

Abstract
Type 1 diabetes (T1D) is an immune-mediated disease resulting in destruction of insulin-producing pancreatic beta cells. Mesenchymal stem cells (MSCs) possess potent immunomodulatory properties, garnering increasing attention as cellular therapy for T1D and other immunologic diseases. However, MSCs generally lack homing molecules, hindering their colonization at inflammatory sites following intravenous (IV) administration. Here we analyzed whether enforced E-selectin ligand expression on murine MSCs could impact their effect in reversing hyperglycemia in non-obese diabetic (NOD) mice. Though murine MSCs natively do not express the E-selectin binding determinant sialyl Lewisx (sLex), we found that fucosyltransferase-mediated α(1,3)-exofucosylation of murine MSCs resulted in sLex display uniquely on cell surface CD44 thereby creating HCELL, the E-selectin-binding glycoform of CD44. Following IV infusion into diabetic NOD mice, allogeneic HCELL+ MSCs showed 3-fold greater peri-islet infiltrates compared to buffer-treated (i.e., HCELL−) MSCs, with distribution in proximity to E-selectin-expressing microvessels. Exofucosylation had no effect on MSC immunosuppressive capacity in in vitro assays, however, though engraftment was temporary for both HCELL+ and HCELL− MSCs, administration of HCELL+ MSCs resulted in durable reversal of hyperglycemia, whereas only transient reversal was observed following administration of HCELL− MSCs. Notably, exofucosylation of MSCs generated from CD44−/− mice induced prominent membrane expression of sLex, but IV administration of these MSCs into hyperglycemic NOD mice showed no enhanced pancreatotropism or reversal of hyperglycemia. These findings provide evidence that glycan engineering to enforce HCELL expression boosts trafficking of infused MSCs to pancreatic islets of NOD mice and substantially improves their efficacy in reversing autoimmune diabetes.
Introduction
Despite significant advances in the pharmacotherapy of glycemia control, T1D is still associated with significant morbidity and mortality, and it continues to pose a major public health burden demanding innovative treatment strategies [1,2]. Cell-based immunomodulatory therapy has emerged as a promising approach in the treatment of T1D [3]. Because of their immunomodulatory properties, safety profile, easy acquisition, and robust ex vivo expansion, mesenchymal stem cells (MSCs) have become the most rapidly growing cell therapy for the treatment of various refractory immune-mediated diseases including T1D [4–7]. In preclinical models using NOD mice, we and others have recently reported that systemically-administered MSCs have utility in dampening autoimmune diabetes [8–13]. However, the benefits of MSC therapy in reversal of hyperglycemia were temporary, highlighting a pressing need to develop strategies to improve the effectiveness of MSC-based therapy for T1D [6].
The efficacy of immunomodulatory cell therapy is closely related to the ability of the infused cells to traffic to the inflamed tissue [14,15]. For some organs (e.g., the heart), direct (local) injection of cells into the affected site can achieve requisite colonization for physiologic benefit [16]. However, for treatment of T1D, the vascular route of cell delivery is mandated, as direct injection of cells into the pancreatic parenchyma would trigger release of proteases and other enzymes that could induce profound, life-threatening pancreatic inflammation.
The migration of blood-borne cells into tissues is initiated by tethering/rolling adhesive interactions on target tissue endothelium. The most potent mediators of these binding interactions are the selectins, a family of three Ca++-dependent lectins (E-, P- and L-selectin (also known as CD62E, CD62P, and CD62L, respectively)) that bind to sialofucosylated glycan determinants expressed on their respective ligands [17]. Importantly, within the microvasculature at all inflammatory sites, the endothelial selectin, E-selectin, is inducibly expressed in response to inflammatory cytokines such as TNF-α [17,18]. E-selectin binds to membrane glycoproteins and/or glycolipids on circulating cells that prototypically display the sialofucosylated tetrasaccharide known as “sialylated Lewis X” (sLex). However, MSCs do not natively express E-selectin ligands [19]. This deficit in trafficking limits the engraftment of MSCs in inflamed peripheral tissues following intravenous administration [17,20], constraining the utility of MSC-based therapeutics. Accordingly, we sought to investigate whether MSC trafficking to inflamed pancreas could be licensed via cell surface glycan modification to enforce E-selectin ligand expression, and whether this would impact MSC therapeutic effect(s) in new onset autoimmune diabetes in NOD mice. Our findings provide new insights on the biology of MSC effects in diabetes, highlighting a unique and prominent role for enforced expression of the E-selectin ligand HCELL in enhancing the capacity of murine MSCs to reverse hyperglycemia in diabetic NOD mice.
MATERIALS AND METHODS
Mice
C57BL/6, B6.129(Cg)-Cd44tm1Hbg/J (CD44 on C57BL/6 genetic background; CD44-knock out (“CD44KO”)), BALB/c, and NOD mice were purchased from Jackson Laboratories and were housed and/or bred in a pathogen-free environment at the Harvard Medical School Facilities for Animal Care and Housing. Experiments requiring the use of mice were approved by the Institutional Animal Care and Use Committee.
MSC culture
Bone marrow MSCs were derived as described previously [9, 10]. In brief, isolated marrow cells from C57BL/6 (wild-type) or from CD44KO mice were flask-seeded in culture medium consisting of Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum (Lonza), 10 ng/ml fibroblast growth factor (PeproTech), 100 U/ml penicillin and 100 ug/ml streptomycin (Gibco). MSCs in culture passage 4 to 6 were used for experiments.
MSC characterization and differentiation
MSCs were characterized by flow cytometry using anti-mouse antibodies (all from eBioscience) directed to cell surface markers Sca1, CD44, CD73, CD45, CD29, and CD 105, together with relevant isotype controls. Recombinant mouse E-selectin (CD62E)-human Fc chimera (“mE-Ig”) was purchased from R&D Systems. MSCs were tested for their differentiation capacity into various mesodermal lineages as previously described [21, 22]. Briefly, chondrogenic differentiation of MSCs was induced with ascorbic acid (50 μg/ml) and TGFβ1 (1 ng/ml) in culture medium. After 3 weeks of culture, plates were washed with PBS, fixed with 4% paraformaldehyde, and stained with 0.05% alcian blue for light microscopic visualization of cartilage. Osteogenic differentiation was induced with ascorbic acid (50 μg/ml), sodium β-glycerophosphate (10 mM), and dexamethasone (10 nM) in culture medium. Two weeks later, plates were washed, fixed with 4% paraformaldehyde, and stained with 2% alizarin red for visualization of characteristic calcium deposits. Adipogenic differentiation was induced by the addition of dexamethasone (100 nM) and insulin (6 ng/ml) in F12 medium supplemented with 1% fetal calf serum, 1% glutamine, 1% penicillin-streptomycin. Cells were fixed in 4% parafomaldehyde and stained with 0.3% Oil Red solution in 60% isopropanol to assess lipid-laden vacuoles.
