

Abstract
Through-space metal/hydrogen shift is an important strategy for transition metal-catalyzed C-H bond activation. Here we describe the synthesis and characterization of a Rh(I) 2,6-dimethoxybenzoate complex that underwent stoichiometric rearrangement via a highly unusual 1,3- rhodium migration. This aryl-to-aryl 1,3-Rh/H shift was also demonstrated in a Rh(I)-catalyzed decarboxylative conjugate addition to form a C-C bond at a meta position instead of the ipso-carboxyl position. A deuterium-labeling study under the conditions of Rh(I)-catalyzed protodecarboxylation revealed the involvement of an ortho-methoxy group in a multi-step pathway of consecutive sp3 and sp2 C-H bond activations.
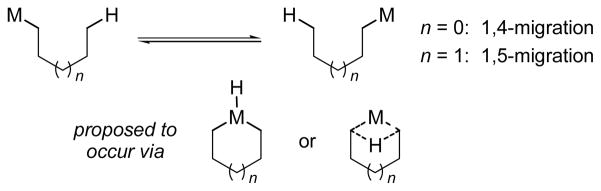
Transition metal-catalyzed direct functionalization of C-H bonds has become a powerful tool for organic synthesis.1 An important method for intramolecular C-H bond activation is the “through-space” metal/hydrogen shift, most commonly at 1,4- and 1,5-positions of a hydrocarbon backbone (1,4- and 1,5-migrations, Scheme 1).2 These rearrangement processes allow functionalization of C-H bonds that are difficult to activate directly. Catalytic 1,4-rhodium migration was first reported in 2000 by Miura and coworkers in the reaction between arylboronic acids and norbornenes.3a In the same year, Larock and coworkers reported the first catalytic 1,4- palladium migration in coupling between aryl iodides and alkynes.4a Since these pioneering studies, catalytic 1,4- migrations of various late transition metal centers such as Rh(I),3 Pd(II),4 Pt(II)5a,b and Ni(I)5c have been successfully explored for selective functionalization of sp2 and sp3 C-H bonds to form carbon-carbon and carbon-heteroatom bonds. Several examples of catalytic 1,5-migrations have also been reported for Rh(I)6a and Pd(II)6b–d intermediates.
Scheme 1.
Intramolecular C-H bond activation via metal-hydrogen shifts.
In contrast to the well established 1,4- and 1,5-migrations, other forms of metal/hydrogen shifts are very rare. In particular, 1,3-migration has not been reported with any transition metal species. From the reaction mechanism perspective, 1,4- and 1,5-migrations have been proposed to be facilitated by stabilized 5- or 6-membered metallacycle intermediates and transition states (Scheme 1).2 In comparison, DFT calculations suggest that direct 1,3-metal/hydrogen shifts would require highly strained 4-member cyclic transition states with prohibitively high activation energies.6c,6d,7 On the other hand, the classic 1,3-shifts of transition metal allyl species only involve migration of metal centers but not hydrogen atoms.8 We herein describe stoichiometric and catalytic rearrangement processes that occur by a formal aryl-to-aryl 1,3- rhodium migration in Rh(I)-mediated decarboxylation. Mechanistic results from deuterium labeling studies suggest a highly unusual, “double 1,4-Rh migration” pathway that involves sp3 C-H bond activation at the methoxy group.9
Over the past decade, late transition metal-mediated decarboxylation of benzoic acids has generated much interest as a non-conventional approach towards reactive metal aryl intermediates in catalysis.10–12 A very important structural motif for decarboxylation is ortho-substitution of benzoic acids. In particular, ortho-methoxy and ortho-fluorine groups have been shown to significantly promote decarboxylation reactivity with various transition metal catalysts.10 We have previously reported Rh(I)-catalyzed decarboxylative transformations of 2,6-difluorobenzoic acids including conjugate addition, oxidative olefination,12a and protodecarboxylation.13 As part of our efforts to gain mechanistic insights into Rh(I)-mediated decarboxylation, we have synthesized (bis)phosphine-ligated Rh(I) benzoate complexes for direct observation of stoichiometric decarboxylation. As described in Scheme 2, κ2-carboxylates 2a and 2b were prepared by reactions between [(cod)Rh(μ-OH)]2 (cod: 1,4- cyclooctadiene), BIPHEP (2,2′-bis(diphenylphosphino)-1,1′-biphenyl), and 2,6-difluorobenzoic acid (1a) or 2,6- dimethoxybenzoic acid (1b) respectively. As we reported previously,13a 2,6-difluorobenzoate 2a underwent stoichiometric decarboxylation at 120 °C with 1 equiv of added pyridine in toluene, giving the corresponding arylrhodium(I) complex 4a in quantitative conversion.
