

Abstract
The response of cardiac muscle to an insult such as myocardial infarction includes changes in the expression of numerous signaling proteins and modulation of gene expression, as well as post-translational modifications of existing proteins. Most studies to date have defined these in end-stage cardiac muscle thus obviating consideration of the temporal progression that causes the heart to transition from a compensated to a decompensated phenotype. To explore these transitions, we examined contractile protein biochemistry in a mouse MI model at two early time points: two days and two weeks post infarct and at two later time points: two and four months post infarct. Phosphorylation of myofilament proteins was analyzed using phosphospecific staining of polyacrylamide gels, and whenever possible, phosphospecific antibodies. Phosphorylation of myosin binding protein c, the myosin regulatory light chain and troponin I were all decreased relative to sham operated animals at both early time points. However, by 2 months, total phosphorylation of all the major myofilament proteins normalized and at both 2 and 4 months, there was a significant increase in troponin I phosphorylation. One-dimensional IEF of troponin I coupled with phospho-specific antibody analysis demonstrated a redistribution of phosphorylation sites with a significant initial decline at the putative PKA sites, Serine 22,23, and a subsequent increase at the putative PKC site, serine 43,45. These data suggest that temporal changes in myofilament protein phosphorylation contribute both to the initial compensatory hyperdynamic response to myocardial infarction and subsequently to the gradual progression to myocardial failure.
Keywords: myocardial infarction, myocardium, proteins, signal transduction, phosphorylation
Introduction
Myocardial infarction is the major cause of heart failure in Western society. Acutely, the loss of functional cardiac muscle causes decreased cardiac output, arrhythmias, increased loading of the remaining viable myocardium, release of inflammatory mediators and the consequent activation of compensatory pathways. Compensatory pathways include neurohumoral responses to restore blood pressure, altered myocardial protein expression, altered myofilament protein phosphorylation and altered myofilament function, all of which have characteristic time domains -- the phosphorylation of proteins can happen in seconds, but significant changes in protein concentration requires days. We believe this results in sequential and sometimes transient activation of diverse pathways to produce and maintain a specific functional compensation. Consequently a full understanding of the compensatory responses to myocardial infarction requires mapping temporal responses in multiple pathways as the mechanical responses progress. In this study, we have focused on the time course of myofilament protein phosphorylation.
Previous work in this area has produced varying results. A study by Li et al [1] examined myocytes from a viable portion of an infarcted rat heart 7 days after the infarction. At 7 days, cardiac systole and diastole remained markedly impaired by the infarction. At the myocyte level they described an increased resting tension and a decrease in calcium sensitivity although there was no change in the maximum force. The phosphorylation of TnI was increased consistent with the decrease in pCa50. In a similar experiment, van der Velden [2] looked at porcine myocyte function 21 days after the infarct when the cardiac output and arterial pressure had been restored to normal levels. They observed an increase in calcium sensitivity and a reduction in maximum force. The change in calcium sensitivity was consistent with a decrease in TnI phosphorylation. While the differences could be due in part to species differences and strategies of sample collection, it is certainly plausible that these differing TnI phosphorylation findings could represent discrete points in a time course of compensatory change or that they may reflect differences in site-specific phosphorylation of troponin I.
Troponin I (TnI) is the inhibitory unit of the troponin complex and inhibits actomyosin interactions at diastolic levels of intracellular calcium and is the target of PKA, PKC, PKG (and likely others) mediated phosphorylation, most of which have overlapping targets within the molecule. Phosphorylation of TnI at serine 22,23 in the unique amino-terminal end molecule [3-5] decreases the calcium sensitivity of the sarcomere, promotes calcium dissociation from troponin C and by extension enhances rates of cross-bridge cycling and diastolic relaxation [5, 6]. Historically, this site has been felt to be PKA dependent however recent data have also shown that it is susceptible to PKCα, PKCβ, and PKG dependent phosphorylation. Moreover, phosphoantibodies directed against this site have shown a high degree (>70%) of in situ phosphorylation. There are at least three other potential kinase dependent sites in the molecule (largely presumed to be PKC specific), at Ser 43,45 and Thr 144 [7-9]. Studies using reconstituted fibers and mutational analysis have shown that PKC phosphorylation of TnI (largely at Ser 43,45) inhibits the actin-cross bridge reaction and reduces the Ca++ dependent actomyosin ATPase rate as well as the calcium sensitivity of force generation [3, 8, 10]. Phosphorylation at Thr 144 (mediated by several PKC isoforms) reduces maximal tension development and cross-bridge cycling rates [11]. Despite relatively clear in vitro data, establishing the physiologic significance of these effects in vivo has been difficult [12]. Moreover, the sites clearly have some interdependence. For example, we have characterized a murine model in which the Ser 43,45 site was mutated to alanines [13-16] rendering them non-phosphorylatable. These animals show enhanced in vivo contractility and a blunted reduction of maximal tension following phorbol treatment that reflects not only loss of the PKC effect at S43, 45 but also the superimposition of the impact of enhanced phosphorylation of the Ser22, 23 site.