MSC Exofucosylation
MSCs in suspension (10 × 106 per 200 ul reaction) were treated for 90 min at 37°C with 60 mU/mL fucosyltransferase VI (FTVI) in reaction buffer consisting of Ca2+ – and Mg2+ -free Hanks Balanced Salt Solution (HBSS) containing 20 mM HEPES, 0.1% human serum albumin and 1 mM GDP-fucose (“FTVI-modified MSC”), or with reaction buffer alone (unmodified MSC). Following treatment, MSCs were washed with HBSS containing 0.2% HSA and 20 mM HEPES. Cell viability was assessed by trypan blue exclusion and propidium iodide staining (consistently >85% viability 24 hours post-treatment). FTVI modification was assessed by flow cytometry. FTVI enzyme was provided by Dr. Roland Wohlgemuth (Sigma Chemical Corporation).
Western blot analysis
FTVI-modified and unmodified MSC lysates were prepared by incubation with 2% Nonidet P-40 (NP-40) in Buffer A (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 20 mg/mL phenylmethylsulfonyl fluoride, 0.02% sodium azide, and protease inhibitor cocktail tablet (Roche Molecular Biochemicals)). All western blots of whole cell lysates or of immunoprecipitated protein were performed under reducing conditions on 7.5% SDS-PAGE gels as described previously [19]. The amount of lysate in each lane was normalized to cell number for each western blot performed. Blots were visualized with chemiluminescence using Lumi-Light Western Blotting Substrate (Roche).
Flow cytometry and Immunoprecipitation studies
Flow cytometry was performed as described previously [19]. For more details please refer to the Supplementary Methods section.
Parallel Plate Flow Chamber Adhesion Assay
A dynamic flow adhesion assay was performed using a parallel plate flow chamber (250 μm channel depth x 5.0 mm channel width) to assess E-selectin-mediated MSC binding over stimulated human umbilical vein endothelial cells (HUVEC), as previously described [19]. For more details, please refer to the Supplementary Methods section.
Transduction of MSCs with hGH viral vector
MSCs were transduced with the lentivirus containing hGH plasmid construct as described previously [21]. Levels of hGH were measured by enzyme-linked immunosorbent assay (Roche Diagnostics) in MSC supernatants and in serum of injected animals.
Transduction of MSCs with GFP viral vector
MSCs were transduced with a retrovirus plasmid containing GFP (GFP-MSC) as described previously [21]. Transduced MSC were assessed for perinuclear expression of GFP by fluorescent microscopy. In a complementary approach to the use of GFP-MSCs to assess homing, MSCs (non-GFP-transduced) were labeled with the fluorescent dye CFSE.
Immunofluorescence
Pancreata were embedded in Tissue Tek OCT, frozen and sectioned in a cryomicrotome. Immunofluorescence images were acquired using a Nikon E-1000 epifluorescence microscope (X400 total magnification). For more details on specific staining, please refer to the Supplementary Methods section.
Mitogen-stimulation CD3/CD28 T cell proliferation assay
T cell proliferation assay using anti-CD3 and anti-CD28 stimulation was used to assess effects of FTVI modification on immunomodulatory capacity of MSCs. Briefly, 1x105 NOD splenocytes were stimulated with purified mouse anti-CD3e and anti-CD28 (each at 1 μg per well; eBioscience) for 72 hours in RPMI media (Lonza), supplemented with 10% fetal bovine serum (Gemini Bio-Products), 1% penicillin streptomycin (Lonza) and 1% glutamine (Lonza), in the presence of a titrating concentration (5×102 – 5×104) of irradiated (3000 rad) unmodified and FTVI-modified MSCs. In the last 12 hours of the 72 hour incubation period, cells were pulsed with 1 μCi of tritiated thymidine, and lymphocyte proliferation (3H-thymidine incorporation) was measured by scintillation counting. Results are reported as an average of triplicate samples (mean cpm ± SEM).
Reversal of new-onset hyperglycemia studies in NOD mice
Unmodified and FTVI-modified MSCs (0.5 × 106 cells in 200ul) were injected intravenously via tail vein without anesthesia into female NOD mice on the second day of hyperglycemia (>250 mg glucose/dL), and at 7 days and 30 days following onset of hyperglycemia. To assess effect of MSC administration on hyperglycemia, blood was obtained via tail vein and glucose measured as described previously [9, 10].
Statistics
Data from experimental assays and immunohistological experiments were analyzed using student t-test and Mann Whitney tests. Survival data were assessed using Kaplan-Meyer analysis. To perform the analysis and to generate graphs, Prism software was used (GraphPad Software, Inc., San Diego, CA). P value <0.05 was considered significant. Data represent mean ± SEM.
RESULTS
Expression and functional analysis of FTVI-modified MSC
Bone marrow-derived MSCs from C57BL/6 mice, (“wild-type”), a diabetes-resistant strain, and from CD44KO mice (CD44−/− on C57BL/6 background) showed characteristic small spindle fibroblast-like pattern and could be differentiated into mesodermal cell types (Supplemental Figure 1). MSCs expressed cell surface markers characteristic of MSCs (Figure 1A). MSCs did not natively express E-selectin ligands, as shown by absence of reactivity with mAb HECA 452 (which recognizes canonical sialofucosylated E-selectin binding determinants (such as sialylated Lewis X (sLex)) and by absence of binding to mouse E-selectin-Ig chimera (mE-Ig) that serves as a probe for E-selectin ligand activity (Figure 1B).
Figure 1. Effects of Exofucosylation (FTVI treatment) on E-selectin ligand expression by mouse MSC.
