

ABSTRACT
Recent work demonstrated that the Niemann-Pick C1 (NPC1) protein is an essential entry receptor for filoviruses. While previous studies focused on filovirus entry requirements of NPC1 in vitro, its roles in filovirus replication and pathogenesis in vivo remain unclear. Here, we evaluated the importance of NPC1, and its partner in cholesterol transport, NPC2, by using a mouse model of Ebolavirus (EBOV) disease. We found that, whereas wild-type mice had high viral loads and succumbed to EBOV infection, Npc1−/− mice were entirely free of viral replication and completely protected from EBOV disease. Interestingly, Npc1+/− mice transiently developed high levels of viremia, but were nevertheless substantially protected from EBOV challenge. We also found Npc2−/− mice to be fully susceptible to EBOV infection, while Npc1−/− mice treated to deplete stored lysosomal cholesterol remained completely resistant to EBOV infection. These results provide mechanistic evidence that NPC1 is directly required for EBOV infection in vivo, with little or no role for NPC1/NPC2-dependent cholesterol transport. Finally, we assessed the in vivo antiviral efficacies of three compounds known to inhibit NPC1 function or NPC1-glycoprotein binding in vitro. Two compounds reduced viral titers in vivo and provided a modest, albeit not statistically significant, degree of protection. Taken together, our results show that NPC1 is critical for replication and pathogenesis in animals and is a bona fide target for development of antifilovirus therapeutics. Additionally, our findings with Npc1+/− mice raise the possibility that individuals heterozygous for NPC1 may have a survival advantage in the face of EBOV infection.
IMPORTANCE
Researchers have been searching for an essential filovirus receptor for decades, and numerous candidate receptors have been proposed. However, none of the proposed candidate receptors has proven essential in all in vitro scenarios, nor have they proven essential when evaluated using animal models. In this report, we provide the first example of a knockout mouse that is completely refractory to EBOV infection, replication, and disease. The findings detailed here provide the first critical in vivo data illustrating the absolute requirement of NPC1 for filovirus infection in mice. Our work establishes NPC1 as a legitimate target for the development of anti-EBOV therapeutics. However, the limited success of available NPC1 inhibitors to protect mice from EBOV challenge highlights the need for new molecules or approaches to target NPC1 in vivo.
INTRODUCTION
Filoviruses, members of the family Filoviridae of nonsegmented negative-strand RNA viruses, cause sporadic viral hemorrhagic fever outbreaks that primarily affect areas of equatorial Africa (1). Five filoviruses are currently associated with severe disease in humans: Ebola virus (EBOV; formerly termed Zaire ebolavirus), Bundibugyo virus (BDBV), Sudan virus (SUDV), Marburg virus (MARV), and Ravn virus (RAVV) (2). Filovirus virions are enveloped filamentous particles with a uniform diameter of 80 nm and variable lengths. A single transmembrane glycoprotein (GP), consisting of two subunits (GP1 and GP2) and organized into trimeric spikes on the virion surface, mediates viral entry into cells (3, 4). Filovirus virions bind to host cells via several reported attachment proteins (5–8) and are then internalized and delivered to the endosomal pathway (9–11). In late endosomes, host cysteine proteases cleave and remove large C-terminal regions of the GP1 subunit (the mucin domain and glycan cap), thereby unmasking a binding site for the cholesterol transport protein Niemann-Pick C1 (NPC1). NPC1 was recently shown to be an essential host factor (12, 13) and endosomal/lysosomal entry receptor (14, 15) for filoviruses.
NPC1 is a large 13-pass transmembrane protein found in the limiting membrane of late endosomes and lysosomes in all cells (16). According to the current model, NPC1 is proposed to work in cooperation with a small soluble lysosomal protein, Niemann-Pick C2 (NPC2), to mediate transport of luminal cholesterol across the endosomal/lysosomal membrane for dispersal to other cellular compartments (17, 18). Loss-of-function mutations in NPC1 or NPC2 cause a rare and often fatal hereditary neurovisceral disorder in humans (19, 20). Over time, NPC disease patients accumulate cholesterol and glycosphingolipids in various tissues and organs, leading to neurological dysfunction and organ failure. U18666a, an amphipathic steroid, reproduces some features of NPC disease at the cellular level, at least in part by disrupting NPC1 function (21–24). A direct interaction between NPC1 and U18666A is proposed to be responsible for U18666A-mediated lysosomal cholesterol accumulation (23, 25, 26). Imipramine, a hydrophobic amine and FDA-approved antidepressant, and a variety of other cationic amphiphiles also induce accumulation of cholesterol and glycosphingolipids in lysosomes and may directly interfere with NPC1 function (26–28).
Carette et al. used a genetic screen in haploid human cells to identify NPC1 as a critical host factor for filovirus entry and replication in vitro (12). They also reported that U18666a and imipramine significantly inhibited filovirus infection by interfering with viral entry. In a separate chemical screen, Cote et al. identified an EBOV-specific antiviral compound, 3.47, and attributed its antiviral activity in cell culture to its ability to block EBOV GP binding to NPC1-containing membranes (13). Both studies provided evidence that the cholesterol transport function and GP-binding function of NPC1 are separable. More recent work showed that EBOV GP, in its cleaved form, binds directly and specifically to purified NPC1, that GP directly engages the second luminal loop of NPC1 (loop C), and that GP-NPC1 binding is required for filovirus entry and infection in cultured cells (14, 15). While these recently published efforts clearly illustrate the importance of NPC1 for filovirus entry in vitro, the importance of NPC1 in vivo is yet to be investigated in detail. Furthermore, NPC1-targeting compounds have demonstrated potent in vitro antiviral efficacy against filoviruses, but none has been reported to limit filovirus replication or pathogenesis in vivo. Here, we provide evidence that Npc1−/− mice are completely protected from EBOV infection and free of replicating virus. We also show that endosomal/lysosomal cholesterol accumulation plays little or no role in murine susceptibility or resistance to EBOV challenge, strongly implicating the NPC1 protein as a direct mediator of filovirus infection in vivo. Finally, we demonstrate that compounds that disrupt NPC1 function in vitro can inhibit viral replication in a murine model of filovirus pathogenesis.
RESULTS
Npc1−/− mice are completely resistant to EBOV pathogenesis and replication.
We previously demonstrated that Npc1+/− mice were highly resistant to mouse-adapted EBOV and mouse-adapted MARV induced mortality, though the animals did display clinical signs of disease and experienced weight loss (12). Here, we examined the capacity of EBOV to infect Npc1−/− mice. We challenged Npc1−/−, Npc1+/−, and wild-type littermate controls intraperitoneally (i.p.) with 100 PFU (3,000 50% lethal dose units) of mouse-adapted EBOV. Wild-type mice developed clinical signs of disease, including weight loss (Fig. 1A), ruffled fur, hunched posture, subdued activity, and severe dehydration, and all succumbed by day 9 postchallenge (Fig. 1B). As reported previously, nearly all Npc1+/− mice were protected from EBOV infection, though weight loss and ruffled fur were noted between days 6 and 9 postchallenge. Npc1−/− mice were completely protected, with no clinical signs of disease. Subsets of these mice were sacrificed on days 3 and 7 postchallenge, and tissues were collected to assess viral titers, histology and immunohistochemistry, and cytokine expression. Virus was detected in liver, kidney, spleen, and serum of all tested wild-type and Npc1+/− mice euthanized on day 3 postchallenge (Fig. 1C). While viral titers remained elevated in wild-type mice on day 7, titers were significantly lower in Npc1+/− mice euthanized on day 7 (Fig. 1D). No virus was detected in tissues collected from Npc1−/− mice at either time point (Fig. 1C and D).