Scheme 2.

Synthesis and thermal transformation of 2,6-disubstituted Rh(I) benzoates.
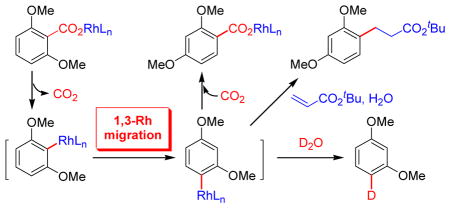
We envisioned that the reaction between Rh(I) κ2- benzoates (2) and pyridine would lead to the formation of pyridine-ligated κ1-benzoate complexes (3). Indeed, we have observed clean formation of 3a and 3b by 31P NMR (Scheme 2). The in situ formed 3a underwent quantitative decarboxylation that was consistent with our previous observation.13a In sharp contrast, thermolysis of in situ formed κ1-2,6- dimethoxybenzoate 3b at 120 °C in toluene did not generate the expected Rh(I) 2,6-dimethoxyphenyl complex by decarboxylation. 14 Instead, a novel “1,3-carboxylate migration” appeared to occur, leading to the formation of κ1-2,4- dimethoxybenzoate 4b in 34% yield as the only detectable Rh(I) species by 31P NMR analysis. Interestingly, the yield of 4b was improved to 71% when the thermolysis was carried out under 1 atm of CO2 instead of N2. Structures of isolated 2b, 3b and 4b were determined by single crystal X-ray diffraction (see Supporting Information for details). In the solid state, the chelating carboxylato ligand in 2b led to a significantly distorted square planar geometry. In comparison, 3b and 4b adopt near square-planar geometry with monodentate carboxylato ligands.
Based on the yield improvement of 4b under CO2 atmosphere, we propose a multi-step pathway for the 1,3-carboxyl migration as described in Scheme 3. Decarboxylation of 3b was expected to generate a Rh(I) 2,6-dimethoxyphenyl intermediate 5a,14 which underwent rearrangement by 1,3-Rh/H shift (1,3-Rh migration) to form Rh(I) 2,4-dimethoxyphenyl complex 5b. With the reduced steric crowding around Rh center in 5b compared to 5a, the decarboxylation/ carboxylation thermodynamics was shifted to favor CO2 insertion into the Rh-aryl linkage15 to give carboxylation product 4b as the most stable Rh(I) species in the reaction system. With lower CO2 concentration in a non-CO2 atmosphere, 5b underwent competitive protonation of the Rh-C bond to generate 1,3-dimethoxybenzene that was detected as the major byproduct.
Scheme 3.
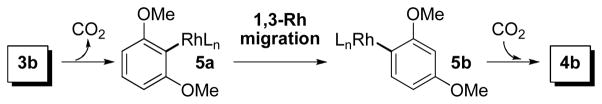
Proposed pathway for isomerization of Rh(I) carboxylates 3b to form 4b.
