

Abstract
The ability of nonsteroidal anti-inflammatory drugs (NSAIDs) to inhibit cyclooxygenase (Cox)-1 and Cox-2 underlies the therapeutic efficacy of these drugs, as well as their propensity to damage the gastrointestinal (GI) epithelium. This toxic action greatly limits the use of NSAIDs in inflammatory bowel disease (IBD) and other chronic pathologies. Fatty acid amide hydrolase (FAAH) degrades the endocannabinoid anandamide, which attenuates inflammation and promotes GI healing. Here, we describe the first class of systemically active agents that simultaneously inhibit FAAH, Cox-1, and Cox-2 with high potency and selectivity. The class prototype 4 (ARN2508) is potent at inhibiting FAAH, Cox-1, and Cox-2 (median inhibitory concentration: FAAH, 0.031 ± 0.002 µM; Cox-1, 0.012 ± 0.002 µM; and Cox-2, 0.43 ± 0.025 µM) but does not significantly interact with a panel of >100 off targets. After oral administration in mice, ARN2508 engages its intended targets and exerts profound therapeutic effects in models of intestinal inflammation. Unlike NSAIDs, ARN2508 causes no gastric damage and indeed protects the GI from NSAID-induced damage through a mechanism that requires FAAH inhibition. Multitarget FAAH/Cox blockade may provide a transformative approach to IBD and other pathologies in which FAAH and Cox are overactive.—Sasso, O., Migliore, M., Habrant, D., Armirotti, A., Albani, C., Summa, M., Moreno-Sanz, G., Scarpelli, R., Piomelli, D. Multitarget fatty acid amide hydrolase/cyclooxygenase blockade suppresses intestinal inflammation and protects against nonsteroidal anti-inflammatory drug-dependent gastrointestinal damage.
Keywords: anandamide, cannabinoid receptor, inflammatory bowel disease, multitarget inhibitors
Nonsteroidal anti-inflammatory drugs (NSAIDs), the most widely used first-line therapy for pain and inflammation, exert their therapeutic effects by inhibiting cyclooxygenase (Cox)-1 and Cox-2, 2 intracellular enzymes that initiate the conversion of membrane-derived arachidonic acid (AA) into inflammatory prostanoids such as prostaglandin E2 (PGE2) and prostacyclin (PGI2) (1). Along with these agents, cells in inflamed tissues can also generate lipid mediators that oppose the inflammatory response (2, 3). One such mediator is anandamide (AEA), an endogenous cannabinoid receptor agonist that is released from macrophages and T lymphocytes following activation of pattern recognition receptors (4, 5) and engages a combination of cannabinoid receptor 1 (CB1)- and cannabinoid receptor 2 (CB2)-dependent mechanisms to suppress neutrophil migration (6) and prevent dendritic cell and T-cell recruitment (7, 8). Consistent with a regulatory role of AEA in inflammation, genetic or pharmacologic interventions that block the deactivating hydrolysis of this compound into AA and ethanolamine, catalyzed by fatty acid amide hydrolase (FAAH) (9), attenuate inflammatory responses in animal models (6, 10). Despite their opposing effects on inflammation, AEA and prostanoids exert similar protective actions on the gastrointestinal (GI) mucosa (11, 12). Indeed, blockade of PGE2 formation is thought to be the primary, albeit not only, cause of NSAID-induced damage to the GI tract, a common and serious side effect of this drug class (13).
Heightened expression of Cox-2 contributes to pathology in chronic inflammation (1). In some inflammatory conditions, FAAH-mediated AEA degradation is also enhanced (8, 14). For example, in the intestinal mucosa of subjects with inflammatory bowel disease (IBD), both Cox-2 (15) and FAAH are expressed at abnormally high levels (16, 17). Elevated FAAH activity weakens AEA’s ability to temper inflammation and protect the GI mucosa and concomitantly makes more AA available for prostanoid biosynthesis. On the other hand, increased Cox-2 activity strengthens prostanoid signaling as well as formation of inflammatory mediators generated by Cox-2-dependent oxygenation of AEA (18). Thus, the concurrent up-regulation of FAAH and Cox-2 may set in motion a loop that exacerbates inflammation by amplifying inflammatory Cox-dependent signals at the expense of defensive AEA-dependent responses (Fig. 1).