Phosphorylation of troponin T (TnT) in the Ca2+-sensitive C-terminal region results in a decrease in the Mg2+-ATPase activity [4], a decrease in Ca2+-sensitivity[11] and a decrease in the affinity of TnT for both tropomyosin and F-actin [17]. Phosphorylation of MyBP-C has been demonstrated at numerous sites [18] and has been shown to result in structural movement of the myofilament, bringing the thick filament into closer proximity with the thin filament [19]. Increased phosphorylation of MyBP-C results in acceleration of the kinetics of force development [20] whereas transgenic mice expressing non-phosphorylatable MyBP-C have a decrease in contractility and a propensity toward cardiac hypertrophy [21].
The function of myosin light chain phosphorylation is even less well understood although a central role for the regulatory light chain (MLC2) in the regulation of cross-bridge cycling is increasingly acknowledged. Morano et al [22] have shown that alterations in the myosin light chain-actin interaction have a profound effect upon tension development and calcium responsiveness in isolated human myocardium. Likewise, MLC2 extraction from skinned myocardium results in decreased velocity of shortening as well as decreased calcium sensitivity [23]. Phosphorylation of MLC2 increases myofilament calcium sensitivity (and transgenic replacement with a non-phosphorylatable MLC2 isoform eliminates the MLCK effect on the tension:calcium relationship) with only a modest effect on tension development [24]. It has also been shown that the gradient of tension development across the myocardium correlates with an analogous gradient of MLC2 phosphorylation [25]. Importantly, our group and others have published that ablation of MLC2 phosphorylation is associated with a reduction in calcium activated tension development (but not calcium sensitivity) and also with a marked reduction in phosphorylation of other contractile proteins including TnI and MyBP-C [26].
Therefore, the aim of this study was to carefully define the temporal changes in protein phosphorylation of the major myofilament proteins following a myocardial infarct and to correlate those changes with changes in contractile function and ventricular remodeling. The results of this study identify changes in the phosphorylation state of MyBP-C, MLC-2 and TnI that are temporally regulated and may, in combination, represent a “phosphofingerprint” that can be correlated with degree and stage of cardiac dysfunction.
Materials and methods
Surgical Induction of Myocardial Infarction in Mice
Eleven week-old C57Bl/6J mice were anesthetized with 2,2,2-tribromoethanol, intubated, and mechanically ventilated with 90% O2. The heart was accessed via an intercostal thoracotomy and the left anterior descending coronary artery (LAD) was permanently occluded by tying off a 7-0 suture passed under the artery. The chest was closed and the mouse removed from the ventilator. The mouse was allowed to recover on a warmed surface, with supplemental oxygen delivered through a nose-cone. Sham-operated animals went through all procedures described, except actual occlusion of the LAD. Ischemia was verified by three-lead electrocardiograms (ECGs), which were obtained pre-operatively and immediately following the end of the surgical procedure. Post-surgical pain was controlled with buprenorphine for the first 48 hrs following surgery and acetaminophen for 7 days.
Echocardiography
Cardiac function was assessed by 2D-transthoracic echocardiography (echo) using a VisualSonics Vevo 770 high resolution ultrasound imager equipped with a 35 Mhz transducer. The mice were lightly sedated with isoflurane and heart rates were maintained above 500 beats/min. Parasternal long axis and multiple short axis B-mode videos and M-modes images (at the level of the mid-papillary short axis) were routinely acquired. Echo images were obtained on the mice 2 days post surgery to determine infarct size and then again immediately prior to animal sacrifice (2 days, 2 weeks, 2 months or 4 months). Infarct size was determined by wall motion score index (Zhang, et al. 2007) and by tracing the infarct during diastole in the B-mode long axis view. Determinations of infarct size, fractional shortening (FS), and ejection fraction (EF) were performed off-line in a blinded mode.
Tissue Harvest
After collecting the final echo data, mice were given an overdose of pentobarbitol and the chest cavity was immediately opened. The apex of the heart was snipped in situ and immediately frozen in liquid nitrogen. The heart was then removed from the chest and the atria and right ventricle were removed and frozen in liquid nitrogen. The infarcted tissue and tissue immediately surrounding the infarct zone was carefully removed and the rest of the left ventricle was rapidly frozen in liquid nitrogen. All samples were stored in liquid nitrogen until analysis.
Gel Electrophoresis
Small samples of the left ventricle were homogenized in 8 M urea, 2.5 M thiourea, 4% CHAPS, 10 mM EDTA and a cocktail of protease and phosphatase inhibitors. For quantification of phosphorylation, samples were separated by 12% SDS-PAGE, fixed and stained with ProQ Diamond Phosphoprotein Gel Stain (Invitrogen). After destaining, gels were imaged using a Typhoon 9410 Gel Imager (GE Lifesciences). Gels were rinsed with water and stained with BioSafe Coomassie Blue (BioRad) for detection of total proteins. Phosphorylation was calculated by dividing the PQD signal for each protein of interest by the CBB signal for the essential myosin light chain (MLC1). We used MLC1 as a normalization factor since there was no change in either quantity or phosphorylation of this protein.
