

Abstract
Ig heavy chain class switching occurs rapidly after activation of mature naïve B cells, resulting in a switch from expressing IgM and IgD to expression of IgG, IgE, or IgA; this switch improves the ability of antibodies to remove the pathogen that induces the humoral immune response. Class switching occurs by a deletional recombination between two different switch (S) regions, each of which is associated with a heavy chain constant (CH) region gene. Class switch recombination (CSR) is instigated by activation-induced cytidine deaminase (AID), which converts cytosines in S regions to uracils. The uracils are subsequently removed by two DNA repair pathways, resulting in mutations, single-strand DNA breaks, and the double-strand breaks required for CSR. We discuss several aspects of CSR, including how CSR is induced, CSR in B-cell progenitors, the roles for transcription and chromosomal looping in CSR, and the roles of certain DNA repair enzymes in CSR.
Introduction
After immunization or infection, activated naïve B cells can switch from expressing IgM and IgD on their surface to expressing IgG, IgE or IgA. This isotype/class switch changes the effector function of the antibody, and improves its ability to eliminate the pathogen that induced the response. Isotype switching involves a replacement of the μ and δ heavy chain constant (CH) regions of the expressed Ig with γ, ε or α CH regions, and occurs by a DNA recombination event termed class switch recombination (CSR). Fig 1 presents a diagram (not to scale) of the CH genes and CSR in mice; human CH genes are similarly arranged although not identical.
Figure 1. Diagram of the mouse IgH genes in naïve mature B cells expressing IgM and IgD, and CSR to IgG2b.
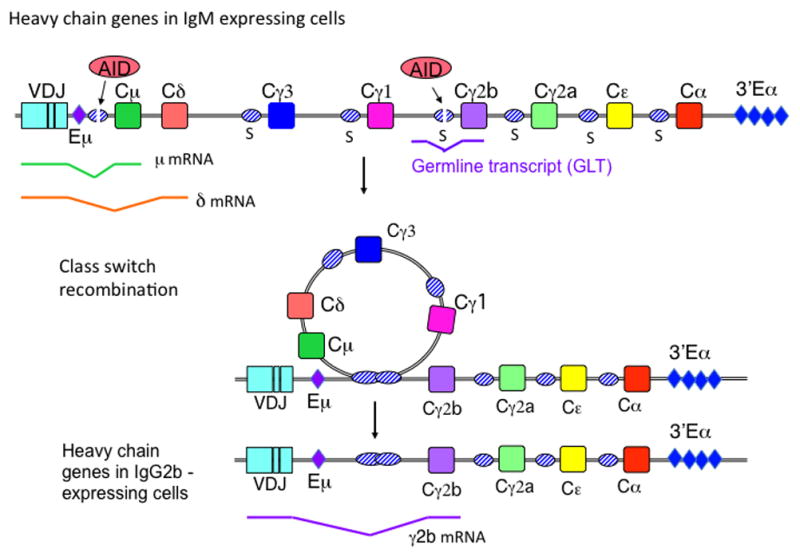
During CSR to IgG2b, AID deaminates the Sμ and Sγ2b regions, instigating DSB formation. The Sμ and Sγ2b regions recombine by an intrachromosomal deletional recombination, which causes the expressed VDJ segment to become associated with the Cγ2b gene. Splicing diagrams of the μ and δ mRNAs, the γ2b germline transcripts (GLTs), and γ2b mRNA are indicated beneath the genes. Eμ and 3′Eα are the two major enhancers that regulate expression of Ig heavy chains and CSR.
CSR is a deletional DNA recombination occurring between switch (S) regions, which are located upstream of all the CH genes except Cδ, and are one to 10 kb in length (1). Recombination occurs between DNA double-strand breaks (DSBs) introduced into the donor Sμ region and a downstream/acceptor S region located from ~65 to 160 kb downstream, although occasionally downstream S regions can subsequently recombine with a S region farther downstream. S regions are G-rich and also have a high density of WGCW (A/T-G-C-A/T) motifs, the preferred target for activation-induced cytidine deaminase (AID), the enzyme that initiates CSR by deaminating cytosines (dC) within S region DNA, converting dC to dU(2, 3). Subsequently, enzymes of the base excision repair (BER) and mismatch repair (MMR) pathways convert the dU’s to DNA double-strand breaks (DSBs), which are required for CSR(4, 5) (Fig 2). The DSBs are subsequently recombined by an end-joining type of DNA recombination, predominantly by non-homologous end-joining (NHEJ). The use of NHEJ rather than homologous recombination is consistent with the facts that S region DSBs are induced and recombined during G1 phase (6–9), and that different S regions do not share long stretches of identity (1), which are required for homologous recombination.
Figure 2. Models for the generation of DNA DSBs during CSR.

(A) Diagram of how the base excision repair (BER) pathway converts AID-induced dUs to DNA breaks. (B) Diagram of a model for how the mismatch repair pathway converts SSBs produced by UNG and APE activity to DSBs appropriate for NHEJ. See text for more information.
CSR occurs very rapidly after infection or immunization, prior to formation of germinal centers, which generally form 7–10 days after exposure to antigen. For example, using mice expressing a transgenic B cell receptor (BCR), both IgM+ and IgG2a+ cells were detected in B cell follicles from days 2–4 after immunization, but only IgG2a+ cells were detected in germinal centers, indicating that CSR occurred prior to germinal center formation (10). Also, CSR was detected in non-transgenic mice 4 days after infection with Salmonella (11). However, CSR is also detected in germinal center B cells from human tonsils (12), and IgA CSR occurs in Peyer’s patch germinal centers (13–15), in which B cells are constantly stimulated by the gut microbiota. CSR also occurs during T-independent responses, which do not induce germinal centers (16). Thus, CSR starts prior to somatic hypermutation (SHM) of variable region [V(D)J] genes, an AID-dependent process which occurs mainly in germinal centers, and which, after selection, can result in antibodies with increased affinity for antigen. The data suggest that CSR may continue as long as B cells are undergoing activation.
Induction of CSR
Many studies of CSR have been performed using cultures of mouse splenic B cells, as these cells can be induced in culture to undergo robust CSR within ~3 days by treatment with the B cell mitogen LPS, acting through the innate receptor TLR4, or by signaling through CD40, the most important receptor for T cell help. LPS alone induces AID expression, but a cytokine, such as IL-4 must also be added to induce AID when antibody to CD40 (αCD40) is used. These ligands also induce cell proliferation, another requirement for CSR(17, 18). Surprisingly, splenic B cells do not require a signal provided by the BCR to switch in culture, as neither LPS nor αCD40 trigger signaling via the BCR. This might be explained by the large amounts of LPS (usually 10–50 μg/ml) used in these cultures, which would not normally be provided to B cells in vivo. Also, in these cultures much more continuous CD40 signaling is provided than normally would be available in vivo. In vivo, CD40 signaling would be delivered to antigen-specific B cells during contacts with antigen-specific TH cells. CD40 signaling is very important for CSR in vivo, as demonstrated by the lack of B cell proliferation and CSR in response to T-dependent antigens in mice and humans deficient in CD40 signaling (19, 20). Also, TLR signaling is important for in vivo immune responses and CSR in response to viruses (21).
