

ABSTRACT
Toll-like receptors (TLRs) are sensors that recognize molecular patterns from viruses, bacteria, and fungi to initiate innate immune responses to invading pathogens. The emergence of highly pathogenic coronaviruses severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) is a concern for global public health, as there is a lack of efficacious vaccine platforms and antiviral therapeutic strategies. Previously, it was shown that MyD88, an adaptor protein necessary for signaling by multiple TLRs, is a required component of the innate immune response to mouse-adapted SARS-CoV infection in vivo. Here, we demonstrate that TLR3−/−, TLR4−/−, and TRAM−/− mice are more susceptible to SARS-CoV than wild-type mice but experience only transient weight loss with no mortality in response to infection. In contrast, mice deficient in the TLR3/TLR4 adaptor TRIF are highly susceptible to SARS-CoV infection, showing increased weight loss, mortality, reduced lung function, increased lung pathology, and higher viral titers. Distinct alterations in inflammation were present in TRIF−/− mice infected with SARS-CoV, including excess infiltration of neutrophils and inflammatory cell types that correlate with increased pathology of other known causes of acute respiratory distress syndrome (ARDS), including influenza virus infections. Aberrant proinflammatory cytokine, chemokine, and interferon-stimulated gene (ISG) signaling programs were also noted following infection of TRIF−/− mice that were similar to those seen in human patients with poor disease outcome following SARS-CoV or MERS-CoV infection. These findings highlight the importance of TLR adaptor signaling in generating a balanced protective innate immune response to highly pathogenic coronavirus infections.
IMPORTANCE
Toll-like receptors are a family of sensor proteins that enable the immune system to differentiate between “self” and “non-self.” Agonists and antagonists of TLRs have been proposed to have utility as vaccine adjuvants or antiviral compounds. In the last 15 years, the emergence of highly pathogenic coronaviruses SARS-CoV and MERS-CoV has caused significant disease accompanied by high mortality rates in human populations, but no approved therapeutic treatments or vaccines currently exist. Here, we demonstrate that TLR signaling through the TRIF adaptor protein protects mice from lethal SARS-CoV disease. Our findings indicate that a balanced immune response operating through both TRIF-driven and MyD88-driven pathways likely provides the most effective host cell intrinsic antiviral defense responses to severe SARS-CoV disease, while removal of either branch of TLR signaling causes lethal SARS-CoV disease in our mouse model. These data should inform the design and use of TLR agonists and antagonists in coronavirus-specific vaccine and antiviral strategies.
INTRODUCTION
The recent emergence of highly pathogenic severe acute respiratory syndrome (SARS; pandemic in 2002 to 2004), Middle East respiratory syndrome (MERS; Arabian Peninsula epidemic from 2012 to the present), and porcine epidemic diarrhea (PEDV; United States porcine epidemic from 2013 to the present) coronavirus (CoV) infections is indicative of a reoccurring global public health vulnerability (1–3). At the end of the SARS-CoV pandemic, of the 8,096 cases confirmed by the WHO, 774 patients died from SARS, a mortality rate of slightly less than 10% (4). Ten years later, the emergence of a novel human coronavirus, Middle East respiratory syndrome coronavirus (MERS-CoV), has been confirmed in more than 1,000 patients, approximately 40% of whom have died, highlighting the need for continued surveillance for emergent human coronaviruses with the potential to cause severe disease (5). The relative ease with which these pathogens have been spread outside the original geographic origins by global travelers is particularly troubling. Furthermore, recent surveys of bat populations, a known reservoir host of zoonotic coronaviruses, have observed that bats harbor myriad novel and potentially emergent coronaviruses with unknown pathogenic potential, indicating that coronavirus spillover into human and livestock populations may continue (6). Despite the importance of SARS-CoV and MERS-CoV as public health threats, there are currently no available antivirals against these pathogens, with current evidence suggesting that the antiviral drugs ribavirin and interferon (IFN) are not efficacious in ameliorating SARS or MERS infections (7–9). While research on MERS-CoV is still in the nascent stages, efforts to develop a vaccine against SARS-CoV have been hindered by the challenges of vaccine-induced immune pathology as well as the likely need for cross-protection against highly variable and antigenically distinct coronaviruses with unknown emergence and pathogenic potential (10–12).
SARS-CoV and MERS-CoV are phylogenetically and antigenically distinct members of the Coronaviridae family (1, 2). Pathogen-associated molecular patterns (PAMPs) that differentiate between viral and host molecules likely traffic within similar locations in coronavirus-infected host cells and may be detected by similar classes of cellular sensors. Innate immune sensors recognize PAMPs specific to viruses and other invading pathogens, triggering transcriptional changes in host cell signaling programs to establish an antiviral state that suppresses viral replication efficiency. Respiratory virus infections are potentially devastating global health concerns, as evidenced by emerging highly pathogenic 1918 and 2009 H1N1, H5N1, and H7N9 influenza A viruses (IAV), as well as the SARS-CoV and MERS-CoV epidemics (13). The human lung has critical functions in gas exchange and represents a large and complex but highly vulnerable mucosal surface that interfaces with multiple microorganisms in the environment. Lung cells, including type II pneumocytes and ciliated cells of the airway epithelium, are the primary targets of SARS-CoV and IAV infection in the lung (13, 14). When these cells are exposed to pathogens, innate immune signaling cascades are initiated by pattern recognition receptors (PRRs), which include multiple classes of cellular sensors distributed at cellular membranes and within the cytosol to ensure maximal detection of viruses at multiple stages of the replication cycle, including viral entry and genome replication (15).
Toll-like receptors (TLRs) are membrane-bound PRRs that detect molecular patterns associated with viruses, bacteria, and fungi at the plasma membrane and within endosomes. TLR3 has been implicated in the detection of many RNA viruses and in altering the pathogenesis of airway disease resulting from respiratory virus infections such as IAV, respiratory syncytial virus (RSV), and rhinovirus infections (16–18). Basal levels of TLR3 expression are detectable in lung tissues such as in human alveolar cells and bronchial epithelial cells, as well as in various immune cell populations (19). In cells, TLR3 is anchored to the membrane of endosomes, where it recognizes double-stranded RNA (dsRNA) motifs from invading pathogens (20). After binding the dsRNA motif, TLR3 dimerizes and recruits the TRIF adaptor protein (21, 22). TRIF recruitment to the endosome results in signaling to activate transcription factors, including IRF3 and NF-κB (23). In addition to TLR3-specific signaling, TRIF has also been described as an adaptor for signaling by DDX1/DDX21/DHX36 complexes as well as an adaptor for TLR4 signaling (22, 24).
TLR4 is expressed at low basal levels in bronchial epithelial cells and alveolar cells, and expression increases upon infiltration of inflammatory cells in response to insults such as viral infections (25, 26). TLR4 signals through either MyD88 or TRIF using two sorting adaptors: MAL (for MyD88-dependent signaling) and TRAM (for TRIF-dependent signaling) (27). The TLR4/TRAM/TRIF signaling cascade has been previously implicated in the exacerbation of acute respiratory distress syndrome (ARDS) caused by influenza virus infections and acid damage models (28). Controversially, TLR4 has been identified as potentially mediating immunopathogenesis of influenza virus, and TLR4 antagonist Eritoran has been proposed as an immunomodulatory therapeutic for influenza virus infections (29, 30). The role of TLR4 in highly pathogenic coronavirus infections is unclear, although C3H/HeJ mice that are naturally deficient in TLR4 are more susceptible to mouse hepatitis virus (MHV) infection than C3H/HeN mice with wild-type TLR4 signaling capability (31). TLR signaling via TRIF leads to the activation of type I interferons (IFN-α and IFN-β), proinflammatory cytokines (IL-6, TNF, IFN-γ, and CCL5), and interferon-stimulated genes (ISGs) (RSAD2, IFIT1, and CXCL10) (19, 22). These effector molecules have defined importance in the context of ARDS and respiratory virus infections (13, 32).
TLR agonists and antagonists have been proposed as compounds with broad-spectrum therapeutic potential against a number of respiratory infections in the context of antiviral drugs and vaccine adjuvants (29, 33–35). Both the TLR3 agonist poly(I:C) and the TLR4 agonist lipopolysaccharide (LPS) are protective against SARS-CoV infection in mice when administered prophylactically, although poly(I:C) is more effective than LPS (33). In addition, treatment with poly(I:C), a TLR3 agonist which signals independently of MyD88, has protective effects in mouse models of infections by highly pathogenic coronavirus species, including group 2c (MERS-like) coronaviruses (36). There is a need to understand how TLR signaling and effector networks may regulate coronavirus pathogenesis, given the diverse pool of zoonotic precursors with potential for spillover into human and livestock populations. Previous data from our laboratory had indicated a protective role for the TLR adaptor protein MyD88, which facilitates downstream signaling through a large number of TLRs, in our mouse model of SARS-CoV disease (37). Here, we present evidence that MyD88-independent signaling operating through TLR3 and TLR4 via the TRIF adaptor protein exerts a powerful protective cell-intrinsic defense network in response to SARS-CoV infection and disease.
RESULTS
Toll-like receptor pathways are key regulators of SARS-CoV pathogenesis.
Using a network integration approach to identify key regulators of SARS-CoV pathogenesis, a previous study that assayed host mRNA responses in C57BL/6J mice infected with 102, 103, 104, or 105 PFU doses of SARS-CoV yielded a highly prioritized list of candidate genes involved in the host response to SARS-CoV (38, 39). From these microarray data derived from the host mRNAs in the lung, network analyses identified host pathways regulating SARS-CoV pathogenesis that were previously uncharacterized (wound repair pathways [38]) as well as pathways that have a well-established foothold in the literature in respect to SARS-CoV pathogenesis (innate immune pathways) (39). Toll-like receptors (TLRs) play a critical role in the recognition of pathogens and induction of the innate immune response to many viruses, but TLR recognition of SARS-CoV is not well characterized. Two TLR-related genes that were highly ranked on the prioritized list are differentially expressed in response to SARS-CoV infection compared to expression in mock-inoculated mice: MyD88 and TLR3 (Fig. 1A and B).
FIG 1 .