Figure 1.

FAAH and Cox-2 expression is elevated in multiple inflammatory conditions such as IBD. Excess activity of these enzymes may exacerbate inflammation by lowering levels of anti-inflammatory and tissue-protective AEA while concurrently increasing levels of AA and its inflammatory prostanoid (PG) metabolites.
A corollary of this hypothesis is that drugs that target both FAAH and Cox should have substantial anti-inflammatory efficacy combined with reduced GI toxicity. This idea has not been tested yet due to the lack of adequate tools. Indeed, whereas several mixed FAAH/Cox inhibitors have been reported in the literature (19, 20), these compounds generally lack the pharmacologic and pharmacokinetic properties needed for in vivo testing. Here, we describe the first class of designed multiple ligands that target FAAH, Cox-1, and Cox-2 with high potency, selectivity, and oral bioavailability. We show that the prototype member of this class, ARN2508 (Table 1), inhibits FAAH and Cox activities in vivo and suppresses intestinal inflammation without causing GI damage. In fact, ARN2508 protects the GI tract from NSAID-induced injury through a mechanism that depends on FAAH inhibition.
TABLE 1.
Inhibitory potencies (IC50) of compounds on rat FAAH, ovine Cox-1, and human Cox-2
| Compound | Structure | FAAH IC50 (μM) | Cox-1 IC50 (μM) | Cox-1 IC50 (μM) |
|---|---|---|---|---|
| URB597, 1 | 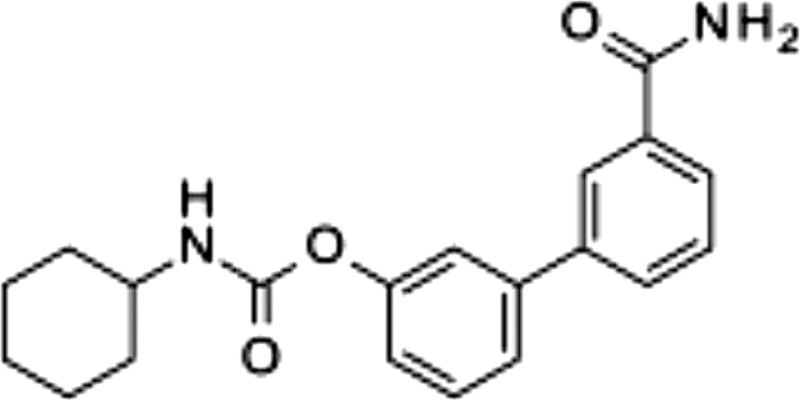 |
0.0017 ± 0.001 | >100 | >100 |
| Flurbiprofen, 2 | 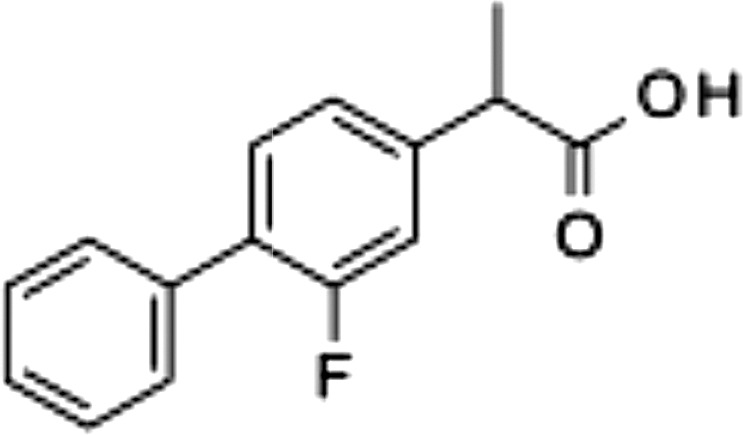 |
>100 | 0.15 ± 0.01 | 1.0 ± 0.5 |
| 3 | 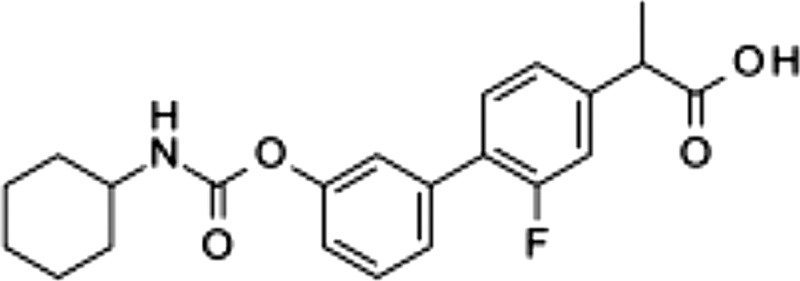 |
8.2 ± 2.4 | 7.9 ± 2.5 | >100 |
| ARN2508, 4 | 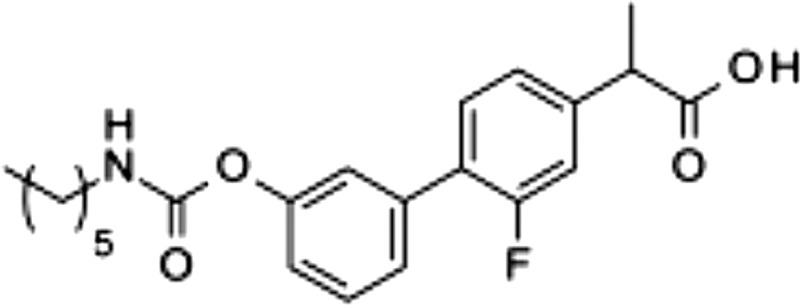 |
0.031 ± 0.002 | 0.012 ± 0.002 | 0.43 ± 0.025 |
| 5 | 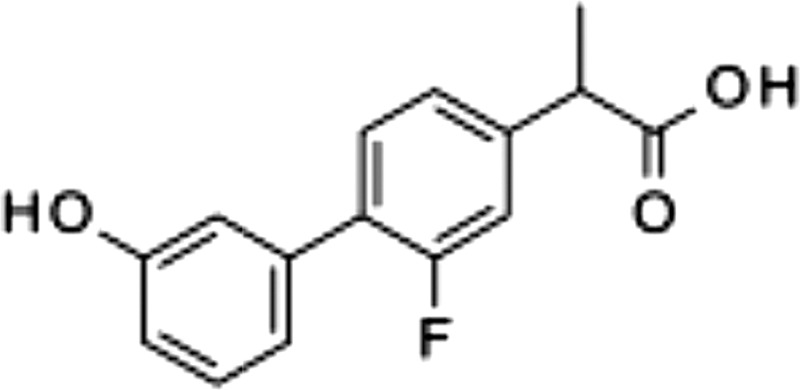 |
>100 | 0.29 ± 0.11 | 3.2 ± 2.5 |
| 6 | 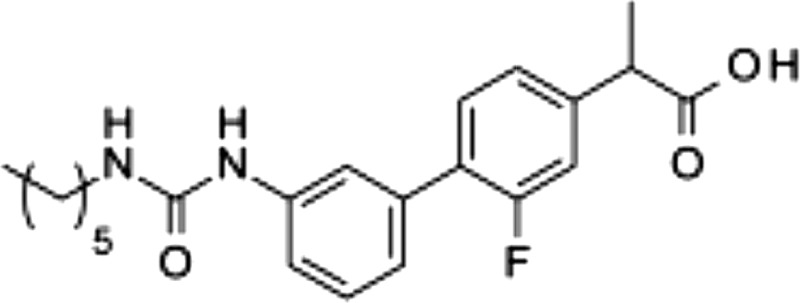 |
88 ± 3.3 | 0.014 ± 0.003 | 0.56 ± 0.15 |
| 7 | 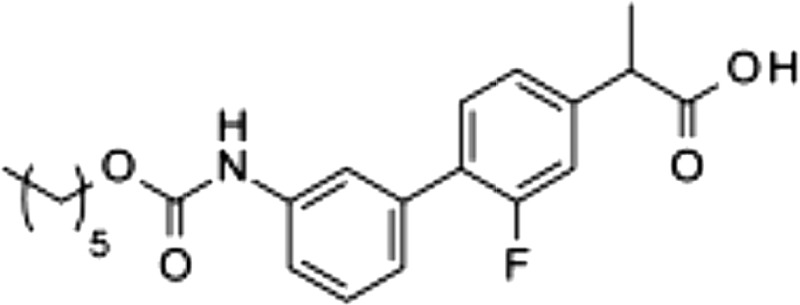 |
14.9 ± 1.9 | 0.030 ± 0.013 | 0.17 ± 0.01 |
| 8 | 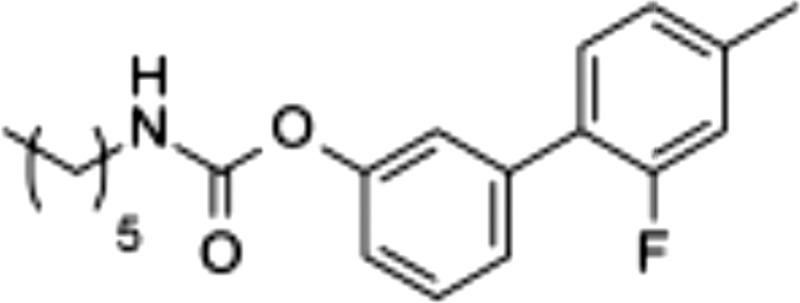 |
0.026 ± 0.011 | >100 | >100 |
MATERIALS AND METHODS
Chemicals and reagents
PF-04457845 and URB597 were prepared following reported procedures (21, 22). Flurbiprofen, AM251, WIN-55212-2, GW-6471, 5-aminosalicylic acid (5-ASA), λ-carrageenan, dextran sodium sulfate (DSS), and 2,4,6-trinitrobenzene sulfonic acid (TNBS) were purchased from Sigma-Aldrich (Milan, Italy). Detailed synthetic procedures for compounds 3–8 will be reported elsewhere (Migliore, Habrant, Sasso, Albani, Tarozzo, Mandrup Bertozzi, Scarpelli and Piomelli, unpublished results).
In vitro metabolic stability
ARN2508 was added to mouse liver microsomes (1.25 mg/ml) in Tris-HCl buffer [0.1 M (pH 7.4), final concentration 5 μM in 0.1% DMSO], and the mixture was incubated at 37°C for 15 minutes with shaking. Reactions were started by adding the following cofactors: NADP+ (1 mM), glucose-6-phosphate (20 mM), magnesium chloride (2 mM), and glucose-6-phosphate dehydrogenase (2 U) (NADPH-dependent oxidation system), or UDP glucaric acid (5 mM), saccharic acid 1,4 lactone (5 mM), and magnesium chloride (2 mM) (glucuronidation system). Samples (50 μl) were collected at various time points and diluted with MeCN (0.15 ml) containing warfarin (0.5 μM) as internal standard. A reference incubation, with microsomes but no cofactors, was sampled at the end of the experiment. After mixing and centrifugation, samples of the supernatants (3 μl) were analyzed by liquid chromatography-mass spectrometry (LC/MS). The following multiple-reaction monitoring (MRM) transitions were monitored: 388 → 215 at 30 electron volts (eV) of collision energy.