FIG 1 .

NPC1 knockout mice are protected from mouse-adapted EBOV infection. Npc1−/− (n = 28), Npc1+/− (n = 21), and wild-type littermate control (n = 19) mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Mice were monitored for weight loss (A) and mortality (B) following challenge. On days 3 (C) and 7 (D) postchallenge, subsets of mice from each group were euthanized and viral titers were determined for the indicated tissues by plaque assay. The dotted line indicates the assay limit of detection. ****, P ≤ 0.0001 compared to wild-type mice.
Histological manifestations within the spleens included lymphocytolysis within germinal centers, splenitis, and fibrin accumulation (see Fig. S1 in the supplemental material). Within the liver, hepatocellular degeneration and necrosis, rare necrotizing hepatitis, and fibrin accumulation were observed (see Fig. S1). Overall, no clear distinctions were noted between tissues collected from wild-type mice and Npc1+/− mice on day 3 postchallenge. Histological abnormalities were more pronounced in the livers of wild-type mice on day 7 postchallenge than in Npc1+/− mice. Npc1−/− mice did not show any notable histological changes in spleen or liver on either day postchallenge. Within the spleen, cells morphologically consistent with fibroblastic reticular cells, dendritic cells, and macrophages were the predominant cell types antigen positive for EBOV (see Fig. S2 in the supplemental material). Within the liver, nonparenchymal cells, i.e., hepatic stellate cells and occasional hepatocytes, were the predominate antigen-positive cell types (see Fig. S2). No notable differences were observed in spleens or livers collected on day 3 from wild-type or Npc1+/− mice. By day 7 postchallenge, antigen-positive cells were widespread in livers of wild-type mice but remained low (<10%) in Npc1+/− mice. Antigen-positive cells in the spleens of both wild-type and Npc1+/− mice were equally abundant on day 7. No EBOV antigen-positive cells were identified in the liver or spleen of Npc1−/− mice on either day postchallenge. No notable changes were observed in kidneys harvested from any group on either day postinfection.
EBOV-induced cytokine expression in wild-type and NPC1 knockout mice.
Despite having similar viral loads on day 3 postinfection and comparable histological findings, Npc1+/− mice were better protected against EBOV infection than their wild-type littermates. To investigate developing immune responses in these groups, we evaluated cytokine levels in serum collected from mouse-adapted EBOV infected Npc1−/−, Npc1+/−, and wild-type littermate controls on days 3 and 7 postinfection. We assessed proinflammatory cytokines and those critical for coordinating innate immunity. By day 3, tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), IL-1α, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and monocyte chemoattractant protein-1 (MCP-1) were elevated in wild-type mice compared to uninfected control mice (Fig. 2A). On day 7, TNF-α, MCP-1, macrophage inflammatory protein 1 alpha (MIP-1α), and regulated on activation, normal T cell expressed and secreted (RANTES) levels were significantly elevated in wild-type mice compared to uninfected mice (Fig. 2B). In Npc1+/− mice on day 3, significant increases in TNF-α, GM-CSF, and MCP-1 expression were observed compared to uninfected control mice, and IL-1α, G-CSF, and MCP-1 levels were significantly lower than in wild-type mice (Fig. 2A). By day 7, TNF-α remained elevated in Npc1+/− mice relative to uninfected control mice, and IL-6 and GM-CSF levels were significantly higher in Npc1+/− mice than in wild-type mice (Fig. 2B). MIP-1α, MCP-1, and RANTES expression was significantly lower in Npc1+/− mice than in wild-type mice (Fig. 2B). On both days 3 (Fig. 2A) and 7 (Fig. 2B), TNF-α and GM-CSF were the only cytokines to be significantly elevated in Npc1−/− mice compared to uninfected controls, while IL-6, IL-1α, G-CSF, MIP-1α, MCP-1, and RANTES remained largely unchanged and significantly lower than in wild-type mice.
FIG 2 .

Serum cytokine levels following mouse-adapted EBOV infection of wild-type and NPC1 knockout mice. Npc1−/−, Npc1+/−, and wild-type littermate control mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Serum collected at day 3 (A and C) or 7 (B and D) postchallenge was used to assess cytokine levels in a BioPlex assay. °, P ≤ 0.05; °°, P ≤ 0.01; °°°, P ≤ 0.001; °°°°, P ≤ 0.0001 compared to uninfected control mice. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001 compared to wild-type mice.
Cytokines important for coordinating adaptive immunity were also evaluated. Wild-type mice had significantly elevated levels of IL-2, IL-4, IL-5, IL-10, and IL-12p70 on day 3 postchallenge compared to uninfected control mice (Fig. 2C). By day 7, only gamma interferon (IFN-γ) and IL-2 remained elevated in wild-type mice compared to uninfected controls (Fig. 2D). IL-2, IL-4, IL-5, IL-10, and IL-12p70 levels were also significantly elevated in Npc1+/− mice relative to levels in uninfected controls on day 3 (Fig. 2C). As in wild-type mice, IL-2 remained elevated in Npc1+/− mice on day 7 compared to uninfected controls. However, unlike wild-type mice, Npc1+/− mice had significantly higher levels of IL-5 and IL-12p70 than in either uninfected controls or infected wild-type mice (Fig. 2D). IL-10 expression was also increased in Npc1+/− mice relative to uninfected control mice. On day 3, Npc1−/− mice expressed elevated levels of IL-10 and IL-12p70 compared to uninfected control mice. IL-10 and IL-2 expression levels were significantly lower than in wild-type mice (Fig. 2C). IL-2 and IFN-γ levels in Npc−/− mice were significantly lower on day 7 compared to levels in wild-type mice (Fig. 2D).
Lysosomal cholesterol accumulation alone does not confer resistance to EBOV infection in vivo, and NPC2 is dispensable.
Similar to NPC1 deficiency, NPC2 deficiency causes cholesterol accumulation in late endosomal/lysosomal compartments of cells in tissue culture and in vivo (20, 29). To evaluate the role of cholesterol accumulation in resistance to EBOV infection, we challenged Npc2−/−, Npc2+/−, and wild-type littermate controls i.p. with 100 PFU of mouse-adapted EBOV and monitored morbidity and mortality. Despite excess cholesterol accumulation, NPC2-deficient mice were not protected against lethal EBOV infection (Fig. 3A and B). All mice developed severe clinical signs of disease, with all groups experiencing ≥90% mortality.
FIG 3 .

Cholesterol accumulation does not confer resistance to mouse-adapted EBOV infection. Npc2−/− (n = 9), Npc2+/− (n = 11), and wild-type littermate control (n = 10) mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Mice were monitored for weight loss (A) and mortality (B) following challenge. Npc1−/− (n = 8), Npc1+/− (n = 11), and wild-type littermate control (n = 11) mice were treated or not with HPBCD starting at 7 days of age and continuing every other day until sacrifice at 7 weeks of age (33 to 36 days old). (C) Filipin staining of unesterified cholesterol was evaluated in the dorsal neocortex of HPBCD-treated and control mice. Cholesterol accumulation is seen as white areas, primarily cytoplasmic, examples of which are denoted by red arrows. Images were taken at 20×. Bar, 50 µm. 2-Hydroxypropyl-β-cyclodextrin-treated mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Mice were monitored for weight loss (D) and mortality (E) following challenge. ***, P ≤ 0.001; ****, P ≤ 0.0001 compared to wild-type mice.