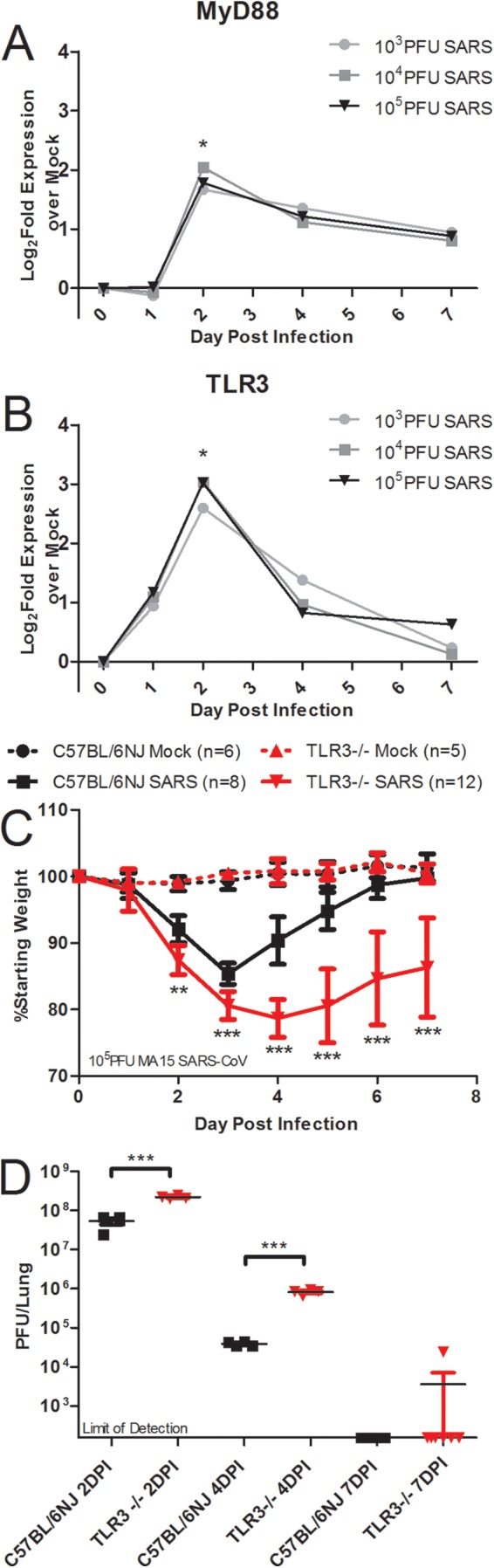
Two discrete TLR pathways regulate SARS-CoV pathogenesis. (A and B) Profiles from microarray analysis of MyD88 (A) and TLR3 (B) RNA expression results in 20-week-old C57BL/6J mice infected with 103, 104, or 105 PFU of SARS-CoV indicate that differential levels of gene expression occurred at day 2 postinfection (a single asterisk [*] indicates differential expression determined by a >1.5 log2-fold increase in expression compared to the results seen with mock infections; P < 0.05). (C and D) Infection of TLR3−/− and C57BL/6NJ mice with SARS-CoV showed significantly greater weight loss in TLR3−/− mice than in wild-type mice (**, P < 0.01; ***, P < 0.001, by nonparametric Mann-Whitney test, where values indicate the mean percent starting weight and error bars indicate standard deviation) (C), and viral titers were significantly higher in the TLR3−/− mice than in wild-type mice (***, P < 0.001 [by Student’s unpaired t test]) (D). TLR3 signaling through the TRIF adaptor protein activated innate immune antiviral signaling programs in a MyD88-independent manner, indicating that at least two discrete TLR signaling pathways are involved in SARS-CoV pathogenesis. DPI, day postinfection.
MyD88 transcripts were significantly upregulated at the doses of 103, 104, and 105 PFU at day 2 postinfection and met the fold change threshold for categorization as representing a differentially expressed gene at that time point but not at day 1, 4, or 7 postinfection (Fig. 1A). SARS-CoV-infected mice had a RNA expression profile for TLR3 similar to that for MyD88: it was differentially expressed at day 2 postinfection with SARS-CoV at the doses of 103, 104, and 105 PFU of SARS-CoV (Fig. 1B). Based on the similarities in gene expression of MyD88 and TLR3, the ranking of the genes by network integration analyses, and the known protective role of MyD88 in SARS-CoV infection, we hypothesized that TLR3 signaling may also be involved in the protective innate immune response to SARS-CoV infection. TLR3 signaling occurs in a MyD88-independent manner via the TRIF adaptor protein, so these data indicate that at least two discrete TLR signaling pathways are involved in the host response to SARS-CoV infection.
To test the hypothesis that TLR3 has a protective role in SARS-CoV infection of mice, 10-week-old female TLR3−/− mice and wild-type C57BL/6NJ mice were infected intranasally with 105 PFU of recombinant mouse-adapted SARS-CoV (rMA15–SARS-CoV) to observe differences in the pathogenesis of SARS-CoV disease. Wild-type mice infected with SARS-CoV experienced transient weight loss that peaked on day 3 postinfection, but all of the wild-type mice began to recover from weight loss on day 4 postinfection and recovered fully from weight loss by 6 to 7 days postinfection (Fig. 1C). TLR3−/− mice lost a greater average percentage of their starting weight, with statistically significant differences in TLR3−/− weight loss, than wild-type mice on days 2 to 7 postinfection (Fig. 1C; **, P < 0.01; ***, P < 0.001). Titers observed in the lungs of TLR3−/− mice infected with SARS-CoV were about 4-fold higher than those observed with wild-type mice at day 2, and titers observed in the lungs of TLR3−/− mice infected with SARS-CoV were about 20-fold higher than those observed with wild-type mice at 4 postinfection (Fig. 1D; ***, P < 0.001). On day 7 postinfection, one TLR3−/− mouse had detectable virus in the lungs, while the rest of the TLR3−/− mice and all of the C57BL/6NJ mice had no detectable virus in the lungs (Fig. 1D). Additionally, TLR3−/− mice showed increased SARS-CoV disease measured by aberrant lung function parameters and histopathology compared to wild-type mice (see Fig. S1 in the supplemental material).
TLR3 regulates downstream responses of several key proinflammatory cytokines, and TLR3 can also regulate the induction of type I interferon and downstream signaling by ISGs (Fig. 2A). Surprisingly, microarray analysis of host gene expression showed few alterations in gene expression downstream of TLR3 in comparisons of TLR3−/− mice to wild-type mice (Fig. 2B to I). There was no change in the levels of IL-6 or TNF, two proinflammatory cytokines downstream of TLR3 signaling (Fig. 2B and C). CCL5 and IFN-γ were differentially expressed in TLR3−/− mice compared to wild-type mice, with higher gene expression in the wild-type mice than in the TLR3−/− mice by greater than a 1.5-fold change difference on day 4 postinfection (Fig. 2D and E; *, >1.5-fold change). No differences were observed in expression of IFN-β, a type I interferon (Fig. 2F), or of RSAD2 (Fig. 2G), CXCL10 (Fig. 2H), and IFIT1 (Fig. 2I), three ISGs. Analysis of microarray results for genes differentially expressed in wild-type C57BL/6NJ mice compared to TLR3−/− mice indicated differences in a number of other genes at days 2 and 4 postinfection (see Table S1 in the supplemental material).
FIG 2 .
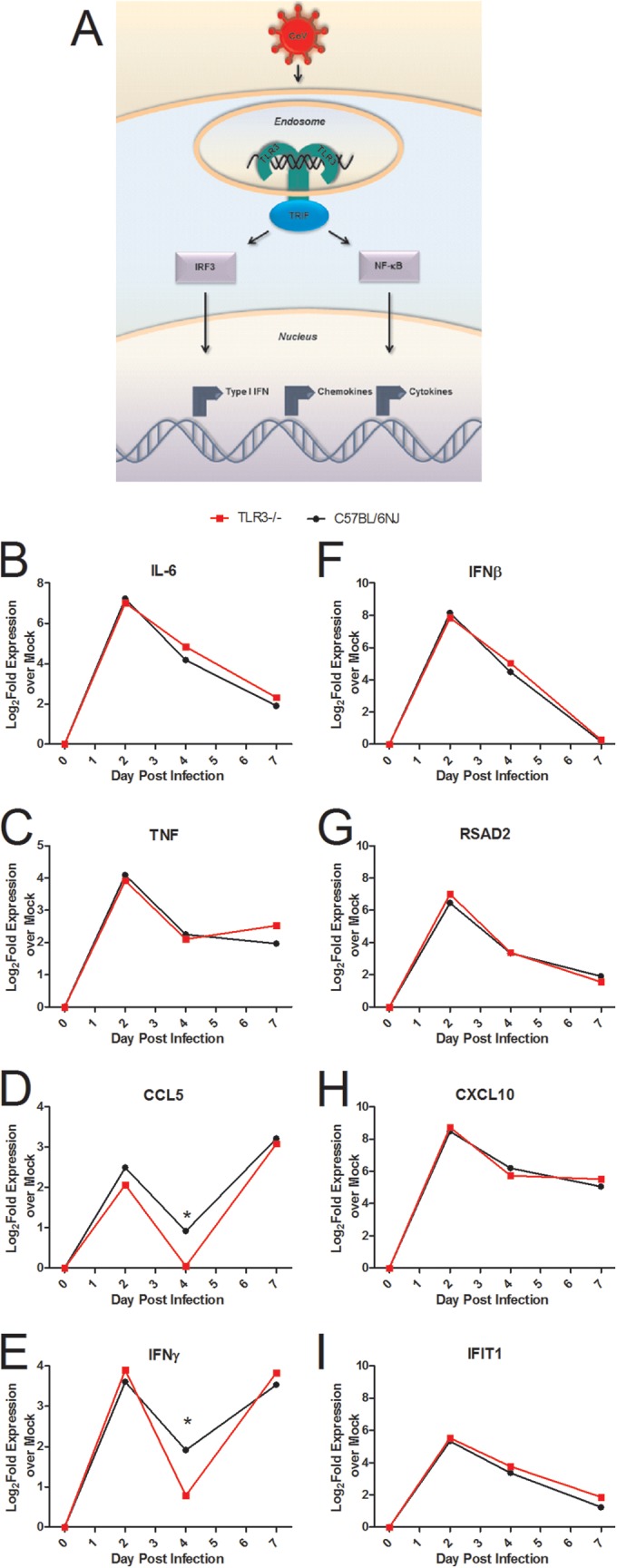
TLR3−/− mice show few alterations in cytokine and IFN signaling responses to SARS-CoV infection compared to wild-type mice. (A) TLR3 is a pattern recognition receptor that recognizes viral pathogen-associated molecular patterns and initiates antiviral signaling programs of IFNs, cytokines, and chemokines via the TRIF adaptor molecule. (B to I) RNA expression profiles of cytokines and ISGs downstream of TLR3 signaling measured by microarray analysis of IL-6 (B), TNF (C), CCL5 (D), IFN-γ (E), IFN-β (F), RSAD2 (G), CXCL10 (H), and IFIT1 (I) from TLR3−/− or C57BL/6NJ mice infected with 105 PFU of SARS-CoV normalized to the corresponding mock-infected, PBS-inoculated TLR3−/− or C57BL/6NJ mice (n = 4 to 5 mice per group). Differentially expressed genes indicated by a single asterisk (*) showed a >1.5-fold change in expression levels between wild-type and knockout mice, with P < 0.05.
Toll-like receptor adaptor TRIF has a protective role in the host response to SARS-CoV.
Because TLR3 utilizes the TRIF adaptor for downstream signaling programs, we infected TRIF−/− mice and wild-type C57BL/6J mice intranasally with 105 PFU of rMA15–SARS-CoV to determine the role of TRIF in SARS-CoV pathogenesis. TRIF−/− mice experienced typical early weight loss and then continued to lose weight on days 4 through 6 postinfection, when wild-type mice were recovering from weight loss (Fig. 3A; ***, P < 0.001). All of the TRIF−/− mice approached 70% of their starting weight on day 6 postinfection, when the experiment was ended according to our humane endpoint animal protocols. At days 2 and 4 postinfection, significantly higher viral loads were observed in the lungs of TRIF−/− mice than in wild-type mice (Fig. 3B; ***, P < 0.001). By day 6 postinfection, wild-type mice had cleared virus to levels below the limit of detection of the plaque assay, but there was still detectable virus in the lungs of TRIF−/− mice (Fig. 3B; ***, P < 0.001). Additionally, at 6 days postinfection, the lungs of TRIF−/− mice infected with SARS-CoV had severe hemorrhage encompassing the entire lung tissue, while little if any hemorrhage was observed in the lungs of wild-type mice (Fig. 3C; ***, P < 0.001). On the basis of these observations, TRIF−/− mice had more-severe SARS-CoV clinical disease signs than TLR3−/− mice, leading to a lethal phenotype from SARS-CoV infection in the TRIF−/− mice (Fig. 1 and 3).
FIG 3 .