Although antigen is not necessary to obtain CSR in culture, addition of low levels (1–100 ng/ml) of soluble hen egg lysozyme (HEL) antigen to LPS- or αCD40-stimulated mouse splenic B cells expressing a HEL-specific BCR increases CSR(22). Increasing the amount of antigen to 1 μg/ml inhibited CSR. Also, low levels (ng amounts) of anti-IgD (anti-δ dextran added to LPS-activated cultures of splenic B cells increases CSR(23–25). The increase in CSR is not due to increased cell proliferation (24, 26). By contrast, addition of large concentrations (10 μg/ml) of anti-IgM to LPS cultures or αCD40 cultures reduces CSR, although it does not inhibit cell proliferation (27, 28). The inhibitory signaling pathway has been studied, and appears to be a feed-back response to extensive BCR cross-linking.
BCR signaling appears to be important for CSR in culture under conditions when signals provided by accessory/secondary signals are limiting. Ligands for the innate receptors TLR1/2, TLR7, and TLR9 have been shown to induce little or no CSR in culture. When anti-δ dextran is added to these cultures, CSR to several isotypes is increased synergistically (24). Likewise, when HEL is added to HEL-specific B cells activated with TLR7 or TLR9, CSR to IgG1 is also greatly increased (22). Unlike mouse B cells, human peripheral blood or tonsillar B cells switch poorly in culture to either CD40 or TLR signaling (29); perhaps activation through BCR signaling would help.
In conclusion, it is likely that CSR in vivo depends upon activation of B cells via their BCR, in addition to secondary signals from CD40 and TLR signaling. It is unclear whether the BCR signaling is required for inducing CSR or whether BCR signaling is required only for initial activation of the B cell and the T cell signals are responsible for inducing CSR. In addition, it appears likely that TLR signaling is important for both T-independent and T–dependent responses in vivo. It is possible that both primary and multiple secondary signals given together increase the robustness of the CSR response.
Regulation of isotype specificity by transcription of unrearranged CH genes
Naïve B cells have the potential to switch to any isotype. Isotype specificity is directed by induction of transcription across S regions, as AID-induced deaminations and CSR are restricted to S regions that are undergoing transcription (5, 30–33). Located upstream of each acceptor S region are transcription promoters, which are activated by cytokines that induce CSR to that specific isotype. Transcription from these promoters produces germline transcripts (GLTs), so-called because they are transcribed from unrearranged genes. GLTs are spliced, as diagrammed in Fig 1 for the γ2bGLT. Due to their unusual high G-content, S region transcripts form R-loops (RNA-DNA hybrids) with the bottom strand DNA, rendering the non-transcribed (top strand) single-stranded (ss)(34–36). The substrate for AID is ss DNA; thus, R-loops cause the top strand to become an extensive AID substrate. However, it is known that AID attacks both the top and bottom strand at S regions nearly equivalently (37). Thus, the R-loop must be removed during CSR to expose the bottom strand to AID.
The R-loop is thought to cause RNA polymerase II (Pol II) to stall during transcription of S regions, resulting in accumulation of Pol II in S regions, although some other feature of S regions might be responsible (38, 39). Interestingly, AID has been demonstrated to be in a complex with Spt5, a protein that associates with Pol II when it is stalled on the DNA template, suggesting that Pol II stalling recruits AID to S regions (40). In addition, stalled Pol II can lead to transcription termination, generating 3′ ends of S region transcripts, which then become substrates for the RNA exosome, a complex which degrades RNA from the 3′ end. The RNA exosome has been shown in biochemical (in vitro) experiments to allow AID to target the bottom strand in transcribed duplex DNA (41), which is otherwise mostly in accessible to AID when associated with newly transcribed RNA. This finding provides an explanation for how AID targets the bottom strand.
These in vitro experiments have been bolstered by in vivo experiments in which CSR was reduced by 50% in cells lacking Nedd4, an E3 ubiquitin ligase. Nedd4 monoubiquitinates paused Pol II, causing an unknown ubiquitin ligase to polyubiquitinate Pol II, leading to degradation of Pol II(42). Pol II degradation leads to transcription termination, creating substrates for the exosome. Interestingly, AID is found associated with polyubiquitinated Pol II, and Nedd4 ubiquitination activity was shown to promote interaction of AID with Spt5 and with the RNA exosome, and to promote binding of the RNA exosome to transcribed S regions. Nedd4 also reduces the level of γ1 GLTs, presumably due to degradation by the exosome. Thus, Nedd4 activity appears to increase the amount of ss bottom strand DNA, and to help recruit AID to S regions. This model has been supported by results indicating that Pol II pausing leads to transcription termination and increased SHM in IgH V genes, dependent on the RNA exosome (43, 44).
GLTs must be spliced to support CSR, although the reason for this is unknown (33, 45–49). As splicing is co-transcriptional, it is possible that splicing factors are involved in recruiting AID to S regions (50), and/or that splicing is required for R-loops to form, perhaps because the RNA must thread back into the DNA to form an R-loop (36). The splicing regulator PTBP2, a protein that binds AID and S region transcripts, is important for efficient CSR. Knockdown of this protein reduces the association of AID to S regions (51, 52). PTBP2 is known to regulate alternative splicing and many aspects of RNA metabolism (53), although its specific role(s) in CSR is not defined. Also, CTNNBL1, a component of the splice some, interacts with AID and is important for both CSR and SHM (50). These interesting studies suggest avenues that might lead to an understanding of why splicing of GLTs is important for CSR, although at this time the role of splicing is not understood.
Regulation of CSR by chromosome looping
Sμ and the acceptor S regions must be in contact in order to recombine. Chromosome-conformation-capture (3C) experiments have shown that in mature naïve splenic B cells prior to CSR, the Eμ intron enhancer (Eμ) and the 3′ Cα enhancer/regulatory region (3′Eα/3′RR), located ~220 kb apart in the genome, are positioned sufficiently near each other in the nucleus to be cross-linked by formaldehyde treatment via proteins bound to them (54–57). Gene targeting experiments have shown that segments of the 3′Eα are essential for CSR to all isotypes (58). This Eμ-3′Eα interaction causes Sμ and the downstream S regions to be located within the same chromosomal loop (Fig 3A). In cells activated to switch, but that have not yet switched to IgG1 by treatment with LPS+ IL-4, the Eμ-Sμ-Cμ and Sγ1-Cγ1 loci are found positioned near each other and the 3′Eα segment (Fig 3B). In cells treated with LPS alone, which induces CSR to IgG3, the Sγ3-Cγ3 locus is associated with Eμ-Sμ-Cμ and with 3′Eα, instead of Sγ1-Cγ1 (54, 55). As LPS+IL-4 induces γ1GLTs, these results suggest that factors that induce transcription from the GLT promoter and/or the GLTs themselves are involved in recruiting the Sγ1-Cγ1 locus to the positions of the Eμ-Sμ-Cμ locus and 3′Eα. Also, it is possible that looping could allow transcriptional activators that bind the enhancers to gain access to GLT promoters, and thus association of a S-CH acceptor locus with the Eμ-Sμ-Cμ locus and with 3′Eα might contribute to transcription or even be required for GL transcription.
Figure 3. Diagrams of models of chromosome looping within the CH gene locus obtained from chromosome conformation capture (3C) assays.

(A) In mature naïve B cells the Eμ and 3′Eα enhancers are interacting. (B) A model for the interactions predicted from 3C assays for splenic B cells induced to switch to IgG1, showing interactions between the two enhancers, and the Em-Sm-Cμ, and Sg1-Cγ1 loci. The S regions are not diagrammed, and the sites of interaction are not precisely known, so the diagram only indicates approximate locations. See text for more explanation.