Recent advances in the field of NPC disease research have highlighted the utility of 2-hydroxypropyl-β-cyclodextrin (HPBCD) treatments for clearing cholesterol and glycosphingolipids from late endosomal/lysosomal compartments, delaying onset of clinical disease, and increasing longevity of NPC1- and NPC2-deficient mice (30, 31). Leveraging these findings, we treated Npc1+/−, Npc1−/−, and wild-type littermate controls with HPBCD every other day to clear cholesterol from late endosomal/lysosomal compartments. HPBCD-treated Npc1+/− and Npc1−/− mice had wild-type phenotypes with respect to late endosomal/lysosomal cholesterol accumulation, as evidenced by filipin labeling in brain sections (Fig. 3C). HPBCD-treated Npc1+/−, Npc1−/−, and wild-type littermate control mice were then exposed i.p. to 100 PFU of mouse-adapted EBOV, and morbidity and mortality were monitored. HPBCD-treated wild-type mice succumbed to EBOV infection with expected clinical signs of disease (Fig. 3D and E). HPBCD-treated Npc1+/− mice were largely protected from EBOV challenge and presented with similar clinical signs of disease and mortality as those observed in untreated Npc1+/− mice. As with untreated Npc1−/− mice, HPBCD-treated Npc1−/− mice were completely protected from EBOV challenge and did not display clinical signs of infection.
NPC1 inhibitors block filovirus replication in vitro.
The amphipathic cationic amine U18666a, a known Niemann-Pick phenotype inducer, was previously shown to block filovirus entry and replication in vitro (12). To determine if imipramine, another reported inducer of the Niemann-Pick phenotype, is capable of inhibiting filovirus replication, we infected human umbilical vein epithelial cells (HUVECs) with MARV or EBOV in the presence or absence of imipramine at nontoxic levels (see Fig. S3 in the supplemental material). Compared to control treated cells, imipramine at 10 µM significantly inhibited both MARV and EBOV replication in HUVECs (Fig. 4A). We also evaluated the antiviral activity of compound 3.47, which was previously shown to block binding of EBOV GP to NPC1-containing membranes (13). Surprisingly, while 20 µM 3.47 strongly inhibited EBOV replication, as previously reported, it had no significant antiviral activity against MARV or SUDV in Vero cells (Fig. 4B). Consistent with these findings, 3.47 potently inhibited EBOV GP-dependent entry and infection by vesicular stomatitis virus (VSV) pseudotypes, but it had little effect on viral entry mediated by SUDV, RESTV, or MARV (Fig. 4C). Thus, U18666A and imipramine broadly inhibit filovirus entry and infection, whereas 3.47 has a more narrow spectrum of action.
FIG 4 .
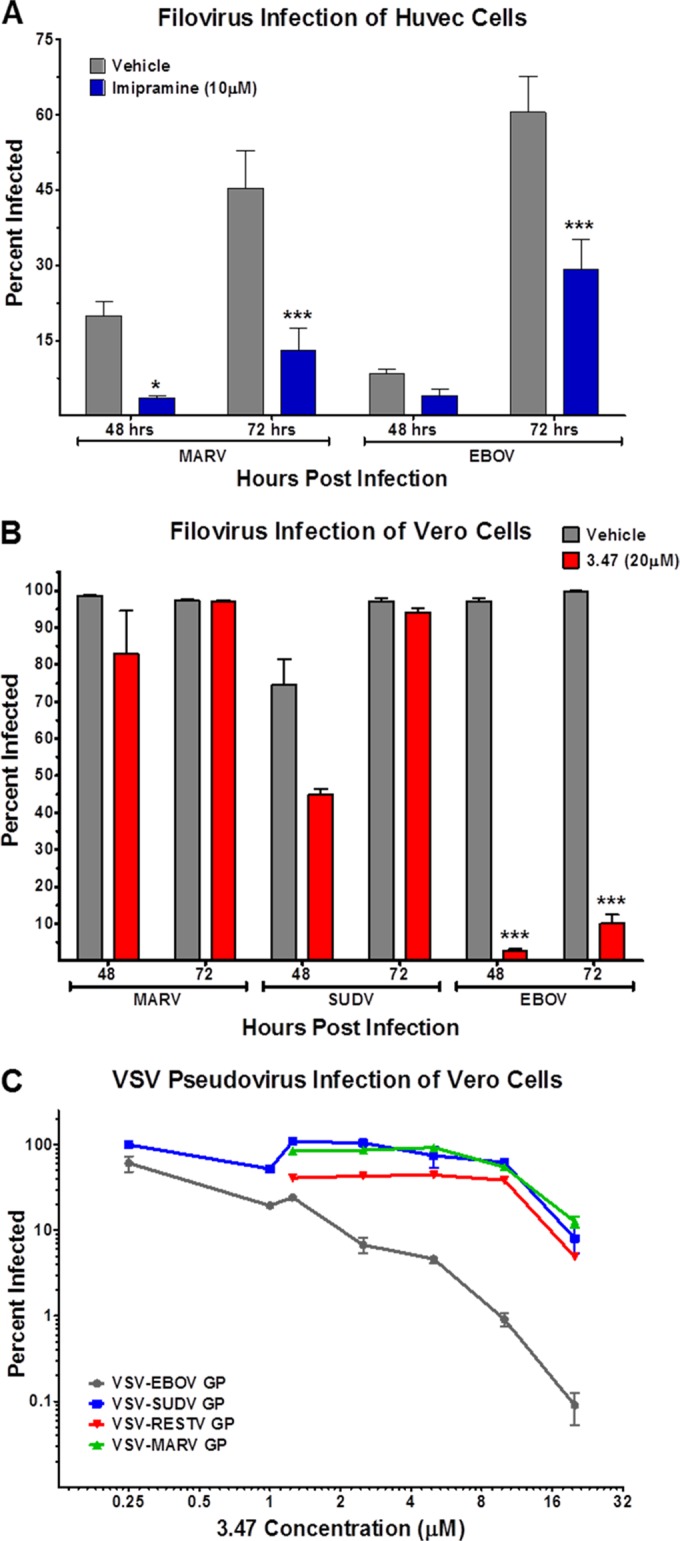
Imipramine and compound 3.47 inhibit filovirus replication and GP-mediated entry in vitro. Filovirus infected HUVEC and Vero cells were treated with or without imipramine (A) or compound 3.47 (B), respectively. Cells were fixed at 48 or 72 h postinfection, and infected cells were enumerated using MARV-, EBOV-, or SUDV-specific monoclonal antibodies and a fluorescently labeled secondary antibody. (C) Vero cells were treated with compound 3.47 at the indicated concentrations prior to infection with GFP-expressing VSV pseudoviruses. Cells were fixed 16 to 24 h postinfection, and infected cells were assessed by eGFP expression analysis. *, P ≤ 0.05; ***, P ≤ 0.001.
Pharmacological induction of the NPC1 phenotype reduces viral replication but does not provide significant protection against filovirus infection in vivo.