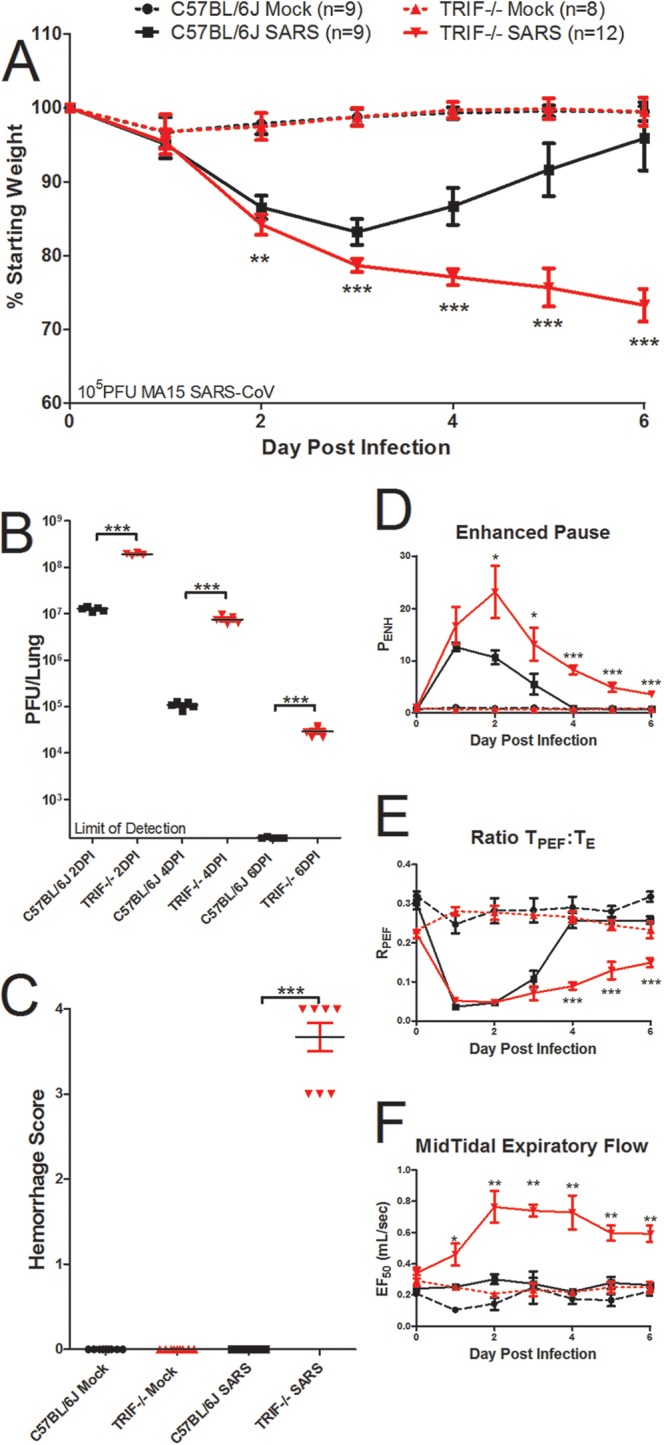
TRIF−/− mice are highly susceptible to SARS-CoV infection. (A to C) TRIF−/− mice infected with SARS-CoV have significantly greater weight loss (**, P < 0.01; ***, P < 0.001 [by a nonparametric Mann-Whitney test, where values indicate mean percent starting weight and error bars indicate standard deviation]) (A), viral titers (***, P < 0.001 [by unpaired Student’s t test]) (B), and lung hemorrhage scores (scored from 0 to 4; ***, P < 0.001 [by unpaired Student’s t test]) (C) than C57BL/6J mice infected with SARS-CoV over a 6-day course of infection. (D to F) Whole-body plethysmography analysis showed that SARS-CoV-infected TRIF−/− mice (solid red line) have alterations in lung functions compared to SARS-CoV-infected C57BL/6J mice (solid black line), including enhanced pause (PENH) (D), TPEF/Te ratio (RPEF) (E), and midtidal expiratory flow (EF50) (F) levels over the course of 6 days (dashed red line, TRIF−/− mock-infected mice; dashed black line, C57BL/6J mock-infected mice) (*, P < 0.05; **, P < 0.01; ***, P < 0.001 [by unpaired Student’s t test]).
To determine if the increased susceptibility of TRIF−/− mice to SARS-CoV infection affects lung function, whole-body plethysmography was used to measure changes in lung function in TRIF−/− mice compared to wild-type mice over the course of the SARS-CoV infection (Fig. 3D to F). TRIF−/− mice had significantly higher levels of enhanced pause (PENH) on days 2 to 6 postinfection (Fig. 3D; *, P < 0.05; ***, P < 0.001) indicative of airway hyperresponsiveness and infection-associated airway obstruction (40, 41). Lower values of the ratio of PENH to midtidal expiratory flow (EF50) (TPEF/Te [RPEF]) in TRIF−/− mice persisted throughout SARS-CoV infection, while wild-type mice RPEF values recovered to basal levels by day 4 postinfection (Fig. 3E; ***, P < 0.001). The EF50 was significantly higher in TRIF−/− mice than in wild-type mice on days 1 to 6 postinfection with SARS-CoV (Fig. 3F; *, P < 0.05; **, P < 0.01), which is consistent with previous data in SARS-CoV infection models and studies of hypoxic conditions (42, 43). These measures indicate that major changes in lung function occurred in the TRIF−/− mice infected with SARS-CoV, potentially due to changes in large-airway debris and denudation, as indicated by the results of histological analysis (see Fig. S2A to I in the supplemental material).
To determine if differences in viral titers in TRIF−/− mice infected with SARS-CoV compared to wild-type mice resulted in increased viral spread or were associated with infection of different cell types, we evaluated lung sections stained by immunohistochemistry (IHC) specific for the SARS-CoV nucleocapsid protein (Fig. 4). Significantly more viral antigen was present in the lungs of TRIF−/− mice than in those of wild-type mice at day 2 postinfection, consonant with the higher viral loads quantified by plaque assay (Fig. 4A and F; *, P < 0.05). In the large airways of TRIF−/− mice infected with SARS-CoV, no difference in the amount of viral antigen present was observed (Fig. 4B and E), but infected cells in both the C57BL/6J and TRIF−/− mice were morphologically consistent with ciliated airway epithelial cells, a primary target of SARS-CoV in humans. Interestingly, significantly more viral antigen staining was observed in the parenchyma of the lungs of TRIF−/− mice than in those of C57BL/6J mice infected with SARS-CoV (Fig. 4A and E; **, P < 0.01). Alveolar spaces showed the presence of viral antigen in cells morphologically consistent with type II pneumocytes, another primary target cell of SARS-CoV (Fig. 4C). Observation of the immunohistochemistry did not reveal any evidence that SARS-CoV infection of TRIF−/− mice occurs in cell types other than those infected in C57BL/6J mice.
FIG 4 .
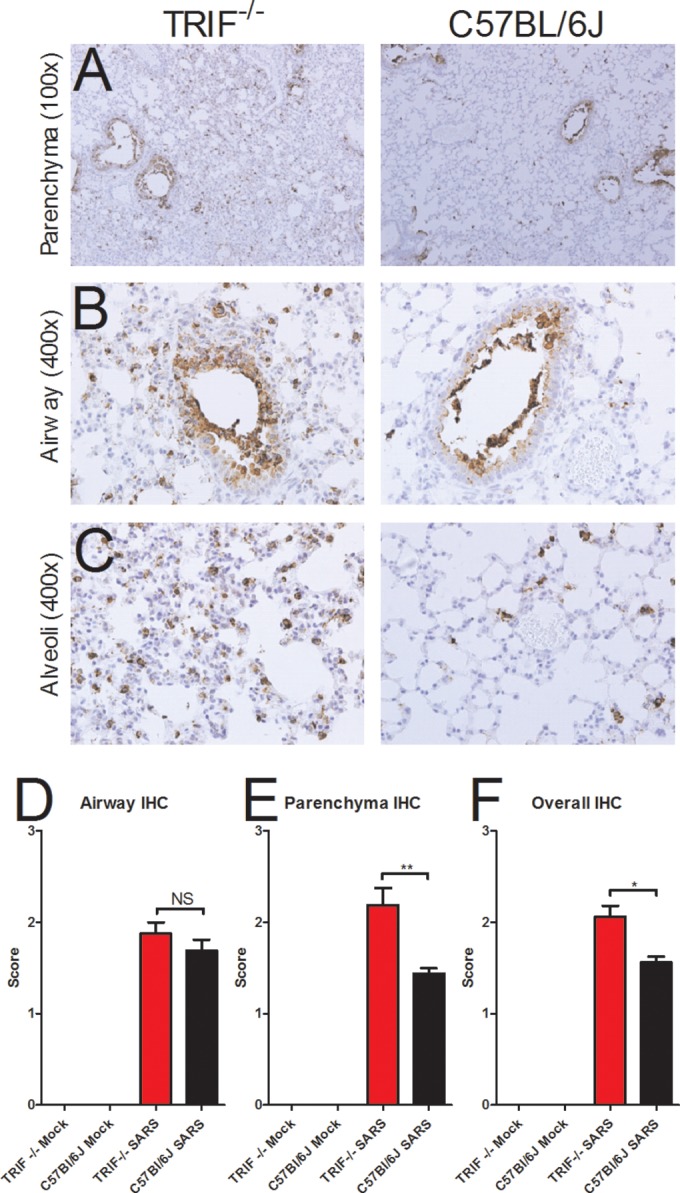
Increased presence of viral antigen in the lungs of TRIF−/− mice. (A to C) Immunohistochemistry was used to stain for SARS-CoV nucleocapsid antigen in the lungs of TRIF−/− SARS-CoV-infected mice (first column in panels A to C) and C57BL/6J SARS-CoV-infected mice (second column in panels A to C) on day 2 postinfection with SARS-CoV. TRIF−/− mock-inoculated mice and C57BL/6J mock-inoculated mice were also evaluated as negative controls and showed no viral antigen staining (data not shown). (D to F) Immunohistochemistry lung sections from day 2 postinfection were scored for the presence of SARS nucleocapsid antigen in the large airways (D) and lung parenchyma (E) and for overall staining (F). Sections were scored in a blinded manner, and scores were evaluated for significance by an unpaired Student’s t test (*, P < 0.05; **, P < 0.01; NS, not significant).
Due to the differences in weight loss and survival between TRIF−/− and TLR3−/− mice infected with SARS-CoV, we hypothesized that TLR4 may also contribute to signaling through TRIF in response to SARS-CoV infection. To determine the role of TLR4 in the pathogenesis of SARS-CoV, we infected TLR4−/− mice and wild-type C57BL6/J mice intranasally with 105 PFU of rMA15–SARS-CoV. TLR4−/− mice lost a greater percentage of their starting weight, with statistically significant differences in weight loss compared to wild-type mice on days 3 to 7 postinfection, but began recovering by 7 days postinfection (Fig. 5A; *, P < 0.05; ***, P < 0.001). TLR4−/− mice had significantly higher titers of virus in the lungs than wild-type mice infected with SARS-CoV at days 2 (3-fold difference; ***, P < 0.001) and 4 (4-fold difference; *, P < 0.05) postinfection but had cleared the virus by day 7 postinfection, similarly to wild-type mice (Fig. 5C). Because TLR4 can signal in either a MyD88-dependent or TRIF-dependent manner via the usage of the sorting adaptor MAL or TRAM, respectively, we infected 10-week-old female TRAM−/− mice and wild-type C57BL6/J mice intranasally with 105 PFU of SARS-CoV in order to discriminate between the effects of MyD88-dependent and TRIF-dependent signaling. TRAM−/− mice infected with SARS-CoV lost significantly more weight than wild-type mice (Fig. 5B; ***, P < 0.001). TRAM−/− mice infected with SARS-CoV demonstrated prolonged weight loss through 10 days postinfection, while wild-type mice recovered from weight loss by day 6 postinfection (Fig. 5B). TRAM−/− mice had titers that were 3-fold higher than those seen with wild-type mice infected with SARS-CoV on day 2 postinfection (**, P < 0.01) but not on day 4 postinfection and cleared the virus by day 7 postinfection similarly to wild-type mice, despite lack of recovery from weight loss (Fig. 5D). The alterations in lung function measured by whole-body plethysmography that were observed in TLR4−/− and TRAM−/− mice infected with SARS-CoV were similar to those observed in wild-type mice (see Fig. S3 in the supplemental material), supporting our hypothesis that TLR4/TRAM/TRIF signaling has a modest protective effect against SARS-CoV infection.