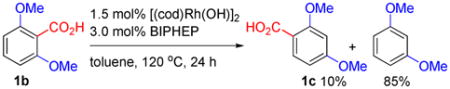
We envisioned that the proposed 1,3-Rh migration could be exploited catalytically to give novel rearrangement products. For example, the 1,3-carboxyl migration of 3b (Scheme 2) could proceed catalytically to allow isomerization of 2,6- dimethoxybenzoic acid (1b) to form 2,4-dimethoxybenzoic acid (1c) (Eq. 1). However, 1,3-dimethoxybenzene was formed as the major product by competitive protodecarboxylation. In comparison, a catalytic decarboxylative 1,4-addition13a of 1b with t-butyl acrylate (6) was successfully carried out to give 1,3-migration product 8a in 71% yield and >20:1 selectivity over the non-rearrangement product 7a (Eq. 2). This reaction was promoted by 1.5 mol% [(cod)Rh(OH)]2, 3.0 mol% BINAP (2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl), 1.0 equiv NaOH additive, and 5:1 toluene/H2O mixed solvent at 120 °C. Notably, this reaction occurred in good selectivity and without the formation of corresponding Heck-Mizoroki olefination products.12
 |
(1) |
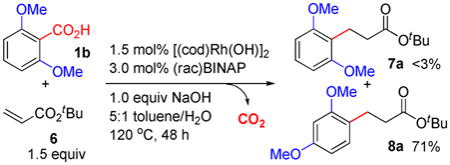 |
(2) |
We have considered several possible pathways for the proposed 1,3-Rh migration with arylrhodium(I) intermediate 5a in decarboxylative transformations of 1b (Scheme 4). A direct 1,3-Rh/H shift (path A) requires a 4-membered cyclometalated Rh(III) hydrido intermediate A or a σ-bond metathesis transition state B.2 Both structures would be extremely strained due to the inherent aromatic planarity and rigidity, making this pathway a highly unlikely scenario.7 Path B involves protonation of the Rh-C bond in 5a by hydrolysis to form 1,3-dimethoxybenzene (9a) and a Rh(I) hydroxo intermediate. 5b is then formed via aromatic C-H bond activation of 9a by Rh(I) hydroxide,16 with the regioselectivity determined by ortho- and para-directing methoxy groups in an electrophilic aromatic substitution (SEAr) mechanism. In path C, 5a undergoes cyclometalation to activate a methoxy sp3 C-H bond at the ortho position and forms a Rh(III) hydrido intermediate C.9 Subsequent C-H reductive elimination at the original ipso position generates a Rh(I) aryloxyalkyl intermediate D, which undergoes further aromatic C-H bond activation at the less hindered meta position to form another cyclometalated Rh(III) intermediate E. E then undergoes C-H reductive elimination at the methoxy position to form 5b. Notably, the proposed transformations of 5a→D and D→5b represent formal 1,4-Rh migrations and could also occur by single-step σ-bond metathesis and without involvement of Rh(III) hydrido intermediates.2 In all three possible pathways, the individual steps are possibly reversible and the driving force for formation of 5b over 5a is most likely the released steric crowding with mono- vs. di-methoxy groups at ortho positions.
Scheme 4.

Proposed pathways for 1,3-Rh migration.
To evaluate the feasibility of path B, we have attempted coupling reaction with t-butyl acrylate (6) using 1,3- dimethoxybenzene (9a) in place of 1b under catalytic conditions shown in Eq. 2. No reaction was observed and 9a was fully recovered, which strongly argues against path B. Regarding path C, our efforts towards a direct observation of the proposed stoichiometric transformations were hampered by failed attempts for an independent synthesis of intermediate 5a. However, the proposed intramolecular transfer of H atoms (Ha and Hb) provides a suitable target for deuterium labeling studies.3b,3c,4g,4h Thus, path C was further evaluated by a catalytic deuterium transfer process described below, using a modified procedure of Rh(I)-catalyzed protodecarboxylation previously reported by our group (Scheme 5).13b,17
Scheme 5.

Deuterium labeling study on Rh(I)- catalyzed hydrodecarboxylation.
aGeneral conditions: ArCO2H (0.225 mmol,1 equiv), [(cod)Rh(OH)]2 (0.015 equiv), DPPP (0.030 equiv), Na2CO3 (1.0 equiv), toluene-H2O (1.50/0.25 mL), 120 °C, 30 h; isolated yields; >90% deuterium transfer by NMR analysis. bD2O was used in place of H2O.