Enzyme assays
FAAH activity was measured in rat brain homogenates (50 µg protein) as previously described (23). Homogenates were incubated in the presence of inhibitors for 10 minutes at 37°C prior to substrate addition. Cox activity was measured using a commercial enzyme immunoassay (EIA) kit (Cayman Chemical, Tallinn, Estonia). Briefly, 50 nM ovine Cox-1 or human Cox-2 was incubated with ARN2508 for 10 minutes in a solution containing 100 mM Tris-HCl (pH 8.0), 5 mM EDTA, and 2 mM phenol. Following the preincubation, dialysis was performed using Slide-A-Lyzer Dialysis Cassettes (0.5–3 ml; 10 kDa molecular weight cut-off; Thermo Fisher Scientific, Illkirch, France) in agitation for 16 hours (overnight) at 4°C. Dialysis buffer was used 1000 times the volume of the sample. After dialysis, AA (5 μM) was added for 2 minutes at 37°C. The reaction was stopped with 1 M HCl, and a stannous chloride solution was added to reduce PGH2 to PGF2α. We measured PGF2α levels using a competitive EIA.
Pharmacokinetic analyses
ARN2508 was dissolved in sterile saline containing PEG-400 (10%) and Tween 80 (10%) and was administered to mice by either intravenous or oral route (n = 3 each). Blood (0.2 ml) was collected from mice decapitated under anesthesia into heparin-containing tubes at various time points after intravenous (5, 15, and 30 minutes; 1, 2, and 4 hours) or oral administration (15 and 30 minutes; 1, 2, 4, 6, and 24 hours). Plasma fractions were prepared by centrifugation (3000 g for 10 minutes at 4°C), frozen in dry ice, and stored at −80°C. After thawing on ice and brief centrifugation, samples (50 μl) were transferred into 96-deep well plates and diluted with MeCN (150 μl) spiked with internal standard (warfarin, 0.5 μM). The plates were stirred for 3 minutes and centrifuged at 3000 g for 10 minutes at 4°C. Supernatants (80 μl) were transferred to 96-well plates and diluted with water (80 μl). Calibration curves were constructed using ARN2508 (1 nM to 10 μM) in PBS (pH 7.4) containing 10% MeCN. There were 3 quality control samples prepared by adding ARN2508 to mouse plasma (final concentrations: 20, 200, and 2000 nM). LC/MS analyses were conducted using an Acquity BEH C18 column (2.1 × 50 mm, 1.7 mm particle size; Waters, Milford, MA, USA) on a Xevo TQ LC-MS/MS system (Waters). A linear gradient was applied from 5 to 100% solvent B over 2 minutes: solvent A, water plus formic acid 0.1%; and solvent B, MeCN plus formic acid 0.1%. Injection volume was 3 μl, and flow rate was 0.5 ml/min. Mass spectrometry parameters were positive ion mode, capillary voltage 3 kV, cone voltage 25 V, source temperature 120°C, cone gas flow 20 L/h, desolvation gas flow 800 L/h, and desolvation temperature 400°C. The following MRM transitions were monitored: ARN2508, 388 → 215 at 30 eV collision energy; and warfarin, 309 → 163 and 309 → 251 at 18 and 16 eV collision energy, respectively. The linear regression of the calibration curve had an R2 value of 0.99. The limit of quantification was 5 nM. Recovery of ARN2508 was 90–95%. Data were analyzed using PKSolutions Excel application (Summit Research Service, Montrose, CO, USA).
Ex vivo lipid analyses
Tissue levels of AEA, palmitoylethanolamide (PEA), oleoylethanolamide (OEA), and 2-arachidonoyl-glycerol (2-AG) were measured by LC/MS as described (24). Briefly, snap-frozen tissue samples were weighed (∼7 mg) and homogenized in MeOH (1 ml) containing [2H4]-AEA, [2H4]-OEA, and [2H8]-2-AG (Cayman Chemical). Lipids were extracted with CHCl3 (2 vol), and the organic phases were washed with water (1 vol), collected, and dried under N2. The organic extracts were fractionated by open-bed silica gel column chromatography. AEA, OEA, PEA, and 2-AG were eluted with CHCl3:MeOH (9:1, vol/vol). Organic phases were evaporated under N2 and reconstituted in MeOH (0.1 ml). LC/MS analyses were conducted on a Xevo TQ LC-MS/MS system equipped with a BEH C18 column, using a linear gradient of MeCN in water. Quantification was performed monitoring the MRM transitions. Analyte peak areas were compared with a standard calibration curve (1 nM to 10 μM). Tissue levels of PGE2 and 6-keto-PGF1α were determined using ELISA kits (Abcam, Cambridge, United Kingdom), following the manufacturer’s instructions.
Animal models
Animals
We used adult male CD1 mice (25–30 g; Charles River Laboratories, Calco, Italy). All procedures were performed in accordance with the Ethical Guidelines of the International Association for the Study of Pain, Italian regulations on the protection of animals used for experimental and other scientific purposes (D.M. 116192), and European Economic Community regulations (O.J. of E.C. L. 358/1 12/18/1986). Mice were group housed in ventilated cages and had free access to food and water. They were maintained under a 12-hour light-dark cycle (lights on at 8:00 AM) at controlled temperature (21 ± 1°C) and relative humidity (55 ± 10%). Behavioral testing was performed during the light cycle (between 9:00 AM and 5:00 PM). Animals were killed by cervical dislocation under anesthesia. Experimenters were blinded to the treatment protocol at the time of the test.
Carrageenan-induced inflammation
We injected λ-carrageenan (1% wt/vol in sterile water, 50 μl) into the left hind paw of lightly restrained CD1 mice and measured edema with a plethysmometer and hyperalgesia with a Hargreaves apparatus (Ugo Basile, Comerio, Italy). Drugs were prepared daily and administered orally in a vehicle of 80% sterile saline/10% PEG-400/10% Tween 80 (10 μl per animal) immediately before carrageenan.
Flurbiprofen-induced gastric toxicity
Food-deprived mice received a single oral dose of flurbiprofen (3–30 mg/kg) (25) preceded by oral administration of vehicle, ARN2508 (3–100 mg/kg) or compound 8 (3–100 mg/kg). The mice were killed 4 h later by CO2 asphyxiation. The stomachs were removed, rinsed with PBS, and the stomach lining was photographed. Scoring was performed as described (26): red coloration, 0.5; spot ulcers, 1; and hemorrhagic streaks, 1.5. If 3 or 4 ulcers were observed, a value of 2 was added to the score. If ≥5 ulcers were observed, a value of 3 was added to the score. The ulceration index was the sum of these scores, with a maximal value of 6.
TNBS-induced colitis
TNBS (4 mg in 0.1 ml of 30% ethanol) was infused into the rectum of lightly anesthetized mice through a catheter inserted 3 cm proximally to the anus. Vehicle or drugs were administered orally (1–30 mg/kg) once daily for 7 days starting immediately before TNBS infusion. The mice were singly housed in wired-bottom cages lined with white absorbent paper, which was used to determine the average number of bowel movements per cage. Body weight changes were measured daily. At the end of the experiments, the animals were killed by cervical dislocation, the colon was removed, rinsed with saline, opened longitudinally, and macroscopic damage was scored under blinded conditions. The following scale was used (10): ulcers, 0.5 points for each 0.5 cm; adhesion, 0 points = absent, 1 point = 1 adhesion, and 2 points = ≥2 adhesions or adhesions to organs; and shortening of the colon, 1 point = >15%, and 2 points = >25% (based on a mean length of the untreated colon of 7.0 ± 0.3 cm; n = 6). The presence of hemorrhage, fecal blood, or diarrhea increased the score by 1 point for each additional feature.