To test the in vivo efficacy of the NPC1 GP-binding inhibitor 3.47, we completed dosing studies in which wild-type mice were exposed i.p. to 100 PFU of mouse-adapted EBOV and treated daily with compound 3.47 at various doses. Compared to vehicle control mice, compound 3.47 did not result in a statistically significant increase in survival following EBOV challenge, as all mice in all dosage groups displayed clinical signs of disease and succumbed by day 10 (see Fig. S4 in the supplemental material). Due to solubility limitations, 25 mg/kg of body weight was the maximum dose attainable for compound 3.47.
To investigate the potential anti-EBOV efficacy of imipramine, we challenged wild-type mice i.p. with 100 PFU of mouse-adapted EBOV and treated mice i.p. daily (sid) or every other day (qod) with imipramine at 20 mg/kg. The dose was based on published reports of the in vivo activity of imipramine (32–36) and on preliminary dosing studies completed by our group (data not shown). Control mice were treated daily with an equal volume of vehicle control i.p. Imipramine treatments did not significantly reduce weight loss attributed to EBOV infection compared to control mice, and daily imipramine treatments did not confer significant protection (P = 0.066) or delay to death (P = 0.067) compared to vehicle-treated control mice (Fig. 5A and B). However, in tissues collected from a subset of mice on days 3 and 5 postchallenge, imipramine treatment did significantly reduce viral titers compared to those in vehicle-treated control mice (Fig. 5C and D).
FIG 5 .

Imipramine reduces EBOV replication in vivo. Wild-type mice were challenged i.p. with 100 PFU of mouse-adapted EBOV and treated either sid (n = 39) or qod (n = 30) with 20 mg/kg of imipramine. Control mice (n = 40) received an equivalent volume of vehicle control sid. Mice were monitored for weight loss (A) and mortality (B) following challenge. On days 3 (C) and 5 (D) postchallenge, subsets of mice from control and qod groups were euthanized, and viral titers were determined for the indicated tissues by plaque assay. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
We next tested the in vivo efficacy of U18666a. Here, we challenged mice i.p. with 100 PFU of mouse-adapted EBOV and treated mice daily, i.p., with U18666a at 2 mg/kg. As with imipramine, the dose was based on the reported in vivo activity of U18666a (37, 38) and results of preliminary studies completed by our group (data not shown). Control mice were treated daily with an equal volume of vehicle control by i.p. injection. Daily treatments with U18666a did not reduce EBOV-induced weight loss, nor did they provide increased protection from mouse-adapted EBOV challenge compared to control treated mice (Fig. 6A and B). Much like imipramine, U18666a treatments did reduce viral titers in serum, liver, and spleen, but only on day 3 postchallenge (Fig. 6C and D). While imipramine and U18666a provided only modest protection against EBOV morbidity, these compounds did significantly reduce viral replication in vivo. These data suggest that NPC1-targeting therapeutics, with enhanced NPC1 specificity and a greater capacity to reduce viral load, could augment survival after EBOV infection.
FIG 6 .

U18666a reduces EBOV replication in vivo. Wild-type mice were challenged i.p. with 100 PFU of mouse-adapted EBOV and treated sid with either 2 mg/kg of U18666a (n = 39) or an equivalent volume of vehicle control (n = 38). Mice were monitored for weight loss (A) and mortality (B) following challenge. On days 3 (C) and 5 (D) postchallenge, subsets of mice from each group were euthanized and viral titers were determined for indicated tissues by plaque assay. *, P ≤ 0.05; **, P ≤ 0.01.
DISCUSSION
The importance of NPC1 for filovirus entry into host cells has been clearly shown in previous work (12, 13, 39, 40). In addition, Npc1+/− mice were reported to have a strong survival advantage, relative to wild-type mice when exposed to mouse-adapted EBOV or MARV; however, these mice did display clinical symptoms of filovirus infection (12). Here we show, for the first time, that Npc1−/− mice are completely resistant to mouse-adapted EBOV infection, with no apparent clinical signs of EBOV infection (Fig. 1). Strikingly, no detectable viral titers were observed in tissues collected from EBOV-challenged Npc1−/− mice (Fig. 1). Furthermore, histological analysis illustrated no virus-induced lesions in liver and spleen tissue, and both tissues were negative for viral antigen by immunohistochemistry staining (see Fig. S1 and S2 in the supplemental material). This is the first evidence that complete loss of NPC1 expression in vivo results in absolute resistance to filovirus replication and pathogenesis. We therefore conclude that NPC1 is a bona fide target for therapeutics aimed at the prevention and control of filovirus infections.
In contrast to the findings with the Npc1−/− mice, we observed high levels of viral replication and weight loss in EBOV-challenged Npc1+/− mice (Fig. 1). Interestingly, however, and consistent with the resistance of the NPC1 heterozygotes to EBOV challenge, both viral titers and clinical signs observed in Npc1+/− mice were short-lived in comparison to those observed in wild-type mice (Fig. 1). The elevated levels of IL-5 and IL-12 at day 7 postinfection (Fig. 2) in Npc1+/− mice and not in wild-type mice strongly suggest that adaptive immune responses, particularly B cell responses, were elicited only in the heterozygotes, a finding that may explain the difference in survival and viral titers between these two groups. Furthermore, proinflammatory cytokines MIP-1α, MCP-1, and RANTES remained elevated in wild-type mice at day 7 postinfection (Fig. 2), when these cytokines are normally downregulated. This unchecked proinflammatory state is a hallmark of EBOV infection, yet Npc1+/− mice seem to more appropriately regulate both innate and adaptive immune responses than their wild-type counterparts, despite having equivalent viral titers at day 3 postinfection (Fig. 1). While the mechanism for this differential regulation of the proinflammatory immune response between wild-type and Npc1+/− mice remains to be fully elucidated, these findings do suggest that complete loss of NPC1 expression or function is not required for survival. Instead, simply lowering NPC1 expression below a yet-to-be defined threshold may be sufficient to provide protection when using NPC1-targeted filovirus therapeutics. Such a possibility also raises the question of whether the partial resistance in Npc1+/− mice could be reflective of a heterozygote advantage in humans exposed to EBOV. While controversial, such conjectures have been made for another lysosomal disease and infectious agent, namely, that of Tay-Sachs disease heterozygosity providing protection against infection by Mycobacterium tuberculosis (41).
Niemann-Pick type C disease caused by abnormality in NPC1 or NPC2 function leads to accumulation of cholesterol and glycosphingolipids in late endosomes (19, 42, 43), the presumed site of filovirus membrane fusion and cytoplasmic escape (44, 45). This raises the possibility that stored lysosomal cholesterol and glycosphingolipids (or their effects on cellular physiology) may inhibit filovirus entry. Carette, Cote, and colleagues previously demonstrated that NPC2-deficient fibroblasts were susceptible to filovirus GP-mediated entry (12, 13), suggesting that late endosomal cholesterol/glycosphingolipid accumulation alone is insufficient to block filovirus entry. Here, we found that NPC2 knockout mice were as susceptible to mouse-adapted EBOV as wild-type mice (Fig. 3), even though these knockout mice have a similar cholesterol/glycosphingolipid accumulation phenotype as filovirus-resistant NPC1 knockout mice (46). Yet, this does not directly address whether the buildup of cholesterol or glycosphingolipids in late endosomes of NPC1-deficient cells contributes to resistance against filovirus infection. To answer this question, we treated NPC1 knockout mice with HPBCD, which successfully cleared cholesterol from the late endosomes of these mice (Fig. 3). Despite the restoration of normal cellular cholesterol levels in these mice, they remained completely resistant to mouse-adapted EBOV challenge (Fig. 3). The converse is true as well. Cote et al. reported that CHO cells expressing NPC1 mutant P692S were still susceptible to EBOV-GP pseudovirus infection despite having a cholesterol trafficking defect that resulted in a Niemann-Pick type C phenotype (13). These findings provide further evidence that the lack of NPC1 in NPC1-deficient cells or mice is solely responsible for filovirus resistance, with little or no role for late endosomal cholesterol or glycosphingolipid accumulation.