For determination of redistribution of phosphorylation of Troponin I, we used non-equilibrium isoelectric focusing gel electrophoresis as previously described [27, 28]. Briefly, homogenates were separated on urea gels consisting of 4% total acrylamide and a 1:4 mixture of 3-10 and 7-9 ampholytes. The cathode and anode buffers were reversed and the gels were run 1 hour at 100 V, 2 hours at 200 V and 20 min at 500 V. Proteins were then transferred to PVDF and the membranes were blocked in 5% BSA for 1 hour, rinsed with TBST and incubated in the appropriate antibody overnight at 4° C. Membranes were washed and incubated with secondary antibody for one hour at room temperature. After washing, the proteins were visualized using enhanced chemiluminescence.
For separation of alpha & beta myosin, samples were run by modified 6% SDS-PAGE (separating acrylamide/bis ratio 1:100; resolving gel buffer pH 9.0; running gel buffer pH 8.2; (β-mercaptoethanol 600 ∝L/L inner gel buffer). Gels were run overnight at 4° C and stained with BioSafe Coomassie Blue protein stain.
Materials
Antibodies: anti-TnI (total); Fitzgerald, Inc 1:2500, 2° anti-mouse, Sigma 1:10,000: anti-pS22/23 Troponin I; Cell Signaling 1:1000, 2° anti-mouse, Sigma 1:10,000.
Statistical Analysis
All data are expressed as mean ± SEM. Comparisons between groups (sham vs. MI) at individual time points were made using Student's t test. Differences in the time course of phosphorylation were assessed by testing for interaction between time and treatment in a multiple regression model. The R statistical language (version 2.4) was used for all statistical tests.
Results
Contractile and Hemodynamic Properties of the Mouse Myocardium
There were no significant differences in the average infarct size amongst the animals in each group. The average infarct sizes in each cohort (2-days, 2-weeks, 2-mo and 4-mo) were 40±5%, 39±5%, 38±3% and 39±7 respectively. Table 1 illustrates changes in ejection fraction in mice following myocardial infarction. Following MI there was a steep decline in ejection fraction at 2-days post MI with a slower, progressive decline in function for the remaining time period. Between 0-2 days, mortality rate was 26% (11% of these deaths were due to surgical complications); between 2 days and 2 weeks, mortality rate was 15% and between 2 weeks and 4 months, mortality rate was 12.5%. At 4-months post MI, only 2 animals remained alive.
Table 1.
Hemodynamic Characteristics of Mouse MI model
| Ejection Fraction | ||||
|---|---|---|---|---|
| 2 days | 2 wks | 2 mo | 4 mo | |
| Sham | 63±5 | 60±14 | 71±2 | 70±3 |
| MI | 49±10 | 39±2 | 33±1 | 30±9 |
Phosphorylation of myofilament proteins
Phosphorylation was assessed in sham-operated controls and in the post-MI mice by phosphoprotein staining of 1-dimensional SDS-PAGE gels of heart tissue extracts at each of the time points (Figure 1). At 2-days post infarct, phosphorylation of myosin-binding protein C (MyBP-C), troponin-I (TnI) and the regulatory myosin light chain (MLC-2) was significantly reduced compared to sham-operated controls. There were no differences in troponin T or desmin phosphorylation. At 14 days there was a trend toward recovery of phosphorylation in each of the proteins examined and MyBP-C phosphorylation was no longer different from control tissues. Phosphorylation of both TnI and MLC-2 was still significantly lower in MI tissues than in the corresponding sham-operated controls. By 2-months post infarct, there were no longer differences in phosphorylation of MyBP-C or MLC-2 between sham-operated controls and post-infarct tissues; phosphorylation of TnI was significantly elevated compared to sham-operated controls and TnI phosphorylation remained elevated at 4-months post infarct in the MI tissues compared to the sham-operated controls. For TnI, a linear multiple regression model showed a significant interaction between time and treatment (p=0.016) indicating that differences in phosphorylation levels between the MI group and the sham-operated control group were due to significantly different temporal profiles of phosphorylation between the two groups (data not shown). Phosphorylation profiling from a region of the left ventricle more distal to the infarcted area showed qualitatively similar changes in phosphorylation at all time points, suggesting that the phosphorylation changes reflect the mechanical and endocrine stresses related to the infarct rather than focal ischemia.
Figure 1.
