

Abstract
Plasmepsin (Plm) is a potential target for new antimalarial drugs, but most reported Plm inhibitors have relatively low antimalarial activities. We synthesized a series of dipeptide-type HIV protease inhibitors, which contain an allophenylnorstatine-dimethylthioproline scaffold to exhibit potent inhibitory activities against Plm II. Their activities against Plasmodium falciparum in the infected erythrocyte assay were largely different from those against the target enzyme. To improve the antimalarial activity of peptidomimetic Plm inhibitors, we attached substituents on a structure of the highly potent Plm inhibitor KNI-10006. Among the derivatives, we identified alkylamino compounds such as 44 (KNI-10283) and 47 (KNI-10538) with more than 15-fold enhanced antimalarial activity, to the sub-micromolar level, maintaining their potent Plm II inhibitory activity and low cytotoxicity. These results suggest that auxiliary substituents on a specific basic group contribute to deliver the inhibitors to the target Plm.
Keywords: Antimalarial drug, Aspartic protease, Plasmepsin inhibitor, Hydoxymethylcarbonyl, Allophenylnorstatine, Peptidomimetics
1. Introduction
The World Health Organization (WHO) estimated the malaria-infected population to have reached around 350–500 million people in 2004.1 Over one million people lose their lives to the disease annually, especially children in sub-Saharan Africa. The growth of malaria’s endemic region by the development of transportation and global warming make the problem even more severe. The most lethal malarial parasite, Plasmodium falciparum, is increasingly building resistance to available drugs such as chloroquine or sulfadoxine- pyrimethamine.2 Therefore, new anti-malarial drugs are needed with different mechanisms of action from those of existing drugs.3
Malaria parasites invade the human body via the bite of the Anopheles mosquito; through hematocytic multiplication, the parasites proliferate by infecting and multiplying inside erythrocytes. During their trophozoite stage, parasites obtain amino acids by hemoglobin digestion, a process essential for their propagation.4 Hemoglobin is transported to the parasite’s acidic food vacuole, where it is cleaved into small peptides by proteases. In the case of P. falciparum, ten aspartic proteases called plasmepsins (Plms) were identified in its genome,5 and four of them participate in hemoglobin degradation.6 Plm I7,8 and II8,9 are known to start the cleavage; recent knockout studies suggested a requisite inhibition of all the four Plms, that is, Plm I, II, III10 (known as histo-aspartic protease, HAP) and IV11, to prevent proliferation.12–15 Treatment of mouse malaria using pepstatin A suggested the potential of Plm inhibitors as antimalarial drugs.16 Several groups reported Plm inhibitors derived from statine, cathepsin D inhibitor libraries, or HIV protease inhibitors.17–20 Most of these compounds, however, possessed low antimalarial activities except aldehyde21 and non-peptidic22 and primaquine-conjugated compounds,23 highlighting a difficulty of the development of antimalarial agents. Analysis of cleaved fragments of hemoglobin has revealed that Plm I and II recognize Phe33 at the P1 position and Leu34 at the P1′ position.24 The Phe-Leu cleavage site is seen also in the HIV protease substrates.25 We previously identified HIV protease inhibitors containing allophenylnorstatine [Apns; (2S,3S)-3-amino-2-hydroxy-4-phenylbutyric acid] with a hydroxymethylcarbonyl (HMC) as an ideal transition state mimic,26,27 which exhibited potent Plm II inhibitory activities.28 Among them, we found some HIV protease inhibitors such as KNI-76429–31 (1, Fig. 1) possessing antimalarial activity in an infected erythrocyte assay. These findings led us to design KNI-10006 (2, Fig. 2) using (1S,2R)-aminoindanol, which resulted in a remarkable improvement in Plm inhibitory activity.32 Compound 2 exhibited potent inhibitory activity against other Plms related to hemoglobin degradation. Despite its prominent molecular potency, however, KNI-10006 exhibited relatively low activity (EC50 = 6.8 μM) against live parasites.33 We hypothesized that this attenuation comes from its unbalanced hydrophobic-hydrophilic nature because the compound must pass through four membranes to bind to its targets in the parasite food vacuole.6 Further extension of the inhibitor size using tripeptidomimetics revealed superior potencies against Plm II but negligible improvement in antimalarial activity.34 In order to overcome the disadvantage of peptidomimetic character, we modified a specific moiety of 2, that is, the 2,6-dimethylphenoxyacetyl group that caused a similar problem in our previous HIV protease inhibitor study.35 An X-ray crystallographic study disclosed the unique binding of 1 to Plm IV from P. malariae, oriented opposite to that of pepstatin A and also to that of 1 in HIV protease (Fig. 1).36 The HMC interacted with two catalytic Asp residues, but the Apns fit in the S1′ pocket, and (R)-5,5-dimethyl-1,3-thiazolidine-4-carboxylic acid (Dmt) was in the S1 space. The 2-methylbenzyl group did not fit in the S1′ pocket. These observations suggested that compound 2 would bind to other Plms in a similar manner to that of 1 in Plm IV.37 Therefore, the dimethylphenoxyacetyl group of 2 is expected to bind to the S1′ pocket of any Plm (Fig. 2). We herein report the structure-activity relationships of dipeptide-type Plm inhibitors and the enhancement of antimalarial activity in these analogues.
Figure 1.
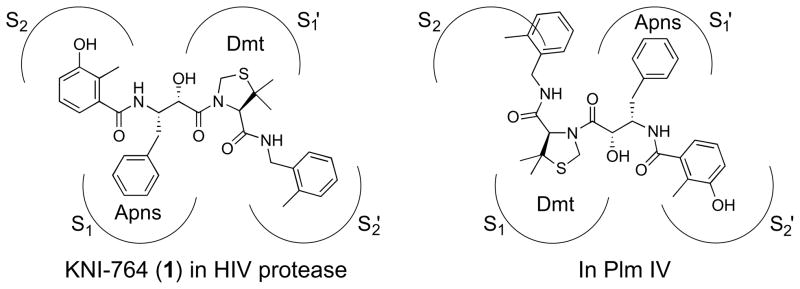
Binding directions of 1 in HIV protease and Plm IV.
Figure 2.