DSS-induced colitis
As previously described (27), we elicited colon inflammation in male CD1 mice by allowing them to drink a 4% (wt/vol) solution of DSS (36–50 kDa) in water for 6 days. DSS-containing water was replaced every other day. At the end of day 6, the mice were given access to regular drinking water for 1 additional day before euthanasia. Control mice received normal drinking water from day 0 to day 7. The mice were examined daily for stool consistency, fecal blood, and weight loss. These features were used to determine the Disease Activity Index, as previously described (28). The scores were as follows: weight loss was graded 0 if body weight increased or remained within 1% of the baseline; 1 for 1–5% loss; 2 for 5–10% loss; 3 for 10–15% loss; or 4 for loss >15%. Stool consistency was graded 0 for no diarrhea, 2 for loose stool that did not stick to the anus, and 4 for liquid stool that did stick to the anus. The presence of fecal blood received a value of 0 when assigned for none, 2 for moderate, and 4 for gross bleeding. Macroscopic colon inflammation and damage were scored using the following 4 items: stool consistency, thickness, length, and length of inflamed area.
Statistical analyses
Data are expressed as the mean ± sem. Differences between groups were assessed by 1- or 2-way ANOVA followed by a Bonferroni post hoc test. Data were analyzed using GraphPad Prism 5.0 for Windows (GraphPad Software Incorporated, San Diego, CA, USA).
RESULTS
Discovery of a potent FAAH/Cox inhibitor
The existence of structural commonalities between the alkyl carbamic acid biphenyl-3yl-ester class of FAAH inhibitors such as URB597 (23) and members of the 2-arylpropionic acid class of NSAIDs such as flurbiprofen (29) allowed us to utilize a framework combination strategy (30) to design ligands that simultaneously target FAAH, Cox-1, and Cox-2. Our approach consisted of integrating key pharmacophoric elements of these 2 inhibitor classes around their common biphenyl core (Fig. 2). We first inserted the carbamate group of URB597, which is obligatory for FAAH inhibition (22), at the 3′ position of the distal phenyl group of flurbiprofen (Fig. 2), which prior studies have shown to be available for modulation (29). The compound obtained (compound 3; Table 1) was a moderate inhibitor of FAAH [median inhibitory concentration (IC50), 8.2 ± 2.4 μM; mean ± sem; n = 3] and Cox-1 (IC50, 7.9 ± 2.5 μM) and showed no activity toward Cox-2 (IC50, >100 μM). Nevertheless, systematic modification of the alkyl substituent of the carbamate group progressively enhanced potency at the 3 targets, eventually yielding a balanced multiple ligand that inhibited each of the enzymes with high potency (ARN2508; Table 1). In the lead optimization process, which will be reported elsewhere, we pursued concomitant Cox-1/Cox-2 inhibition because both isoforms contribute to inflammatory hyperalgesia (31).
Figure 2.
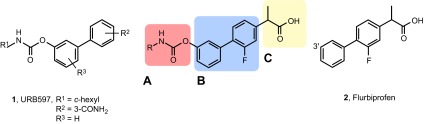
Design of multitarget inhibitors of FAAH, Cox-1, and Cox-2. General structure is shown of alkyl carbamic acid biphenyl-3yl-ester inhibitors (URB597, compound 1) and 2-arylpropionic acid Cox-1/Cox-2 inhibitors (flurbiprofen, compound 2). The new inhibitors integrate the alkyl carbamic acid group needed for FAAH inhibition (A) and the propionic acid group needed for Cox-1/Cox-2 inhibition (C) around a common biphenyl core (B).
Mechanism of inhibition
The carbamate group of URB597 binds covalently to the catalytic serine of FAAH, interrupting its activity (22, 32). Dialysis experiments showed that ARN2508 also irreversibly interacts with FAAH (Fig. 3A). Moreover, focused structure-activity relationship studies demonstrated that the ability of ARN2508 to inhibit FAAH depends on the presence of the carbamate moiety because removal of such moiety (as in compound 5) abrogated FAAH inhibition while preserving Cox inhibition (Table 1). The key role of the carbamate was confirmed by the finding that analogs of ARN2508 in which this group was replaced with a urea (compound 6) or was reversed (compound 7) showed a significant decrease in potency against FAAH, while retaining Cox-1 and Cox-2 inhibitory activities (Table 1).
Figure 3.

Effects of overnight dialysis on FAAH inhibition (A), Cox-1 inhibition (B), or Cox-2 inhibition (C) by ARN2508 (1 μM, 100 nM and 10 μM, respectively). Open bars, vehicle; closed bars, ARN2508. Results are expressed as means ± SEM of 3 independent determinations.
Flurbiprofen is a tight-binding inhibitor of Cox-1 and Cox-2, and its free carboxylate end, which takes part in a network of polar interactions in the active sites of these enzymes (33), is essential for this property (34). Consistent with those data, we found that 1) ARN2508 blocks both Cox-1 and Cox-2 in a functionally irreversible manner (Fig. 3B, C), and 2) removal of its carboxylate moiety (as in compound 8) caused a complete loss of activity toward both Cox isoforms, but not toward FAAH (Table 1).
Target selectivity
When tested at a concentration that fully inhibits FAAH, Cox-1, and Cox-2 (10 μM), ARN2508 had no effect on a panel of 30 biologically relevant targets, including lipid-metabolizing enzymes that contribute to the inflammatory response, such as 5-lipoxygenase, 12-lipoxygenase, 15-lipoxygenase, and secreted phospholipase A2 (type V) (Supplemental Table S1). Furthermore, ARN2508 had no effect on the activity of monoacylglycerol lipase, the primary enzyme involved in the deactivation of the endocannabinoid 2-arachidonoyl-glycerol (2-AG), N-acylethanolamide amide hydrolase, which preferentially hydrolyzes PEA and OEA in macrophages, and did not significantly affect other inflammation-related targets, such as iNOS, histone deacetylase (types 3, 4, 6, and 11), sirtuin 1 and 2, and phosphodiesterases 1B, 2A1, 3A, 4D2, and 5 (Supplemental Table S1). Even further, ARN2508 did not interact with 75 common receptors, ion channels, and neurotransmitter transporters, comprising cannabinoid CB1 and CB2; prostaglandin EP1, EP3, EP4, and prostacyclin receptor; adenosine A1, A2A, and A3; adrenergic α1, α2, β1, and β2; bradykinin B1 and B2; endothelin A and B; histamine H1 and H2; leukotriene BLT1 and cysteinyl leukotriene receptor 1; and protease-activated receptor-2 (Supplemental Table S2). Collectively, the results outlined above identify ARN2508 as the first highly potent and selective inhibitor of FAAH, Cox-1, and Cox-2.
Target engagement
The data reported in Table 2 show that ARN2508 is stable in mouse plasma and liver microsomes supplemented with an NADPH-regenerating system but is metabolized in the presence of cofactors that sustain glucuronic acid conjugation. Pharmacokinetic experiments in mice support the possibility that ARN2508 undergoes glucuronidation in the liver: the compound was absorbed well after oral administration but was quickly eliminated from the circulation (Fig. 4A) and found in bile as glucuronic acid conjugate (Fig. 4B; structural identification in Supplemental Fig. S1). The time course of this response suggests that ARN2508 is subjected to substantial enterohepatic cycling, as previously documented for flurbiprofen (35, 36) and consistent with the compound’s high fractional absorption (Table 2) (37).
TABLE 2.
Metabolic and pharmacokinetic properties of ARN2508
| Mouse plasma (t1/2= 106 ± 17 min) | Mouse microsome fractions |
|
|---|---|---|
| NADP 102 ± 1 min | UDPG 18 ± 3 min | |
| Cmax (ng/ml) | 2915 | 1616 |
| Tmax (min) | 5 | 15 |
| AUC (ng × min/ml) | 102,273 | 105,290 |
| Volume of distribution (ml/kg) | 1929 | 1703 |
| Clearance (ml/min/kg) | 28.8 | 25.2 |
| t1/2 (min, elimination phase) | 46 | 47 |
| F% | 103 | |
AUC, area under the curve; Cmax, maximal concentration in plasma; F%, fractional absorption; NADP, NADPH-regenerating system; Tmax, time (min) at which Cmax is reached; UDPG, glucuronidation-enabling system (see Materials and Methods).