Class 2 amphiphiles U18666a and imipramine are known to interfere with intracellular cholesterol trafficking, possibly through antagonizing NPC1 function, though the mechanism is not well understood (47). Cells treated with either U18666a or imipramine accumulate cholesterol and glycosphingolipids in late endosomes in much the same way NPC1- or NPC2-deficient cells do (26–28). Carette et al. demonstrated that treating cells with U18666a severely limited filovirus replication (12); however, the antifilovirus activity of U18666a did not seem to be a result of direct inhibition of GP-NPC1 binding (13). Here, we have illustrated a similar antiviral activity of imipramine against filovirus using physiologically relevant HUVECs (Fig. 4), which is in agreement with previously reported antiviral activity in VSV pseudovirus assays (12) and authentic EBOV infection in HeLa cells (48). Compound 3.47 has previously been shown to inhibit EBOV GP-mediated entry of VSV pseudotypes by blocking GP-NPC1 binding (13). We determined that compound 3.47 was efficient at limiting EBOV infection in vitro but that the antiviral effect seemed to be limited to EBOV (Fig. 4). Compound 3.47 had little or no antiviral effect against MARV or SUDV, suggesting a mechanistic difference between MARV and SUDV GP engagement with NPC1 compared to that of EBOV GP. This is somewhat surprising, given that presumptive NPC1-binding sequences in the GP1 head subdomain are highly conserved among ebolaviruses (marburgviruses are more divergent) (49). Moreover, infection by EBOV, MARV, or SUDV requires the same loop C region of NPC1 (39). Uncovering the molecular basis of the GP-loop C binding interaction and delineating GP strain-dependent differences in this interaction may help explain the filovirus species selectivity of 3.47.
NPC1 represents a promising target for the development of filovirus therapeutics. Given the in vitro efficacy of U18666a, imipramine, and compound 3.47, we chose to investigate the therapeutic potential of these compounds by using a mouse model of EBOV disease. We were unable to provide protection to EBOV-infected mice with compound 3.47 (see Fig. S4 in the supplemental material), even when we treated them with the maximal allowable dose, dictated by solubility limitations. As for U18666a, lower doses seemed to be better tolerated and more efficacious against EBOV infection, though at no dose of U18666a was there a statistically significant enhancement in survival compared to control mice (Fig. 6). Lower doses of imipramine also seemed to be more protective against EBOV infection (Fig. 5). Imipramine treatment afforded a moderate increase in survival that approached statistical significance (P = 0.066), and both U18666a and imipramine treatment did significantly reduce viral replication in liver, spleen, and serum compared to control mice (Fig. 5 and 6). Other cationic amphiphiles reported to induce an NPC1-deficient phenotype have also demonstrated potent antiviral activity against Ebolavirus replication in cells (50), though no direct perturbance of GP-NPC1 binding could be established. In addition to in vitro antiviral activity, preliminary mouse studies suggest that two of these cationic amphiphiles, clomiphene and toremifene, can provide protection against EBOV infection (51). Collectively, these data suggest that therapies targeting the NPC1 pathway, either directly or indirectly, can effectively limit filovirus replication and that therapies more efficient at blocking NPC1-GP binding may prove to be potent antifilovirus therapies. In the case of cationic amphiphiles, the lack of a mechanistic understanding makes it difficult to decipher why exactly these compounds are more or less effective. One possible explanation is their limited NPC1 specificity, which may result in only reduced, yet sufficient, NPC1 availability for filovirus engagement (27, 52, 53). Alternatively, these compounds may be hindered by pharmacokinetic limitations such that therapeutic concentrations may not be without adverse side effects. Developing NPC1-directed therapies that limit filovirus replication and pathogenesis more efficiently than U18666a and imipramine may tip the balance in favor of the immune system, allowing the host to clear the virus and survive.
MATERIALS AND METHODS
Cells and viruses.
Vero E6 cells (ATCC) were maintained in Eagle’s minimal essential medium (EMEM) supplemented with 5% heat-inactivated fetal bovine serum (ΔFBS) and gentamicin (50 µg/ml) at 37°C, 5% CO2, and 80% humidity. HUVECs (ATCC) were maintained in endothelial cell Basal Medium-2 (Lonza) supplemented with 5% ΔFBS and penicillin-streptomycin (100 U; 100 µg/ml) at 37°C, 5% CO2, and 80% humidity. SUDV Boniface, EBOV Kikwit, and MARV Ci67 viruses were used for all in vitro experiments. Mouse-adapted EBOV was used for all mouse challenge studies (54). VSV pseudotypes bearing glycoproteins derived from VSV, EBOV, SUDV, RESTV, and MARV were generated as described previously (55).
Drugs and compounds.
Imipramine-HCl stock (catalog number I7379; Sigma-Aldrich) used for both in vitro and in vivo studies was reconstituted in sterile 0.9% sodium chloride solution (0409-4888; Hospira) at 50 mg/ml and stored at room temperature. Imipramine working stocks used for in vitro studies were diluted at the indicated concentrations in appropriate culture media. Imipramine working stocks used for daily in vivo treatments were diluted to appropriate concentrations in sterile 0.9% sodium chloride solution. U18666a stock (U3633; Sigma-Aldrich) was reconstituted in sterile water at 10 mg/ml and stored at 4°C. U18666a working stocks used for daily in vivo treatments were diluted to appropriate concentrations in sterile phosphate-buffered saline (PBS). 3.47 stock (MBX2545; Microbiotix) used for in vitro studies was reconstituted in dimethyl sulfoxide (DMSO) at 20 mM and stored at −20°C. 3.47 stock used for in vivo studies was reconstituted in sterile 20% HPBCD solution (H107; Sigma-Aldrich) at 5 mg/ml and stored at 4°C. 3.47 working stocks used for daily in vivo treatments were diluted to appropriate concentrations in sterile 40% HPBCD solution.
In vitro infection of Vero cells and HUVECs.
Vero E6 cells or HUVECs were seeded in 96-well poly-d-lysine-coated black plates (Greiner Bio-OneCellcoat) at 5 × 104 cells per well in culture medium. Cells were pretreated with culture medium containing imipramine (10 µM), 3.47 (20 µM), or culture medium alone for 1 h at 37°C, 5% CO2, and 80% humidity prior to infection with MARV, EBOV, or SUDV at a multiplicity of infection of 1. Following a 1-h incubation with virus at 37°C, 5% CO2, and 80% humidity, fresh culture medium with or without imipramine (10 µM) or 3.47 (20 µM) was added to the cells. Uninfected cells with or without imipramine (10 µM) or 3.47 (20 µM) served as negative controls. Cells were incubated at 37°C, 5% CO2, and 80% humidity and subsequently fixed with 10% formalin at 48 and 72 h postinfection. Infectivities of VSV pseudotypes in Vero cells were measured by manually counting enhanced green fluorescent protein (eGFP)-positive cells using fluorescence microscopy at 16 to 24 h postinfection, as described previously (44).
