Lecture. Invest. Ophthalmol. Vis. Sci. 38, 1911–1921 [PubMed] [Google Scholar]
- 51.Gilmour D. T., Lyon G. J., Carlton M. B., Sanes J. R., Cunningham J. M., Anderson J. R., Hogan B. L., Evans M. J., Colledge W. H. (1998) Mice deficient for the secreted glycoprotein SPARC/osteonectin/BM40 develop normally but show severe age-onset cataract formation and disruption of the lens. EMBO J. 17, 1860–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ralston E., Lu Z., Biscocho N., Soumaka E., Mavroidis M., Prats C., Lømo T., Capetanaki Y., Ploug T. (2006) Blood vessels and desmin control the positioning of nuclei in skeletal muscle fibers. J. Cell. Physiol. 209, 874–882 [DOI] [PubMed] [Google Scholar]
- 53.Willmann R., Possekel S., Dubach-Powell J., Meier T., Ruegg M. A. (2009) Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul. Disord. 19, 241–249 [DOI] [PubMed] [Google Scholar]
- 54.Lambrechts A., Kwiatkowski A. V., Lanier L. M., Bear J. E., Vandekerckhove J., Ampe C., Gertler F. B. (2000) cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J. Biol. Chem. 275, 36143–36151 [DOI] [PubMed] [Google Scholar]
- 55.Narindrasorasak S., Lowery D. E., Altman R. A., Gonzalez-DeWhitt P. A., Greenberg B. D., Kisilevsky R. (1992) Characterization of high affinity binding between laminin and Alzheimer’s disease amyloid precursor proteins. Lab. Invest. 67, 643–652 [PubMed] [Google Scholar]
- 56.Beyreuther K., Multhaup G., Mönning U., Sandbrink R., Beher D., Hesse L., Small D. H., Masters C. L. (1996) Regulation of APP expression, biogenesis and metabolism by extracellular matrix and cytokines. Ann. N. Y. Acad. Sci. 777, 74–76 [DOI] [PubMed] [Google Scholar]
- 57.Hoe H. S., Lee K. J., Carney R. S., Lee J., Markova A., Lee J. Y., Howell B. W., Hyman B. T., Pak D. T., Bu G., Rebeck G. W. (2009) Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 29, 7459–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cáceres J., Brandan E. (1997) Interaction between Alzheimer’s disease beta A4 precursor protein (APP) and the extracellular matrix: evidence for the participation of heparan sulfate proteoglycans. J. Cell. Biochem. 65, 145–158 [DOI] [PubMed] [Google Scholar]
- 59.Frederikse P. H., Ren X. O. (2002) Lens defects and age-related fiber cell degeneration in a mouse model of increased AbetaPP gene dosage in Down syndrome. Am. J. Pathol. 161, 1985–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Da Cunha R. P., Moreira J. B. (1996) Ocular findings in Down’s syndrome. Am. J. Ophthalmol. 122, 236–244 [DOI] [PubMed] [Google Scholar]
- 61.Jun G., Moncaster J. A., Koutras C., Seshadri S., Buros J., McKee A. C., Levesque G., Wolf P. A., St George-Hyslop P., Goldstein L. E., Farrer L. A. (2012) δ-Catenin is genetically and biologically associated with cortical cataract and future Alzheimer-related structural and functional brain changes. PLoS ONE 7, e43728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kerbage C., Sadowsky C. H., Tariot P. N., Agronin M., Alva G., Turner F. D., Nilan D., Cameron A., Cagle G. D., Hartung P. D. (In press) Detection of amyloid β signature in the lens and its correlation in the brain to aid in the diagnosis of Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Michael R., Rosandić J., Montenegro G. A., Lobato E., Tresserra F., Barraquer R. I., Vrensen G. F. (2013) Absence of beta-amyloid in cortical cataracts of donors with and without Alzheimer’s disease. Exp. Eye Res. 106, 5–13 [DOI] [PubMed] [Google Scholar]