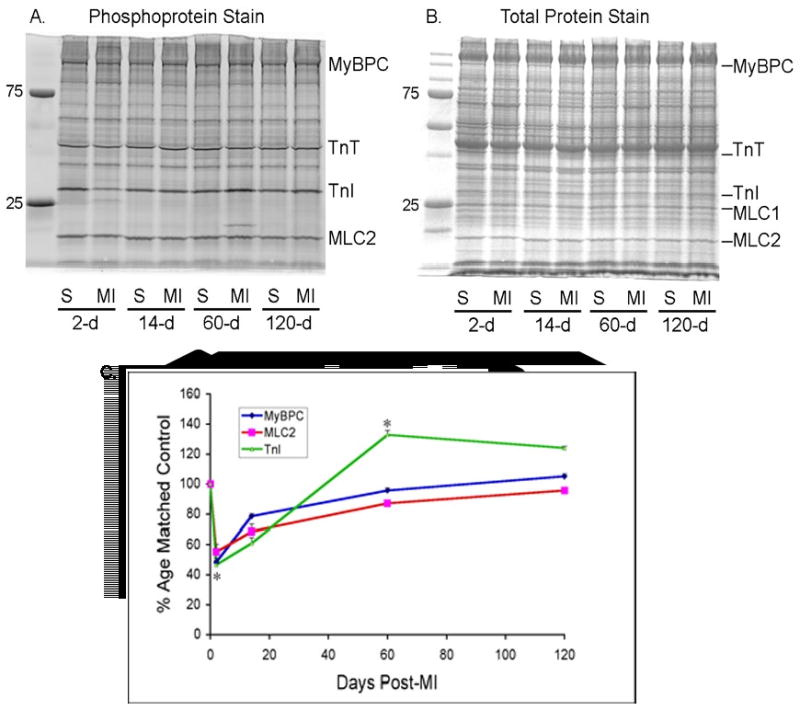
A. Representative SDS-PAGE of sham operated (S) or infarcted animals (MI) at each time point. ProQ Diamond staining demonstrates phosphoprotein signal. B. Coomassie Brilliant Blue staining of the same gel for total protein signal. C. Summary data from sham operated and infarcted mice. Normalized signal was derived by dividing the ProQ Diamond phosphoprotein signal by the total protein (Coomassie blue) signal for the essential myosin light chain and multiplying by 100. Data are expressed as percentage change from matched control. Sham; n=4 at each time point; MI; n=12 (2 days), n=12 (12 days), n=4 (60 days), n=2 (120 days). *P<0.05
Site-specific changes in Troponin I Phosphorylation
Cardiac troponin I can be phosphorylated at a number of distinct sites, each of which has unique effects on contractile function (see Introduction). In order to better understand the consequences of the temporal changes in TnI phosphorylation, we characterized the site-specific changes in TnI phosphorylation. First, using one-dimensional isoelectric focusing where the anode and cathode buffers were reversed [27], we separated the TnI into distinct phosphospecies. In the 2-day post-MI samples, there was a significant increase in the percentage of unphosphorylated TnI, consistent with the phosphorylation data generated with ProQ Diamond signaling. The 1-dimensional IEF gels show that the increase in unphosphorylated TnI results from a decrease in higher level phosphorylation (P4), rather than dephosphorylation of the P2 species. By 2-months post-MI, there is a increase in TnI phosphorylation which resides in the higher level phosphospecies. That is, the amount of P4 TnI is significantly increased, but P2 is not different from control tissues. Using phosphospecific antibodies generated against the three different phosphorylation sites in TnI, the PKA sites (serines 22, 23), and the PKC sites serine43 and threonine143, we found that at 2 days post MI, there was a significant decrease in S22/23 phosphorylation which returned to control levels by 14 days post MI and remained unchanged thereafter. Interestingly, phosphorylation of serine43 was elevated at 2-days post MI and at 14 days, and returned to sham operated control levels by 2 months. Threonine143 phosphorylation was unchanged at every time point (data not shown). Since total TnI phosphorylation is reduced immediately post-MI (Figure 1 and Figure 2), quantitatively the predominant effect is the dephosphorylation of the PKA sites, serine 22/23.
Figure 2.
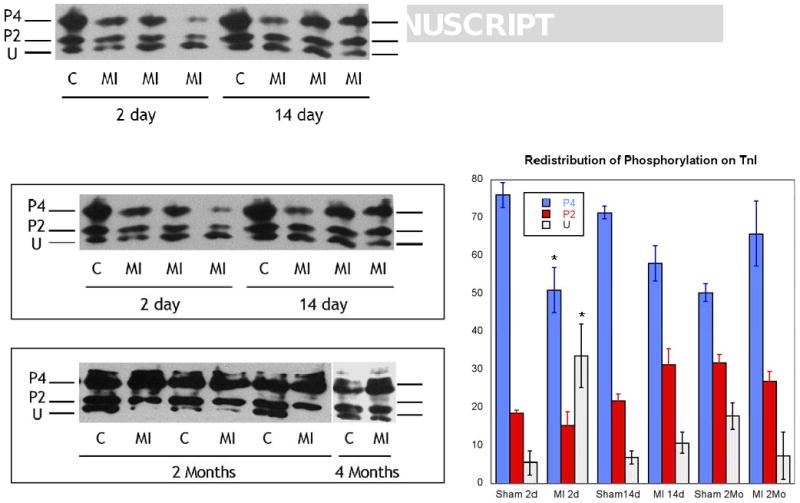
Representative 1-dimensional isoelectric focusing of Troponin I in sham operated control (C) and infracted animals (MI) at 2d, 14d, 2 mo, and 4 mo. The major bands seen in these tissues are represented as U: unphosphorylated TnI; P2: phosphorylated at 2-sites; and P4: phosphorylated at 4-sites. Right panel: Summary of data for each time point. N=4. *P<0.05 compared to paired control.