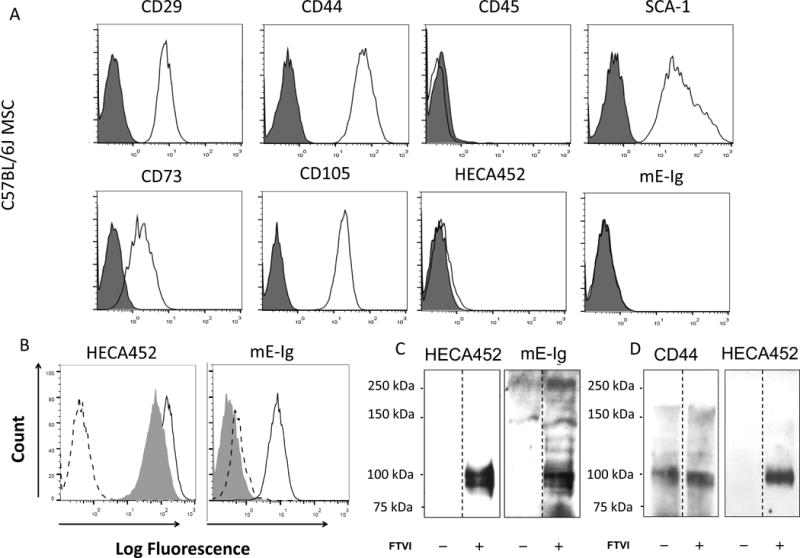
(A) MSC derived from C57BL/6 marrow lacked expression of CD45 and expressed characteristic mouse MSC markers Sca-1, CD29, CD44, CD73 and CD105. Cells lacked reactivity with mAb HECA452 and with E-selectin-Ig chimera (mE-Ig) (istoype= red color and antibody = blue color). (B) FTVI-modified MSC (solid line) stained positive for mAbs HECA452 and were reactive with mE-Ig. Digestion of FTVI-modified MSC with bromelain and proteinase K (shaded histogram) significantly reduced mE-Ig reactivity, but not HECA452 staining, indicating that protease-sensitive glycoproteins serve as the principal E-selectin ligand(s). Dashed line represents staining controls (isotype control for HECA452 staining and calcium chelation with EDTA for mE-Ig staining). (C) Western blot analysis of HECA452 (left) and mE-Ig (right) reactivity of cell lysates of unmodified MSC (−) and FTVI-modified MSC (+). FTVI modification induced HECA452- and mE-Ig-reactive moieties predominantly on a doublet glycoprotein band of ~100 kDa. (D) CD44 was immunoprecipitated from equivalent amounts of cell lysate from FTVI-modified (+) or unmodified (−) MSCs. Immunoprecipitates were then electrophoresed and blotted with anti-CD44 mAbs (KM114 and IM7; left) and with mAb HECA452 (right).
A prior study of human MSCs showed that cell surface glycoengineering (i.e., Glycosyltransferase-Programmed Stereosubstitution (GPS)) using the α-(1,3)-fucosyltransferase VI (FTVI) induces E-selectin adherence via conversion of native membrane CD44 into the molecule known as Hematopoietic Cell E-/L-selectin Ligand (HCELL), a fucosylated sialyllactosaminyl glycovariant of CD44 that potently binds E-selectin [19]. To determine whether cell surface exofucosylation could program E-selectin ligand activity on murine MSCs, cells were treated with FTVI using reaction conditions that were optimized to preserve cell viability. FTVI modification of MSCs markedly induced staining with mAb HECA452, as well as binding with a murine E-selectin-Ig chimera (mE-Ig) (Figure 1B). Notably, protease digestions of MSCs prior to FTVI modification markedly reduced mE-Ig reactivity, indicating that glycoproteins, not glycolipids, were predominant carriers of E-selectin binding determinants (Figure 1B). Following exofucosylation of mouse MSC, western blot of whole cell lysates (Figure 1C) and of immunoprecipitated CD44 (Figure 1D) revealed that the principal membrane glycoprotein decorated with the sialofucosylations recognized by mE-Ig and HECA-452 was the “standard” (i.e., containing no splice variant exons) form of CD44 (~100 kDa). These data indicate that FTVI modification converts murine MSC surface CD44 into HCELL.
FTVI-modified mouse MSCs have increased binding to E-selectin under physiologic shear stress conditions
To assess the capacity of FTVI-modified mouse MSCs to bind E-selectin under physiologic shear stress conditions, we performed parallel plate flow chamber assays of both unmodified and FTVI-modified MSCs. To this end, HUVEC monolayers were first stimulated with TNF-α to upregulate E-selectin expression. As shown in Figure 2, FTVI-modification enabled MSC adhesion to HUVEC at shear stress levels of 0.5 dynes/cm to as much as 20 dynes/cm2. In flow chamber studies, MSC adherence to stimulated HUVEC was strictly dependent on E-selectin receptor/ligand interactions as incubation of HUVEC with function-blocking E-selectin mAb and sialidase treatment of MSCs (to eliminate sLex display) each profoundly reduced adhesion of FTVI-modiied MSCs, to levels similar to that of unmodified MSCs (Figure 2). Altogether, these findings indicate that FTVI modification of murine MSC induces potent E-selectin binding activity, capable of sustaining E-selectin adherence at hemodynamic shear stress levels well beyond those typical of post-capillary venules [17].
Figure 2. Parallel plate flow chamber assay of FTVI-modified and unmodified wild type MSC adherence to TNF-α treated HUVEC.