Lead compound 2 and its phenoxyacetyl derivatives.
2. Synthesis
Phenoxyacetic acid derivatives 4a–w were synthesized from commercially available phenols 3, following the general procedure in Scheme 1. o-Nitro derivative 4m was hydrogenated to form the amino group, which was further protected by tert-butyloxycarbonyl (Boc) to obtain 4p. Other amino derivatives 4q and 4r were prepared without protection from the corresponding nitro derivatives 4n and 4o. Some of the p-substituted 2,6-dimethylphenoxy-acetic acids were synthesized from 2,6-dimethyl-4-nitrophenol 5, followed by alkylation and hydrogenation to obtain 6 (Scheme 2). The intermediate was protected with Boc, then hydrolyzed to result in Boc-amino derivative 8. Intermediate 8 was transformed to the hydroxyl derivative 9 via diazonium salt formation. Dimethylamino analogue 10 was prepared by hydrogenation with formaldehyde and saponification. Mono-alkylamino derivatives 11–15 were synthesized using iodomethane or alkylbromides with NaH, hydrolyzed simultaneously. Intermediate 18 was prepared as described in previous reports33,34 from Boc-Dmt-OH 16 with subsequent couplings of aminoindanol and Boc-Apns-OH using benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP) or N-ethyl-N′-[3-(dimethylamino)propyl] carbodiimide hydrochloride (EDC·HCl) plus 1-hydroxybenzotriazole (HOBt) methods and deprotections of the Boc groups using HCl-dioxane, shown in Scheme 3. Phenoxyacetic acid derivatives were then condensed with 18 using BOP. Additional deprotection of the Boc group was performed in the case of compounds 31, 43, 44, and 46–49. p-Butylamino and p-pentylamino derivatives 50 and 51 were synthesized from compound 43 by reductive alkylation with aldehydes and NaBH3CN (Scheme 4). All of the final products were identified by MALDI-TOF MS and FAB-MS after purifications by preparative HPLC.
Scheme 1.

Reagents: (a) BrCH2COOEt, K2CO3, DMF; (b) 1M-NaOH, MeOH; (c) 10% Pd–C, H2, MeOH; (d) (Boc)2O, Et3N, THF–H2O.
Scheme 2.

Reagents and condition: (a) BrCH2COOEt, K2CO3, DMF; (b) 10% Pd–C, H2, MeOH; (c) (Boc)2O, Et3N, THF–H2O; (d) 1 MNaOH, MeOH; (e) 4 MHCl/dioxane; (f) NaNO2, H2SO4, then H2O, reflux 3 h; (g) HCHO aq, 10% Pd–C, H2, MeOH; (h) NaH, CH3I or R–Br, THF.
Scheme 3.

Reagents: (a) (1S,2R)-1-amino-2-indanol, BOP, Et3N, DMF; (b) 4 MHCl/dioxane, anisole; (c) Boc-Apns-OH, EDC·HCl, HOBt·H2O, Et3N, DMF; (d) 4a–4w or 8–15, BOP, Et3N, DMF.
Scheme 4.