It is clearly important to identify the proteins regulating loop formation between the Eμ and 3′Eα enhancers and CH genes in B cells before and during CSR. YY-1, a protein that binds both the 3′Eα and Eμ enhancers is a candidate (59, 60). Better evidence is available for PTIP, which has been shown to interact with the B cell specific protein Pax5, and to be important for the interaction between the 3′Eα and the γ1 GLT promoter (61). Mice deficient in PTIP have reduced γ3, γ2b, and γ1GLTs and reduced CSR to these isotypes (62), but it is unknown how much of this effect is due to reduced interaction between the CH loci and the 3′Eα, and how much is due to the fact that PTIP is important for recruiting the histone methyl transferase MLL, which produces the activating histone modification H3K4me3. It is possible that MLL and/or H3K4me3 is important for the looping. CTCF and cohesin, two proteins involved in contraction/looping of the VH and Vκ gene loci (63), do not appear to bind within the 180kb region encoding the CH genes nor to the 3′Eα. Cohesin, however, has been shown to be required for optimum CSR, but the mechanism of its contribution is unknown (64, 65).
CSR in B cell progenitors
Pre-B cells purified from mouse bone marrow express low amounts of AID and have been shown to have ongoing H chain switch recombination (μ>γ2b), despite the fact that they do not express light chains and IgM (66). This was determined by two molecular assays for switch recombination events at the DNA level, detection of RNA transcripts from the excised DNA circles (circle transcripts), and also post-switch transcripts produced by transcription of the recombined genes. In a different study, pro-B cells from Rag1-deficient mice could be induced in culture to switch at the DNA level from μ>γ2b and from μ>ε, although these cells do not express either μ chains or light chains (67). The physiological role of this H chain switching is unknown, although it might contribute to autoimmunity (68).
Interestingly, differing from CSR regulation in mature B cells, μ>γ2b but not μ>γ3 switching occurs in pro-B cells activated with LPS+CD40 ligand (CD40L), and μ>ε but not μ>γ1 switching occurs in cells treated with LPS+CD40L+IL-4(67). GLT expression correlates with CSR, i.e. γ2b and ε GLTs, but not γ3 or γ1 GLTs, are detected in pro-B cells, activated without or with IL-4, respectively. The explanation for this restricted choice of isotypes appears to be that pro-B cells have a chromosomal loop between the Cγ3 and Cγ1 loci that sequesters these genes away from the Eμ and 3′Eα enhancers (67). This loop was not detected in mature splenic B cells. Likewise, ex vivo bone marrow pre-B cells from C57BL/6 mice switch to γ2b but not to γ3; surprisingly, however, BALB/c pre-B cells switch to γ2b and γ3(66). The pre-B cells in both mouse strains also switched to α, consistent with results indicating that mice that cannot express IgM can undergo CSR to IgA (69). There are two mature mouse B cell lines (CH12F3 and I.29μ) that can be induced to switch in culture, but the switching is restricted to IgA, or rarely, in I.29μ, to IgE or IgG2a. Perhaps these cell lines have a chromosomal loop that sequesters the other CH loci from the enhancers. Much more research is needed to understand how specific chromosomal loops are regulated and the roles of chromosomal loops in regulating CSR.
Introduction of DSBs in S regions by AID involves the BER and mismatch repair pathways
Although AID is rapidly induced after B cell activation, the enzymes and proteins that convert the AID-induced dUs to DSBs are constitutively expressed, as the lesions these proteins repair occur in all types of cells. Uracil, whether due to AID activity or caused by spontaneous hydrolysis of cytosine, can be excised by the BER enzyme uracil DNA glycosylase (UNG), which leaves an a basic (apyrimidinic/apurinic) (AP) site (Fig 2A). Although cells express 4 different uracil DNA glycosylases (UNG, Smug1, MBD4 and TDG), deficiency of UNG alone results in a 95–99% reduction in CSR in mice and humans (4, 70, 71), and reduces S region DSBs detected during CSR in cultured splenic B cells nearly to levels observed in aid−/− B cells (72). Recently, it was found that in ung−/− mice, Smug1 can partially substitute, i.e., in smug1−/−ung−/− splenic B cells, CSR is reduced another 5-fold (71). Smug1-deficiency by itself has no effect on CSR, probably due to its low abundance (73)and low activity on dU in ss DNA, whereas UNG is abundant and more active on ss DNA than on ds DNA(74). As AID only has activity on ss DNA, it is possible that UNG excises most dU’s prior to reformation of the DNA duplex.
A basic (AP) sites are subsequently cut by AP endonuclease, APE1 and/or APE2, creating a single-strand DNA break (SSB) (Fig 2A). If SSBs are sufficiently near each other on both DNA strands, DSBs are produced. APE1 is essential for cell viability, and although ape1−/− mice have not been produced, ape1+/− mice have DNA repair defects. Both APE1 and APE2 contribute to CSR in splenic B cells induced to switch in culture (75). S region DSBs are greatly reduced in ape1+/−ape2−/− splenic B cells, but only marginally reduced in ape1+/− or ape2−/− cells, indicating these enzymes are partially redundant. However, deletion of these enzymes in CH12F3 B lymphoma cells gave somewhat different results. APE1-deficient cells have an 80% reduction in CSR, but APE2-deficiency has no effect on CSR (76). The difference between results in splenic B cells and CH12F3 cells remains a puzzle.
APE2 is a very inefficient endonuclease, ~1000-fold less active than APE1. APE2 has stronger exonuclease activity than APE1 and thus one could envision APE1 producing the SSB and APE2 creating a gap at the SSB, which could increase the probability that a SSB might become a DSB. However, evidence suggesting that APE2 does act as an endonuclease at AID-induced AP sites in splenic B cells was obtained by determining the locations of S region DSBs in APE-deficient mice compared to WT mice. APE2-deficiency reduced their preference for G:C bp so that the proportion of breaks at G:C bp were not significantly different from their proportion in the sequence itself, whereas DSBs in ape1+/− cells were similar to WT(75). It is not understood why APE2 is important for CSR, especially since APE1 is a much more efficient endonuclease.
A major question in the field is why AID-induced lesions are not accurately repaired despite the fact that BER is a highly active and error-free repair pathway. In most cells, DNA Pol β accurately replaces the excised nucleotide, and Ligase III-XRCC1 seals the phosphodiester backbone, and DSBs are not generated (See Fig 2A). In fact, DNA Pol β modestly inhibits S region DSBs and CSR, suggesting that it competes with DSB formation, but is overwhelmed by the numbers of AID-induced SSBs (77). Another study suggested an additional possibility. In this study, SHM of antibody variable region genes was examined in germinal center B cells from Peyer’s patches (78). These cells are undergoing constant stimulation by gut microbes and undergo robust SHM. The results suggest that the use of APE2 instead of APE1 in repair of AID-induced lesions in germinal centers converts BER to an error-prone repair pathway (78). Germinal center B cells express very low levels of APE1 protein and mRNA compared to APE2. APE2 was found to be important for mutations at A:T bp, whereas ape1+/− B cells have unperturbed SHM (78). Since a SSB is required as an entry point for the error-prone trans lesion polymerase DNA Pol η to introduce mutations at A:T bp, this suggests that APE2 is indeed acting as an endonuclease during SHM, creating SSBs. It is possible that the use of APE2 might inhibit error-free repair of AID-induced lesions. Interestingly, APE2, but not APE1, interacts with PCNA a protein that recruits DNA Pol η to DNA (79).