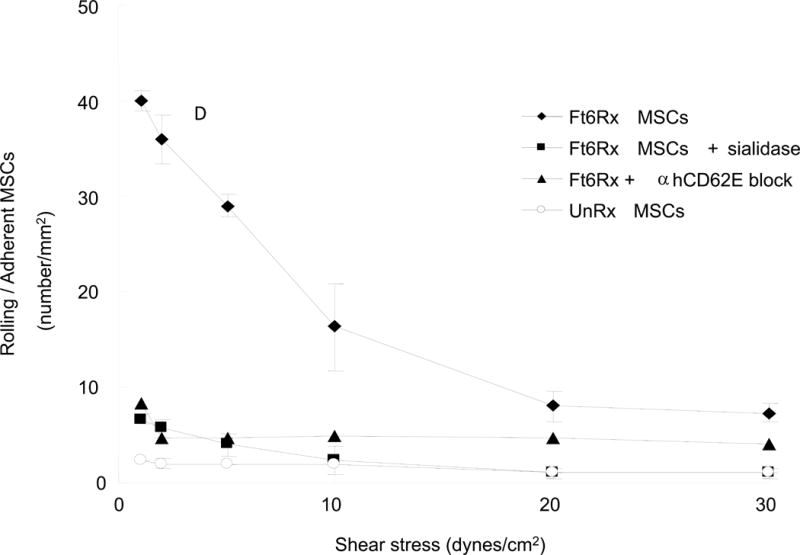
FTVI modification markedly improved MSC adhesion to HUVEC at 0.5 dynes/cm2. Treatment of HUVEC with anti-E-selectin (anti-CD62E) function-blocking mAb reduced rolling adhesive interactions of FTVI-modified MSC to levels similar to that of unmodified MSC. Similarly, removal of sLex determinants by sialidase treatment of FTVI-modified MSC reduced adhesion to levels equivalent to that of unmodified MSC.
Systemically-administered FTVI-modified MSCs potently reverse new onset hyperglycemia in NOD mice
To assess whether FTVI-modification affected the capacity of MSCs to modulate diabetes in NOD mice, new onset diabetic NOD mice either received no cells (untreated control), or received 5×105 FTVI-modified or 5×105 unmodified C57BL/6 MSCs intravenously (via tail vein) on day 2 of hyperglycemia (glucose >250 mg/dL), followed by intravenous injections at days 7 and 30 after onset of hyperglycemia. As shown in Figure 3A, untreated NOD mice showed a rapid increase in their blood glucose levels and, with exception of one animal, died within a few weeks of the onset of hyperglycemia. As compared to the administration of unmodified MSCs (Figure 3B), which resulted in a temporary reversal of autoimmune diabetes (i.e., 3 of 9 mice were normoglycemic at 3 weeks, 2 of 9 animals were normoglycemic at 6 weeks, and all animals were hyperglycemic by 12 weeks), the infusion of FTVI-modified MSCs robustly and durably reversed hyperglycemia (Figure 3C). Strikingly, 7 out of 8 mice were normoglycemic for 3 weeks, 6 of 8 mice remained normoglycemic for 6 weeks, and 5 of 8 mice were free of diabetes for upwards of 12 weeks following initial treatment (Figure 3C). While the majority of the mice in the unmodified and FTVI-modified MSC group which had blood glucose levels above 600 mg/dl survived up to 90 days, only one (out of 7 mice) in the untreated group (i.e., receiving no MSCs) survived 90 days.
Figure 3. Effects of intravenous administration of unmodified and FTVI-modified MSC on hyperglycemia in new onset diabetic NOD mice.
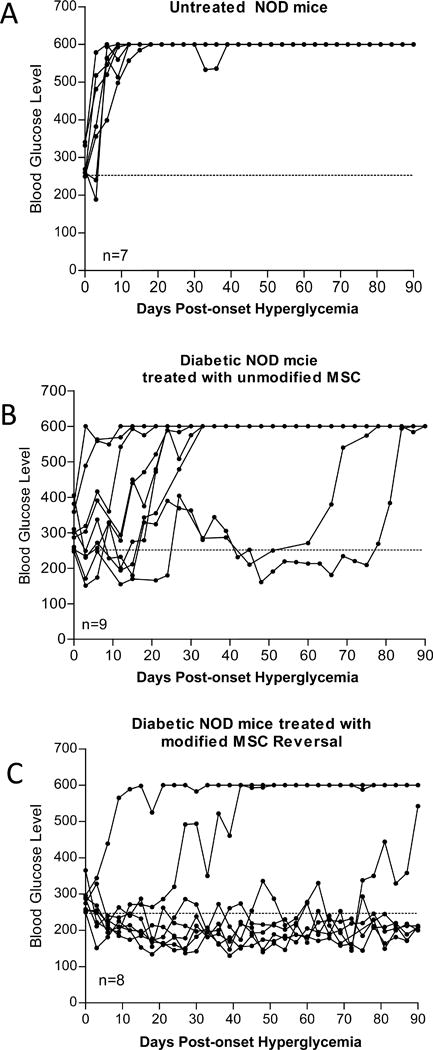
(A) Hyperglycemic NOD injected with PBS (untreated control) showed no reversal of hyperglycemia (i.e., glucose levels consistently above 600 mg glucose/dL). As compared to infusion of unmodified MSCs (B), infusion of FTVI-modified MSCs (C) resulted in a marked increase in number of mice with reversion to normoglycemia and in the durability of diabetes reversal.
E-selectin expression and MSC localization in the pancreas of NOD mice
To examine the effect of FTVI-modification on MSC trafficking to the pancreas, we first assessed the expression of E-selectin within microvasculature of the pancreas of 14-week-old NOD mice using immunofluorscence microscopy. Peri-islet microvessels are reported to be the primary site of inflammatory cell trafficking which results in insulitis [23, 24]. There was no E-selectin expression in pancreata of diabetic-resistant mice (Figure 4A), whereas peri-islet microvessels of the pancreas of NOD mice expressed E- selectin (Figure 4B), which co-localized with CD31 staining in sequential sections (Figures 4C and D). Infiltration of T-cells into the diabetic islet margins was confirmed by CD3 staining of NOD pancreata during diabetic onset (Figure 4E). To assess pancreatic infiltration of systemically administered FTVI-modified MSCs in NOD mice, we stained frozen sections of pancreata with antibody HECA452. As shown in Figures 4F and G, HECA452+ MSCs were observed in the peri-islet area within the vicinity of E-selectin-expressing microvasculature 1 day after transplantation. To assess the extent of homing of FTVI-modified and unmodified MSCs after systemic administration, GFP-transduced FTVI-modified and unmodified MSCs were intravenously injected into NOD mice, and their pancreata, pancreatic and mesenteric lymph nodes and spleen were harvested at day 4 post-injection, sectioned, and assessed for presence of MSCs by fluorescence microscopy. FTVI-modified MSCs showed 3-fold higher infiltration of pancreas as compared to unmodified MSCs, whereas no difference in pancreatic infiltrates of infused FTVI-modified and unmodified MSCs were seen in diabetes-resistant BALB/c mice (Figure 4H). However, no difference was noted in the extent of homing of FTVI-modified and unmodified MSCs into lymphoid tissues of treated NOD mice.