Protodecarboxyation of 2,6-dimethoxybenzoic acid (1b) was effectively promoted by a catalyst system of 1.5 mol% [(cod)Rh(OH)]2, 3.0 mol% DPPP ligand (1,2-bis(diphenylphosphino) ethane), 1 equiv of Na2CO3 additive in 6:1 toluene/ H2O at 120 °C to give 1,3-dimethoxybenzene (9a) in 64% isolated yield. Using D2O in place of H2O in the solvent system led to the exclusive formation of 4-d-1,3-dimethoxybenzene (9b) in 61% yield. Such regioselective deuterium incorporation confirmed the involvement of 1,3-Rh migration to form intermediate 5b (Scheme 3), which underwent subsequent deuteration of the Rh-aryl bond with D2O. The catalytic protodecarboxylation was then studied with two siteselective deuterium-labeled derivatives of 2,6-dimethoxybenzoic acid (1b), and both results supported the proposed intramolecular H atom transfers by path C: (1) Substrate d6- 1b (fully deuterium-labeled methoxy groups) underwent intramolecular deuterium transfer from a OCD3 group to the original ipso position, forming hydrodecarboxylation product 9c in 67% yield. This result was consistent with the proposed (ipso)aryl/methoxy 1,4-Rh/H shift in path C (Scheme 4, 5a→D). (2) Substrate 3,5-d2-1b (deuterium-labeling at both meta positions relative to the carboxyl group) underwent deuterium transfer from one of the meta positions to the nearby methoxy group, forming hydrodecarboxylation product 9d in 59% yield. This result was consistent with the proposed methoxy/(meta)aryl 1,4-Rh/H shift in path C (Scheme 4, D→5b). It is noteworthy that the individual steps of 5a→D and D→5b have been reported for Pd(II)-catalyzed rearrangement processes by aryl-to-alkyl4d,4h,4i and alkyl-to-aryl4c 1,4-Pd migrations respectively. However, a formal 1,3- migration by two consecutive 1,4-migrations has not been reported. The highly selective formation of 9b suggested that both steps of 1,4-migration were impressively rapid processes that effectively prevented competitive protonation of intermediates 5a or D, which would allow incorporation of external deuteriums at ortho and methoxy positions. In addition, catalytic hydrodecarboxylation of 2,6-diethoxybenzoic acid (10) in toluene/D2O did lead to exclusive ipso-deuteration to form 2-d-1,3-diethoxybenzene (11) as the only detectable product. Thus, the target 1,3-Rh migration process appears to rely on a delicate balance on steric effects of the orthosubstituents: significant steric crowding (OMe vs. F) is needed to slow down ipso-functionalization and promote rapid, consecutive Rh/H shifts, whereas too much steric crowding (OEt vs. OMe) inhibits the first Rh/H shift step and shuts down the overall migration process.
Based on the proposed mechanism, we envisioned that 1,3- Rh migration is not limited to decarboxylation process and could occur with analogous Rh(I) aryl species generated by other transformations. Indeed, preliminary results showed that methoxy-directed 1,3-migration also occurred in Rh(I)- catalyzed coupling of arylboronic acids with olefins (Eq. 3), where arylrhodium(I) species were formed by B-to-Rh transmetalation.18,19 A catalyst system of [(cod)Rh(OH)]2 precursor and racemic BINAP ligand promoted the reaction between 2,6-dimethoxyphenylboronic acid (12) and t-butyl acrylate (6) at 120 °C to form a mixture of ipso products (7a, 7b) and meta products (8a, 8b) by competitive 1,4-addition and Heck-Mizoroki olefination. The tandem 1,3- migration/1,4-addition product 8a was isolated as the major component in 45% yield. Despite the moderate selectivity, this result serves as a proof-of-concept for methoxy-directed 1,3-Rh migration in general coupling reactions that may be exploited for site-selective arene functionalization.
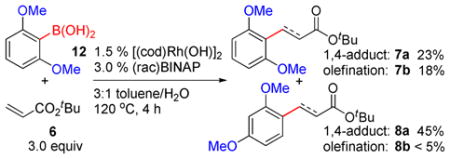 |
(3) |
In summary, we report a novel 1,3-migration of rhodium that was demonstrated in several stoichiometric and catalytic isomerization processes involving proposed Rh(I) 2,6- dimethoxyphenyl intermediates. Mechanistic results from a deuterium-labeling study support a highly unusual, “consecutive 1,4-migration” pathway via sp3 C–H bond activation of the methoxy group. With ongoing studies on further mechanistic details, we aim to better understand structurereactivity correlations in this novel isomerization process and seek broader applications in synthetic chemistry.