When the AID-UNG-APE pathway induces SSBs that are too far apart on opposite DNA strands to produce DSBs, MMR can convert the SSBs to a DSB(5) (See Fig 2B). The Msh2-Msh6 heterodimer binds to U:G mismatches in duplex DNA, recruits Mlh1-Pms2 and Exonuclease 1 (Exo1), which initiates resection from a SSB located 5′ to the mismatch (80, 81). Pms2 also has endonuclease activity and can create additional SSBs on the previously nicked strand, providing additional entry sites for Exo1 or other nucleases (82, 83). As diagrammed in Fig 2B, this resection should result in creation of a DSB with a long ss tail, which when filled in by DNA Pol creates a blunt, or nearly blunt, DSB appropriate for NHEJ(5). Several types of data support this model, including the facts that S region DSBs and CSR are reduced by 2–7 fold in MMR-deficient B cells (7, 84, 85), and that in the absence of Sμ tandem repeats, with their numerous AID target hotspots, CSR is absolutely dependent upon Msh2 (86).
Recombination of S region DSBs by NHEJ
S-S recombination occurs by NHEJ, which involves binding of the abundant toroidal heterodimer KU70/80 to each DSB, forming a platform for nucleases Artemis and PALF, and for DNA polymerases, and greatly stimulating the ligation activity of DNA ligase IV-XRCC4-XLF (reviewed in (87–89)). The Mre11-Nbs1-Rad50 (MRN) complex also rapidly binds DSBs, but it is unclear whether KU and MRN compete or cooperate at S region DSBs. Rad50 has a long coiled-coil domain with a hook at the end by which MRN complexes bound at different DSBs can interact and tether the DSBs to each other (90). MRN recruits additional factors, including the kinase ataxia-telangiectasia-mutant (ATM), which activates and coordinates the cellular response to DSBs. KU and MRN have both been shown to bind S region DSBS (6, 91–93) and contribute to CSR (94–98).
There is a great deal of mechanistic flexibility in NHEJ, which is necessary because DSBs can differ greatly, having 3′ or 5′ ss tails of different lengths, and possibly even having hairpins, in addition to being blunt. Generally, the junctions formed between two S regions have 0 or 1 bp of microhomology between Sμ and the acceptor S region, i.e., one cannot discern whether the 1 bp came from Sμ or the acceptor S region, although this microhomology can increase up to 6 bp or more. In cells deficient in a NHEJ protein, e.g. KU or Ligase IV, or deficient in Mlh1 or Pms2, S-S junctions show further increased lengths of microhomology, especially if Sμ-Sα junctions are analyzed, as these S regions have the most homology with each other (99). To explain the increased lengths of junctional microhomology, investigators have thought that an alternate end-joining pathway, (A)-EJ or microhomology-mediated (MM)-EJ, substitutes for NHEJ, resulting in these longer microhomologies (100–102). However, it is not clear whether MM-EJ is really a defined pathway or instead that NHEJ has partially redundant components. For example, in the absence of Ligase IV, Ligase I and III can perform end-joining, but not as efficiently, and the other components of NHEJ can still participate (88, 103). Deficiencies in a NHEJ component might expose the DSBs to end-resection activities; also, the less efficient recombination might allow time for increased end-resection prior to ligation, and result in increased use of microhomology, which can help stabilize the junctions. In situations in which fewer S region DSBs are induced, e.g. in human patients heterozygous for AID with a C terminal deletion (104) or in mlh1−/− or pms2−/− mouse and human B cells (7, 99, 105), junctional microhomology is also increased. Perhaps this occurs because DSBs are limiting, thus delaying recombination. Note that although Msh2-deficient cells have reduced DSBs (7), they do not have increased junctional microhomology, indicating that reduced DSB frequency does not always result in increased use of microhomology. The explanation for this difference from Mlh1- and Pms2-deficient cells is unknown.
The AID C terminus is essential for CSR but not for SHM
The fact that AID lacking the C terminal 8–17 amino acids (ΔAID) cannot support CSR but appears to support normal SHM of V(D)J segments has been known for several years but the explanation is still unclear (106, 107). It is not due to aberrant targeting of AID to S regions, as cells expressing ΔAID appear to have approximately normal levels of Sμ and Sγ DSBs; thus, the problem is subsequent to break formation, despite the evidence that recruitment of UNG and Msh2-Msh6 to S regions is impaired in these cells (93, 108, 109). The DSB repair process appears to be aberrant, as there is reduced recruitment of NHEJ proteins to Sμ regions and increased lengths of S-S junctional microhomology in cells expressing ΔAID (93, 104, 110, 111). However, how the C terminus recruits NHEJ proteins, and if this is important for the greatly reduced CSR is still unknown.
The role of ATM during CSR and phosphorylation of AID
ATM kinase is a major regulator of the DNA damage response. ATM is rapidly activated by the binding of MRN to DSBs, and in turn, ATM phosphorylates MRN and several downstream effectors of DSB repair (112). Humans with ATM mutations have numerous problems due to poor DNA repair functions. Mouse splenic B cells lacking ATM have reduced CSR (30% of WT B cells) (113, 114), and impaired V(D)J recombination (115). In response to S region DSBs, ATM induces phosphorylation of AID at S38 by an undefined pathway, and this increases the ability of AID to induce S region DSBs, and to bind to APE1 via an unknown protein (116). APE2 has not been tested. Thus, AID is activated by a feed-forward mechanism involving ATM. Phosphorylation of AID at S38 is not necessary for its deaminase activity in cell-free experiments, although activated cultured splenic B cells expressing AIDS38A have greatly reduced Sμ DSBs and CSR (116–118). It is hypothesized that this feed-forward mechanism increases AID activity at localized regions that have DSBs, such as at the IgH S regions, helping to explain why AID is so much more active at S regions than at off-target sites (91, 119, 120).
Interestingly, atm−/−B cells induced to switch show decreased DSBs at Sγ regions, but have increased DSBs at Sμ(9). These data have been interpreted to suggest that the feed-forward activation of AID by ATM is very important for AID activity at acceptor S regions, and that when DSBs are poorly induced in the acceptor S region, Sμ DSBs accumulate due to lack of a downstream partner. These data also suggest that acceptor S region DSBs are limiting for CSR. It had previously been found that AID is more active at Sμ than at Sγ regions (37, 121), suggesting that AID attacks the Sμ region first (121). Taken together, these data lead to the hypothesis that ATM is activated by Sμ DSBs, resulting in phosphorylation of AID, which increases its activity at the downstream S region that is co-localized due to chromosome looping. The studies of chromosomal looping described above suggest that Sμ and acceptor S regions are located near each other prior to induction of AID and DSBs, as the looping might coincide with expression of GLTs. Thus, when AID is activated by phosphorylation, the acceptor S region might be localized sufficiently near to be attacked. Close localization of the two S regions also decreases the likelihood of aberrant recombination events. Surprisingly, ATM deficiency has no effect on cell cycle regulation of S region DSBs, as they are restricted to G1 phase in atm−/− cells, just as in WT cells, indicating that Sμ DSBs only accumulate during G1 phase and are repaired before S phase, even if the DSBs do not undergo Sμ-Sx recombination (9).