Figure 4.
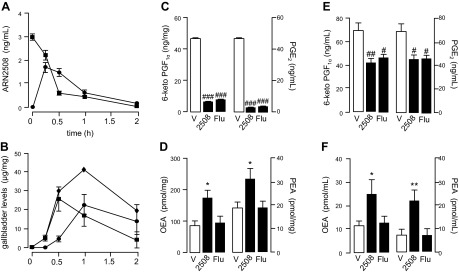
Pharmacokinetic and pharmacodynamic profile of ARN2508 in mice. Plasma concentrations of ARN2508 after intravenous (squares, 3 mg/kg) or oral (circles, 3 mg/kg) administration (A). Levels of ARN2508 (squares), conjugate of ARN2508 with glucuronic acid (circles) and sum of the 2 (diamonds) in bile after oral administration of ARN2508 (30 mg/kg) (B). Levels of Cox and FAAH metabolites in gastric mucosa after oral administration of ARN2508 (30 mg/kg): 6-keto-PGF1a (left) and PGE2 (right) (C); OEA (left) and PEA (right) (D). Plasma levels of Cox and FAAH metabolites after oral administration of ARN2508 (30 mg/kg): 6-keto-PGF1a (left) and PGE2 (right) (E); OEA (left) and PEA (right) (F). Results are expressed as the mean ± sem of 3–6 independent determinations. *P < 0.05, **P < 0.01 vs. vehicle, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. vehicle.
Despite its efficient elimination, ARN2508 strongly engaged its molecular targets in vivo. Oral administration of a 30 mg/kg dose in mice suppressed Cox-mediated biosynthesis of PGE2 and PGI2 (measured as its stable derivative, 6-keto-PGF1α) in stomach mucosa, along with hydrolysis of FAAH substrates OEA and PEA (Fig. 4C, D). Furthermore, ARN2508 decreased PGE2 and 6-keto-PGF1α levels and increased OEA and PEA levels in circulation (Fig. 4E, F). Those effects were accompanied by attenuation of hyperalgesia and edema elicited by paw injection of carrageenan (Supplemental Fig. S2). By contrast, as expected of an NSAID, flurbiprofen lowered prostanoid content in stomach and blood (Fig. 4C, E) without affecting OEA and PEA in either compartment (Fig. 4D, F). These findings indicate that ARN2508 is orally available and systemically active.
Gastroprotective effects
Because flurbiprofen and ARN2508 abolish PGE2 biosynthesis in gastric mucosa (Fig. 4C), we expected both compounds to damage the epithelial lining of the stomach (11). However, whereas flurbiprofen caused pronounced dose-dependent gastric injury, ARN2508 had no such effect (Fig. 5A) and in fact protected the stomach of mice exposed to flurbiprofen (Fig. 5B). This protective action was likely due to the ability of ARN2508 to enhance, via FAAH blockade, an intrinsic defensive mechanism mediated by the endocannabinoid AEA because 1) damage induced by flurbiprofen was accompanied by increased local formation of AEA (Fig. 5C), but not OEA, PEA, or 2-AG, another endocannabinoid substance present in the gut (Fig. 5D); 2) coadministration of the CB1 receptor antagonist AM251 (1 mg/kg, oral) heightened the gastrotoxic effects of flurbiprofen (Fig. 5E); 3) coadministration of AM251 unmasked a similar toxic action of ARN2508 (Fig. 5E), whereas combining AM251 with compound 8 (30 mg/kg), a closely related analog of ARN2508 that inhibits FAAH but not Cox-1 or Cox-2 (Table 1), did not cause stomach injury and blunted damage evoked by flurbiprofen (Fig. 5F); and finally 4) administration of the cannabinoid agonist WIN 55,212-2 (1 mg/kg, oral) (Fig. 5G) or deletion of the Faah gene by homologous recombination (Fig. 5H) protected mice against the effects of flurbiprofen. An economic interpretation of these data is that gastric damage caused by the NSAID flurbiprofen engages a defensive CB1-dependent mechanism, mediated by AEA, which is magnified by FAAH blockade.
Figure 5.

Gastroprotective properties of ARN2508 in mice. A) Effects of vehicle (open bar), ARN2508 (closed bars), or flurbiprofen (shaded bars) on stomach integrity. B) Effects of ARN2508 on gastric damage caused by flurbiprofen. C) Levels of AEA in gastric mucosa 4 hours after administration of flurbiprofen. D) Levels of PEA (left), OEA (center), and 2-AG (right) in gastric mucosa 4 hours after administration of flurbiprofen. E) Effects of vehicle (open bar) or CB1 antagonist AM251 on stomach integrity: AM251 alone (open bar), AM251 plus flurbiprofen (shaded bars), and AM251 plus ARN2508 (closed bars). F) Effects of vehicle (open bar) or single-target FAAH inhibitor 8 on stomach integrity: compound 8 alone (closed bars), and compound 8 plus flurbiprofen (shaded bars). G) Effects of vehicle (open bar) or cannabinoid agonist WIN 55,212-2 (WIN) on stomach integrity: WIN or flurbiprofen (closed bars), and WIN plus flurbiprofen (shaded bar). H) Effects of flurbiprofen on stomach integrity in wild-type (open bar) or FAAH-null mice (closed bar). Compounds were administered orally; doses are in milligrams per kilograms. Results are expressed as the mean ± sem (n = 6). *P < 0.05, ***P < 0.001 vs. vehicle; ○P < 0.05, ○○P < 0.01, ○○○P < 0.001 vs. vehicle.
Anti-inflammatory effects in models of colon inflammation
Heightened Cox-2 expression may contribute to pathology in IBD (15, 38, 39). Nevertheless, NSAIDs cannot be used in this disease due to their propensity to cause enteric injury through a mechanism that partially depends on Cox inhibition (40). We modeled this effect in mice by administering flurbiprofen along with the inflammatory agent, TNBS (41).
Rectal instillation of TNBS produced intestinal inflammation and increased mortality (Fig. 6). Administration of flurbiprofen exacerbated these effects (Fig. 6A–C). By contrast, confirming previous studies with single-target FAAH inhibitors (10), treatment with compound 8 attenuated the response elicited by TNBS alone (Fig. 6D–F) or by the combination of TNBS plus flurbiprofen (Fig. 6G–I). In contrast with flurbiprofen, ARN2508 was highly potent and effective at reducing TNBS-induced inflammation and mortality, fully suppressing both responses at the maximal dose tested (Fig. 7A–C). Notably, ARN2508 was more efficacious than 5-ASA, which is clinically used to treat IBD (Supplemental Fig. S3), and of 2 structurally distinct single-target FAAH inhibitors (URB597 and PF-04457845) (Supplemental Fig. S4). Investigations into the mechanism of action of ARN2508 showed that the effects of this compound were blunted, but not completely eliminated, by CB1 blockade (AM251, 1 mg/kg, oral) (Fig. 7D, E). This suggests that the compound acts in part by enhancing AEA activity at CB1 receptors. Supporting this idea, biochemical analyses of colon mucosa showed that AEA levels, which were lower in TNBS-treated mice than controls, were normalized by ARN2508 (Fig. 7F). In addition to AEA, ARN2508 reinstated normal mucosal levels of the bioactive FAAH substrates PEA and OEA, which were reduced by TNBS (Fig. 7F), as previously shown for other inflammatory stimuli (42, 43). Because PEA and OEA attenuate inflammation by engaging peroxisome proliferator-activated receptor-α (PPAR-α) (17, 42–44), we asked whether this nuclear receptor might also contribute to the effects of ARN2508. Figure 7D, E shows that the PPAR-α antagonist GW6471 (1 mg/kg, oral) reduced the anti-inflammatory actions of ARN2508 to a similar extent as CB1 blockade. The findings suggest that ARN2508 prevents TNBS-induced inflammation by enhancing the activity of an intrinsic defensive mechanism mediated by AEA (acting at CB1) and PEA/OEA (acting at PPAR-α).