Supplementary Material
Acknowledgments
This work was supported by ND EPSCoR (EPS-0447679), the NSF (CHE-1301409 to P.Z.), and the NIH National Center for Research Resources (Grant 2P20 RR015566). Z.-M.S. thanks Nature Science Fund of China (21171162), Jilin Province Youth Foundation (20130522132JH), and SRF for ROCS (State Education Ministry of China) for financial support. We also thank NSF-CRIF (CHE-0946990) for funding the purchase of departmental XRD instrumentation.
Footnotes
Experimental procedures, spectral data, and structural parameters of compounds 2b, 3b and 4b (CIF and PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Leading reviews on C–H bond activations: Goldman AS, Goldberg KI, editors. Activation and Functionalization of C-H Bonds. Washington, DC: 2004. ACS Symposium Series 885.Dyker G, editor. Handbook of C–H Transformations. Vol. 1 Wiley-VCH; 2005. Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem, Int Ed. 2009;48:5094. doi: 10.1002/anie.200806273.Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058.Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.Sun CL, Li BJ, Shi ZJ. Chem Rev. 2011;111:1293. doi: 10.1021/cr100198w.Arockiam PB, Bruneau C, Dixneuf PH. Chem Rev. 2012;112:5879. doi: 10.1021/cr300153j.
- 2.Leading reviews on Pd and Rh migrations: Ma S, Gu Z. Angew Chem, Int Ed. 2005;44:7512. doi: 10.1002/anie.200501298.Shi F, Larock RC. Top Curr Chem. 2010;292:123. doi: 10.1007/128_2008_46.
- 3.Representative studies on 1,4-Rh migration: Oguma K, Miura M, Satoh T, Nomura M. J Am Chem Soc. 2000;122:10464.Hayashi T, Inoue K, Taniguchi N, Ogasawara M. J Am Chem Soc. 2001;123:9918. doi: 10.1021/ja0165234.Zhao J, Yue D, Campo MA, Larock RC. J Am Chem Soc. 2007;129:5288. doi: 10.1021/ja070657l.Matsuda T, Shigeno M, Murakami M. J Am Chem Soc. 2007;129:12086–12087. doi: 10.1021/ja075141g.Seiser T, Roth OA, Cramer N. Angew Chem, Int Ed. 2009;48:6320. doi: 10.1002/anie.200903189.
- 4.Representative studies on 1,4-Pd migration: Tian Q, Larock RC. Org Lett. 2000;2:3329. doi: 10.1021/ol000220h.Karig G, Moon MT, Thasana N, Gallagher T. Org Lett. 2002;4:3115. doi: 10.1021/ol026426v.Huang Q, Fazio A, Dai G, Campo MA, Larock RC. J Am Chem Soc. 2004;126:7460. doi: 10.1021/ja047980y.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j.Hitce J, Retailleau P, Baudoin O. Chem Eur J. 2007;13:792. doi: 10.1002/chem.200600811.Zhao J, Yue D, Campo MA, Larock RC. J Am Chem Soc. 2007;129:5288. doi: 10.1021/ja070657l.Campo MA, Zhang H, Yao T, Ibdah A, McCulla RD, Huang Q, Zhao J, Jenks WS, Larock RC. J Am Chem Soc. 2007;129:6298. doi: 10.1021/ja069238z.Kesharwani T, Larock RC. Tetrahedron. 2008;64:6090.Pan J, Su M, Buchwald SL. Angew Chem Int Ed. 2011;50:8647. doi: 10.1002/anie.201102880.
- 5.Selected studies on 1,4-migration of other transition metals: (Pd and Pt) Singh A, Sharp PR. J Am Chem Soc. 2006;128:5998. doi: 10.1021/ja060159x.(Pt) Crosby SH, Clarkson GJ, Rourke JP. J Am Chem Soc. 2009;131:14142. doi: 10.1021/ja905046n.(Ni) Keen AL, Doster M, Johnson SA. J Am Chem Soc. 2007;129:810. doi: 10.1021/ja067112w.
- 6.Representative studies on 1,5-Rh and Pd migrations: Tobisu M, Hyodo I, Onoe M, Chatani N. Chem Commun. 2008:6013. doi: 10.1039/b806285d.Bour C, Suffert J. Org Lett. 2005;7:653. doi: 10.1021/ol047537s.Mota AJ, Dedieu A, Bour C, Suffert J. J Am Chem Soc. 2005;127:7171. doi: 10.1021/ja050453+.Mota AJ, Dedieu A. J Org Chem. 2007;72:9669. doi: 10.1021/jo701701s.