Function of 53BP1 in CSR
One of the DNA repair proteins that binds DSBs in response to the phosphorylation activities of ATM is 53BP1. CSR is reduced by 90% in cells deficient in 53BP1, but its roles in CSR are still not clear. ATM phosphorylates H2AX, converting it to γH2AX, which in turn interacts with 53BP1 (122), although additional kinases, other enzymes, and histone modifications also recruit and activate 53BP1 (123, 124). Resection at S region DSBs during G1 phase is normally inhibited by 53BP1 and its effector protein Rif1, two proteins very important for CSR that function epistatically, and whose activities favor recombination by NHEJ (120, 123, 125–131). Surprisingly, S-S junctions in 53bp1−/− cells do not have increased microhomology (126). Another activity of 53BP1 essential for CSR is its ability to oligomerize (127, 132). 53BP1 likely binds DSBs in both Sμ and downstream S regions, and most likely the oligomerization of 53BP1 helps hold two different S regions together in the proper conformation. This might explain why inverted S region sequences and a segment from the IgH Eμ enhancer have been detected between the recombining S region segments in 53bp1−/− cells (126). In addition, in 53bp1−/− cells induced to undergo CSR there are numerous deletions within Sμ, suggesting that DSBs within Sμ failed to find an acceptor S region recombination partner. Thus, 53BP1 is essential for CSR, and appears to have multiple roles, although they are still not entirely understood.
Questions remaining
There are many more interesting issues to discuss about CSR than can even be briefly discussed here due to space limitations. We recommend several recent reviews of CSR that include additional topics: (33, 49, 103, 133–135).
Although much is now known about CSR, there are still many questions left unanswered, some of which have been mentioned in this review. Although AID deaminates off-target genomic sites, and also induces DSBs at off-target sites, most of its activity is directed toward Ig loci. How AID is targeted to S regions is mostly unknown. We know that transcription, and probably RNA stalling and RNA splicing are important, but of course there are numerous transcribed genes with spliced transcripts and stalled Pol II. What causes the stalling of Pol II at S regions? S regions and S region transcripts are likely to bind specific proteins, for examples 14-3-3 (136) and PTBP2, that help recruit AID, but are there others? How are the looping of the CH gene loci and enhancers regulated? What causes AID-induced S region DSBs to be restricted to G1 phase? Is AID activity restricted to G1 phase, as is UNG activity? What signaling mechanism causes the S region DSBs to be repaired prior to S phase even in the absence of ATM or p53(9)? What is the role APE2 in S region DSBs, and why isn’t APE1 sufficient? Although APE1 is expressed as well as APE2 in splenic B cells induced to switch in culture, it is poorly expressed in germinal center B cells. Does the low expression of APE1 reduce CSR in germinal center cells? Is APE1 well-expressed during CSR in vivo in non-germinal center cells? How does phosphorylation of AID at S38 cause increased S region DSBs? Is this specific for S region DSBs? What does the C terminus of AID do during CSR? Although it might be involved in recruitment of NHEJ proteins, its deletion has a much greater effect on CSR than loss of NHEJ functions. What is the importance and what are the roles of histone modifications of IgH genes for CSR? What are the functions of AID expression in pro- and pre-B cells? is this advantageous or only deleterious?
Acknowledgments
We thank Drs Lyne Khair, Patricia Mongini, and Amy Kenter for very helpful discussions. J.S. and C.E.S. are supported by grants from the NIH: RO1-AI23283 and R21-AI99988.
Abbreviations
- AID
activation-induced cytidine deaminase
- AP
apyrimidinic/apurinic
- APE
AP endonuclease
- ATM
ataxia telangiectasia mutant
- BER
base excision repair
- CSR
class switch recombination
- DSB
double-strand break
- NHEJ
non-homologous end-joining
- SHM
somatic hypermutation
- S region
switch region
- ss
single-stranded
- UNG
uracil DNA glycosylase
- 53BP1
p53-binding protein
Footnotes
The authors declare no conflicts of interest.
References
- 1.Dunnick W, Hertz GZ, Scappino L, Gritzmacher C. DNA sequences at immunoglobulin switch region recombination sites. Nuc Acids Res. 1993;21:365–372. doi: 10.1093/nar/21.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 3.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 4.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 5.Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Ann Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol. 2007;179:6064–6071. doi: 10.4049/jimmunol.179.9.6064. [DOI] [PubMed] [Google Scholar]
- 8.Sharbeen G, Yee CW, Smith AL, Jolly CJ. Ectopic restriction of DNA repair reveals that UNG2 excises AID-induced uracils predominantly or exclusively during G1 phase. J Exp Med. 2012;209:965–974. doi: 10.1084/jem.20112379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khair L, Guikema JE, Linehan EK, Ucher AJ, Leus NG, Ogilvie C, Lou Z, Schrader CE, Stavnezer J. ATM Increases Activation-Induced Cytidine Deaminase Activity at Downstream S Regions during Class-Switch Recombination. J Immunol. 2014;192:4887–4896. doi: 10.4049/jimmunol.1303481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pape KA, Kouskoff V, Nemazee D, Tang HL, Cyster JG, Tze LE, Hippen KL, Behrens TW, Jenkins MK. Visualization of the genesis and fate of isotype-switched B cells during a primary immune response. J Exp Med. 2003;197:1677–1687. doi: 10.1084/jem.20012065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, Kenny SM, Khan M, Toellner KM, Lane PJ, Maclennan IC. Salmonella Induces a Switched Antibody Response without Germinal Centers That Impedes the Extracellular Spread of Infection. J Immunol. 2007;178:6200–6207. doi: 10.4049/jimmunol.178.10.6200. [DOI] [PubMed] [Google Scholar]
- 12.Liu YJ, Malisan F, de Bouteiller O, Guret C, Lebecque S, Banchereau J, Mills FC, Max EE, Martinez-Valdez H. Within germinal centers, isotype switching of immunoglobulin genes occurs after the onset of somatic mutation. Immunity. 1996;4:241–250. doi: 10.1016/s1074-7613(00)80432-x. [DOI] [PubMed] [Google Scholar]
- 13.Butcher EC, Rouse RV, Coffman RL, Nottenburg CN, Hardy RR, Weissman IL. Surface phenotype of Peyer’s patch germinal center cells: implications for the role of germinal centers in B cell differentiation. J Immunol. 1982;129:2698–2707. [PubMed] [Google Scholar]
- 14.Cebra JJ, Shroff KE. Peyer’s patches as inductive sites for IgA commitment. Handbook of Mucosal Immunology. 1994:151–157. [Google Scholar]
- 15.Lin M, Du L, Brandtzaeg P, Pan-Hammarstrom Q. IgA subclass switch recombination in human mucosal and systemic immune compartments. Mucosal immunology. 2014;7:511–520. doi: 10.1038/mi.2013.68. [DOI] [PubMed] [Google Scholar]
- 16.Bergqvist P, Gardby E, Stensson A, Bemark M, Lycke NY. Gut IgA class switch recombination in the absence of CD40 does not occur in the lamina propria and is independent of germinal centers. J Immunol. 2006;177:7772–7783. doi: 10.4049/jimmunol.177.11.7772. [DOI] [PubMed] [Google Scholar]
- 17.Hodgkin PD, Lee J-H, Lyons AB. B cell differentiation and isotype switching is related to division cycle number. J Exp Med. 1996;184:277–281. doi: 10.1084/jem.184.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rush JS, Liu M, Odegard VH, Unniraman S, Schatz DG. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc Natl Acad Sci U S A. 2005;102:13242–13247. doi: 10.1073/pnas.0502779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van-Kooten C, Banchereau J. CD40-CD40 ligand: A mulifunctional receptor-ligand pair. Adv Immunol. 1996;61:1–77. doi: 10.1016/s0065-2776(08)60865-2. [DOI] [PubMed] [Google Scholar]