Alpha-Beta myosin expression
Heart failure has been shown to be associated with an increase in β-myosin expression in rodents and this increase in β-myosin is associated with a decrease in myofibrillar ATPase seen in failing hearts. To examine the potential contribution of myosin isoform switching in our model, we examined the ratio of alpha to beta myosin at each time point, using6% SDS-PAGE gels to separate α & β-myosin. There was no detectable β-myosin in the sham-operated animals at any time point. Consistent with other findings, there was a small shift in myosin isoform expression in our model. That is, two-days post MI, there was no detectable β-myosin, but by 14-days post-MI, β-myosin was increased to 9.8% and at 2-months and 4 months was 10% and 9.5% respectively.
Discussion
The major finding of this study is the demonstration of distinct temporal changes in the phosphorylation of both the regulatory thin filament and thick filament sarcomeric proteins. Specifically, we found that acutely following MI there was a marked decrease in phosphorylation of the regulatory myosin light chain, myosin binding protein C and troponin I. The phosphorylation of MLC2 and MBP-C recovered to near sham levels by 2 weeks post MI and was not different from sham by 2 months post MI. Similarly, Troponin I phosphorylation was significantly reduced acutely following MI, returning to sham levels by 2 weeks. However, phosphorylation of troponin I at 2 and 4-months was significantly elevated compared to sham operated controls. Phosphospecific antibodies showed that the dephosphorylation of troponin I was at the PKA sites serine 22 & serine 23 whereas the subsequent increase in phosphorylation was at the PKC serine 43 site.
Enhanced β-adrenergic stimulation following MI may be expected to have 2 seemingly opposing effects; 1) enhanced calcium handling mediated by phospholamban and 2) decreased calcium sensitivity of the myofilament by phosphorylation of serine 22 and 23 of troponin I. However, we describe decreased phosphorylation of serine 22/23, almost certainly due to preferential activation of phosphatase activity which targets these sites[29]. Dephosphorylation of troponin I at serines 22 and 23, results in an increase in calcium sensitivity of the myofilament, which presumably would allow the β-adrenergic stimulation seen immediately post MI to enhance calcium handling and contractility simultaneously as an immediate compensatory response to muscle loss. However, it is possible that early down regulation of β1-adrenergic receptor density and/or receptor uncoupling [30, 31] may contribute to the phosphorylation changes seen immediately post-MI and that the persistent decrement in β1-adrenergic receptor function (proportional to infarct size [32]) in the absence of pharmacotherapy may partially account for the shift from PKA to PKC dependent phosphorylation.
Additionally, we determined that the phosphorylation of the PKC site, serine 43 is increased immediately following MI and returns to sham levels by 2 months. Walker and colleagues recently showed that phosphorylation of isolated skinned cardiac myocytes with PKC-βII resulted in an increase in calcium sensitivity that correlated with an increase in troponin I phosphorylation [33]. However, the increase in calcium sensitivity was only seen in a mouse line that had the PKA sites, serine 22 & 23, mutated to alanines, suggesting that phosphorylation at serine 22 & 23 masked the effect of phosphorylation by PKC-βII. These investigators suggested that it was most likely that the PKC-βII phosphorylation of TnI was at threonine-144 however they were not able to unequivocally exclude serine 43 or 45 as the relevant site of phosphorylation mediating the increase in calcium sensitivity. Taken together, our data demonstrating both a decrease in serine22/23 phosphorylation and an increase in serine43 phosphorylation at early time points following MI suggest that regulation of troponin I phosphorylation is a dynamic and sensitive way of increasing contractility immediately following injury.
Functionally, decreases in myosin light chain 2 phosphorylation are associated with decreases in contractility as Morano and colleagues [22] have shown that MLC2 dephosphorylation has profound effects on both tension development and calcium responsiveness in the myocardium. Additionally, our group and others have published that ablation of MLC2 phosphorylation is associated with a reduction in ventricular ejection time and calcium activated tension development (but not calcium sensitivity) and also with a marked reduction in phosphorylation of other contractile proteins including TnI and MyBP-C [34]. Moreover, we have shown that phosphorylation of MLC2 is enhanced by dobutamine treatment (other adrenergic agonists were not tested) in isolated muscle. These data suggest a potential role for MLC2 phosphorylation in the regulation of tension development and myofilament calcium sensitivity and the decrease in MLC2 phosphorylation may, in part, contribute to the initial steep decline in contractile function.