- 7.Mota AJ, Dedieu A. Inorg Chem. 2009;48:11131. doi: 10.1021/ic901506v. [DOI] [PubMed] [Google Scholar]
- 8.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; 2010. Section 3.5. [Google Scholar]
- 9.Pd-catalyzed C-H activation of methoxy groups by aryl-tomethoxy 1,4-migration: Dyker G. Angew Chem, Int Ed. 1992;31:1023.Dyker G. J Org Chem. 1993;58:6426.
- 10.Leading reviews on transition metal-catalyzed decarboxylation: Gooßen LJ, Rodríguez N, Gooßen K. Angew Chem Int Ed. 2008;47:3100. doi: 10.1002/anie.200704782.Rodríguez N, Gooßen LJ. Chem Soc Rev. 2011;40:5030. doi: 10.1039/c1cs15093f.
- 11.Transition metal-catalyzed decarboxylative cross-coupling was pioneered almost a half-century by Nilsson and coworkers with studies on Cu(I)-catalyzed decarboxylative biaryl synthesis using arene- and heteroarenecarboxylic acids and aryl iodides: Nilsson M. Acta Chem Scand. 1966;20:423.Nilsson M, Ullenius C. Acta Chem Scand. 1968;22:1998.
- 12.Representative recent studies on transition metal-catalyzed decarboxylative couplings for C-C bond formation: (a) Pd(II)- catalyzed decarboxylative Heck-Mizoroki olefination by Myers and coworkers: Myers AG, Tanaka D, Mannion MR. J Am Chem Soc. 2002;124:11250. doi: 10.1021/ja027523m.Pd(II)-catalyzed decarboxylative biaryl synthesis by Gooßen and coworkers: Gooßen LJ, Deng G, Levy LM. Science. 2006;313:662. doi: 10.1126/science.1128684.
- 13.(a) Sun ZM, Zhao P. Angew Chem Int Ed. 2009;48:6726. doi: 10.1002/anie.200901097. [DOI] [PubMed] [Google Scholar]; (b) Sun ZM, Zhang J, Zhao P. Org Lett. 2010;12:992. doi: 10.1021/ol100001b. [DOI] [PubMed] [Google Scholar]
- 14.An Au(I) 2,6-dimethoxyphenyl complex with IPr carbene ligand has been reported to form by stoichiometric decarboxylation of 2,6-dimethoxybenzoic acid: Dupuy S, Lazreg F, Slawin AMZ, Cazin CSJ, Nolan SP. Chem Commun. 2011;47:5455. doi: 10.1039/c1cc10917k.
- 15.Leading studies on stoichiometric and catalytic carboxylation of Rh(I) aryl species: Darensbourg DJ, Groetsch G, Wiegreffe P, Rheingold AL. Inorg Chem. 1987;26:3827.Ukai K, Aoki M, Takaya J, Iwasawa N. J Am Chem Soc. 2006;128:8706. doi: 10.1021/ja061232m.
- 16.Examples of Rh(I) hydroxide-mediated stoichiometric and catalytic C-H bond activation: Kloek SM, Heinekey DM, Goldberg KI. Angew Chem Int Ed. 2007;46:4736. doi: 10.1002/anie.200700270.Sun ZM, Zhang J, Manan RS, Zhao P. J Am Chem Soc. 2010;132:6935. doi: 10.1021/ja102575d.
- 17.For a recent report on Ag- and Cu-catalyzed decarboxylative deuteration, see: Rudzki M, Alcalde-Aragones A, Dzik WI, Rodriguez N, Gooßen LJ. Synthesis. 2012;44:184.
- 18.Leading reviews on Rh(I)-catalyzed addition reactions with organoboron species: Hayashi T, Yamasaki K. Chem Rev. 2003;103:2829. doi: 10.1021/cr020022z.Miyaura N. Bull Chem Soc Jpn. 2008;81:1535.
- 19.Zhao P, Incarvito CD, Hartwig JF. J Am Chem Soc. 2007;129:1876. doi: 10.1021/ja068587q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.