- 20.Foy TM, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Immune regulation by CD40 and its ligand gp39. Ann Rev Immunol. 1996;14:591–618. doi: 10.1146/annurev.immunol.14.1.591. [DOI] [PubMed] [Google Scholar]
- 21.Hou B, Saudan P, Ott G, Wheeler ML, Ji M, Kuzmich L, Lee LM, Coffman RL, Bachmann MF, DeFranco AL. Selective utilization of Toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity. 2011;34:375–384. doi: 10.1016/j.immuni.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turner ML, Corcoran LM, Brink R, Hodgkin PD. High-affinity B cell receptor ligation by cognate antigen induces cytokine-independent isotype switching. J Immunol. 2010;184:6592–6599. doi: 10.4049/jimmunol.0903437. [DOI] [PubMed] [Google Scholar]
- 23.McIntyre TM, Kehry MR, Snapper CM. Novel in vitro model for high-rate IgA class switching. J Immunol. 1995;154:3156–3161. [PubMed] [Google Scholar]
- 24.Pone EJ, Zhang J, Mai T, White CA, Li G, Sakakura JK, Patel PJ, Al-Qahtani A, Zan H, Xu Z, Casali P. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nature communications. 2012;3:767. doi: 10.1038/ncomms1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaminski DA, Stavnezer J. Stimuli that enhance IgA class switching increase histone 3 acetylation at S alpha, but poorly stimulate sequential switching from IgG2b. Eur J Immunol. 2007;37:240–251. doi: 10.1002/eji.200636645. [DOI] [PubMed] [Google Scholar]
- 26.Kaminski DA, Stavnezer J. Antibody class switching differs among SJL, C57BL/6 and 129 mice. Int Immunol. 2007;19:545–556. doi: 10.1093/intimm/dxm020. [DOI] [PubMed] [Google Scholar]
- 27.Heltemes-Harris LM, Gearhart PJ, Ghosh P, Longo DL. Activation-induced deaminase-mediated class switch recombination is blocked by anti-IgM signaling in a phosphatidylinositol 3-kinase-dependent fashion. Mol Immunol. 2008;45:1799–1806. doi: 10.1016/j.molimm.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jabara HH, Chaudhuri J, Dutt S, Dedeoglu F, Weng Y, Murphy MM, Franco S, Alt FW, Manis J, Geha RS. B-cell receptor cross-linking delays activation-induced cytidine deaminase induction and inhibits class-switch recombination to IgE. J Allergy Clin Immunol. 2008;121:191–196. e192. doi: 10.1016/j.jaci.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Borte S, Pan-Hammarstrom Q, Liu C, Sack U, Borte M, Wagner U, Graf D, Hammarstrom L. Interleukin-21 restores immunoglobulin production ex vivo in patients with common variable immunodeficiency and selective IgA deficiency. Blood. 2009;114:4089–4098. doi: 10.1182/blood-2009-02-207423. [DOI] [PubMed] [Google Scholar]
- 30.Gu H, Zou Y-R, Rajewsky K. Independent control of immunoglobulin switch recombinaiton at individual switch regions evidenced through Cre-lox P-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- 31.Stavnezer J. Antibody Class Switching. Adv Immunol. 1996;61:79–146. doi: 10.1016/s0065-2776(08)60866-4. [DOI] [PubMed] [Google Scholar]
- 32.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 33.Matthews AJ, Zheng S, DiMenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Advances in immunology. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 35.Huang FT, Yu K, Balter BB, Selsing E, Oruc Z, Khamlichi AA, Hsieh CL, Lieber MR. Sequence-Dependence of Chromosomal R-loops at the Immunoglobulin Heavy Chain S{mu} Class Switch Region. Mol Cell Biol. 2007;27:5921–5932. doi: 10.1128/MCB.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol Cell Biol. 2010;30:146–159. doi: 10.1128/MCB.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med. 2006;203:2085–2094. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, Gearhart PJ. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med. 2009;206:1237–1244. doi: 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009;206:1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, San-Martin BR, Barreto V, Nieland TJ, Root DE, Casellas R, Nussenzweig MC. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, Alt FW. The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun J, Keim CD, Wang J, Kazadi D, Oliver PM, Rabadan R, Basu U. E3-ubiquitin ligase Nedd4 determines the fate of AID-associated RNA polymerase II in B cells. Genes Dev. 2013;27:1821–1833. doi: 10.1101/gad.210211.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Fan M, Kalis S, Wei L, Scharff MD. A source of the single-stranded DNA substrate for activation-induced deaminase during somatic hypermutation. Nature communications. 2014;5:4137. doi: 10.1038/ncomms5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kodgire P, Mukkawar P, Ratnam S, Martin TE, Storb U. Changes in RNA polymerase II progression influence somatic hypermutation of Ig-related genes by AID. J Exp Med. 2013;210:1481–1492. doi: 10.1084/jem.20121523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bottaro A, Lansford R, Xu L, Zhang J, Rothman P, Alt F. I region transcription (per se) promotes basal IgE class switch recombination but additional factors regulate the efficiency of the process. EMBO J. 1994;13:665–674. doi: 10.1002/j.1460-2075.1994.tb06305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267:1825–1828. doi: 10.1126/science.7892607. [DOI] [PubMed] [Google Scholar]
- 47.Hein K, Lorenz MG, Siebenkotten G, Petry K, Christine R, Radbruch A. Processing of switch transcripts is required for targeting of antibody class switch recombination. J Exp Med. 1998;188:2369–2374. doi: 10.1084/jem.188.12.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu G, Harriman GR, Stavnezer J. Iα exon-replacement mice synthesize a spliced HPRT-Ca transcript which may explain their ability to switch to IgA: Inhibition of switching to IgG in these mice. Int Immunol. 1999;11:37–46. doi: 10.1093/intimm/11.1.37. [DOI] [PubMed] [Google Scholar]
- 49.Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev. 2013;27:1–17. doi: 10.1101/gad.200014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell. 2008;31:474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 51.Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matthews AJ, Husain S, Chaudhuri J. Binding of AID to DNA does not correlate with mutator activity. J Immunol. 2014;193:252–257. doi: 10.4049/jimmunol.1400433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romanelli MG, Diani E, Lievens PM. New insights into functional roles of the polypyrimidine tract-binding protein. International journal of molecular sciences. 2013;14:22906–22932. doi: 10.3390/ijms141122906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, Alt FW, Cogne M, Pinaud E, Kenter AL. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chatterjee S, Ju Z, Hassan R, Volpi SA, Emelyanov AV, Birshtein BK. Dynamic changes in binding of immunoglobulin heavy chain 3′ regulatory region to protein factors during class switching. J Biol Chem. 2011;286:29303–29312. doi: 10.1074/jbc.M111.243543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med. 2009;206:1073–1087. doi: 10.1084/jem.20082311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kenter AL, Feldman S, Wuerffel R, Achour I, Wang L, Kumar S. Three- dimensional architecture of the IgH locus facilitates class switch recombination. Ann N Y Acad Sci. 2012;1267:86–94. doi: 10.1111/j.1749-6632.2012.06604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vincent-Fabert C, Fiancette R, Pinaud E, Truffinet V, Cogne N, Cogne M, Denizot Y. Genomic deletion of the whole IgH 3′ regulatory region (hs3a, hs1,2, hs3b, and hs4) dramatically affects class switch recombination and Ig secretion to all isotypes. Blood. 2010;116:1895–1898. doi: 10.1182/blood-2010-01-264689. [DOI] [PubMed] [Google Scholar]