Recent studies suggest that the phosphorylation of myosin binding protein C may play a major role in regulating both the function and the structure of the thick filament. MyBP-C can be phosphorylated by PKA [35, 36], Ca2+-camodulun-dependent kinase [36, 37] and protein kinase C [38, 39]. Phosphorylation is associated with increases in calcium sensitization of the thick filament, while decreases in phosphorylation are associated with decreases in contractility [21], and it has been suggested that phosphorylation of MyBP-C is necessary for normal cardiac function [40]. Furthermore, phosphorylation of MyBP-C accelerates the kinetics of force development [20] and dephosphorylation of MyBP-C immediately post-MI likely contributes to the initial rapid decline in contractile function.
Chronically, the changes in regulatory protein phosphorylation evolve following the acute insult, so that phosphorylation of MLC2 and MyBP-C return to pre-infarction levels whereas phosphorylation of TnI increases, driven predominately by an increase in the PKC dependent site, Ser43,45, which functionally decreases the calcium sensitivity of contraction. Mechanical performance of the heart and of the non-infarcted myocardium in particular declines over this interval a finding which is consistent with previous animal studies from our lab and others showing that PKC dependent phosphorylation of TnI is associated with reduced sarcomeric calcium sensitivity and maximal tension development and in transgenic animals with the development of a dilated cardiomyopathy.
The contribution of myosin heavy chain gene and protein expression to cardiac muscle dysfunction in rodent models (and in particular the α to β isoform switch) has been extensively reviewed [41-43]. However, the increase in β-myosin heavy chain is small and constant after the initial switch at 2 weeks and it seems likely that cross bridge cycling and ATPase activity in this context are more strongly influenced by changes in the regulatory elements described above than by MHC.
Taken together these data underscore the dynamic and complex post-translational responses of the sarcomeric regulatory proteins that contribute to ventricular remodeling in response to an acquired insult such as myocardial infarction. These would appear to reflect not only a balance between kinase and phosphatase activation, the later dominating the acute response to injury, but also a balance amongst various kinase activities which appears to dominate the chronic (mal) adaptation seen as overall contractility declines and the heart remodels. In this later circumstance it would appear as if PKC (and perhaps CAMK) activity is more potent that PKA in defining the dilated, hypocontractile phenotype. How the various post-translational modifications of the regulatory proteins cooperatively interact to define overall sarcomeric dynamics remains to be defined, but regardless it does appear as if a sarcomeric “phosphofingerprint” can be defined that establishes stage specific responses to a pathologic insult.
Figure 3.
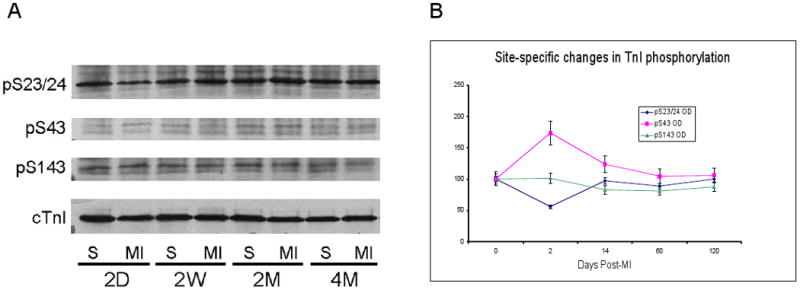
Western blots of phosphorylated troponin I. Samples were separated on 12.5% SDS-PAGE and probed with antibodies against A: phospho-serine 22/23, phosphoserine43, phosphoS143; and cTnI (for total protein). Panel B: relative changes in phosphorylation at each site, compared to matched control. N=4 (2, 14, 60 days); N=2 (120 days).
Figure 4.
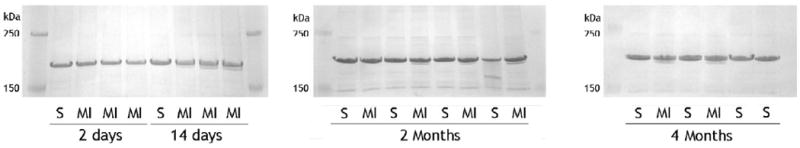
SDS-PAGE separation of α/β myosin in sham operated (S) or infracted (MI) animals at each time point. Extracts were separated using 6% SDS-PAGE and gels were stained with colloidal Coomassie blue.