Figure 6.

Effects of test compounds on TNBS-induced colon inflammation in mice. Effects of flurbiprofen (closed bars or symbols) on colon length (A), macroscopic inflammation score (B), and animal survival (C). Effects of compound 8 (closed bars or symbols) on colon length (D), macroscopic score (E), and survival (F). Effects of coadministration of compound 8 plus flurbiprofen on colon length (G), macroscopic score (H), and survival (I). Shaded bars indicate naive mice; open bars represent mice treated with vehicle. Compounds were administered orally; doses are in milligrams per kilograms. Results are expressed as the mean ± sem (n = 6). *P < 0.05, **P < 0.01 vs. vehicle; ○P < 0.05, ○○P < 0.01 vs. vehicle.
Figure 7.
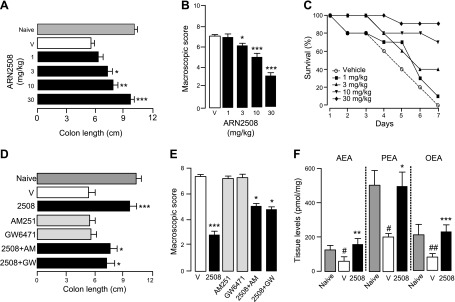
Anti-inflammatory effects of ARN2508 on TNBS-induced colon inflammation in mice. Effects of ARN2508 (closed bars or symbols) on colon length (A), macroscopic score (B), and animal survival (C). Effects of CB1 antagonist AM251 (AM) or PPAR-α antagonist GW-6471 (GW), administered alone (shaded bars) or in combination with ARN2508 (closed bars), on colon length (D) and macroscopic score (E). F) Levels of AEA, PEA, and OEA in colon from naive mice, mice treated with TNBS plus vehicle, and mice treated with TNBS plus ARN2508. All compounds were administered orally; doses are in milligrams per kilograms. Shaded bars indicate naive mice; open bars represent vehicle-treated mice. Results are expressed as the mean ± sem (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle; #P < 0.05, ##P < 0.01 vs. vehicle.
There is no animal model of IBD. Nevertheless, the ability to reverse the inflammatory state established following rectal infusion of TNBS or oral administration of DSS is suggestive of therapeutic efficacy in this disease (45, 46). When administered after induction of inflammation, ARN2508 was highly effective in both the TNBS and DSS models. A 6-day regimen with ARN2508, starting on the second day after TNBS instillation, significantly decreased inflammatory symptoms and mortality, compared to vehicle-treated mice (Fig. 8). Similarly, a 4-day regimen with ARN2508 attenuated inflammation and weight loss elicited after exposure to DSS for 3 d (Supplemental Fig. S5). The findings suggest that ARN2508 is able to suppress established colon inflammation produced by mechanistically different insults.
Figure 8.
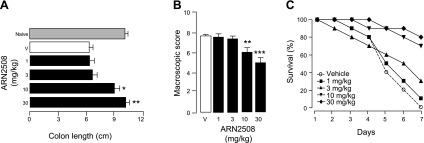
Therapeutic treatment of TNBS-induced colon inflammation in mice with ARN2508. Anti-inflammatory effects of 4-day regimen with ARN2508 (closed bars or symbols) on colon length (A), macroscopic score (B), and animal survival (C). ARN2508 was administered orally starting on the second day after TNBS instillation; doses are in milligrams per kilogram. Shaded bars indicate naive mice; open bars represent vehicle-treated mice. Results are expressed as the mean ± sem (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle.
DISCUSSION
The results presented here support the hypothesis that pharmacologic interventions aimed at restoring normal levels of FAAH and Cox-1/Cox-2 activities provide a rational therapeutic approach to disease conditions in which the expression of these enzymes is abnormally elevated (Fig. 1). We have shown that oral administration of ARN2508—the first highly potent and selective inhibitor of FAAH, Cox-1, and Cox-2—lowers systemic levels of Cox-derived inflammatory prostanoids, while raising levels of AEA and other anti-inflammatory lipid amides that are degraded by FAAH (i.e., PEA and OEA). These simultaneous effects are likely to underpin the marked efficacy displayed by ARN2508 in models of intestinal inflammation, though high target exposure due to enterohepatic cycling might also contribute. Our findings further indicate that ARN2508 protects both upper and lower GI tract from NSAID-induced injury through a mechanism that depends on FAAH blockade.
Previous work has shown that FAAH inhibitors enhance the analgesic effects of the NSAIDs in a synergistic manner (47, 48). By interrupting the degradation of AEA, these agents heighten its ability to control peripheral nociceptive signals (24, 49) resulting in a superadditive potentiation of NSAID-mediated analgesia. These and other findings (50) have led to propose that dual FAAH/Cox inhibitors might be better than current nonnarcotic analgesics at providing pain control (19).
Although this hypothesis remains to be tested, the present study demonstrates that simultaneous blockade of FAAH and Cox-1/Cox-2 results in a combination of profound anti-inflammatory and tissue-protective actions, which clearly distinguish dual FAAH/Cox inhibitors from agents that individually target FAAH or Cox enzymes. Models of intestinal inflammation provide a striking illustration of this difference. In those models, the dual FAAH/Cox inhibitor ARN2508 is markedly anti-inflammatory, whereas the NSAID flurbiprofen aggravates inflammation (as observed in the clinical setting) (40), and 2 structurally different single-target FAAH inhibitors are marginally effective. The molecular basis for this dramatic shift in pharmacology is still unclear, but the ability of ARN2508 to elevate tissue levels of 3 key FAAH substrates—the endocannabinoid AEA and the endogenous PPAR-α agonists PEA and OEA—is likely to play an important role. These lipid mediators are known to exert protective effects on mucosal epithelia (12, 51) and to act synergistically to reduce pain (49, 52). Other anti-inflammatory lipid mediators that might be affected by FAAH inhibition, such as N-acyl-taurines (53) or N-acyl-glycines (54), might also contribute to ARN2508 pharmacology.
In the optimization process that led to the identification of ARN2508, we pursued concomitant Cox-1/Cox-2 inhibition because both isoforms of this enzyme are known to contribute to the development of inflammatory hyperalgesia (31). Nevertheless, dual FAAH/Cox ligands that selectively recognize either isoform would be experimentally useful to elucidate possible select functional interactions of FAAH substrates with Cox-1- or Cox-2-derived prostanoids. This objective should be attainable, given the significant structural differences existing between the 2 isoforms (33). Another important aim for future work is the characterization of the pharmacologic properties of the (S) and (R) enantiomers of ARN2508, which might be as different as those of (S) and (R) profens (55).
IBD, which encompasses Crohn’s disease and ulcerative colitis, is a chronic intestinal inflammation of unknown cause affecting 1–1.3 million people in the United States alone (56). Despite its prevalence, IBD remains a medical challenge due to the limited efficacy and frequent side effects of existing therapies (45, 46). Our results suggest that multitarget FAAH/Cox inhibitors such as ARN2508 might fill this gap. Additional work will be needed to determine whether these agents could also find application in other inflammatory and neuroinflammatory states in which FAAH and Cox are overexpressed (8, 14).
Supplementary Material
Acknowledgments
The authors thank Drs. Angelo Reggiani and Tiziano Bandiera for helpful discussions; Ms. Sine Mandrup-Bertozzi, Ms. Silvia Venzano, and Mr. Luca Goldoni for technical help; and the U.S. National Institutes of Health National Institute on Drug Abuse (Grant DA012413 to D.P.) for financial support. D.P., R.S., M.M., and D.H. are inventors on patent application WO2014023643, filed by the University of California–Irvine (Irvine, CA, USA) and the Istituto Italiano di Tecnologia, which protects novel compounds disclosed in this paper.