FIG 5 .
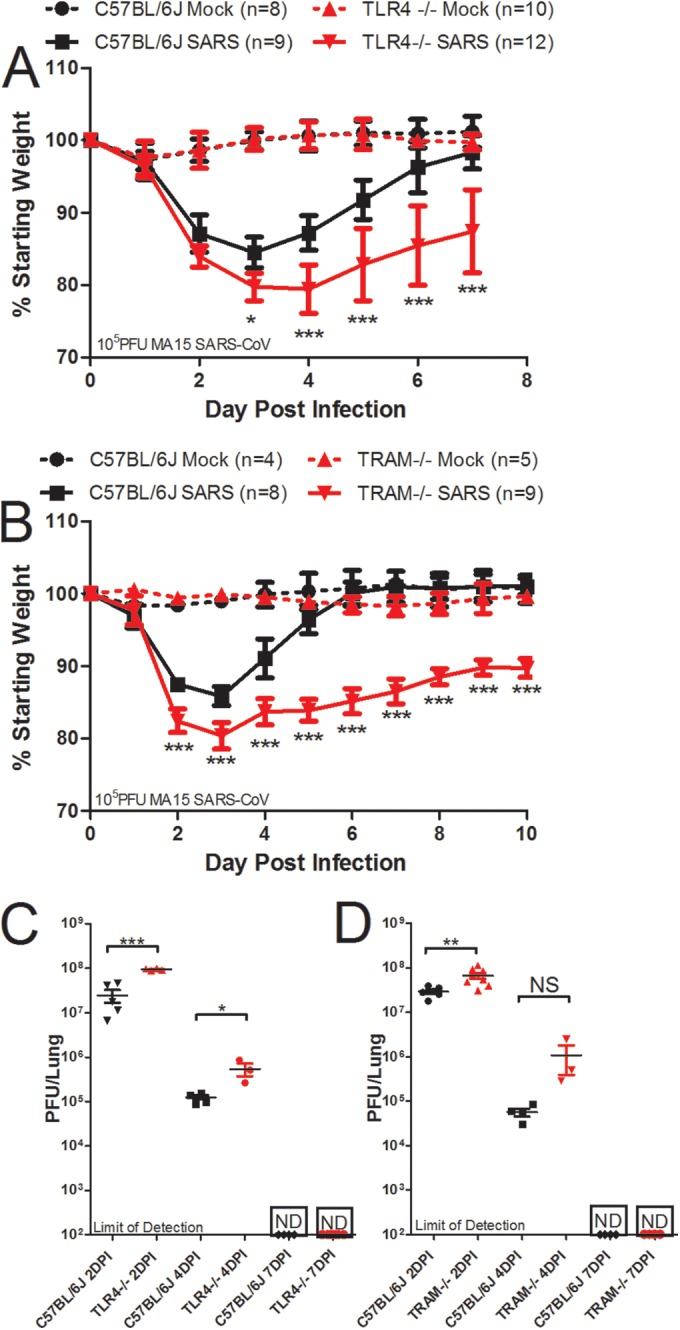
TLR4−/− and TRAM−/− mice are more susceptible to SARS-CoV infection than wild-type mice. (A) TLR4−/−, TRAM−/−, and wild-type mice were infected intranasally with 105 PFU of SARS-CoV. Weight loss was measured each day postinfection, and TLR4−/− mice lost significantly more weight than wild-type mice on days 3 to 7 postinfection (*, P < 0.05; ***, P < 0.001 [by nonparametric Mann-Whitney test; values indicate mean percent starting weight and error bars indicate standard deviation]). (B) TRAM−/− mice lost significantly more weight than wild-type mice on days 2 to 10 postinfection (***, P < 0.001 [by nonparametric Mann-Whitney test, values indicate the mean percent starting weight and error bars indicate standard deviation]). (C) Virus titer in the lung was measured by plaque assay, and TLR4−/− mice had significantly higher virus titers in the lungs at days 2 and 4 postinfection but had cleared the virus by day 7 postinfection, similarly to wild-type mice. (D) TRAM−/− mice had significantly higher virus titers in the lungs at day 2 postinfection as measured by plaque assay, but the differences were not significant at day 4 postinfection, and both TRAM−/− mice and wild-type mice had no detectable virus in the lungs by day 7 postinfection (*, P < 0.05; ***, P < 0.001 [by unpaired Student’s t test]; ND, not detected by plaque assay).
Aberrant cellular and signaling responses in TRIF−/− mice in response to SARS-CoV infection.
Because TRIF acts as a TLR adaptor protein that leads to activation of transcription factors that transcribe cytokines and chemokines, we hypothesized that differences in chemokines downstream of TRIF−/− signaling would be altered in TRIF−/− mice compared to wild-type mice. Measurements of proinflammatory cytokines IL-6, TNF, and IFN-γ and chemokines CCL2, CCL3, CCL5, CCL7, and CCL8 showed reduced expression of all these genes at day 2 postinfection in TRIF−/− mice, while these transcripts are highly induced in wild-type mice infected with SARS-CoV (Fig. 6A to D [*, P < 0.05]; see also Fig. S4A to D in the supplemental material [***, P < 0.001]). At day 4 postinfection, significantly more expression of IL-6, TNF, CCL5, and IFN-γ was observed in TRIF−/− mice, while expression was diminished in wild-type animals infected with SARS-CoV (Fig. 6A to D; *, P < 0.05; ***, P < 0.001). On day 4 postinfection, there was no significant difference in the levels of chemokines CCL2, CCL3, CCL7, and CCL8 in the TRIF−/− and wild-type mice infected with SARS-CoV (Fig. S4A to D). In contrast, increased protein levels of IFN-β were observed in TRIF−/− mice on day 2 and day 4 postinfection compared to wild-type mice infected with SARS-CoV (Fig. 6E; *, P < 0.05); this result was likely due to the increased viral loads in the lungs of TRIF−/− mice at those time points. Expression of ISGs downstream of IFN, including RSAD2, CXCL10, and IFIT1, was greatly reduced in TRIF−/− mice infected with SARS-CoV on day 2 postinfection compared to wild-type mice (Fig. 6F to H; *, P < 0.05; **, P < 0.01; ***, P < 0.001) but was induced to very high levels in the TRIF−/− mice infected with SARS-CoV on day 4 postinfection while the ISG response was waning in wild-type mice (*, P < 0.05; **, P < 0.01).
FIG 6 .

Aberrant proinflammatory cytokine, interferon, and interferon-stimulated gene signaling responses in TRIF−/− mice infected with SARS-CoV. (A to D) RNA expression profiles of cytokines and chemokines downstream of TRIF and TLR signaling programs measured by qPCR analysis of IL-6 (A), TNF (B), CCL5 (C), and IFN-γ (D) from TRIF−/− mice (red bars) or wild-type C57BL/6J mice (black bars) infected with 105 PFU of SARS-CoV normalized to mock-infected TRIF−/− or wild-type mice (n = 4 mice per group) at day 2 and day 4 postinfection. (E) TRIF−/− mice infected with SARS-CoV have significantly higher protein levels of IFN-β measured by ELISA on day 2 and day 4 postinfection in lung homogenates. (F to H) RNA expression profiles of ISGs measured by qPCR analysis of RSAD2 (F), CXCL10 (G), and IFIT1 (H) in TRIF−/− mice (red bars) or wild-type C57BL/6J mice (black bars) infected with 105 PFU of SARS-CoV normalized to mock-infected TRIF−/− mice or wild-type mice at day 2 and day 4 postinfection. Significant differences between groups were evaluated by an unpaired Student’s t test, bar graphs show the mean normalized fold change on the day postinfection, and the error bars indicate 1 standard deviation from the mean (*, P < 0.05, **, P < 0.01; ***, P < 0.001; NS, not significant).
Because differences in inflammation were observed in TRIF−/− mouse lung sections stained with hematoxylin and eosin (H&E) (see Fig. S2 in the supplemental material) and because differences in chemokines that recruit inflammatory cells were observed in TRIF−/− mice compared to wild-type mice (Fig. 6; see also Fig. S4 in the supplemental material), we hypothesized that differences in infiltrating immune cell populations would be observed in TRIF−/− mice compared to wild-type mice infected with SARS-CoV. Both wild-type and TRIF−/− lungs from mice infected with SARS-CoV had larger numbers of cells due to infiltrating cell populations than lungs from mock-inoculated mice, but there were no significant differences in the overall numbers of infiltrating cells of TRIF−/− lungs on day 4 or 6 postinfection compared to those of lungs from wild-type mice infected with SARS-CoV (Table 1). There were significantly more neutrophils in the lungs of TRIF−/− mice infected with SARS-CoV than in lungs of wild-type mice on day 4 postinfection (Table 1; *, P < 0.05); however, by day 6 postinfection there were no longer significant differences in neutrophils. The levels of chemokines that are chemotactic for neutrophil recruitment (CXCL1, CXCL2, and CXCL3) were significantly lower on day 2 postinfection in TRIF−/− mice infected with SARS-CoV but were higher in TRIF−/− mice on day 4 postinfection, coinciding with the increased recruitment of neutrophils in the TRIF−/− mice infected with SARS-CoV (Fig. S4E to G; *, P < 0.05; **, P < 0.01; ***, P < 0.05).
TABLE 1 .
Quantitation of infiltrating cells in the lungs of TRIF−/− and C57BL/6J mice following SARS-CoV infectiona
| Cell category | No. of cells (×104) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Day 4 |
Day 6 |
|||||||
| Mock |
SARS |
Mock |
SARS |
|||||
| WT | TRIF−/− | WT | TRIF−/− | WT | TRIF−/− | WT | TRIF−/− | |
| Total | 1,430 | 1,490 | 13,100 | 16,500 | 813 | 1,620 | 7,240 | 7,880 |
| Neutrophil | 54.6 | 49.9 | 272 | 892* | 35.4 | 45.8 | 44.5 | 252 |
| Monocyte | 91.4 | 111 | 1,860 | 2,470 | 85.8 | 110 | 1,360 | 877 |
| Ly6Chi Mono | 11.0 | 24.3 | 250 | 1,070* | 47.9 | 27.9 | 394 | 276 |
| Ly6Clo Mono | 80.8 | 87.6 | 1,620 | 1,430 | 28.9 | 63.5 | 951 | 466* |
| Alveolar Mac | 66.1 | 80.5 | 178 | 338 | 34.0 | 59.4 | 119 | 229* |
| pDC | 1.27 | 4.86 | 13.2 | 18.7 | 4.74 | 5.92 | 5.81 | 36.4*** |
| Viable lymphocyte | 1,080 | 1,080 | 10,800 | 10,800 | 586 | 1,110 | 5,950 | 5,000 |
| Total T cells | 180 | 287 | 1,930 | 1,800 | 120 | 294 | 1,910 | 972* |
| CD4+ T cells | 67.6 | 108 | 689 | 625 | 51.4 | 159 | 693 | 476 |
| CD8+ T cells | 71.2 | 119 | 622 | 642 | 52.4 | 118 | 1,000 | 398** |
Flow cytometry was used to measure cell populations in the lungs of mice; numbers indicate mean counts of cells from lungs. Numbers in bold indicate TRIF−/− mice infected with SARS-CoV whose results were significantly different from those determined for wild-type (WT) mice infected with SARS-CoV (*, P < 0.05; **, P < 0.01; ***, P < 0.001 [by Student’s unpaired t test]). There were no significant differences between TRIF−/− mice and wild-type mice in the numbers of cells from mock-inoculated mice.