Acknowledgments
Funding Sources: This work was supported by NIH grants HL-62426 and the Temple-Hoyne Buell Endowment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li P, Hofmann PA, Li B, Malhotra A, Cheng W, Sonnenblick EH, et al. Myocardial infarction alters myofilament calcium sensitivity and mechanical behavior of myocytes. Am J Physiol. 1997 Jan;272(1 Pt 2):H360–70. doi: 10.1152/ajpheart.1997.272.1.H360. [DOI] [PubMed] [Google Scholar]
- 2.van der Velden J, Merkus D, Klarenbeek BR, James AT, Boontje NM, Dekkers DH, et al. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res. 2004 Nov 26;95(11):e85–95. doi: 10.1161/01.RES.0000149531.02904.09. [DOI] [PubMed] [Google Scholar]
- 3.Noland TA, Jr, Guo X, Raynor RL, Jideama NM, Averyhart-Fullard V, Solaro RJ, et al. Cardiac troponin I mutants. Phosphorylation by protein kinases C and A and regulation of Ca(2+)-stimulated MgATPase of reconstituted actomyosin S-1. J Biol Chem. 1995 Oct 27;270(43):25445–54. doi: 10.1074/jbc.270.43.25445. [DOI] [PubMed] [Google Scholar]
- 4.Noland TA, Jr, Raynor RL, Kuo JF. Identification of sites phosphorylated in bovine cardiac troponin I and troponin T by protein kinase C and comparative substrate activity of synthetic peptides containing the phosphorylation sites. J Biol Chem. 1989 Dec 5;264(34):20778–85. [PubMed] [Google Scholar]
- 5.Solaro RJ. Modulation of cardiac myofilament activity by protein phosphorylation. In: Page EFH, Solaro RJ, editors. Handbook of Physiology: The Heart. New York: Oxfor University Press; 2002. pp. 264–300. [Google Scholar]
- 6.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997 Sep 2;96(5):1495–500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- 7.Jideama NM, Noland TA, Jr, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, et al. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem. 1996 Sep 20;271(38):23277–83. doi: 10.1074/jbc.271.38.23277. [DOI] [PubMed] [Google Scholar]
- 8.Noland TA, Jr, Raynor RL, Jideama NM, Guo X, Kazanietz MG, Blumberg PM, et al. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996 Nov 26;35(47):14923–31. doi: 10.1021/bi9616357. [DOI] [PubMed] [Google Scholar]
- 9.Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, et al. Tyrosine phosphorylation modifies protein kinase C delta-dependent phosphorylation of cardiac troponin I. J Biol Chem. 2008 Aug 15;283(33):22680–9. doi: 10.1074/jbc.M802396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noland TA, Jr, Kuo JF. Protein kinase C phosphorylation of cardiac troponin I and troponin T inhibits Ca(2+)-stimulated MgATPase activity in reconstituted actomyosin and isolated myofibrils, and decreases actin-myosin interactions. J Mol Cell Cardiol. 1993 Jan;25(1):53–65. doi: 10.1006/jmcc.1993.1007. [DOI] [PubMed] [Google Scholar]
- 11.Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem. 2003 Sep 12;278(37):35135–44. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 12.McDonough JL, Van Eyk JE. Developing the next generation of cardiac markers: disease-induced modifications of troponin I. Prog Cardiovasc Dis. 2004 Nov-Dec;47(3):207–16. doi: 10.1016/j.pcad.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 13.MacGowan GA, Du C, Cowan DB, Stamm C, McGowan FX, Solaro RJ, et al. Ischemic dysfunction in transgenic mice expressing troponin I lacking protein kinase C phosphorylation sites. Am J Physiol Heart Circ Physiol. 2001 Feb;280(2):H835–43. doi: 10.1152/ajpheart.2001.280.2.H835. [DOI] [PubMed] [Google Scholar]
- 14.Montgomery DE, Wolska BM, Pyle WG, Roman BB, Dowell JC, Buttrick PM, et al. alpha-Adrenergic response and myofilament activity in mouse hearts lacking PKC phosphorylation sites on cardiac TnI. Am J Physiol Heart Circ Physiol. 2002 Jun;282(6):H2397–405. doi: 10.1152/ajpheart.00714.2001. [DOI] [PubMed] [Google Scholar]
- 15.Pyle WG, Sumandea MP, Solaro RJ, De Tombe PP. Troponin I serines 43/45 and regulation of cardiac myofilament function. Am J Physiol Heart Circ Physiol. 2002 Sep;283(3):H1215–24. doi: 10.1152/ajpheart.00128.2002. [DOI] [PubMed] [Google Scholar]
- 16.Roman BB, Goldspink PH, Spaite E, Urboniene D, McKinney R, Geenen DL, et al. Inhibition of PKC phosphorylation of cTnI improves cardiac performance in vivo. Am J Physiol Heart Circ Physiol. 2004 Jun;286(6):H2089–95. doi: 10.1152/ajpheart.00582.2003. [DOI] [PubMed] [Google Scholar]
- 17.Noland TA, Jr, Kuo JF. Protein kinase C phosphorylation of cardiac troponin T decreases Ca(2+)-dependent actomyosin MgATPase activity and troponin T binding to tropomyosin-F-actin complex. Biochem J. 1992 Nov 15;288(Pt 1):123–9. doi: 10.1042/bj2880123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlender KK, Bean LJ. Phosphorylation of chicken cardiac C-protein by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 1991 Feb 15;266(5):2811–7. [PubMed] [Google Scholar]