Figure 4. Immunofluorescence staining of islets to assess expression of E-selectin and localization of MSC in the pancreas.
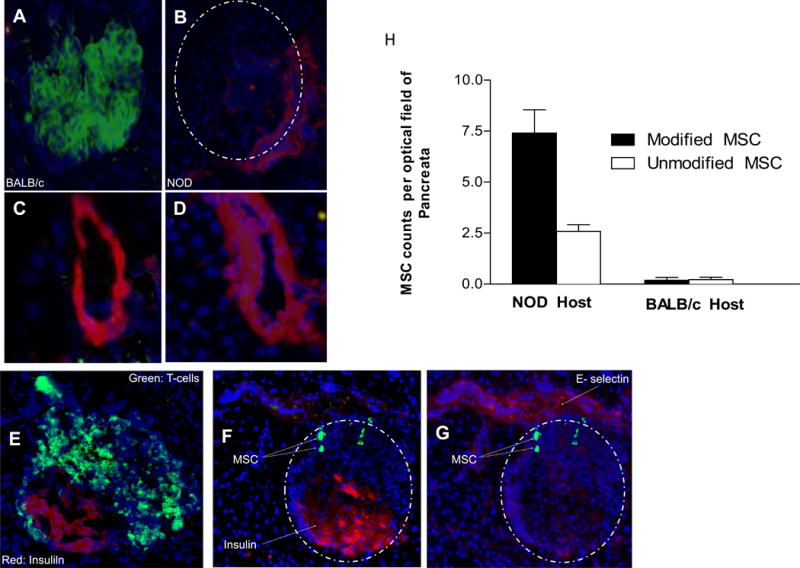
Figures (A) and (B): Pancreatic islets of (A) diabetic-resistant BALB/c mice and (B) NOD mice were stained for expression of insulin (green) and E-selectin (red). Islets are demarcated by dashed line. Compared to BALB/c (A), NOD mice (B) show diminished insulin production due to insulitis. In Figures (C)–(G), cryostat sections of pancreas from MSC-treated NOD mice are stained with DAPI to identify nuclei (blue). Figures (C) and (D): Staining of sequential sections of NOD pancreas demonstrates co-localization of endothelial marker CD31 (C) and E-selectin (D), confirming the presence of E-selectin on peri-islet endothelial cells. Figure (E): Co-staining of NOD islet with T-cell marker CD3 (green) and insulin (red) reveals characteristic T-cell infiltration at the margins of the islet. Figures (F) and (G): Immunofluorescence images of a cryostat section stained for infiltrating MSC (visualized with FITC-conjugated anti-sLex mAb HECA452; green), islet (F) (visualized by APC-conjugated anti-insulin mAb; red) and E-selectin-expressing microvessel (G) (visualized with PE-conjugated anti-E-selectin mAb; red). Staining identifies HECA452+ MSC in zones of insulitis, in proximity to E-selectin-expressing microvessels in the peri-islet area. Figure (H): Pancreatic infiltration of intravenously administered MSC into NOD and BALB/c hosts. Accumulation of FTVI-modified MSC into pancreata of NOD mice is 3-fold higher compared to that of unmodified MSC (p<0.01), whereas no difference in pancreatic infiltrates is observed in BALB/c host (n=3 mice per group; minimum 30 fields counted per group at 60X magnification).
Exofucosylation has no effect on MSC immunosuppressive capacity or on in vivo survival
We have previously reported that measuring human growth hormone (hGH) produced by hGH-transduced MSCs is a sensitive method to ascertain longevity of administered MSCs [21]. To assess if the enhanced reversal of hyperglycemia of administered FTVI-modified MSCs was consequent to a survival advantage, 5×105 of hGH-transduced FTVI-modified and unmodified MSCs were injected into NOD mice at day 2 and 7 following onset of hyperglycemia, and serum hGH was measured at various time-points postinjection. As shown in Figure 5A, there was no difference in the pattern of hGH production, indicating that FTVI modification did not affect MSC persistence in vivo. To evaluate whether exofucosylation modifies MSC immunomodulatory capabilities, we performed co-culture T cell suppression assays. T cell proliferation was equally dampened by FTVI-modified and unmodified MSC (Figure 5B), indicating that exofucosylation does not impart additional immunomodulation properties on MSCs.
Figure 5. FTVI-modification of MSC does not affect cell survival or immunosuppressive capacity.
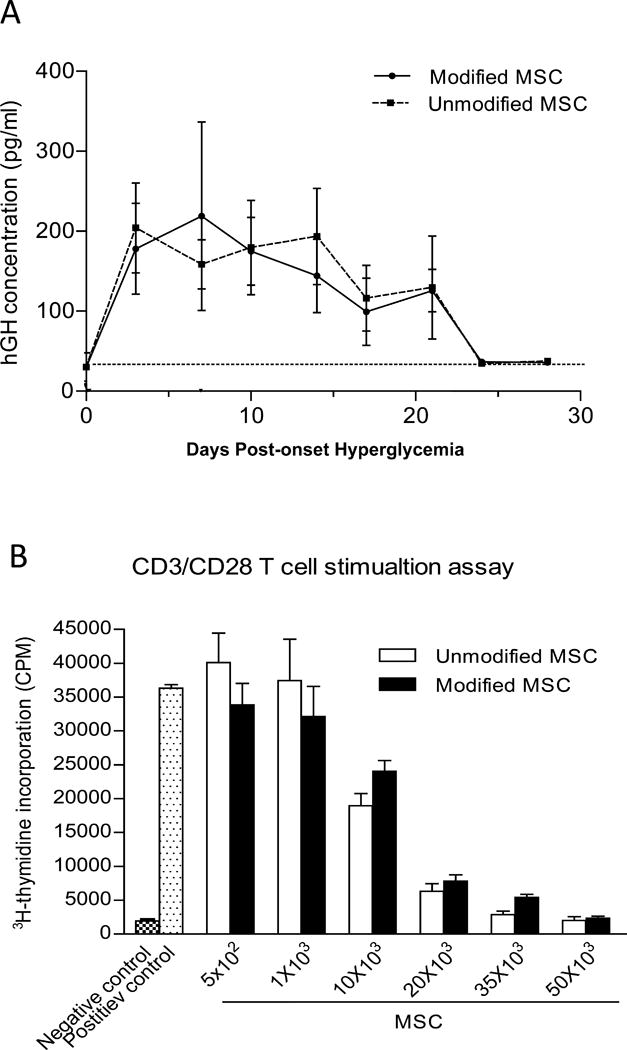
(A) Similar levels of hGH were detected in the serum of NOD mice at different time points following injection with pHRST-hGH-transduced FTVI-modified or unmodified MSC. (B) FTVI-modified and unmodified MSC equally suppressed proliferation of NOD CD4+ T cells stimulated with CD3/CD28.