Differences in monocytic subpopulations were observed on days 4 and 6 postinfection with SARS-CoV (Table 1). There were significantly more Ly6Chi inflammatory monocytes in the TRIF−/− mice infected with SARS-CoV on day 4 postinfection (Table 1; *, P < 0.05), but there was no difference between wild-type and TRIF−/− mice in the Ly6Clo population of monocytes on day 4 postinfection. In contrast, there was no difference in the levels of Ly6Chi monocytes between TRIF−/− and wild-type mice infected with SARS-CoV on day 6 postinfection, but wild-type mice infected with SARS-CoV had significantly more Ly6Clo regulatory monocytes on day 6 postinfection (Table 1; *, P < 0.05). On day 4 postinfection, there was no significant difference in the number of alveolar macrophages or plasmacytoid dendritic cells (pDCs) between the lungs of TRIF−/− and wild-type mice infected with SARS-CoV, but at day 6 postinfection there were significantly more alveolar macrophages and pDCs in the lungs of TRIF−/− mice infected with SARS-CoV than in the lungs of wild-type mice (Table 1; *, P < 0.05; ***, P < 0.001). No statistically significant differences were observed in populations of eosinophils or monocyte-derived DCs or in CD11bpos DC populations between TRIF−/− mice and wild-type mice infected with SARS-CoV (data not shown).
The total numbers of lymphocytes were not significantly different between TRIF−/− mice and wild-type mice at day 4 or day 6 postinfection (Table 1). There were no differences in the number of T cells in the lungs of TRIF−/− mice and wild-type mice infected with SARS-CoV on day 4 postinfection, but on day 6 postinfection, there were significantly more T cells in the lungs of wild-type mice infected with SARS-CoV than in the lungs of TRIF−/− mice (Table 1; *, P < 0.05). There were no significant differences in the number of CD4+ T cells on day 4 or day 6 postinfection or the number of CD8+ T cells on day 4 postinfection, but there were significantly more CD8+ T cells observed in wild-type mice at day 6 postinfection than in TRIF−/− mice (Table 1; **, P < 0.01). Aberrant SARS-CoV disease responses were observed in the TRIF−/− mice that led to a lethal phenotype, including increased weight loss, lack of viral clearance, alterations in lung function and pathology, changes in infiltrating cell populations, and aberrant cellular signaling programs, all indicating that TRIF is a master regulator in the protective innate immune response to SARS-CoV disease.
DISCUSSION
The critical importance of TLR signaling programs is demonstrated by the key regulation of host immune responses by TLR adaptor proteins MyD88 and TRIF in controlling respiratory virus infections. In SARS-CoV-infected TRIF−/− mice, there is significantly increased mortality, weight loss, and viral titers (Fig. 3A and B), leading to expression of cytokines, chemokines, and ISGs (Fig. 6; see also Fig. S4 in the supplemental material) consistent with the aberrant cellular signaling programs seen in patients who succumbed to SARS or MERS disease (44, 45). Although MyD88−/− mice infected with SARS-CoV had mortality, weight loss, and viral loads comparable to those of TRIF−/− mice infected with SARS-CoV, the outcomes in downstream cellular signaling programs were very different (37). In MyD88−/− mice infected with SARS-CoV, there was an absence of induction of cytokine and chemokine signaling at days 2, 4, and 6 postinfection compared to wild-type mice; in contrast, TRIF−/− mice infected with SARS-CoV had a similar lack of cytokine and chemokine signaling on day 2 but an increased amount of IFN-β followed by a marked increase in proinflammatory cytokine and ISG signaling on day 4 postinfection (Fig. 6). One explanation of these data may be that an initial lack of innate immune response in TRIF−/− mice infected with SARS-CoV leads to higher viral titers, which in turn leads to compensatory innate immune signaling resulting in the induction of cytokine, chemokine, and ISG expression at day 4 postinfection. Consistent with these data, TRIF−/− mice infected intranasally with herpes simplex virus (HSV-1) have increased mortality rate, significantly greater viral titers in the brain, and increased production of type I IFN (46). In contrast, TRIF−/− mice infected with IAV were not significantly different from wild-type mice in mortality, but one study found that MyD88−/− mice were more susceptible than wild-type mice, while another found there was no difference between the responses of MyD88−/− mice and wild-type mice infected with IAV (16, 47). These data indicate that, although both TLR adaptors (MyD88 and TRIF) are vitally important to a protective immune response to SARS-CoV, differences exist between the cellular signaling programs induced by highly pathogenic respiratory infections caused by coronaviruses and influenza viruses that should be considered prior to the administration of therapeutic regimes (48).
The differences in viral pathogenesis between MyD88−/− and TRIF−/− mice also include major differences in infiltrating cell populations resulting from SARS-CoV infection. MyD88−/− mice had significantly fewer inflammatory monocytes and macrophages at day 2 postinfection than wild-type mice infected with SARS-CoV, but no cellular populations measured were significantly different at day 4 postinfection (37). In addition, despite similarities in infiltrating cell populations in MyD88−/− and wild-type mice infected with SARS-CoV on day 4 postinfection, a lack of cytokine and chemokine signaling persisted, indicating a likely deficiency in the activation of signaling programs of these cells. In TRIF−/− mice, however, many differences in infiltrating cell populations were observed at day 4 and day 6 postinfection, but the increase in transcription of proinflammatory cytokines indicates that a lack of TRIF does not inhibit cell signaling programs by these cell populations. Rather, the large induction of IFN-β expression on day 2 postinfection and the presence of significantly more viral antigen at early times postinfection likely drive the increased stimulation of infiltrating cell types in TRIF−/− mice, contributing to aberrant cellular responses.
The accumulation of neutrophils in TRIF−/− mice on day 4 postinfection with SARS-CoV correlates with increased amounts of neutrophil recruitment chemokines CXCL1 and CXCL2 (IL-8 rodent homologs) and increased levels of proinflammatory cytokines such as TNF and IL-6, mirroring the neutrophil infiltration and cellular responses of ARDS patients (reviewed in reference 49). Similarly indicative of lethal pathogenesis of respiratory viruses, infection of mice with highly pathogenic strains of influenza virus, including 1918 H1N1 and H5N1 IAV, resulted in significantly more recruitment of neutrophils (at levels similar to the levels seen in TRIF−/− mice infected with SARS-CoV) than was observed following infection with seasonal IAV strains of low pathogenicity (50). There is evidence that neutrophils infiltrating the pulmonary compartment produce robust amounts of CXCL10, contributing to the pathogenesis of ARDS from IAV infection, and the induction of large levels of CXCL10 was observed in TRIF−/− mice infected with SARS-CoV on day 4 postinfection, coinciding with the influx of neutrophils (32). In influenza virus infection, infiltration of Ly6Chi monocytes resulting from IFN induction contributes to resistance of influenza virus infection, while significantly more Ly6Chi monocytes were observed in the more susceptible TRIF−/− mice on day 4 postinfection in our model (51). This inflammatory monocyte population can differentiate into macrophage and pDC subsets, which were observed in significantly higher numbers in TRIF−/− mice infected with SARS-CoV on day 6 postinfection (Table 1; reviewed in reference 52).
Because TLR3 senses double-stranded RNAs, an intermediate nucleic acid species present during acute viral infections, it could be predicted that loss of TLR3 signaling would negatively impact the host and alter cellular signaling programs after SARS-CoV infection. Although TLR3−/− mice infected with SARS-CoV experienced greater weight loss, higher viral titers, and more significant alterations in lung function over the course of infection (Fig. 1C and D; see also S1A to C in the supplemental material), relatively few changes in TRIF-dependent downstream cellular signaling programs resulted from the absence of TLR3 (Fig. 2B to I), indicating that additional pathways may compensate for the absence of TLR3 in SARS-CoV infection. Other innate immune sensors, such as MDA5, that detect dsRNA and that have been implicated in the sensing of coronaviruses may be important for compensatory effects in the absence of TLR3 (53). Among the few changes that were observed in TRIF-dependent signaling pathways in TLR3−/− mice on day 4 postinfection, IFN-γ expression was significantly higher in wild-type mice than in TLR3−/− mice; however, absence of the IFN-γ receptor has minimal impact on SARS-CoV pathogenesis in a similar mouse model (54). The relevance of changes in CCL5 is difficult to infer, as CCL5 and other related chemokines signal through multiple receptors, including CCR1 and CCR5, the absence of which modestly increases SARS-CoV pathogenesis in a mouse model (37). In contrast to our results with SARS-CoV, TLR3−/− mice are less susceptible to H3N2 and H5N1 IAV, with a decreased mortality rate compared to lethal infection of wild-type mice, but there is no difference in the survival of TLR3−/− mice compared to wild-type mice infected with a lethal dose of p2009 H1N1 IAV (16, 55). The phenotype of TLR3−/− mice in West Nile virus (WNV) mouse models is somewhat controversial, with one group showing a modest increase in WNV-induced mortality with no differences in type I IFN levels in TLR3−/− mice, while another group showed that TLR3−/− mice have less susceptibility to WNV and reduced proinflammatory cytokine responses compared to wild-type mice (56, 57).
Our study results indicate that TLR4 is also involved in mediating the pathogenesis of SARS-CoV infection, and it is likely that TLR4 signaling occurs in a TRIF-dependent manner through the sorting adaptor TRAM, as TRAM−/− mice recapitulate features of increased SARS-CoV pathogenesis similarly to TLR4−/− mice, including increased weight loss, similar alterations in lung parameters, and higher viral titers at early times postinfection. The involvement of TRAM signaling indicates that the TRIF adaptor protein mediates a large part of TLR4 signaling in response to SARS-CoV. The studies presented here indicated that TLR3 and TLR4 individually mediate a portion of the TRIF-dependent TLR signaling necessary for survival of SARS-CoV in our mouse model. These are among the first observations of the role of individual TLRs in contributing to protection from SARS-CoV disease. Interestingly, the absence of either TLR3 or TLR4 does not lead to lethal SARS-CoV disease similar to that seen with TRIF−/− mice infected with SARS-CoV, likely because an absence of signaling via a single TLR may be compensated for by sensing of viral PAMPs by other PRRs.
In other models of acute lung injury, TLR4−/− mice and TRIF−/− mice are less susceptible to lung injury mediated by the introduction of acid and IAV into the lung (28). Imai et al. observed that oxidized phospholipids, putative PAMPs potentially contributing to acute lung injury by activating TLR4 and signaling through TRIF, were present in ARDS patient samples as a consequence of the presence of infectious diseases such as H5N1 IAV and SARS-CoV infections (28). The finding that TLR4−/− mice are resistant to acute lung injury via IAV and acid models is in contrast to our findings here that TLR4−/− mice have significantly more disease resulting from SARS-CoV infection than wild-type mice (Fig. 5). One explanation for these conflicting data is that activation of TLR4 by oxidized phospholipid PAMPs may be detrimental in acid injury and IAV infection mouse models but that, in the case of SARS-CoV infection, the benefits of TLR4 sensing of PAMPs may outweigh the damaging effects of cellular signaling programs resulting from sensing of oxidized phospholipids by TLR4. Additionally, TLR4−/− mice are less susceptible to influenza virus infection and an immunomodulatory approach using a TLR4 antagonist was proposed to have ameliorative properties for the treatment of influenza virus infections (29). These findings indicate that protective signaling via TLR4/TRAM/TRIF may be a unique feature in the pathogenesis of coronaviruses compared to that of other respiratory pathogens such as influenza viruses and that different cellular sensors recognize pathogens with similar clinical features and infecting similar cell types.