- 59.Guo C, Gerasimova T, Hao H, Ivanova I, Chakraborty T, Selimyan R, Oltz EM, Sen R. Two forms of loops generate the chromatin conformation of the immunoglobulin heavy-chain gene locus. Cell. 2011;147:332–343. doi: 10.1016/j.cell.2011.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Atchison ML. Function of YY1 in Long-Distance DNA Interactions. Frontiers in immunology. 2014;5:45. doi: 10.3389/fimmu.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwab KR, Patel SR, Dressler GR. Role of PTIP in class switch recombination and long-range chromatin interactions at the immunoglobulin heavy chain locus. Mol Cell Biol. 2011;31:1503–1511. doi: 10.1128/MCB.00990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, Filsuf D, Chen HT, Gazumyan A, Yamane A, Cho YW, Sun HW, Ge K, Peng W, Nussenzweig MC, Casellas R, Dressler GR, Zhao K, Nussenzweig A. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329:917–923. doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annual review of genetics. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 64.Enervald E, Du L, Visnes T, Bjorkman A, Lindgren E, Wincent J, Borck G, Colleaux L, Cormier-Daire V, van Gent DC, Pie J, Puisac B, de Miranda NF, Kracker S, Hammarstrom L, de Villartay JP, Durandy A, Schoumans J, Strom L, Pan-Hammarstrom Q. A regulatory role for the cohesin loader NIPBL in nonhomologous end joining during immunoglobulin class switch recombination. J Exp Med. 2013;210:2503–2513. doi: 10.1084/jem.20130168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thomas-Claudepierre AS, Schiavo E, Heyer V, Fournier M, Page A, Robert I, Reina-San-Martin B. The cohesin complex regulates immunoglobulin class switch recombination. J Exp Med. 2013;210:2495–2502. doi: 10.1084/jem.20130166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. Class switch recombination and somatic hypermutation in early mouse B Cells are mediated by B Cell and toll-like receptors. Immunity. 2007;27:64–75. doi: 10.1016/j.immuni.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar S, Wuerffel R, Achour I, Lajoie B, Sen R, Dekker J, Feeney AJ, Kenter AL. Flexible ordering of antibody class switch and V(D)J joining during B-cell ontogeny. Genes Dev. 2013;27:2439–2444. doi: 10.1101/gad.227165.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Umiker BR, McDonald G, Larbi A, Medina CO, Hobeika E, Reth M, Imanishi-Kari T. Production of IgG autoantibody requires expression of activation- induced deaminase in early-developing B cells in a mouse model of SLE. Eur J Immunol. 2014 doi: 10.1002/eji.201344282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Macpherson AJ, Lamarre A, McCoy K, Harriman GR, Odermatt B, Dougan G, Hengartner H, Zinkernagel RM. IgA production without mu or delta chain expression in developing B cells. Nat Immunol. 2001;2:625–631. doi: 10.1038/89775. [DOI] [PubMed] [Google Scholar]
- 70.Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, Durandy A. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- 71.Dingler FA, Kemmerich K, Neuberger MS, Rada C. Uracil excision by endogenous SMUG1 glycosylase promotes efficient Ig class switching and impacts on A:T substitutions during somatic mutation. Eur J Immunol. 2014 doi: 10.1002/eji.201444482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent upon AID and UNG. J Exp Med. 2005;202:561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 74.Doseth B, Ekre C, Slupphaug G, Krokan HE, Kavli B. Strikingly different properties of uracil-DNA glycosylases UNG2 and SMUG1 may explain divergent roles in processing of genomic uracil. DNA Repair (Amst) 2012;11:587–593. doi: 10.1016/j.dnarep.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 75.Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, Schrader CE. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204:3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol Cell Biol. 2013;33:1468–1473. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu X, Stavnezer J. DNA polymerase beta is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J Exp Med. 2007;204:1677–1689. doi: 10.1084/jem.20070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stavnezer J, Linehan EK, Thompson MR, Habboub G, Ucher AJ, Kadungure T, Tsuchimoto D, Nakabeppu Y, Schrader CE. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A:T mutations during somatic hypermutation. Proc Natl Acad Sci U S A. 2014;111:9217–9222. doi: 10.1073/pnas.1405590111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garg P, Burgers PM. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci U S A. 2005;102:18361–18366. doi: 10.1073/pnas.0505949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiricny J. Postreplicative mismatch repair. Cold Spring Harbor perspectives in biology. 2013;5:a012633. doi: 10.1101/cshperspect.a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schaetzlein S, Chahwan R, Avdievich E, Roa S, Wei K, Eoff RL, Sellers RS, Clark AB, Kunkel TA, Scharff MD, Edelmann W. Mammalian Exo1 encodes both structural and catalytic functions that play distinct roles in essential biological processes. Proc Natl Acad Sci U S A. 2013;110:E2470–2479. doi: 10.1073/pnas.1308512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 83.van Oers JM, Roa S, Werling U, Liu Y, Genschel J, Hou H, Jr, Sellers RS, Modrich P, Scharff MD, Edelmann W. PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1008589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ehrenstein MR, Neuberger MS. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. Embo J. 1999;18:3484–3490. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schrader CE, Edelmann W, Kucherlapati R, Stavnezer J. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J Exp Med. 1999;190:323–330. doi: 10.1084/jem.190.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Min I, Schrader C, Vardo J, D’Avirro N, Luby T, Stavnezer J, Selsing E. The Sm tandem repeat region is critical for isotype switching in the absence of Msh2. Immunity. 2003;19:515–524. doi: 10.1016/s1074-7613(03)00262-0. [DOI] [PubMed] [Google Scholar]
- 87.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pannunzio NR, Li S, Watanabe G, Lieber MR. Non-homologous end joining often uses microhomology: implications for alternative end joining. DNA Repair (Amst) 2014;17:74–80. doi: 10.1016/j.dnarep.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annual review of genetics. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- 90.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 91.Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Mol Cell. 2011;41:232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cortizas EM, Zahn A, Hajjar ME, Patenaude AM, Di Noia JM, Verdun RE. Alternative End-Joining and Classical Nonhomologous End-Joining Pathways Repair Different Types of Double-Strand Breaks during Class-Switch Recombination. J Immunol. 2013;191:5751–5763. doi: 10.4049/jimmunol.1301300. [DOI] [PubMed] [Google Scholar]
- 93.Zahn A, Eranki AK, Patenaude AM, Methot SP, Fifield H, Cortizas EM, Foster P, Imai K, Durandy A, Larijani M, Verdun RE, Di Noia JM. Activation induced deaminase C-terminal domain links DNA breaks to end protection and repair during class switch recombination. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1320486111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Manis JP, Gu Y, Lansford R, Sonoda E, Ferrini R, Davidson L, Rajewsky K, Alt FW. Ku70 is required for late B cell development and immunoglobulin heavy chain switching. J Exp Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Casellas R, Nussenzweig A, Wuerffel R, Pelanda R, Reichlin A, Suh H, Qin XF, Besmer E, Kenter A, Rajewsky K, Nussenzweig MC. Ku80 is required for immunoglobulin isotype switching. EMBO J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nature structural & molecular biology. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lahdesmaki A, Taylor AM, Chrzanowska KH, Pan-Hammarstrom Q. Delineation of the role of the Mre11 complex in class switch recombination. J Biol Chem. 2004;279:16479–16487. doi: 10.1074/jbc.M312796200. [DOI] [PubMed] [Google Scholar]