Immunofluorescence assay.
For the immunofluorescence assays, formalin-fixed cells were blocked with 1% bovine serum albumin solution prior to incubation with primary antibodies. MARV-, EBOV-, or SUDV-infected cells and uninfected control cells were incubated with MARV-specific murine monoclonal antibody 9G4, EBOV-specific human monoclonal antibody KZ52, or SUDV-specific murine monoclonal antibody 3C10, respectively. Cells were washed with PBS prior to incubation with either goat anti-mouse IgG–Alexa Fluor 488 or goat anti-human IgG–Alexa Fluor 488 (Molecular Probes). Cells were stained with Hoechst stain (Molecular Probes) and then washed with PBS and stored at 4°C. Images were acquired at 9 fields/well with a 10× objective lens on a Discovery-1 high-content imager (Molecular Devices) or at 6 fields/well with a 20× objective lens on an Operetta (PerkinElmer) high-content device. Discovery-1 images were analyzed with the “live/dead” module in MetaXpress software. Operetta images were analyzed with a customized scheme built from image analysis functions present in Harmony software, and the percentage of infected cells was determined using the analysis functions.
Cholesterol was visualized using 0.005% filipin complex (F9765; Sigma-Aldrich) diluted in DMSO, and staining was carried out as previously described (56). Briefly, 4% paraformaldehyde-fixed hemicoronal sections were cut at 35-µm thickness on a Leica VT1000S Vibratome. Sections were washed two times for 10 min in PBS, then two times for 10 min in 0.02% saponin (S7900; Sigma-Aldrich)–PBS (wash solution), and incubated with DMSO-PBS (control) or filipin complex-PBS for 20 min at room temperature. Sections were then washed two times for 10 min in wash solution and finally two times for 10 min in PBS. Tissue sections were mounted on slides, and coverslips were attached using ProLong Gold Antifade mounting medium (P36930; Life Technologies).
In vivo challenge studies.
Animal research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals of the National Research Council (57). The facility is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International, All challenge studies were conducted under maximum containment in an animal biosafety level 4 (BSL-4) facility at USAMRIID and were approved by the USAMRIID Institutional Animal Care and Use Committee under protocols AP-10-004, AP-10-005, and AP-11-004.
Npc1 and Npc2 heterozygous and homozygous knockout mice (BALB/c background) and wild-type littermates were bred and genotyped at Albert Einstein College of Medicine, as described previously (30), and subsequently shipped to USAMRIID at 3 to 5 weeks of age. Wild-type BALB/c mice used for drug efficacy studies were purchased from the National Cancer Institute, Frederick, MD.
For knockout studies, 3- to 5-week-old Npc1 or Npc2 heterozygous and homozygous knockout mice and wild-type littermates were challenged i.p. with a target dose of 100 PFU of mouse-adapted EBOV diluted in 200 µl of PBS. Mice were monitored for morbidity and mortality, and weight loss was recorded. On days 3 and 7 postchallenge, a subset of Npc1+/−, Npc1−/−, and wild-type mice were euthanized to collect serum, liver, spleen, and kidney tissue for virus titer determinations. Npc1+/−, Npc1−/−, and wild-type control animals that received HPBCD treatments were injected subcutaneously with 20% HPBCD solution (Sigma-Aldrich) at 4,000 mg/kg every other day starting at postnatal day 7 (P7) until the end of the challenge study or death of the animal (30). Titration of challenge inocula determined that the mice received between 92.5 and 135 PFU of mouse-adapted EBOV.
For drug efficacy studies, 8- to 10-week-old female wild-type BALB/c mice were challenged i.p. with a target dose of 100 PFU of mouse-adapted EBOV diluted in 200 µl of PBS. Thirty minutes postchallenge, mice were treated i.p. with imipramine, U18666a, or 3.47 at the indicated concentrations in 100 to 200 µl of diluent. Control mice for each experiment received an equal volume of appropriate vehicle control. Treatments continued daily or every other day as on day 0, until day 10 postinfection. Where indicated, a subset of mice from treatment groups and control groups were euthanized on days 3 and 5 postinfection to collect serum, liver, and spleen tissue for viral titer determinations. Titration of challenge inocula determined that the mice received between 92.25 and 110 PFU of mouse-adapted EBOV.
Plaque assay for viral titers.
For plaque assays to determine viral titers, tissues were weighed and 10% tissue homogenates were generated in RPMI supplemented with 2% ΔFBS using a gentleMACS dissociator (Miltenyi Biotec). Ten-fold serial dilutions of tissue homogenates or serum were prepared in modified Eagle’s medium with Earle’s balanced salts and nonessential amino acids (EMEM/NEAA) supplemented with 5% ΔFBS, 2 mM l-glutamine, and 1% gentamicin. Aliquots (200 μl) of each dilution were inoculated onto 6-well plates of confluent monolayers of Vero E6 cells. After adsorption for 1 h at 37°C, 5% CO2, and 80% humidity, monolayers were overlaid with a mixture of 1 part 1% agarose (SeaKem) and 1 part 2× Eagle basal medium (EBME), 30 mM HEPES buffer, and 5% ΔFBS. After incubation at 37°C, 5% CO2, and 80% humidity for 6 days, a second overlay with 5% neutral red was added. Plaques were counted the following day, and titers are reported as PFU per milliliter of the 10% tissue homogenates.
Pathology.
The liver, spleen, and kidney were collected at the indicated days postinfection and fixed in formalin for histopathology and immunohistochemistry. Each formalin-fixed tissue sample was trimmed and embedded in paraffin. Sections of the paraffin-embedded tissues, 5 µm thick, were cut for histology. The histology slides were deparaffinized, placed on coverslips, and stained with hematoxylin and eosin (H&E).
Immunohistochemistry was performed on all tissue sections by using the UltraVision (mouse on mouse) HRP kit. A mouse monoclonal anti-EBOV antibody (702/703) was used at a dilution of 1:8,000. After deparaffinization and methanol and H2O2 blocking, sections were pretreated with proteinase K (Dako) at room temperature for 6 min. Sections were rinsed with PBS and then covered with serum-free protein block (Dako) plus 10% normal goat serum for 30 min. A rodent block was then applied for 45 min, followed by primary antibody for 30 min. The sections were then rinsed, and peroxidase-labeled polymer (secondary antibody) was applied for 30 min. Slides were rinsed, and a substrate-chromogen solution was applied for 5 min. The substrate-chromogen solution was rinsed off the slides, and slides were stained with H&E and rinsed. The sections were dehydrated, cleared with Xyless, and then placed on coverslips. The intensity of EBOV antigen labeling was semiquantitatively documented using a scale of 1 to 4, with 4 representing the highest concentration of antigen labeling (0, no cells in section were EBOV positive; 1, <10% of cells in section were positive [minimal]; 2, 11 to 25% of cells in section were positive [mild]; 3, 26 to 50% of cells in section were positive [moderate]; 4. >50% of cells in section were positive [marked]).
BioPlex assay for cytokine analysis.
For the BioPlex assay (Bio-Rad Laboratories, Inc.), serum was collected from Npc1 homozygous and heterozygous knockout mice and wild-type littermate controls on day 3 or 7 postinfection with mouse-adapted EBOV. Cytokine concentrations were evaluated in accordance with the manufacturer’s instructions. Serum cytokine levels for each respective cytokine were quantitated using defined standard curves optimized for serum product.