Despite the various outcomes in host survival and morbidity in SARS-CoV, WNV, and IAV infection models, the commonality is that mice deficient in TLR signaling have increased viral loads in the infected tissues, demonstrating that the initial recognition of viral PAMPs by TLRs is necessary for controlling viral replication and that the increased presence of viral antigen could partially drive downstream phenotypes in these systems (Fig. 1D and 5C) (16, 56, 57). Neither MyD88−/− mice nor TRIF−/− mice infected with SARS-CoV efficiently cleared the virus by day 6 postinfection, but both showed increased signs of disease, ultimately leading to death of the TLR adaptor knockout mice from SARS-CoV infection. Our observations support previous findings indicating that signaling through TRIF is critical for CD8+ T cell expansion, a key component of adaptive immunity for viral clearance (58, 59). In contrast to the TLR adaptor knockout mice, RAG1−/− mice with no mature T cells fail to clear SARS-CoV but show no signs of increased disease, as defined by weight loss, indicating that the lack of clearance of virus alone is not responsible for the disease phenotypes seen in the TRIF−/− and MyD88−/− mice (37). In generating a protective immune response to highly pathogenic coronavirus infections, our findings indicate that not only the activation of an adaptive response but also the proper activation of a balanced innate immune response through both adaptor arms of TLR-mediated signaling is required for viral clearance.
TLR agonists have been proposed for usage as respiratory vaccine adjuvants and may also have utility in protection against respiratory virus-induced disease or immunopathology (29, 33–35). TLR3 and TLR4 agonists have protective effects against SARS-CoV infection. In addition, adjuvant approaches that stimulate TLR pathways through MyD88 and TRIF may prove synergistic, especially when both components are critical to the host response, such as in SARS-CoV infection. Our data support the idea that innate immune responses are important for the antiviral state of cells, immune cell recruitment, and expansion of adaptive immune responses. Comparison of these data to those from other models of highly pathogenic respiratory virus infection (particularly influenza virus infection) indicates that although these viruses may be detected by similar pathways, the result of that sensing can lead to differences in disease outcome which should be considered in the design and administration of vaccine and antiviral therapeutics.
MATERIALS AND METHODS
Viruses, cells, and plaque assay.
A mouse model of the recombinant mouse-adapted rMA15–SARS-CoV virus used in this study has been previously described (37, 60). As previously described, virus stocks were propagated on Vero E6 cells (60). Plaque assays to quantify virus in viral stocks and to enumerate the number of viruses in the lower left lobe of lungs from mice were performed in Vero E6 cells, with a limit of detection of 100 PFU (12). Experiments with rMA15–SARS-CoV were performed in a certified biosafety level 3 laboratory, using a class II biological safety cabinet. Laboratory workers were equipped with high-efficiency particulate air (HEPA)-filtered powered air-purifying respirators (PAPRs), Tyvek suits, hoods, aprons, booties, and personal protective equipment.
Ethics statement.
The ethical treatment of mice in this study was in accordance with the guidelines of The University of North Carolina (UNC) at Chapel Hill Institutional Animal Care and Use Committee (IACUC), and approved documentation registered under protocol no. 11-213 was used in the experiments described. The guidelines for care of the mice are from the Guide for the Care and Use of Laboratory Animals, 8th ed., published by the U.S. National Institutes of Health (NIH). All animal housing and care was conducted according to Institutional Animal Care and Use Committee (IACUC)-approved protocols of the University of North Carolina, Chapel Hill (NIH/PHS Animal Welfare Assurance no. a3410-01).
Animals.
Animals were maintained in HEPA-filtered Sealsafe cages (Techniplast) during experiments performed with rMA15–SARS-CoV. The following strains of age-matched female mice were obtained from Jackson Laboratory: C57B/6NJ (stock no. 005304), TLR3−/− (stock no. 009675), C57BL/6J (stock no. 000664), and TRIF−/− (stock no. 005307). At 10 weeks of age, mice were anesthetized with a mixture of ketamine and xylazine and inoculated intranasally with either 50 µl of phosphate-buffered saline (PBS) (for mock-inoculated controls) or 105 PFU of rMA15–SARS-CoV–PBS. Animals were weighed daily, and lung tissues from days 2, 4, and 7 postinfection (C57BL/6NJ and TLR3−/− mice) or days 2, 4, and 6 postinfection (C57BL/6J and TRIF−/− mice) were collected for downstream analyses by plaque assay, histology, and RNA analysis.
Flow cytometry.
On days 4 and 6 postinfection, whole lungs were harvested from groups of 3 to 4 mock-treated wild-type and TRIF−/− mice and 4 to 5 SARS-CoV-infected wild-type and TRIF−/− mice. Whole lungs were prepared for flow cytometry analysis by collagenase digestion and tissue disruption into cell suspensions as previously described (11). Antibody staining panels were used to stain cell preparations (see Text S1 in the supplemental material). Analysis was performed with Summit software (Beckman-Coulter) for sorting into defined subpopulations (see Text S1).
Hemorrhage scores, histological analysis, and immunohistochemistry.
Scores for gross hemorrhage were recorded by observation of the lung during necropsy using a scale ranging from a value of zero, indicating no hemorrhage, to a value of 4, indicating severe hemorrhage in all lobes of the lung, as previously described (38). For histological analysis, the entire right lobe of lungs from infected or mock-treated wild-type and knockout mice was fixed in 10% formalin, embedded in paraffin, and prepared in 5-µm-thick sections for hematoxylin and eosin (H&E) staining by the UNC Histopathology core facility. For immunohistochemistry (IHC), formalin-fixed and paraffin-embedded histology samples from C57BL/6J and TRIF−/− mice were sectioned (thickness, 5 µm) and stained for viral antigen using a commercially available polyclonal SARS-CoV anti-nucleocapsid antibody (Imgenex) following the manufacturer’s protocols (61). Slides for IHC and H&E histology were scored in a blinded manner (n = 4 to 5 mice per group) for metrics of inflammation, and images were captured using an Olympus BX41 microscope with an Olympus DP71 camera.
Whole-body plethysmography.
Lung function was measured by unrestrained whole-body plethysmography using IACUC-approved protocols as has been previously described (42). Briefly, animals were introduced into randomized individual plethysmography chambers following calibration according to protocols of the manufacturer (Buxco). After a 30-min acclimation period, data on lung function parameters were collected during a 5-min measurement period. Data were analyzed by Finepoint software (Buxco) for established metrics of airway hyperresponsiveness and virus infection-associated airway obstruction, including enhanced pause (PENH), tidal midexpiratory flow (EF50), and ratio of TPEF to Te (RPEF). PENH is an empirical measure calculated by , where PEF is the peak expiratory flow, PIF is the peak inspiratory flow, Te represents the time of expiration, and Tr represents the relaxation time to 36% of the peak expiration pressure (62). PENH has been controversially linked to airway hyperresponsiveness, obstruction, and bronchoconstriction but has been used in several viral models of airway infection, where increases in the PENH value correlate with increased lung pathology following respiratory viral infection (40, 42, 62–64). EF50 indicates the flow rate at 50% of the tidal volume. RPEF is calculated by , where TPEF is the time to peak expiratory flow and Te is the time of expiration, and the ratio may be interpreted as an indication of the shape of the exhalation curve.
Real-time qPCR analysis.
The upper left lobe of rMA15–SARS-CoV-infected mice was stored in RNAlater solution (Life Technologies) for 48 h at 4°C and then stored at −80°C. Lung sections were thawed and then homogenized in TRIzol (Life Technologies) for 60 s at 6,000 rpm using a Magna Lyzer instrument (Roche). Following chloroform-isopropanol extraction of RNA from TRIzol homogenates, cDNA was generated by reverse transcription-PCR (RT-PCR) using a SuperScript III First-Strand synthesis kit (Life Technologies). Quantitative PCR (qPCR) was performed using TaqMan gene expression assays (Life Technologies) for cytokines or chemokines normalized to 18-s expression. For each cytokine or chemokine, expression from groups of 4 rMA15–SARS-CoV-infected mice was normalized to mock (PBS)-inoculated mice (either wild-type or knockout mice). Normalized fold change was calculated using the threshold cycle (ΔΔCT) method as has been previously described (11).
ELISA.
Levels of IFN-β were quantified using a VeriKine mouse IFN-β enzyme-linked immunosorbent assay (ELISA) kit (RD Systems) according to manufacturer protocols. The lower left lobe of lungs from TRIF−/− or C57BL/6J mice from the mock-inoculated or SARS-CoV-infected group (n = 4 each group) was homogenized in 1 ml of PBS using a Magna Lyzer instrument (Roche). One hundred microliters of cleared homogenate was used for the ELISA assay sample. A seven-point standard curve was prepared using the manufacturer’s protocol, and interferon titers in the samples were determined by plotting the standards using a 4-parameter fit. Optical densities were read at an absorbance of 450 nm.
Differentially expressed gene identification.
Differentially expressed gene targets were selected from data collected in a previously described study performed with transcriptomics data banked at NCBI Gene Expression Omnibus (accession no. gse33266) (38). Briefly, that study was performed using microarray analysis of RNA from lungs of 20-week-old female C57BL/6J mice infected with doses of 102, 103, 104, or 105 PFU of mouse-adapted SARS-CoV to identify genes that were differentially expressed compared to those of mock (PBS)-inoculated mice. A linear fit model was used to determine differential expression (DE) for each transcript, requiring an absolute log2 (fold change) value of >1.5 as well as an adjusted false-discovery-rate (FDR) P value of <0.05. In a separate study, TLR3−/− lung homogenates in TRIzol were analyzed by microarray, and the full data set has been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database and is available at GEO accession number GSE68820.