Absence of MSC CD44 expression abrogates the anti-diabetic effect of FTVI-modified MSC
To examine whether CD44 expression is requisite for the enhanced anti-diabetic potency of FTVI-modified MSCs, we generated MSCs from CD44KO mice (CD44−/− on C57BL/6 genetic background). With exception of CD44, MSCs generated from marrow of CD44KO mice showed identical levels of cell surface adhesion molecules. By flow cytometry, exofucosylated CD44KO MSCs showed HECA452-reactivity equivalent to that of exofucosylated wild-type MSCs (Supplemental Figure 2), indicating that sialofucosylated determinants were created on alternative (i.e., non-CD44) scaffolds. To assess whether administration of CD44KO MSCs conferred anti-diabetic activity, 5×105 cells each of FTVI-modified and unmodified CD44KO and of C57BL/6 wild-type MSCs were injected into new-onset diabetic NOD mice at days 2, 7 and 30 post-hyperglycemia onset. Compared to wild-type MSCs, CD44 deficiency of MSCs resulted in a significant impairment in their anti-diabetic effect, as demonstrated by failure in reversing autoimmune diabetes in 5 out 6 mice receiving unmodified CD44KO MSCs, and in 6 out 7 mice receiving FTVI-modified CD44KO MSCs (Figures 6 A and B, respectively). Following injection in NOD mice, CFSE-labeled FTVI-modified and unmodified CD44KO MSCs showed no difference in trafficking to pancreata (Figure 6C), with levels similar to that observed for wild-type MSCs (Figure 4H). MSC suppressive capacity in vitro was not abrogated by lack of MSC CD44 expression nor by exofucosylation of CD44KO MSCs (Figure 6D), suggesting that the absence of euglycemic effect(s) of administered FTVI-treated CD44−/− MSCs is not attributable to inherent deficits in immunoregulation.
Figure 6. Lack of CD44 expression abrogates the anti-diabetic effect of systemically administered FTVI-modified MSC.
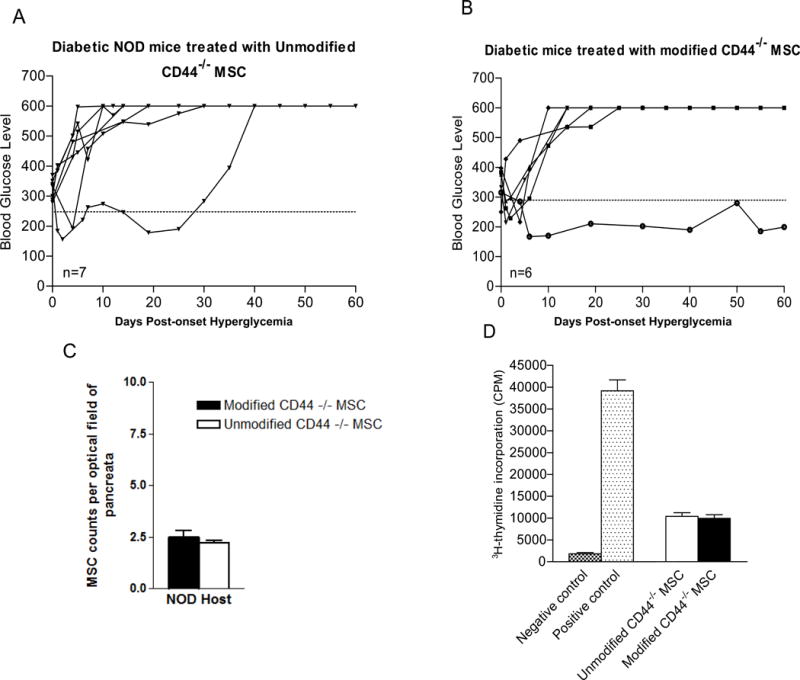
(A) As compared to unmodified wild type MSC (Figure 3B), administration of CD44-deficient MSC shows modest anti-diabetic effect. Only 1 NOD mouse (out of 7) receiving unmodified CD44KO MSC showed reversal of hyperglycemia which was transient (diabetes recurrence at ~day 30), and 6 out of 7 diabetic NOD mice remained hyperglycemic. (B) As compared to results in mice receiving FTVI-modified wild-type MSC (Figure 3C), administration of FTVI-modified CD44KO MSC conferred minimal anti-diabetic effects, with only 1 out of 6 NOD mice showing reversal of hyperglycemia. (C) Accumulation of MSC in NOD pancreata was no different in mice receiving FTVI-modified CD44KO MSC (white bar) compared to mice receiving unmodified CD44KO MSC (black bar) (p<0.01), and in each case was similar to that receiving unmodified MSC (Figure 4H). MSC infiltrates were quantified at X60 magnification. (D) Both FTVI-modified and unmodified CD44KO MSC possess immunosuppressive capacity, similarly dampening T cell proliferation in the CD3/CD28 T cell stimulation assay.
DISCUSSION
Because of their immunomodulatory properties, safety profile, and robust expansion ex vivo, MSCs have become the focus of several human trials to treat various refractory immune-mediated diseases including T1D [6]. MSCs have been found to suppress insulitis and autoimmune diabetes via multifaceted immunomodulatory effects on pathogenic components critical to elaboration of T1D [25, 26]. These effects include the capacity to modulate expression of inflammatory vs. regulatory cytokines thereby promoting expansion of regulatory DC and T cells, and MSC expression of negative costimulatory molecules (e.g., PDL-1) which can directly inhibit autoreactive T cells [8–10,13]. Hence, MSC therapy has great potential to be an exciting and unique strategy for T1D treatment. Nevertheless, there is a pressing need for studies to improve the efficacy MSC-based therapy in T1D [6].