Statistical analysis.
Survival curves were analyzed using the Fisher’s exact test (two tailed) with a step-down Bonferroni adjustment for survival rate comparisons. Time-to-death comparisons were completed using the t test with Dunnett’s correction. All cytokine and tissue titration data were analyzed via group comparisons using linear regression of the response variable with the main effects for day and group interactions. Cytokine variables were untransformed, while all the titer variables were based on log10 transformations.
SUPPLEMENTAL MATERIAL
H&E staining of tissues from mouse-adapted EBOV-infected wild-type and NPC1 knockout mice. Npc1−/−, Npc1+/−, and wild-type littermate control mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Tissues were harvested and fixed in formalin prior to processing for H&E staining. (A) At day 3, cells were relatively normal histologically. (B) By day 7, splenitis and hepatitis (asterisks) were associated with fibrin and hemorrhage, respectively, in wild-type mice; individual cell apoptosis/necrosis was observed in Npc1+/− mice, while histology in Npc−/− mice remained normal. Download
Immunohistochemistry staining of tissues from mouse-adapted EBOV-infected wild-type and NPC1 knockout mice. Npc1−/−, Npc1+/−, and wild-type littermate control mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Tissues were harvested at days 3 (A) and 7 (B) and fixed in formalin prior to processing for immunohistochemical staining for EBOV antigen. Overall, the strongest and most diffusely present immunopositivity was noted in wild-type mice (asterisk). Npc1+/− mice displayed mild to multifocal positivity with diminished intensity compared to wild-type mice and Npc1−/− mice remained negative at both time points. Download
Cell cytotoxicity of imipramine treatment of HUVECs. HUVECs were seeded in 96-well plates and treated with the indicated concentrations of imipramine. At 72 h posttreatment, CellTiter-Glo reagent (Promega) was added to the cells and luminescence was measured. Luminescent values for untreated control cells were used to calculate the percentage of control viability for each treatment condition. Download
Compound 3.47 treatment does not protect against mouse-adapted EBOV infection. Wild-type mice were challenged i.p. with 100 PFU of mouse-adapted EBOV and treated daily with either 1 (n = 10), 5 (n = 10), or 25 (n = 10) mg/kg of compound 3.47. Control mice (n = 10) received an equivalent volume of vehicle control daily. Mice were monitored for weight loss (A) and mortality (B) following challenge. Download
ACKNOWLEDGMENTS
We thank Sam Dickson for help with statistical analyses.
Opinions, conclusions, interpretations, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army. The mention of trade names or commercial products does not constitute endorsement or recommendation for use by the Department of the Army or the Department of Defense.
Footnotes
Citation Herbert AS, Davidson C, Kuehne AI, Bakken R, Braigen SZ, Gunn KE, Whelan SP, Brummelkamp TR, Twenhafel NA, Chandran K, Walkley SU, Dye JM. 2015. Niemann-Pick C1 is essential for Ebolavirus replication and pathogenesis in vivo. mBio 6(3):e00565-15. doi:10.1128/mBio.00565-15.
REFERENCES
- 1.Hoenen T, Groseth A, Falzarano D, Feldmann H. 2006. Ebola virus: unravelling pathogenesis to combat a deadly disease. Trends Mol Med 12:206–215. doi: 10.1016/j.molmed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Kuhn JH, Becker S, Ebihara H, Geisbert TW, Johnson KM, Kawaoka Y, Lipkin WI, Negredo AI, Netesov SV, Nichol ST, Palacios G, Peters CJ, Tenorio A, Volchkov VE, Jahrling PB. 2010. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch Virol 155:2083–2103. doi: 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez CP, Lasala F, Carrillo J, Muñiz O, Corbí AL, Delgado R. 2002. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J Virol 76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker S, Spiess M, Klenk HD. 1995. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J Gen Virol 76:393–399. doi: 10.1099/0022-1317-76-2-393. [DOI] [PubMed] [Google Scholar]
- 7.Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, Meyerholz DK, Rennert P, Mullins RF, Brindley M, Sandersfeld LM, Quinn K, Weller M, McCray PB Jr., Chiorini J, Maury W. 2011. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc Natl Acad Sci U S A 108:8426–8431. doi: 10.1073/pnas.1019030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimojima M, Takada A, Ebihara H, Neumann G, Fujioka K, Irimura T, Jones S, Feldmann H, Kawaoka Y. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J Virol 80:10109–10116. doi: 10.1128/JVI.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. 2010. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog 6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog 6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulherkar N, Raaben M, de la Torre JC, Whelan SP, Chandran K. 2011. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 419:72–83. doi: 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan A, Miller EH, Herbert AS, Ng M, Ndungo E, Whelan SP, Dye JM, Chandran K. 2012. Niemann-Pick C1 (NPC1)/NPC1-like chimeras define sequences critical for NPC1’s function as a flovirus entry receptor. Viruses 4:2471–2484. doi: 10.3390/v4112471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. 2012. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J 31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies JP, Ioannou YA. 2000. Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J Biol Chem 275:24367–24374. doi: 10.1074/jbc.M002184200. [DOI] [PubMed] [Google Scholar]
- 17.Cruz JC, Sugii S, Yu C, Chang TY. 2000. Role of Niemann-Pick type C1 protein in intracellular trafficking of low density lipoprotein-derived cholesterol. J Biol Chem 275:4013–4021. doi: 10.1074/jbc.275.6.4013. [DOI] [PubMed] [Google Scholar]
- 18.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. 2008. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A 105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF III, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O’Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. 1997. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 20.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. 2000. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 21.Karten B, Vance DE, Campenot RB, Vance JE. 2003. Trafficking of cholesterol from cell bodies to distal axons in Niemann-Pick C1-deficient neurons. J Biol Chem 278:4168–4175. doi: 10.1074/jbc.M205406200. [DOI] [PubMed] [Google Scholar]
- 22.Ko DC, Gordon MD, Jin JY, Scott MP. 2001. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol Biol Cell 12:601–614. doi: 10.1091/mbc.12.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higgins ME, Davies JP, Chen FW, Ioannou YA. 1999. Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab 68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- 24.Cenedella RJ. 2009. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 44:477–487. doi: 10.1007/s11745-009-3305-7. [DOI] [PubMed] [Google Scholar]
- 25.Sugimoto Y, Ninomiya H, Ohsaki Y, Higaki K, Davies JP, Ioannou YA, Ohno K. 2001. Accumulation of cholera toxin and GM1 ganglioside in the early endosome of Niemann-Pick C1-deficient cells. Proc Natl Acad Sci U S A 98:12391–12396. doi: 10.1073/pnas.221181998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lange Y, Ye J, Rigney M, Steck T. 2000. Cholesterol movement in Niemann-Pick type C cells and in cells treated with amphiphiles. J Biol Chem 275:17468–17475. doi: 10.1074/jbc.M000875200. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Lafrasse C, Rousson R, Bonnet J, Pentchev PG, Louisot P, Vanier MT. 1990. Abnormal cholesterol metabolism in imipramine-treated fibroblast cultures. Similarities with Niemann-Pick type C disease. Biochim Biophys Acta 1043:123–128. doi: 10.1016/0005-2760(90)90284-5. [DOI] [PubMed] [Google Scholar]
- 28.Underwood KW, Andemariam B, McWilliams GL, Liscum L. 1996. Quantitative analysis of hydrophobic amine inhibition of intracellular cholesterol transport. J Lipid Res 37:1556–1568. [PubMed] [Google Scholar]