SUPPLEMENTAL MATERIAL
Supplemental text. Additional data on the methods used are provided for flow cytometry procedures. Download
TLR3−/− mice have aberrant lung function and pathology resulting from SARS-CoV infection. (A to C) To determine if there were differences in lung function associated with the increased viral titer and weight loss observed in TLR3−/− versus wild-type mice infected with SARS-CoV, whole-body plethysmography was used to measure parameters indicative of airway disease in SARS-CoV-infected mice. Whole-body plethysmography analysis showed that SARS-CoV-infected TLR3−/− mice (solid red line) had alterations in lung functions compared to C57BL/6NJ mice (solid black line) over the course of 7 days (the dashed red line indicates data from TLR3−/− mock-infected mice, and the dashed black line indicates data from C57BL/6NJ mock-infected mice; *, P < 0.05; **, P < 0.01; ***, P < 0.001 [by unpaired Student’s t test]). (A) A rise to similar peak levels of PENH, a measure indicative of airway resistance and obstruction, was seen in both groups of SARS-CoV-infected mice by day 2 postinfection. However, while wild-type mice infected with SARS-CoV returned to basal PENH values of by day 4 postinfection, TLR3−/− mice infected with SARS-CoV retained significantly higher PENH values on days 3 to 7 postinfection (*, P < 0.05; ***, P < 0.001). (B) TLR3−/− mice maintained a lower TPEF/Te (RPEF) ratio at days 4 to 7 postinfection, while wild-type mice resolved to the levels seen with mock-infected mice. (D) The midtidal expiratory flow (EF50) was significantly higher in the TLR3−/− mice on days 4 through 7 postinfection (Fig. S1C; ***, P < 0.001). Histopathology analysis of formalin-fixed H&E-stained lung tissues indicated that whereas the TLR3−/− (first panel) and C57BL/6NJ (second panel) mock-inoculated mice had normal alveolar architecture, TLR3−/− mice infected with SARS-CoV (third panel) showed signs of alveolar exudates not present in C57BL/6NJ mice infected with SARS-CoV (last panel) on day 7 postinfection. (E to H) Evaluation of H&E-stained lung tissue samples from TLR3−/− and wild-type mice infected with SARS-CoV at day 7 postinfection revealed no statistically significant differences in inflammation surrounding the large airways (E), vasculature (F), or alveoli (G and H). (D and I) However, significantly more exudates and debris were present in the alveolar spaces of TLR3−/− mice infected with SARS-CoV than in wild-type mice on day 7 postinfection, indicating that alveolar fluid may have been limiting lung function in the TLR3−/− mice even after the mice had begun to recover from weight loss due to SARS-CoV infection. Histology sections were scored in a blinded manner, and scores were evaluated for significance by unpaired Student’s t test (*, P < 0.05; NS, not significant). Download
Increased pathology of SARS-CoV in the lungs of TRIF−/− mice. (A to D) Because TRIF−/− mice infected with SARS-CoV showed signs of severe lung dysfunction by whole-body plethysmography, H&E-stained lung sections from TRIF−/− and C57BL6/J mice were scored for signs of SARS-CoV disease on day 6 postinfection. Although mock-inoculated wild-type and TRIF−/− mice show no signs of abnormal pathology (A to D, left panels), wild-type and TRIF−/− mice infected with SARS-CoV displayed hallmarks of SARS-CoV disease, including airway inflammation (C and E), perivascular cuffing (B and F), and alveolar inflammation (D and G to I). TRIF−/− mice infected with SARS-CoV had significantly more accumulation of inflammatory cells surrounding the large airways than wild-type mice (A and C, right panels; E, *, P < 0.05). Similar observations revealed increased levels of infiltrating cells surrounded the vasculature in the lungs of TRIF−/− mice infected with SARS-CoV compared to wild-type mice (C and F; *, P < 0.05). In the alveolar spaces of TRIF−/− mice, increased thickening of the septa between alveoli (D and G; *, P < 0.05) was observed in addition to greater numbers of infiltrating cells (D and H; *, P < 0.05). Histology sections were scored in a blinded manner, and scores were evaluated for significance by unpaired Student’s t test (*, P < 0.05; **, P < 0.01). Download
TLR4−/− mice and TRAM−/− mice have similar lung dysfunctions in response to SARS-CoV infection. Following SARS-CoV infection, the lung function parameters of TLR4−/− mice (A to C) and TRAM−/− mice (D to F) were measured daily along with those of wild-type mice and mock-inoculated controls. Differences from wild-type mice infected with SARS-CoV as determined by the lung parameters of PENH (A and D), RPEF (B and E), and EF50 (C and F) were observed in TLR4−/− mice and TRAM−/− mice infected with SARS-CoV but not in mock-inoculated controls (*, P < 0.05; **, P < 0.01 [by unpaired Student’s t test]). Download
Aberrant chemokine signaling programs in TRIF−/− mice infected with SARS-CoV. RNA expression profiles of chemokine signaling programs downstream of TLRs were measured by qPCR analysis of lung homogenates from TRIF−/− mice (red bars) or wild-type C57BL/6J mice (black bars) infected with 105 PFU of SARS-CoV normalized to the results seen with mock-infected TRIF−/− or wild-type mice (n = 4 mice per group). The levels of chemokines CCL2 (A), CCL3 (B), CCL7 (C), and CCL8 (D) as well as chemokines that influence neutrophil chemotaxis, including CXCL1 (E), CXCL2 (F), and CXCL3 (G), were measured at day 2 and day 4 postinfection. Significant differences between groups were evaluated by an unpaired Student’s t test, bar graphs show the mean normalized fold change on the indicated day postinfection, and the error bars indicate 1 standard deviation from the mean (*, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant). Download
Genes differentially expressed from TLR3−/− mice compared to wild-type C57BL/6NJ mice.
ACKNOWLEDGMENTS
We acknowledge the Systems Virology Center (NIAID contract no. HHSN272200800060C) for transcriptomics data used from a previous study.
This work was supported by NIH grant no. U19 AI100625 to R.S.B. and M.T.H. and NIH grant no. U19 AI106772 to R.S.B.
Footnotes
Citation Totura AL, Whitmore A, Agnihothram S, Schaefer A, Katze MG, Heise MT, Baric RS. 2015. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 6(3):e00638-15. doi:10.1128/mBio.00638-15.
REFERENCES
- 1.Drosten C, Günther S, Preiser W, van der Werf S, Brodt H-R, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguière A-M, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra J-C, Müller S, Rickerts V, Stürmer M, Vieth S, Klenk H-D, Osterhaus ADME, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 3.Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ. 2013. Emergence of porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Invest 25:649–654. doi: 10.1177/1040638713501675. [DOI] [PubMed] [Google Scholar]
- 4.WHO. 31 December 2003, posting date Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. WHO, Geneva, Switzerland: http://www.who.int/csr/sars/country/table2004_04_21/en/. [Google Scholar]
- 5.Ohnmeiss DD. 2015. ECDC Communicable Disease Threats Report (CDTR) European Center for Disease Control. [Google Scholar]
- 6.Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A, Ojeda-Flores R, Arrigo NC, Islam A, Ali Khan S, Hosseini P, Bogich TL, Olival KJ, Sanchez-Leon MD, Karesh WB, Goldstein T, Luby SP, Morse SS, Mazet JAK, Daszak P, Lipkin WI. 2013. A strategy to estimate unknown viral diversity in mammals. mBio 4:e00598-13. doi: 10.1128/mBio.00598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. 2014. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis 20:42–46. doi: 10.1016/j.ijid.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stockman LJ, Bellamy R, Garner P. 2006. SARS: systematic review of treatment effects. PLoS Med 3:e343. doi: 10.1371/journal.pmed.0030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falzarano D, de Wit E, Martellaro C, Callison J, Munster VJ, Feldmann H. 2013. Inhibition of novel β coronavirus replication by a combination of interferon-α2b and ribavirin. Sci Rep 3:1686. doi: 10.1038/srep01686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge X-Y, Li J-L, Yang X-L, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang Y-J, Luo C-M, Tan B, Wang N, Zhu Y, Crameri G, Zhang S-Y, Wang L-F, Daszak P, Shi Z-L. 2013. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolles M, Deming D, Long K, Agnihothram S, Whitmore A, Ferris M, Funkhouser W, Gralinski L, Totura A, Heise M, Baric RS. 2011. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virol 85:12201–12215. doi: 10.1128/JVI.06048-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deming D, Sheahan T, Heise M, Yount B, Davis N, Sims A, Suthar M, Harkema J, Whitmore A, Pickles R, West A, Donaldson E, Curtis K, Johnston R, Baric R. 2006. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med 3:e525. doi: 10.1371/journal.pmed.0030525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Short KR, Kroeze EJ, Fouchier RA, Kuiken T. 2014. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis 14:57–69. doi: 10.1016/S1473-3099(13)70286-X. [DOI] [PubMed] [Google Scholar]
- 14.Chow K, Hsiao C, Lin T, Chen C, Chiou S. 2004. Detection of severe acute respiratory syndrome associated coronavirus in pneumocytes of the lung. Am J Clin Pathol 121:574–580. doi: 10.1309/C0EDU0RAQBTXBHCE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoo J-K, Kim TS, Hufford MM, Braciale TJ. 2013. Viral infection of the lung: host response and sequelae. J Allergy Clin Immunol 132:1263–1276. doi: 10.1016/j.jaci.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M. 2006. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus–induced acute pneumonia. PLoS Pathog 2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudd BD, Smit JJ, Flavell RA, Alexopoulou L, Schaller MA, Gruber A, Berlin AA, Lukacs NW. 2006. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J Immunol 176:1937–1942. doi: 10.4049/jimmunol.176.3.1937. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, Miller DJ, Bowman ER, Nagarkar DR, Schneider D, Zhao Y, Linn MJ, Goldsmith AM, Bentley JK, Sajjan US, Hershenson MB. 2011. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog 7:e1002070. doi: 10.1371/journal.ppat.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. 2005. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 20.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-[kappa]B by Toll-like receptor 3. Nature 413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 21.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-[beta] induction. Nat Immunol 4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J Immunol 169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu Y-J. 2011. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34:866–878. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. 2004. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem 279:2712–2718. doi: 10.1074/jbc.M305790200. [DOI] [PubMed] [Google Scholar]
- 26.Monick MM, Yarovinsky TO, Powers LS, Butler NS, Carter AB, Gudmundsson G, Hunninghake GW. 2003. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem 278:53035–53044. doi: 10.1074/jbc.M308093200. [DOI] [PubMed] [Google Scholar]
- 27.O’Neill LA, Bowie AG. 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 28.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. 2008. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J, Chen WH, Ernst RK, Rossignol DP, Gusovsky F, Blanco JC, Vogel SN. 2013. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497:498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morales-Nebreda L, Mutlu GM, Scott Budinger GR, Radigan KA. 2014. Loss of TLR4 does not prevent influenza A-induced mortality. Am J Respir Crit Care Med 189:1280–1281. doi: 10.1164/rccm.201401-0193LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khanolkar A, Hartwig SM, Haag BA, Meyerholz DK, Harty JT, Varga SM. 2009. Toll-like receptor 4 deficiency increases disease and mortality after mouse hepatitis virus type 1 infection of susceptible C3H mice. J Virol 83:8946–8956. doi: 10.1128/JVI.01857-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, Sasaki T, Ohteki T, Ranieri VM, dos Santos CC, Kawaoka Y, Akira S, Luster AD, Lu B, Penninger JM, Uhlig S, Slutsky AS, Imai Y. 2013. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am J Respir Crit Care Med 187:65–77. doi: 10.1164/rccm.201203-0508OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao J, Wohlford-Lenane C, Zhao J, Fleming E, Lane TE, McCray PB, Perlman S. 2012. Intranasal treatment with poly(I·C) protects aged mice from lethal respiratory virus infections. J Virol 86:11416–11424. doi: 10.1128/JVI.01410-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwata-Yoshikawa N, Uda A, Suzuki T, Tsunetsugu-Yokota Y, Sato Y, Morikawa S, Tashiro M, Sata T, Hasegawa H, Nagata N. 2014. Effects of Toll-like receptor stimulation on eosinophilic infiltration in lungs of BALB/c mice immunized with UV-inactivated severe acute respiratory syndrome-related coronavirus vaccine. J Virol 88:8597–8614. doi: 10.1128/JVI.00983-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pérez-Girón JV, Belicha-Villanueva A, Hassan E, Gómez-Medina S, Cruz JL, Lüdtke A, Ruibal P, Albrecht RA, García-Sastre A, Muñoz-Fontela C. 2014. Mucosal polyinosinic-polycytidylic acid improves protection elicited by replicating influenza vaccines via enhanced dendritic cell function and T cell immunity. J Immunol 193:1324–1332. doi: 10.4049/jimmunol.1400222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao J, Li K, Wohlford-Lenane C, Agnihothram SS, Fett C, Zhao J, Gale MJ, Baric RS, Enjuanes L, Gallagher T, McCray PB, Perlman S. 2014. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc Natl Acad Sci U S A 111:4970–4975. doi: 10.1073/pnas.1323279111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheahan T, Morrison TE, Funkhouser W, Uematsu S, Akira S, Baric RS, Heise MT. 2008. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog 4:e1000240. doi: 10.1371/journal.ppat.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gralinski LE, Bankhead A, Jeng S, Menachery VD, Proll S, Belisle SE, Matzke M, Webb-Robertson BJ, Luna ML, Shukla AK, Ferris MT, Bolles M, Chang J, Aicher L, Waters KM, Smith RD, Metz TO, Law GL, Katze MG, McWeeney S, Baric RS. 2013. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio 4:e00271-13. doi: 10.1128/mBio.00271-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitchell HD, Eisfeld AJ, Sims AC, McDermott JE, Matzke MM, Webb-Robertson BJ, Tilton SC, Tchitchek N, Josset L, Li C, Ellis AL, Chang JH, Heegel RA, Luna ML, Schepmoes AA, Shukla AK, Metz TO, Neumann G, Benecke AG, Smith RD, Baric RS, Kawaoka Y, Katze MG, Waters KM. 2013. A network integration approach to predict conserved regulators related to pathogenicity of influenza and SARS-CoV respiratory viruses. PLoS One 8:e69374. doi: 10.1371/journal.pone.0069374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamelin ME, Prince GA, Gomez AM, Kinkead R, Boivin G. 2006. Human metapneumovirus infection induces long-term pulmonary inflammation associated with airway obstruction and hyperresponsiveness in mice. J Infect Dis 193:1634–1642. doi: 10.1086/504262. [DOI] [PubMed] [Google Scholar]
