

Many physiological and behavioral events exhibit circadian rhythms, which are driven by internal circadian “clocks” that coordinate biological functions through the cyclic expression of at least 10 to 20% of the genes in any given tissue (1). The robustness of circadian rhythms deteriorates with age, and circadian perturbation results in the development of disorders such as diabetes, obesity, and brain dysfunction. Central to the mammalian clock is the complex of transcriptional regulatory proteins CLOCK and BMAL1 (2). A recent study by Masri et al. (3) proposes that SIRT1 and SIRT6, two sirtuin family members with nicotinamide adenine dinucleotide (NAD+)–dependent deacetylase activity, regulate different facets of the CLOCK-BMAL1 network, and more surprisingly, control distinct classes of hepatic circadian genes. Partitioning circadian transcription by sirtuins suggests that in response to internal and external stimuli, circadian clocks selectively control sirtuin-dependent functions that are broadly associated with metabolism, stress resistance, inflammation, aging, and tissue regeneration, to provide organisms with plasticity to adapt to changing environments.
The molecular basis of circadian rhythms is a transcriptional-translational feedback loop (2). The CLOCK-BMAL1 complex induces the expression of a number of genes, including the negative regulators of CLOCK-BMAL1. CLOCK has acetyltransferase activity toward BMAL1 and histones (4), implicating chromatin remodeling in regulating circadian transcription. Acetylated BMAL1 appears more stable, and histone acetylation is associated with a relaxed chromatin state that is more permissive to gene transcription in eukaryotic cells. Masri et al. show that SIRT6, a histone deacetylase (5), associates with CLOCK-BMAL1 and reduces their chromatin binding. This finding provides a critical piece in the circadian clock puzzle. CLOCK-BMAL1 induces the expression of the gene Nampt, which encodes an enzyme that catalyzes a rate-limiting step in NAD+ biosynthesis (6, 7). The resulting increase in the cellular NAD+ concentrations activates SIRT1 and SIRT6. The deacetylase activity of SIRT1 counteracts CLOCK to drive the cyclic acetylation/deacetylation of BMAL1 and histones and orchestrate their function along the circadian cycle (8, 9). By contrast, SIRT6 activation reduces chromatin binding of CLOCK-BMAL1, tipping the balance toward the establishment of a repressive chromatin state.
Masri et al. further took a systems biology approach to study hepatic circadian transcription regulated by SIRT1 and SIRT6. Ablation of the genes encoding SIRT1 or SIRT6 specifically in the mouse liver disrupted the expression of a large number of genes whose expression normally oscillates over a 24-hour period. This supports an essential role for these sirtuins in regulating CLOCK-BMAL1 activity. Surprisingly, the absence of hepatic SIRT1 or SIRT6 also caused a widespread oscillatory transcription of genes that was not observed in the livers of wild-type mice. Such large-scale de novo oscillating transcripts can also be triggered by nutritional challenge (10). These findings highlight the existence of numerous molecular pathways that influence circadian clocks, which may serve to systematically reprogram biological functions in a cell in response to changing environments.
A key discovery of Masri et al. is that SIRT1 and SIRT6 regulate distinct classes of circadian genes (see the figure). Comparison of SIRT1- and SIRT6-dependent oscillating transcripts revealed remarkably little overlap. Genomic partitioning by sirtuins has physiological consequences. By integrating high-throughput circadian transcriptomics with circadian metabolomics data, Masri et al. found that SIRT1 and SIRT6 control different classes of circadian metabolites, reflecting their differential regulation of circadian transcription. Whereas SIRT1 preferentially controls peptide and cofactor metabolism, SIRT6 preferentially regulates fatty acid and carbohydrate metabolism.
Figure. Clock control.
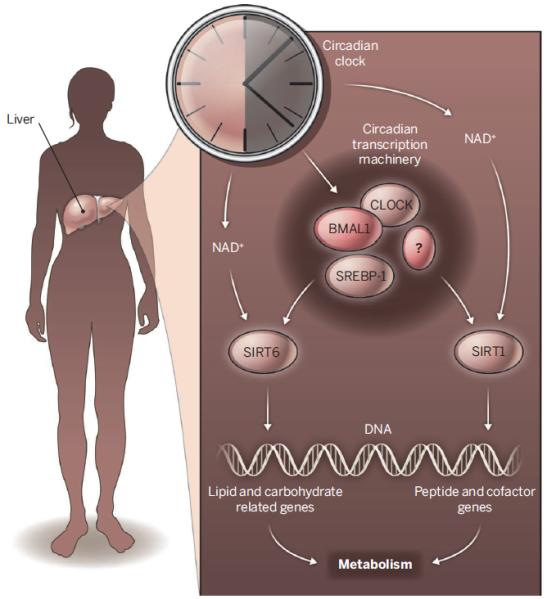
SIRT1 and SIRT6 regulate CLOCK-BMAL1 activity through different mechanisms and control distinct classes of hepatic circadian transcription and metabolic outputs.
How does a deficiency in SIRT6 result in de novo rhythmic expression of a large number of transcripts and their related metabolites? Sterol regulatory element–binding protein 1 (SREBP-1), a transcription factor that controls fatty acid metabolism, may play an essential role. Masri et al. found that SREBP-1 binding sites are highly enriched at the promoters of circadian genes that respond to SIRT6. Circadian recruitment of SREBP-1 to the promoter of its target gene increased in the absence of SIRT6. The livers of SREBP-1–deficient mice displayed disrupted circadian expression of SREBP-1 target genes. How SIRT6 specifically influences the circadian chromatin recruitment of SREBP-1 remains an open question.
The findings by Masri et al. have many important implications. The high-resolution systems approach used in their study, integrating circadian transcriptome and circadian metabolome, contrasts with current metabolic and physiological studies that sample gene expression and metabolites at one nonspecified time point, which may inevitably miss important information and generate inconsistency. The systems approach provides a new framework for future physiological studies. The discovery that circadian genes can be differentially controlled by sirtuins will also initiate further studies into signals that differentially activate sirtuins. This may yield new insights about the reorganization of circadian rhythms by environmental stimuli.
A closer connection between sirtuins and circadian clocks is likely to enrich the understanding of both sirtuin biology and circadian regulation. Elucidating the circadian regulation by SIRT2 and SIRT7, two other sirtuin family members with nuclear localization and important physiological functions (11, 12), may provide insights into the etiology of metabolic diseases and aging.
ACKNOWLEDGMENTS
This work was supported by NIH (AG040990), Ellison Medical Foundation, and American Heart Association.
REFERENCES AND NOTES
- 1.Masri S, Sassone-Corsi P. Nat. Neurosci. 2010;13:1324. doi: 10.1038/nn.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dibner C, Schibler U, Albrecht U. Annu. Rev. Physiol. 2010;72:517. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- 3.Masri S, et al. Cell. 2014;158:659. doi: 10.1016/j.cell.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doi M, Hirayama J, Sassone-Corsi P. Cell. 2006;125:497. doi: 10.1016/j.cell.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 5.Michishita E, et al. Nature. 2008;452:492. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Science. 2009;324:654. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramsey KM, et al. Science. 2009;324:651. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asher G, et al. Cell. 2008;134:317. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 9.Nakahata Y, et al. Cell. 2008;134:329. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckel-Mahan KL, et al. Cell. 2013;155:1464. doi: 10.1016/j.cell.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim HS, et al. Cancer Cell. 2011;20:487. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin J, et al. Cell Reports. 2013;5:654. doi: 10.1016/j.celrep.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]