Glossary
- 2-AG
2-arachidonoyl-glycerol
- 5-ASA
5-aminosalicylic acid
- AA
arachidonic acid
- AEA
anandamide
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- Cox
cyclooxygenase
- DSS
dextran sodium sulfate
- EIA
enzyme immunoassay
- eV
electron volt
- FAAH
fatty acid amide hydrolase
- GI
gastrointestinal
- IBD
inflammatory bowel disease
- IC50
median inhibitory concentration
- LC/MS
liquid chromatography-mass spectrometry
- MRM
multiple-reaction monitoring
- NSAID
nonsteroidal anti-inflammatory drug
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- PGE2
prostaglandin E2
- PGI2
prostacyclin
- PPAR-α
peroxisome proliferator-activated receptor-α
- TNBS
2,4,6-trinitrobenzene sulfonic acid
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Ricciotti E., FitzGerald G. A. (2011) Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serhan C. N., Chiang N., Van Dyke T. E. (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morisseau C., Hammock B. D. (2013) Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 53, 37–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maccarrone M., Bari M., Battista N., Di Rienzo M., Finazzi-Agrò A. (2001) Endogenous cannabinoids in neuronal and immune cells: toxic effects, levels and degradation. Funct. Neurol. 16(4, Suppl)53–60 [PubMed] [Google Scholar]
- 5.Liu J., Wang L., Harvey-White J., Osei-Hyiaman D., Razdan R., Gong Q., Chan A. C., Zhou Z., Huang B. X., Kim H. Y., Kunos G. (2006) A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 103, 13345–13350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Massa F., Marsicano G., Hermann H., Cannich A., Monory K., Cravatt B. F., Ferri G. L., Sibaev A., Storr M., Lutz B. (2004) The endogenous cannabinoid system protects against colonic inflammation. J. Clin. Invest. 113, 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cencioni M. T., Chiurchiù V., Catanzaro G., Borsellino G., Bernardi G., Battistini L., Maccarrone M. (2010) Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS One 5, e8688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiurchiù V., Cencioni M. T., Bisicchia E., De Bardi M., Gasperini C., Borsellino G., Centonze D., Battistini L., Maccarrone M. (2013) Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide in multiple sclerosis. Ann. Neurol. 73, 626–636 [DOI] [PubMed] [Google Scholar]
- 9.Piomelli D., Sasso O. (2014) Peripheral gating of pain signals by endogenous lipid mediators. Nat. Neurosci. 17, 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Storr M. A., Keenan C. M., Emmerdinger D., Zhang H., Yüce B., Sibaev A., Massa F., Buckley N. E., Lutz B., Göke B., Brand S., Patel K. D., Sharkey K. A. (2008) Targeting endocannabinoid degradation protects against experimental colitis in mice: involvement of CB1 and CB2 receptors. J. Mol. Med. 86, 925–936 [DOI] [PubMed] [Google Scholar]
- 11.Wilson D. E. (1991) Role of prostaglandins in gastroduodenal mucosal protection. J. Clin. Gastroenterol. 13(Suppl 1), S65–S71 [DOI] [PubMed] [Google Scholar]
- 12.Wright K., Rooney N., Feeney M., Tate J., Robertson D., Welham M., Ward S. (2005) Differential expression of cannabinoid receptors in the human colon: cannabinoids promote epithelial wound healing. Gastroenterology 129, 437–453 [DOI] [PubMed] [Google Scholar]
- 13.Patrono C., Baigent C. (2009) Low-dose aspirin, coxibs, and other NSAIDS: a clinical mosaic emerges. Mol. Interv. 9, 31–39 [DOI] [PubMed] [Google Scholar]
- 14.Richardson D., Pearson R. G., Kurian N., Latif M. L., Garle M. J., Barrett D. A., Kendall D. A., Scammell B. E., Reeve A. J., Chapman V. (2008) Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res. Ther. 10, R43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singer I. I., Kawka D. W., Schloemann S., Tessner T., Riehl T., Stenson W. F. (1998) Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 115, 297–306 [DOI] [PubMed] [Google Scholar]
- 16.Di Sabatino A., Battista N., Biancheri P., Rapino C., Rovedatti L., Astarita G., Vanoli A., Dainese E., Guerci M., Piomelli D., Pender S. L., MacDonald T. T., Maccarrone M., Corazza G. R. (2011) The endogenous cannabinoid system in the gut of patients with inflammatory bowel disease. Mucosal Immunol. 4, 574–583 [DOI] [PubMed] [Google Scholar]
- 17.Suárez J., Romero-Zerbo Y., Márquez L., Rivera P., Iglesias M., Bermúdez-Silva F. J., Andreu M., Rodríguez de Fonseca F. (2012) Ulcerative colitis impairs the acylethanolamide-based anti-inflammatory system reversal by 5-aminosalicylic acid and glucocorticoids. PLoS One 7, e37729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rouzer C. A., Marnett L. J. (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 111, 5899–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fowler C. J., Naidu P. S., Lichtman A., Onnis V. (2009) The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or TRPV1. Br. J. Pharmacol. 156, 412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Favia A. D., Habrant D., Scarpelli R., Migliore M., Albani C., Bertozzi S. M., Dionisi M., Tarozzo G., Piomelli D., Cavalli A., De Vivo M. (2012) Identification and characterization of carprofen as a multitarget fatty acid amide hydrolase/cyclooxygenase inhibitor. J. Med. Chem. 55, 8807–8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson D. S., Stiff C., Lazerwith S. E., Kesten S. R., Fay L. K., Morris M., Beidler D., Liimatta M. B., Smith S. E., Dudley D. T., Sadagopan N., Bhattachar S. N., Kesten S. J., Nomanbhoy T. K., Cravatt B. F., Ahn K. (2011) Discovery of PF-04457845: a highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med. Chem. Lett. 2, 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarzia G., Duranti A., Tontini A., Piersanti G., Mor M., Rivara S., Plazzi P. V., Park C., Kathuria S., Piomelli D. (2003) Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J. Med. Chem. 46, 2352–2360 [DOI] [PubMed] [Google Scholar]
- 23.Kathuria S., Gaetani S., Fegley D., Valiño F., Duranti A., Tontini A., Mor M., Tarzia G., La Rana G., Calignano A., Giustino A., Tattoli M., Palmery M., Cuomo V., Piomelli D. (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 9, 76–81 [DOI] [PubMed] [Google Scholar]
- 24.Clapper J. R., Moreno-Sanz G., Russo R., Guijarro A., Vacondio F., Duranti A., Tontini A., Sanchini S., Sciolino N. R., Spradley J. M., Hohmann A. G., Calignano A., Mor M., Tarzia G., Piomelli D. (2010) Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 13, 1265–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace J. L., Muscará M. N., de Nucci G., Zamuner S., Cirino G., del Soldato P., Ongini E. (2004) Gastric tolerability and prolonged prostaglandin inhibition in the brain with a nitric oxide-releasing flurbiprofen derivative, NCX-2216 [3-[4-(2-fluoro-alpha-methyl-[1,1′-biphenyl]-4-acetyloxy)-3-methoxyphenyl]-2-propenoic acid 4-nitrooxy butyl ester]. J. Pharmacol. Exp. Ther. 309, 626–633 [DOI] [PubMed] [Google Scholar]
- 26.Chan C. C., Boyce S., Brideau C., Ford-Hutchinson A. W., Gordon R., Guay D., Hill R. G., Li C. S., Mancini J., Penneton M. (1995) Pharmacology of a selective cyclooxygenase-2 inhibitor, L-745,337: a novel nonsteroidal anti-inflammatory agent with an ulcerogenic sparing effect in rat and nonhuman primate stomach. J. Pharmacol. Exp. Ther. 274, 1531–1537 [PubMed] [Google Scholar]