- 41.Schwarze J, Hamelmann E, Bradley KL, Takeda K, Gelfand EW. 1997. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J Clin Invest 100:226–233. doi: 10.1172/JCI119516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menachery VD, Yount BL, Josset L, Gralinski LE, Scobey T, Agnihothram S, Katze MG, Baric RS. 2014. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J Virol 88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer LA, May WJ, deRonde K, Brown-Steinke K, Gaston B, Lewis SJ. 2013. Hypoxia-induced ventilatory responses in conscious mice: gender differences in ventilatory roll-off and facilitation. Respir Physiol Neurobiol 185:497–505. doi: 10.1016/j.resp.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cameron MJ, Ran L, Xu L, Danesh A, Bermejo-Martin JF, Cameron CM, Muller MP, Gold WL, Richardson SE, Poutanen SM, Willey BM, DeVries ME, Fang Y, Seneviratne C, Bosinger SE, Persad D, Wilkinson P, Greller LD, Somogyi R, Humar A, Keshavjee S, Louie M, Loeb MB, Brunton J, McGeer AJ; Canadian SARS Research Network, Kelvin DJ. 2007. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol 81:8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faure E, Poissy J, Goffard A, Fournier C, Kipnis E, Titecat M, Bortolotti P, Martinez L, Dubucquoi S, Dessein R, Gosset P, Mathieu D, Guery B. 2014. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS One 9:e88716. doi: 10.1371/journal.pone.0088716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menasria R, Boivin N, Lebel M, Piret J, Gosselin J, Boivin G. 2013. Both TRIF and IPS-1 adaptor proteins contribute to the cerebral innate immune response against herpes simplex virus 1 infection. J Virol 87:7301–7308. doi: 10.1128/JVI.00591-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seo S-U, Kwon H-J, Song J-H, Byun Y-H, Seong BL, Kawai T, Akira S, Kweon M-N. 2010. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J Virol 84:12713–12722. doi: 10.1128/JVI.01675-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menachery VD, Eisfeld AJ, Schäfer A, Josset L, Sims AC, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Williams CM, Weiss J, Matzke MM, Webb-Robertson B-J, Schepmoes AA, Shukla AK, Metz TO, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS. 2014. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 5:e01174-14. doi: 10.1128/mBio.01174-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams AE, Chambers RC. 2014. The mercurial nature of neutrophils: still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol 306:L217–L230. doi: 10.1152/ajplung.00311.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perrone LA, Plowden JK, García-Sastre A, Katz JM, Tumpey TM. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seo S-U, Kwon H-J, Ko H-J, Byun Y-H, Seong BL, Uematsu S, Akira S, Kweon M-N. 2011. Type I interferon signaling regulates Ly6Chi monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog 7:e1001304. doi: 10.1371/journal.ppat.1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi C, Pamer EG. 2011. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Züst R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2[prime]-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frieman MB, Chen J, Morrison TE, Whitmore A, Funkhouser W, Ward JM, Lamirande EW, Roberts A, Heise M, Subbarao K, Baric RS. 2010. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog 6:e1000849. doi: 10.1371/journal.ppat.1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leung YH, Nicholls JM, Ho C-K, Sia S-F, Mok CK, Valkenburg SA, Cheung P, Hui KP, Chan RW, Guan Y, Akira S, Peiris JS. 2014. Highly pathogenic avian influenza A H5N1 and pandemic H1N1 virus infections have different phenotypes in Toll-like receptor (TLR) 3 knock-out mice. J Gen Virol 95:1870–1879. doi: 10.1099/vir.0.066258-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daffis S, Samuel MA, Suthar MS, Gale M, Diamond MS. 2008. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol 82:10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 58.Kumar H, Koyama S, Ishii KJ, Kawai T, Akira S. 2008. Cutting edge: cooperation of IPS-1- and TRIF-dependent pathways in poly IC-enhanced antibody production and cytotoxic T cell responses. J Immunol 180:683–687. doi: 10.4049/jimmunol.180.2.683. [DOI] [PubMed] [Google Scholar]
- 59.Mandraju R, Murray S, Forman J, Pasare C. 2014. Differential ability of surface and endosomal TLRs to induce CD8 T cell responses in vivo. J Immunol 192:4303–4315. doi: 10.4049/jimmunol.1302244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L, Herman BD, Sheahan T, Heise M, Genrich GL, Zaki SR, Baric R, Subbarao K. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog 3:e5. doi: 10.1371/journal.ppat.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Agnihothram S, Yount BL, Donaldson EF, Huynh J, Menachery VD, Gralinski LE, Graham RL, Becker MM, Tomar S, Scobey TD, Osswald HL, Whitmore A, Gopal R, Ghosh AK, Mesecar A, Zambon M, Heise M, Denison MR, Baric RS. 2014. A mouse model for betacoronavirus subgroup 2c using a bat coronavirus strain HKU5 variant. mBio 5:e00047–14. doi: 10.1128/mBio.00047-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. 1997. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Q, Lai K, Xie J, Chen G, Zhong N. 2009. Does unrestrained single-chamber plethysmography provide a valid assessment of airway responsiveness in allergic BALB/c mice? Respir Res 10:61. doi: 10.1186/1465-9921-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Julander JG, Kesler K, Van Wettere AJ, Morrey JD, Smee DF. 2014. The use of plethysmography in determining the severity of lung pathology in a mouse model of minimally lethal influenza virus infection. Antiviral Res 108:10–13. doi: 10.1016/j.antiviral.2014.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental text. Additional data on the methods used are provided for flow cytometry procedures. Download
TLR3−/− mice have aberrant lung function and pathology resulting from SARS-CoV infection. (A to C) To determine if there were differences in lung function associated with the increased viral titer and weight loss observed in TLR3−/− versus wild-type mice infected with SARS-CoV, whole-body plethysmography was used to measure parameters indicative of airway disease in SARS-CoV-infected mice. Whole-body plethysmography analysis showed that SARS-CoV-infected TLR3−/− mice (solid red line) had alterations in lung functions compared to C57BL/6NJ mice (solid black line) over the course of 7 days (the dashed red line indicates data from TLR3−/− mock-infected mice, and the dashed black line indicates data from C57BL/6NJ mock-infected mice; *, P < 0.05; **, P < 0.01; ***, P < 0.001 [by unpaired Student’s t test]). (A) A rise to similar peak levels of PENH, a measure indicative of airway resistance and obstruction, was seen in both groups of SARS-CoV-infected mice by day 2 postinfection. However, while wild-type mice infected with SARS-CoV returned to basal PENH values of by day 4 postinfection, TLR3−/− mice infected with SARS-CoV retained significantly higher PENH values on days 3 to 7 postinfection (*, P < 0.05; ***, P < 0.001). (B) TLR3−/− mice maintained a lower TPEF/Te (RPEF) ratio at days 4 to 7 postinfection, while wild-type mice resolved to the levels seen with mock-infected mice. (D) The midtidal expiratory flow (EF50) was significantly higher in the TLR3−/− mice on days 4 through 7 postinfection (Fig. S1C; ***, P < 0.001). Histopathology analysis of formalin-fixed H&E-stained lung tissues indicated that whereas the TLR3−/− (first panel) and C57BL/6NJ (second panel) mock-inoculated mice had normal alveolar architecture, TLR3−/− mice infected with SARS-CoV (third panel) showed signs of alveolar exudates not present in C57BL/6NJ mice infected with SARS-CoV (last panel) on day 7 postinfection. (E to H) Evaluation of H&E-stained lung tissue samples from TLR3−/− and wild-type mice infected with SARS-CoV at day 7 postinfection revealed no statistically significant differences in inflammation surrounding the large airways (E), vasculature (F), or alveoli (G and H). (D and I) However, significantly more exudates and debris were present in the alveolar spaces of TLR3−/− mice infected with SARS-CoV than in wild-type mice on day 7 postinfection, indicating that alveolar fluid may have been limiting lung function in the TLR3−/− mice even after the mice had begun to recover from weight loss due to SARS-CoV infection. Histology sections were scored in a blinded manner, and scores were evaluated for significance by unpaired Student’s t test (*, P < 0.05; NS, not significant). Download
Increased pathology of SARS-CoV in the lungs of TRIF−/− mice. (A to D) Because TRIF−/− mice infected with SARS-CoV showed signs of severe lung dysfunction by whole-body plethysmography, H&E-stained lung sections from TRIF−/− and C57BL6/J mice were scored for signs of SARS-CoV disease on day 6 postinfection. Although mock-inoculated wild-type and TRIF−/− mice show no signs of abnormal pathology (A to D, left panels), wild-type and TRIF−/− mice infected with SARS-CoV displayed hallmarks of SARS-CoV disease, including airway inflammation (C and E), perivascular cuffing (B and F), and alveolar inflammation (D and G to I). TRIF−/− mice infected with SARS-CoV had significantly more accumulation of inflammatory cells surrounding the large airways than wild-type mice (A and C, right panels; E, *, P < 0.05). Similar observations revealed increased levels of infiltrating cells surrounded the vasculature in the lungs of TRIF−/− mice infected with SARS-CoV compared to wild-type mice (C and F; *, P < 0.05). In the alveolar spaces of TRIF−/− mice, increased thickening of the septa between alveoli (D and G; *, P < 0.05) was observed in addition to greater numbers of infiltrating cells (D and H; *, P < 0.05). Histology sections were scored in a blinded manner, and scores were evaluated for significance by unpaired Student’s t test (*, P < 0.05; **, P < 0.01). Download
TLR4−/− mice and TRAM−/− mice have similar lung dysfunctions in response to SARS-CoV infection. Following SARS-CoV infection, the lung function parameters of TLR4−/− mice (A to C) and TRAM−/− mice (D to F) were measured daily along with those of wild-type mice and mock-inoculated controls. Differences from wild-type mice infected with SARS-CoV as determined by the lung parameters of PENH (A and D), RPEF (B and E), and EF50 (C and F) were observed in TLR4−/− mice and TRAM−/− mice infected with SARS-CoV but not in mock-inoculated controls (*, P < 0.05; **, P < 0.01 [by unpaired Student’s t test]). Download
Aberrant chemokine signaling programs in TRIF−/− mice infected with SARS-CoV. RNA expression profiles of chemokine signaling programs downstream of TLRs were measured by qPCR analysis of lung homogenates from TRIF−/− mice (red bars) or wild-type C57BL/6J mice (black bars) infected with 105 PFU of SARS-CoV normalized to the results seen with mock-infected TRIF−/− or wild-type mice (n = 4 mice per group). The levels of chemokines CCL2 (A), CCL3 (B), CCL7 (C), and CCL8 (D) as well as chemokines that influence neutrophil chemotaxis, including CXCL1 (E), CXCL2 (F), and CXCL3 (G), were measured at day 2 and day 4 postinfection. Significant differences between groups were evaluated by an unpaired Student’s t test, bar graphs show the mean normalized fold change on the indicated day postinfection, and the error bars indicate 1 standard deviation from the mean (*, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant). Download
Genes differentially expressed from TLR3−/− mice compared to wild-type C57BL/6NJ mice.