- 19.Weisberg A, Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc Natl Acad Sci U S A. 1996 Aug 20;93(17):8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stelzer JE, Patel JR, Walker JW, Moss RL. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res. 2007 Aug 31;101(5):503–11. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- 21.Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, et al. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res. 2005 Nov 25;97(11):1156–63. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morano I, Ritter O, Bonz A, Timek T, Vahl CF, Michel G. Myosin light chain-actin interaction regulates cardiac contractility. Circ Res. 1995 May;76(5):720–5. doi: 10.1161/01.res.76.5.720. [DOI] [PubMed] [Google Scholar]
- 23.Morano I. Effects of different expression and posttranslational modifications of myosin light chains on contractility of skinned human cardiac fibers. Basic Res Cardiol. 1992;87(1):129–41. doi: 10.1007/978-3-642-72474-9_11. [DOI] [PubMed] [Google Scholar]
- 24.Sanbe A, Fewell JG, Gulick J, Osinska H, Lorenz J, Hall DG, et al. Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J Biol Chem. 1999 Jul 23;274(30):21085–94. doi: 10.1074/jbc.274.30.21085. [DOI] [PubMed] [Google Scholar]
- 25.Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, et al. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001 Nov 30;107(5):631–41. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 26.Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, et al. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem. 2009 Feb 20;284(8):5097–106. doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi T, Yang X, Walker LA, Van Breemen RB, Solaro RJ. A non-equilibrium isoelectric focusing method to determine states of phosphorylation of cardiac troponin I: identification of Ser-23 and Ser-24 as significant sites of phosphorylation by protein kinase C. J Mol Cell Cardiol. 2005 Jan;38(1):213–8. doi: 10.1016/j.yjmcc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 28.Scruggs SB, Walker LA, Lyu T, Geenen DL, Solaro RJ, Buttrick PM, et al. Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCepsilon phosphorylation. J Mol Cell Cardiol. 2006 Apr;40(4):465–73. doi: 10.1016/j.yjmcc.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Jideama NM, Crawford BH, Hussain AK, Raynor RL. Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T. Int J Biol Sci. 2006;2(1):1–9. doi: 10.7150/ijbs.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinberg SF, Zhang H, Pak E, Pagnotta G, Boyden PA. Characteristics of the beta-adrenergic receptor complex in the epicardial border zone of the 5-day infarcted canine heart. Circulation. 1995 Jun 1;91(11):2824–33. doi: 10.1161/01.cir.91.11.2824. [DOI] [PubMed] [Google Scholar]
- 31.Vleeming W, van der Wouw PA, te Biesebeek JD, van Rooij HH, Wemer J, Porsius AJ. Density of beta adrenoceptors in rat heart and lymphocytes 48 hours and 7 days after acute myocardial infarction. Cardiovasc Res. 1989 Oct;23(10):859–66. [PubMed] [Google Scholar]
- 32.Sanbe A, Takeo S. Diminished responsiveness to cardiac beta 1-adrenoceptor agonists in rats with chronic heart failure following myocardial infarction. Biol Pharm Bull. 1995 Oct;18(10):1362–6. doi: 10.1248/bpb.18.1362. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Grant JE, Doede CM, Sadayappan S, Robbins J, Walker JW. PKC-betaII sensitizes cardiac myofilaments to Ca2+ by phosphorylating troponin I on threonine-144. J Mol Cell Cardiol. 2006 Nov;41(5):823–33. doi: 10.1016/j.yjmcc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, et al. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem. 2008 Dec 23; doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garvey JL, Kranias EG, Solaro RJ. Phosphorylation of C-protein, troponin I and phospholamban in isolated rabbit hearts. Biochem J. 1988 Feb 1;249(3):709–14. doi: 10.1042/bj2490709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction. EMBO J. 1995 May 1;14(9):1952–60. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartzell HC, Glass DB. Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. J Biol Chem. 1984 Dec 25;259(24):15587–96. [PubMed] [Google Scholar]
- 38.Mohamed AS, Dignam JD, Schlender KK. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys. 1998 Oct 15;358(2):313–9. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 39.Xiao L, Zhao Q, Du Y, Yuan C, Solaro RJ, Buttrick PM. PKCepsilon increases phosphorylation of the cardiac myosin binding protein C at serine 302 both in vitro and in vivo. Biochemistry. 2007 Jun 12;46(23):7054–61. doi: 10.1021/bi700467k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Dijk SJ, Dooijes D, dos Reedios C, Michels M, Lamers JM, Winegrad S, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009 Mar 24;119(11):1473–83. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 41.Pette D, Staron RS. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech. 2000 Sep 15;50(6):500–9. doi: 10.1002/1097-0029(20000915)50:6<500::AID-JEMT7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 42.de Tombe PP. Congestive heart failure: role of cross-bridge cycle kinetics. Cardiovasc Res. 1998 Dec;40(3):440–3. doi: 10.1016/s0008-6363(98)00247-8. [DOI] [PubMed] [Google Scholar]
- 43.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999 Jan;79(1):215–62. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]