In prior studies, we observed that intravenous administration of fully allogeneic MSCs was associated with a transient reversal of hyperglycemia in diabetic NOD mice [9]. We hypothesized that a proximate hurdle in realizing a more robust anti-diabetic effect might be the relative paucity of MSC homing to inflamed pancreas. Here, we sought to assess whether systemic administration of MSCs with enhanced homing capacity via enforced HCELL expression would influence reversal of diabetes in the NOD model. To this end, we utilized the C57BL/6 strain as the allogeneic source of MSCs since the CD44KO phenotype was available on this genetic background. We focused on metabolic control as the primary end-point for assessing the efficacy of MSC therapy, as we had previously examined the immune mechanisms mediating anti-diabetic effects of MSC [9, 10].
In contrast to other studies which characteristically assess the effects of anti-diabetic interventions for relatively short time periods (typically <30 days), in this study the effects of MSC on reversal of autoimmune diabetes were assessed over a prolonged observation period (90 days). Our data here indicate that conversion of native membrane CD44 into HCELL by cell surface α-(1,3)-exofucosylation (“FTVI-modification”) confers high efficiency mouse MSC binding to E-selectin. We observed E-selectin expression in the peri-islet area of pancreata of NOD mice with insulitis, but not in pancreata of diabetic-resistant mice. Consistent with these findings, we did not observe differences in the level of homing of injected allogeneic C57BL/6 FTVI-modified and unmodified MSCs into pancreata of diabetic-resistant mice (BALB/c hosts), but, compared with unmodified MSCs, there was heightened pancreatic infiltration of systemically administered C57BL/6 (allogeneic) FTVI-modified MSCs in diabetic NOD mice, with concomitant durable reversal of diabetes. Notably, we did not observe differences in infiltration of lymphoid tissues between animals receiving FTVI-modified and unmodified MSCs. Collectively, these data draw attention to a primary role of MSC pancreatic colonization in suppressing autoimmune diabetes, but do not exclude the potential contribution of extrapancreatic effects of MSCs.
Compared to the anti-diabetic effects observed using intravenously administered fully allogeneic unmodified MSCs in this study and in prior studies [9], the marked improvement in T1D reversal using fully allogeneic FTVI-modified (HCELL+) MSCs observed here does not appear to be attributable to an enhanced immunosuppressive capacity of the glycoengineered MSC itself or to in vivo survival advantage endowed by glycoengineering of MSCs. In a prior study, we observed improved anti-diabetic effects in NOD mice following intravenous administration of semi-allogeneic MSCs obtained from NOR mice [10], likely a reflection of decreased rejection of the semi-allogeneic MSCs yielding improved in vivo tissue colonization/persistence of cells. However, in clinical applications, it would be advantageous to employ strategies to augment potency of fully allogeneic-source MSCs, especially as manufacturing and regulatory issues support the use of “off-the-shelf” (i.e., culture-expanded, pooled) allogeneic MSC products. The finding that exofucosylated CD44KO MSCs lack the capacity to induce durable reversal of hyperglycemia, despite evident creation of FTVI-dependent surface sialofucosylated determinants detectable by HECA452 reactivity, indicates that E-selectin binding determinants displayed expressly on the CD44 scaffold (i.e., elaboration of HCELL) are required to achieve potent anti-diabetic effects. This may be related to a key role for HCELL expression to attain requisite tissue homing and extravasation [27] and/or could reflect a requirement for HCELL/CD44 expression to achieve appropriate lodgment in relevant microenvironmental sites [28]. A role for in situ tissue lodgment to achieve MSC effect(s) has been highlighted by studies of MSC co-transplantation with islet allografts engendering immunoprivileged zones [29–31]. While further studies of these MSC effects in vivo are warranted, the data here highlight the potential of enhanced pancreatotropism via systemic administration of HCELL+ MSCs in the therapy of T1D. More broadly, the fact that E-selectin is upregulated by cytokines such as TNF and IL-1 within endothelial beds at all inflammatory sites [17,18] offers the opportunity to exploit enforced MSC HCELL expression to improve the efficacy of MSC-based therapy for a wide variety of inflammatory conditions.
Supplementary Material
Supplemental Figure 1. Characterization of bone marrow derived MSC. MSC were differentiated into osteocytes, adipocytes and chondrocytes (X200 magnification).
Supplemental Figure 2. FTVI modification of CD44−/− MSC results in similar increase in HECA452 reactivity as WT MSC. Unmodified MSC (dotted line) show no reactivity with HECA452, while FTVI-modified MSC (solid line) and FTVI-modified CD44KO MSC (filled grey) showed similar levels of staining with HECA452, indicating that exofucosylation creates sialofucosylated epitopes on alternative (i.e., non-CD44) scaffolds in CD44KO MSC.