- 98.Kracker S, Bergmann Y, Demuth I, Frappart PO, Hildebrand G, Christine R, Wang ZQ, Sperling K, Digweed M, Radbruch A. Nibrin functions in Ig class-switch recombination. Proc Natl Acad Sci U S A. 2005;102:1584–1589. doi: 10.1073/pnas.0409191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stavnezer J, Bjorkman A, Du L, Cagigi A, Pan-Hammarstrom Q. Mapping of switch recombination junctions, a tool for studying DNA repair pathways during immunoglobulin class switching. Adv Immunol. 2010;108:45–109. doi: 10.1016/B978-0-12-380995-7.00003-3. [DOI] [PubMed] [Google Scholar]
- 100.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, Alt FW. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 101.Han L, Yu K. Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV--deficient B cells. J Exp Med. 2008;205:2745–2753. doi: 10.1084/jem.20081623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boboila C, Yan C, Wesemann DR, Jankovic M, Wang JH, Manis J, Nussenzweig A, Nussenzweig M, Alt FW. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med. 2010;207:417–427. doi: 10.1084/jem.20092449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kracker S, Imai K, Gardes P, Ochs HD, Fischer A, Durandy AH. Impaired induction of DNA lesions during immunoglobulin class-switch recombination in humans influences end-joining repair. Proc Natl Acad Sci U S A. 2010;107:22225–22230. doi: 10.1073/pnas.1012591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peron S, Metin A, Gardes P, Alyanakian MA, Sheridan E, Kratz CP, Fischer A, Durandy A. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. J Exp Med. 2008;205:2465–2472. doi: 10.1084/jem.20080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell. 2003;12:501–508. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 107.Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, Nonoyama S, Tashiro J, Ikegawa M, Ito S, Kinoshita K, Muramatsu M, Honjo T. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol. 2003;4:843–848. doi: 10.1038/ni964. [DOI] [PubMed] [Google Scholar]
- 108.Doi T, Kato L, Ito S, Shinkura R, Wei M, Nagaoka H, Wang J, Honjo T. The C-terminal region of activation-induced cytidine deaminase is responsible for a recombination function other than DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A. 2009;106:2758–2763. doi: 10.1073/pnas.0813253106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ranjit S, Khair L, Linehan EK, Ucher AJ, Chakrabarti M, Schrader CE, Stavnezer J. AID binds cooperatively with UNG and Msh2-Msh6 to Ig switch regions dependent upon the AID C terminus. J Immunol. 2011;187:2464–2475. doi: 10.4049/jimmunol.1101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sabouri S, Kobayashi M, Begum NA, Xu J, Hirota K, Honjo T. C-terminal region of activation-induced cytidine deaminase (AID) is required for efficient class switch recombination and gene conversion. Proc Natl Acad Sci U S A. 2014;111:2253–2258. doi: 10.1073/pnas.1324057111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ucher AJ, Ranjit S, Kadungure T, Linehan EK, Khair L, Xie E, Limauro J, Rauch KS, Schrader CE, Stavnezer J. Mismatch Repair Proteins and AID Activity Are Required for the Dominant Negative Function of C-Terminally Deleted AID in Class Switching. J Immunol. 2014:193. doi: 10.4049/jimmunol.1400365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 113.Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med. 2004;200:1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, Morse HC, 3rd, Gearhart PJ, Wynshaw-Boris A, Max EE, Hodes RJ. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J Exp Med. 2004;200:1111–1121. doi: 10.1084/jem.20041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Callen E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, Pellegrini M, McBride K, Wangsa D, Bredemeyer AL, Sleckman BP, Ried T, Nussenzweig M, Nussenzweig A. ATM Prevents the Persistence and Propagation of Chromosome Breaks in Lymphocytes. Cell. 2007;130:63–75. doi: 10.1016/j.cell.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 116.Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella JN, Ucher AJ, Donghia NM, Gu X, Nicolas L, Nowak U, Rahman N, Strout MP, Mills KD, Stavnezer J, Chaudhuri J. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat Immunol. 2013;14:1183–1189. doi: 10.1038/ni.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med. 2008;205:2585–2594. doi: 10.1084/jem.20081319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cheng HL, Vuong BQ, Basu U, Franklin A, Schwer B, Astarita J, Phan RT, Datta A, Manis J, Alt FW, Chaudhuri J. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proc Natl Acad Sci U S A. 2009;106:2717–2722. doi: 10.1073/pnas.0812304106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 120.Yamane A, Robbiani DF, Resch W, Bothmer A, Nakahashi H, Oliveira T, Rommel PC, Brown EJ, Nussenzweig A, Nussenzweig MC, Casellas R. RPA accumulation during class switch recombination represents 5′-3′ DNA-end resection during the S-G2/M phase of the cell cycle. Cell reports. 2013;3:138–147. doi: 10.1016/j.celrep.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schrader CE, Bradley SP, Vardo J, Mochegova SN, Flanagan E, Stavnezer J. Mutations occur in the Ig Sμ region but rarely in Sγ regions prior to class switch recombination. Embo J. 2003;22:5893–5903. doi: 10.1093/emboj/cdg550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 123.Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol. 2014;34:1380–1388. doi: 10.1128/MCB.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee DH, Acharya SS, Kwon M, Drane P, Guan Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto JA, Chowdhury D. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell. 2014;54:512–525. doi: 10.1016/j.molcel.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, Nussenzweig MC, Chen J. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Reina-San-Martin B, Chen J, Nussenzweig A, Nussenzweig MC. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1−/− B cells. Eur J Immunol. 2007;37:235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- 127.Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, Nussenzweig A, Nussenzweig MC. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Daley JM, Sung P. RIF1 in DNA break repair pathway choice. Mol Cell. 2013;49:840–841. doi: 10.1016/j.molcel.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 131.Bothmer A, Rommel PC, Gazumyan A, Polato F, Reczek CR, Muellenbeck MF, Schaetzlein S, Edelmann W, Chen PL, Brosh RM, Jr, Casellas R, Ludwig T, Baer R, Nussenzweig A, Nussenzweig MC, Robbiani DF. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J Exp Med. 2013;210:115–123. doi: 10.1084/jem.20121975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc Natl Acad Sci U S A. 2013;110:2146–2151. doi: 10.1073/pnas.1222617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol. 2012;12:517–531. doi: 10.1038/nri3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32:194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Storck S, Aoufouchi S, Weill JC, Reynaud CA. AID and partners: for better and (not) for worse. Curr Opin Immunol. 2011;23:337–344. doi: 10.1016/j.coi.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 136.Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, Steinacker P, Li Z, Yates J, 3rd, Herron B, Otto M, Zan H, Fu H, Casali P. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nature structural & molecular biology. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]