- 29.Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P. 2004. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci U S A 101:5886–5891. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. 2009. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM. 2009. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the Npc1−/− mouse. Proc Natl Acad Sci U S A 106:2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Llacuna L, Marí M, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. 2006. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology 44:561–572. doi: 10.1002/hep.21285. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Qu JM, Summah H, Zhang J, Zhu YG, Jiang HN. 2010. Protective effects of imipramine in murine endotoxin-induced acute lung injury. Eur J Pharmacol 638:128–133. doi: 10.1016/j.ejphar.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Hendriksen H, Prins J, Olivier B, Oosting RS. 2010. Environmental enrichment induces behavioral recovery and enhanced hippocampal cell proliferation in an antidepressant-resistant animal model for PTSD. PLoS One 5:e11943. doi: 10.1371/journal.pone.0011943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isacson R, Nielsen E, Dannaeus K, Bertilsson G, Patrone C, Zachrisson O, Wikström L. 2011. The glucagon-like peptide 1 receptor agonist exendin-4 improves reference memory performance and decreases immobility in the forced swim test. Eur J Pharmacol 650:249–255. doi: 10.1016/j.ejphar.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Need AB, Baez M, Witkin JM. 2006. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther 319:254–259. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]
- 37.Hagiwara K, Nakamura Y, Nishijima M, Yamakawa Y. 2007. Prevention of prion propagation by dehydrocholesterol reductase inhibitors in cultured cells and a therapeutic trial in mice. Biol Pharm Bull 30:835–838. doi: 10.1248/bpb.30.835. [DOI] [PubMed] [Google Scholar]
- 38.Cenedella RJ. 1983. Source of cholesterol for the ocular lens, studied with U18666A: a cataract-producing inhibitor of lipid metabolism. Exp Eye Res 37:33–43. doi: 10.1016/0014-4835(83)90147-1. [DOI] [PubMed] [Google Scholar]
- 39.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. 2012. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J 31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haines KM, Vande Burgt NH, Francica JR, Kaletsky RL, Bates P. 2012. Chinese hamster ovary cell lines selected for resistance to ebolavirus glycoprotein mediated infection are defective for NPC1 expression. Virology 432:20–28. doi: 10.1016/j.virol.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rotter JI, Diamond JM. 1987. What maintains the frequencies of human genetic diseases? Nature 329:289–290. doi: 10.1038/329289a0. [DOI] [PubMed] [Google Scholar]
- 42.Vanier MT, Duthel S, Rodriguez-Lafrasse C, Pentchev P, Carstea ED. 1996. Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am J Hum Genet 58:118–125. [PMC free article] [PubMed] [Google Scholar]
- 43.Walkley SU, Suzuki K. 2004. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta 1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 44.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White JM, Schornberg KL. 2012. A new player in the puzzle of filovirus entry. Nat Rev Microbiol 10:317–322. doi: 10.1038/nrmicro2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen GK, Dagnaes-Hansen F, Holm IE, Meaney S, Symula D, Andersen NT, Heegaard CW. 2011. Protein replacement therapy partially corrects the cholesterol-storage phenotype in a mouse model of Niemann-Pick type C2 disease. PLoS One 6:e27287. doi: 10.1371/journal.pone.0027287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koh CH, Cheung NS. 2006. Cellular mechanism of U18666A-mediated apoptosis in cultured murine cortical neurons: bridging Niemann-Pick disease type C and Alzheimer’s disease. Cell Signal 18:1844–1853. doi: 10.1016/j.cellsig.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 48.Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. 2012. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol 86:7473–7483. doi: 10.1128/JVI.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhn JH, Radoshitzky SR, Guth AC, Warfield KL, Li W, Vincent MJ, Towner JS, Nichol ST, Bavari S, Choe H, Aman MJ, Farzan M. 2006. Conserved receptor-binding domains of Lake Victoria Marburgvirus and Zaire Ebolavirus bind a common receptor. J Biol Chem 281:15951–15958. doi: 10.1074/jbc.M601796200. [DOI] [PubMed] [Google Scholar]
- 50.Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, White JM. 2013. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, Hoffstrom BG, Dewald LE, Schornberg KL, Scully C, Lehár J, Hensley LE, White JM, Olinger GG. 2013. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med 5:190ra179. doi: 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sobo K, Le Blanc I, Luyet PP, Fivaz M, Ferguson C, Parton RG, Gruenberg J, van der Goot FG. 2007. Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS One 2:e851. doi: 10.1371/journal.pone.0000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neufeld EB, Wastney M, Patel S, Suresh S, Cooney AM, Dwyer NK, Roff CF, Ohno K, Morris JA, Carstea ED, Incardona JP, Strauss JF III, Vanier MT, Patterson MC, Brady RO, Pentchev PG, Blanchette-Mackie EJ. 1999. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J Biol Chem 274:9627–9635. doi: 10.1074/jbc.274.14.9627. [DOI] [PubMed] [Google Scholar]
- 54.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. 1998. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis 178:651–661. doi: 10.1086/515386. [DOI] [PubMed] [Google Scholar]
- 55.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A 94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McGlynn R, Dobrenis K, Walkley SU. 2004. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol 480:415–426. doi: 10.1002/cne.20355. [DOI] [PubMed] [Google Scholar]
- 57.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
H&E staining of tissues from mouse-adapted EBOV-infected wild-type and NPC1 knockout mice. Npc1−/−, Npc1+/−, and wild-type littermate control mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Tissues were harvested and fixed in formalin prior to processing for H&E staining. (A) At day 3, cells were relatively normal histologically. (B) By day 7, splenitis and hepatitis (asterisks) were associated with fibrin and hemorrhage, respectively, in wild-type mice; individual cell apoptosis/necrosis was observed in Npc1+/− mice, while histology in Npc−/− mice remained normal. Download
Immunohistochemistry staining of tissues from mouse-adapted EBOV-infected wild-type and NPC1 knockout mice. Npc1−/−, Npc1+/−, and wild-type littermate control mice were challenged i.p. with 100 PFU of mouse-adapted EBOV. Tissues were harvested at days 3 (A) and 7 (B) and fixed in formalin prior to processing for immunohistochemical staining for EBOV antigen. Overall, the strongest and most diffusely present immunopositivity was noted in wild-type mice (asterisk). Npc1+/− mice displayed mild to multifocal positivity with diminished intensity compared to wild-type mice and Npc1−/− mice remained negative at both time points. Download
Cell cytotoxicity of imipramine treatment of HUVECs. HUVECs were seeded in 96-well plates and treated with the indicated concentrations of imipramine. At 72 h posttreatment, CellTiter-Glo reagent (Promega) was added to the cells and luminescence was measured. Luminescent values for untreated control cells were used to calculate the percentage of control viability for each treatment condition. Download
Compound 3.47 treatment does not protect against mouse-adapted EBOV infection. Wild-type mice were challenged i.p. with 100 PFU of mouse-adapted EBOV and treated daily with either 1 (n = 10), 5 (n = 10), or 25 (n = 10) mg/kg of compound 3.47. Control mice (n = 10) received an equivalent volume of vehicle control daily. Mice were monitored for weight loss (A) and mortality (B) following challenge. Download