Acknowledgments
This work was supported by the Edward and Dana Slatkin Research Fund (to RS), and by National Heart Lung Blood Institute grants PO1 HL107146 (Program of Excellence in Glycosciences), RO1 HL60528 and RO1 HL73714 (each to RS), and by the American Diabetes Association Basic Science Grant 110151 and the Harvard Stem Cell Institute Grant 109210 (each to RA). SS was supported by a postdoctoral fellowship from the Japan Society for the Promotion of Science. According to National Institutes of Health policies and procedures, the Brigham & Women’s Hospital has assigned intellectual property rights regarding HCELL and GPS to the inventor (RS), who may benefit financially if the technology is licensed. RS’s ownership interests were reviewed and are managed by the Brigham & Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policy. RA and RS are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Bresson D, von Herrath M. Immunotherapy for the prevention and treatment of type 1 diabetes: optimizing the path from bench to bedside. Diabetes Care. 2009;32:1753–1768. doi: 10.2337/dc09-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludvigsson J, Krisky D, Casas R, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. 2012;366:433–442. doi: 10.1056/NEJMoa1107096. [DOI] [PubMed] [Google Scholar]
- 3.Bonifacio E. Immunotherapy in type 1 diabetes: a shorter but more winding road? Diabetes. 2012;61:2214–2215. doi: 10.2337/db12-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prockop DJ, Brenner M, Fibbe WE, et al. Defining the risks of mesenchymal stromal cell therapy. Cytotherapy. 2010;12:576–578. doi: 10.3109/14653249.2010.507330. [DOI] [PubMed] [Google Scholar]
- 5.Dazzi F, van Laar JM, Cope A, et al. Cell therapy for autoimmune diseases. Arthritis Res Ther. 2007;9:206. doi: 10.1186/ar2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdi R, Fiorina P, Adra CN, et al. Immunomodulation by mesenchymal stem cells: a potential therapeutic strategy for type 1 diabetes. Diabetes. 2008;57:1759–1767. doi: 10.2337/db08-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tyndall A, Houssiau FA. Mesenchymal stem cells in the treatment of autoimmune diseases. Ann Rheum Dis. 2010;69:1413–1414. doi: 10.1136/ard.2010.132639. [DOI] [PubMed] [Google Scholar]
- 8.Madec AM, Mallone R, Afonso G, et al. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia. 2009;52:1391–1399. doi: 10.1007/s00125-009-1374-z. [DOI] [PubMed] [Google Scholar]
- 9.Fiorina P, Jurewicz M, Augello A, et al. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol. 2009;183:993–1004. doi: 10.4049/jimmunol.0900803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurewicz M, Yang S, Augello A, et al. Congenic mesenchymal stem cell therapy reverses hyperglycemia in experimental type 1 diabetes. Diabetes. 2010;59:3139–3147. doi: 10.2337/db10-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bassi EJ, Moraes-Vieira PM, Moreira-Sa CS, et al. Immune regulatory properties of allogeneic adipose-derived mesenchymal stem cells in the treatment of experimental autoimmune diabetes. Diabetes. 2012;61:2534–2545. doi: 10.2337/db11-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kota DJ, Wiggins LL, Yoon N, et al. TSG-6 produced by hMSCs delays the onset of autoimmune diabetes by suppressing Th1 development and enhancing tolerogenicity. Diabetes. 2013;62:2048–2058. doi: 10.2337/db12-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borg DJ, Weigelt M, Wilhelm C, et al. Mesenchymal stromal cells improve transplanted islet survival and islet function in a syngeneic mouse model. Diabetologia. 2014;57:522–531. doi: 10.1007/s00125-013-3109-4. [DOI] [PubMed] [Google Scholar]
- 14.Lee RH, Seo MJ, Reger RL, et al. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci U S A. 2006;103:17438–17443. doi: 10.1073/pnas.0608249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montane J, Bischoff L, Soukhatcheva G, et al. Prevention of murine autoimmune diabetes by CCL22-mediated Treg recruitment to the pancreatic islets. J Clin Invest. 2011;121:3024–3028. doi: 10.1172/JCI43048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amado LC, Saliaris AP, Schuleri KH, et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci U S A. 2005;102:11474–11479. doi: 10.1073/pnas.0504388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sackstein R. Glycosyltransferase-programmed stereosubstitution (GPS) to create HCELL: engineering a roadmap for cell migration. Immunol Rev. 2009;230:51–74. doi: 10.1111/j.1600-065X.2009.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bevilacqua MP, Stengelin S, Gimbrone MA, Jr, Seed B. Endothelial leukocyte adhesion molecule 1: an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160–5. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- 19.Sackstein R, Merzaban JS, Cain DW, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 20.Kang SK, Shin IS, Ko MS, et al. Journey of mesenchymal stem cells for homing: strategies to enhance efficacy and safety of stem cell therapy. Stem Cells Int. 2012 doi: 10.1155/2012/342968. Article ID 342968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Haddad N, Heathcote D, Moore R, et al. Mesenchymal stem cells express serine protease inhibitor to evade the host immune response. Blood. 2011;117:1176–1183. doi: 10.1182/blood-2010-06-287979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee RH, Kim B, Choi I, et al. Characterization and expression analysis of mesenchymal stem cells from human bone marrow and adipose tissue. Cell Physiol Biochem. 2004;14:311–324. doi: 10.1159/000080341. [DOI] [PubMed] [Google Scholar]
- 23.Stranford S, Ruddle NH. Follicular dendritic cells, conduits, lymphatic vessels, and high endothelial venules in tertiary lymphoid organs: Parallels with lymph node stroma. Front Immunol. 2012;3:350. doi: 10.3389/fimmu.2012.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korpos E, Kadri N, Kappelhoff R, et al. The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes. 2013;62:531–542. doi: 10.2337/db12-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dazzi F, Lopes L, Weng L. Mesenchymal stromal cells: a key player in ‘innate tolerance’? Immunology. 2012;137:206–213. doi: 10.1111/j.1365-2567.2012.03621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–36. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 27.Thankamony SP, Sackstein R. Enforced hematopoietic cell E- and L-selectin ligand (HCELL) expression primes transendothelial migration of human mesenchymal stem cells. Proc Natl Acad Sci U S A. 2011;108:2258–2263. doi: 10.1073/pnas.1018064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu H, Mitsuhashi N, Klein A, et al. The role of the hyaluronan receptor CD44 in mesenchymal stem cell migration in the extracellular matrix. Stem Cells. 2006;24:928–935. doi: 10.1634/stemcells.2005-0186. [DOI] [PubMed] [Google Scholar]
- 29.Ding Y, Xu D, Feng G, et al. Mesenchymal stem cells prevent the rejection of fully allogenic islet grafts by the immunosuppressive activity of matrix metalloproteinase-2 and -9. Diabetes. 2009;58:1797–1806. doi: 10.2337/db09-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berman DM, Willman MA, Han D, et al. Mesenchymal stem cells enhance allogeneic islet engraftment in nonhuman primates. Diabetes. 2010;59:2558–2568. doi: 10.2337/db10-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeung TY, Seeberger KL, Kin T, et al. Human mesenchymal stem cells protect human islets from pro-inflammatory cytokines. PLoS ONE. 2012;7:e38189. doi: 10.1371/journal.pone.0038189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Characterization of bone marrow derived MSC. MSC were differentiated into osteocytes, adipocytes and chondrocytes (X200 magnification).
Supplemental Figure 2. FTVI modification of CD44−/− MSC results in similar increase in HECA452 reactivity as WT MSC. Unmodified MSC (dotted line) show no reactivity with HECA452, while FTVI-modified MSC (solid line) and FTVI-modified CD44KO MSC (filled grey) showed similar levels of staining with HECA452, indicating that exofucosylation creates sialofucosylated epitopes on alternative (i.e., non-CD44) scaffolds in CD44KO MSC.