- 27.Cluny N. L., Keenan C. M., Duncan M., Fox A., Lutz B., Sharkey K. A. (2010) Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone (SAB378), a peripherally restricted cannabinoid CB1/CB2 receptor agonist, inhibits gastrointestinal motility but has no effect on experimental colitis in mice. J. Pharmacol. Exp. Ther. 334, 973–980 [DOI] [PubMed] [Google Scholar]
- 28.Bento A. F., Claudino R. F., Dutra R. C., Marcon R., Calixto J. B. (2011) Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J. Immunol. 187, 1957–1969 [DOI] [PubMed] [Google Scholar]
- 29.Bayly C. I., Black W. C., Léger S., Ouimet N., Ouellet M., Percival M. D. (1999) Structure-based design of COX-2 selectivity into flurbiprofen. Bioorg. Med. Chem. Lett. 9, 307–312 [DOI] [PubMed] [Google Scholar]
- 30.Morphy R., Rankovic Z. (2006) The physicochemical challenges of designing multiple ligands. J. Med. Chem. 49, 4961–4970 [DOI] [PubMed] [Google Scholar]
- 31.Araldi D., Ferrari L. F., Lotufo C. M., Vieira A. S., Athié M. C., Figueiredo J. G., Duarte D. B., Tambeli C. H., Ferreira S. H., Parada C. A. (2013) Peripheral inflammatory hyperalgesia depends on the COX increase in the dorsal root ganglion. Proc. Natl. Acad. Sci. USA 110, 3603–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mileni M., Kamtekar S., Wood D. C., Benson T. E., Cravatt B. F., Stevens R. C. (2010) Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 400, 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurumbail R. G., Stevens A. M., Gierse J. K., McDonald J. J., Stegeman R. A., Pak J. Y., Gildehaus D., Miyashiro J. M., Penning T. D., Seibert K., Isakson P. C., Stallings W. C. (1996) Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 384, 644–648 [DOI] [PubMed] [Google Scholar]
- 34.Blobaum A. L., Marnett L. J. (2007) Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 50, 1425–1441 [DOI] [PubMed] [Google Scholar]
- 35.Risdall P. C., Adams S. S., Crampton E. L., Marchant B. (1978) The disposition and metabolism of flurbiprofen in several species including man. Xenobiotica 8, 691–703 [DOI] [PubMed] [Google Scholar]
- 36.Wallace J. L. (2012) NSAID gastropathy and enteropathy: distinct pathogenesis likely necessitates distinct prevention strategies. Br. J. Pharmacol. 165, 67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts M. S., Magnusson B. M., Burczynski F. J., Weiss M. (2002) Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin. Pharmacokinet. 41, 751–790 [DOI] [PubMed] [Google Scholar]
- 38.Lauritsen K., Laursen L. S., Bukhave K., Rask-Madsen J. (1988) Use of colonic eicosanoid concentrations as predictors of relapse in ulcerative colitis: double blind placebo controlled study on sulphasalazine maintenance treatment. Gut 29, 1316–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costa-Romero M., Coto-Segura P., Suarez-Saavedra S., Ramos-Polo E., Santos-Juanes J. (2008) Guttate psoriasis induced by infliximab in a child with Crohn’s disease. Inflamm. Bowel Dis. 14, 1462–1463 [DOI] [PubMed] [Google Scholar]
- 40.Blackler R. W., Gemici B., Manko A., Wallace J. L. (2014) NSAID-gastroenteropathy: new aspects of pathogenesis and prevention. Curr. Opin. Pharmacol. 19, 11–16 [DOI] [PubMed] [Google Scholar]
- 41.Shi Y., Qi L., Wang J., Xu M. S., Zhang D., Wu L. Y., Wu H. G. (2011) Moxibustion activates mast cell degranulation at the ST25 in rats with colitis. World J. Gastroenterol. 17, 3733–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo Verme J., Fu J., Astarita G., La Rana G., Russo R., Calignano A., Piomelli D. (2005) The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 67, 15–19 [DOI] [PubMed] [Google Scholar]
- 43.Riccardi L., Mazzon E., Bruscoli S., Esposito E., Crisafulli C., Di Paola R., Caminiti R., Riccardi C., Cuzzocrea S. (2009) Peroxisome proliferator-activated receptor-alpha modulates the anti-inflammatory effect of glucocorticoids in a model of inflammatory bowel disease in mice. Shock 31, 308–316 [DOI] [PubMed] [Google Scholar]
- 44.Solorzano C., Zhu C., Battista N., Astarita G., Lodola A., Rivara S., Mor M., Russo R., Maccarrone M., Antonietti F., Duranti A., Tontini A., Cuzzocrea S., Tarzia G., Piomelli D. (2009) Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. USA 106, 20966–20971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffmann J. C., Pawlowski N. N., Kuhl A. A., Hohne W., Zeitz M. (2002) Animal models of inflammatory bowel disease: an overview. Pathobiology 70, 121–130 [DOI] [PubMed] [Google Scholar]
- 46.Strober W., Fuss I. J., Blumberg R. S. (2002) The immunology of mucosal models of inflammation. Annu. Rev. Immunol. 20, 495–549 [DOI] [PubMed] [Google Scholar]
- 47.Naidu P. S., Booker L., Cravatt B. F., Lichtman A. H. (2009) Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J. Pharmacol. Exp. Ther. 329, 48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasso O., Bertorelli R., Bandiera T., Scarpelli R., Colombano G., Armirotti A., Moreno-Sanz G., Reggiani A., Piomelli D. (2012) Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol. Res. 65, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calignano A., La Rana G., Giuffrida A., Piomelli D. (1998) Control of pain initiation by endogenous cannabinoids. Nature 394, 277–281 [DOI] [PubMed] [Google Scholar]
- 50.Holt S., Paylor B., Boldrup L., Alajakku K., Vandevoorde S., Sundström A., Cocco M. T., Onnis V., Fowler C. J. (2007) Inhibition of fatty acid amide hydrolase, a key endocannabinoid metabolizing enzyme, by analogues of ibuprofen and indomethacin. Eur. J. Pharmacol. 565, 26–36 [DOI] [PubMed] [Google Scholar]
- 51.Di Paola R., Esposito E., Mazzon E., Paterniti I., Galuppo M., Cuzzocrea S. (2010) GW0742, a selective PPAR-beta/delta agonist, contributes to the resolution of inflammation after gut ischemia/reperfusion injury. J. Leukoc. Biol. 88, 291–301 [DOI] [PubMed] [Google Scholar]
- 52.Russo R., LoVerme J., La Rana G., D’Agostino G., Sasso O., Calignano A., Piomelli D. (2007) Synergistic antinociception by the cannabinoid receptor agonist anandamide and the PPAR-alpha receptor agonist GW7647. Eur. J. Pharmacol. 566, 117–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saghatelian A., McKinney M. K., Bandell M., Patapoutian A., Cravatt B. F. (2006) A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry 45, 9007–9015 [DOI] [PubMed] [Google Scholar]
- 54.Bradshaw H. B., Rimmerman N., Hu S. S., Burstein S., Walker J. M. (2009) Novel endogenous N-acyl glycines identification and characterization. Vitam. Horm. 81, 191–205 [DOI] [PubMed] [Google Scholar]
- 55.Duggan K. C., Hermanson D. J., Musee J., Prusakiewicz J. J., Scheib J. L., Carter B. D., Banerjee S., Oates J. A., Marnett L. J. (2011) (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 7, 803–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kappelman M. D., Rifas-Shiman S. L., Kleinman K., Ollendorf D., Bousvaros A., Grand R. J., Finkelstein J. A. (2007) The prevalence and geographic distribution of Crohn's disease and ulcerative colitis in the United States. Clin. Gastroenterol. Hepatol. 5, 1424–1429 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.