Reagents: (a) Et3N, MeOH, butanal or pentanal, NaBH3CN.
3. Results and discussion
Various substituents were attached at each position of the phenoxyacetyl group of KNI-10006 without the inherent dimethyl groups. The inhibitory activity results are summarized in Table 1. Methyl substitution showed Plm II’s preference of the m- and p-positions (20 and 21). Particularly noteworthy is the large difference in Ki value between o-methyl and o-dimethyl groups, 145 (19) and 0.5 nM (2), respectively. The result indicates the importance of the o-dimethyl structure to fit in the pocket of Plm II with some hydrophobic interactions. A methoxy group was preferred only at the m-position (compound 23). Less adaptation of o-methoxy (22) was similar to that of the methyl group. Compound 24 with a p-methoxy group was the worst among these analogues. The extension of the methyl group may cause a hydrophobic repulsion that affects the binding of the whole ligand. Modification of the methyl with a hydroxyl, hydroxymethyl, or amino group resulted in similar potency to methyl substitution with a slight improvement of Plm II inhibitory activity (compounds 25–33). In the case of nitro substitutions (34–36), similar preference at the m- and p-positions was observed, but the difference between these and the o-position was not considerable. We also tested the HIV-1 protease inhibition of these analogues. Preferences of o-substitutions such as methyl (19), hydroxymethyl (28), or amino group (31) were specific for HIV protease compared to Plm II, in which only the nitro group (34) was accepted for Plm II, highlighting the differences of the Plm and HIV protease binding pockets. Compounds with relatively potent Plm II inhibitory activity, with Ki values less than 100 nM, were tested against P. falciparum. Among them, compound 30 with a p-hydroxymethyl group exhibited relatively potent inhibition, EC50 = 1.1 μM, more potent than 1 and 2. Compound 29, with a slightly different substituent position from 30, did not show antimalarial activity. The TD50 value of 30 was >120 μM with more than 100-fold selectivity. Compounds 35 and 36, with nitro substituents, also exhibited moderate antimalarial activity, but they showed some cytotoxicity. These results suggest that small changes in chemical properties affect a compound’s ability to reach the Plms in the food vacuole. The mono-substitution studies resulted in an attenuation of Plm inhibition, suggesting an importance of 2,6-disubstitution for inhibitory activity. Therefore, we shifted our focus to 2,6-substituted derivatives of KNI-10006.
Table 1.
Inhibitory activity of phenoxyacetyl derivatives

| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Plm II Ki (nM) | HIV-1 PR % inhibition (at 50 nM) | Anti-P. falciparum EC50 (μM) | TD50 (μM) |
| 19 (KNI-10079) | CH3 | H | H | 145 | 72 | ||
| 20 (KNI-10080) | H | CH3 | H | 42 | 13 | >8 | |
| 21 (KNI-10081) | H | H | CH3 | 55 | 27 | >8 | |
| 22 (KNI-10092) | OCH3 | H | H | 153 | 27 | ||
| 23 (KNI-10093) | H | OCH3 | H | 4 | 10 | >8 | |
| 24 (KNI-10094) | H | H | OCH3 | 479 | 38 | ||
| 25 (KNI-10369) | OH | H | H | 112 | 34 | ||
| 26 (KNI-10216) | H | OH | H | 10 | 13 | >8 | |
| 27 (KNI-10217) | H | H | OH | 10 | 31 | >8 | |
| 28 (KNI-10113) | CH2OH | H | H | 132 | 61 | ||
| 29 (KNI-10124) | H | CH2OH | H | 9 | 24 | >8 | |
| 30 (KNI-10125) | H | H | CH2OH | 10 | 37 | 1.1 | >120 |
| 31 (KNI-10106) | NH2 | H | H | 126 | 62 | ||
| 32 (KNI-10087) | H | NH2 | H | 29 | 29 | >8 | |
| 33 (KNI-10088) | H | H | NH2 | 28 | 27 | >8 | |
| 34 (KNI-10095) | NO2 | H | H | 55 | 9 | ||
| 35 (KNI-10255) | H | NO2 | H | 38 | 12 | 2.8 | 58 |
| 36 (KNI-10256) | H | H | NO2 | 25 | 53 | 4.2 | 63 |
We changed the dimethyl moiety to difluoro, dichloro, dimethoxy, or diisopropyl groups (37–40). All of the modifications failed to improve Plm II inhibitory activity; in particular, dimethoxy substitutions resulted in 1500-fold decrease in potency (Table 2). The o-disubstituents moderately affected HIV protease inhibition, except diisopropyl, which had no activity. Besides the lower potency against Plm II compared to 2, the antimalarial activity of difluoro, dichloro, and diisopropyl derivatives were improved. Because the dimethyl groups of KNI-10006 appeared indispensable for Plm II inhibition, we decided to attach an additional substituent at the 4-position of the phenyl group. Although fluoro derivative 41 exhibited a moderate decrease in Plm II inhibition, the antimalarial activity was improved to 1.4 μM. This small modification with fluorine yielded fourfold improvement of activity against the parasite. Attachment of a hydroxyl group at the 4-position resulted in slight improvement in Ki compared to fluorine but less antimalarial activity (compound 42). The HIV protease inhibitory activity showed no difference without the hydroxyl group (42 and 2), suggesting no interference to its binding.
Table 2.
Inhibitory activity of 2,6- and 2,4,6-substituted phenoxyacetyl derivatives

| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Plm II Ki (nM) | HIV-1 PR % inhibition (at 50 nM) | Anti-P. falciparum EC50 (μM) | TD50 (μM) |
| 2 | CH3 | H | CH3 | 0.5 | 98 | 6.8 | |
| 37 (KNI-10265) | F | H | F | 14 | 58 | 3.3 | 55 |
| 38 (KNI-10074) | Cl | H | Cl | 39 | 95 | 5.2 | |
| 39 (KNI-10260) | OCH3 | H | OCH3 | 782 | 54 | ||
| 40 (KNI-10152) | CH(CH3)2 | H | CH(CH3)2 | 23 | 0 | 2.9 | 6.3 |
| 41 (KNI-10266) | CH3 | F | CH3 | 19 | 87 | 1.4 | 29 |
| 42 (KNI-10155) | CH3 | OH | CH3 | 5 | 98 | 3.3 | |
| 43 (KNI-1293) | CH3 | NH2 | CH3 | 23 | 96 | ||
| 44 (KNI-10283) | CH3 | NHCH3 | CH3 | 25 | 91 | 0.45 | >120 |
| 45 (KNI-10282) | CH3 | N(CH3)2 | CH3 | 21 | 86 | 1.2 | 45 |
p-Amino substitution on 2,6-dimethylphenoxyacetyl moiety was previously studied with HIV protease inhibitors to improve cellular activity.38,39 Plm II inhibition of free amino and its methylated analogues, compounds 43–45, was moderate around 20 nM with about a 40-fold decrease compared to 2. Besides its moderate activity, an attachment of a monomethylamino group, compound 44, dramatically enhanced antimalarial activity, 0.45 μM, with 15-fold improvement compared to that of 2. The dimethylamino derivative also exhibited relatively potent antimalarial activity. These results suggested that addition of some basicity would contribute to inhibitory activity in the parasite-infected erythrocyte assay. The low cytotoxicity of 44 led us to vary the monomethylamino moiety (Table 3).
Table 3.
Inhibitory activity of monosubstituted-amino KNI-10006 derivatives

| |||||
|---|---|---|---|---|---|
| Compound | R | Plm II Ki (nM) | HIV-1 PR % inhibition (at 50 nM) | Anti-P. falciparum EC50 (μM) | TD50 (μM) |
| 44 |
|
25 | 91 | 0.45 | >120 |
| 46 (KNI-10529) |
|
7 | 84 | 0.72 | 64 |
| 50 (KNI-10539) |
|
30 | 85 | 0.54 | 22 |
| 51 (KNI-10541) |
|
26 | 78 | 0.43 | 12 |
| 47 (KNI-10538) |

|
10 | 91 | 0.30 | >120 |
| 48 (KNI-10526) |

|
21 | 91 | 0.50 | >120 |
| 49 (KNI-10527) |

|
6 | 91 | 0.49 | >120 |
We elongated the carbon chain length with ethyl, butyl, and pentyl (compounds 46, 50 and 51). Among them, compound 46 with the ethyl moiety exhibited the best Plm II inhibition. Antimalarial activity increased along with the chain length; however, cytotoxicity increased as well. Therefore, the first methylamino analogue was the best among the compounds modified in this manner.
Addition of a pyridine group is an option to reduce hydrophobicity, and its basicity would contribute enhancement of antimalarial activity. Therefore, we decided to incorporate a pyridine group on the methyl of 44. Resultant three pyridinylmethyl analogues 47–49 exhibited potent Plm II inhibition, especially 49, with a Ki value of 6 nM.
To explore the moderate attenuation in Plm II inhibitory activity of 49 compared to 2, we constructed a binding model of 49 with Plm II (PDB ID, 1SME) using the molecular modeling package MOE 2007.09 (Chemical Computing Group, Inc., Montreal, Canada). Pepstatin A was replaced with compound 49. The X-ray crystallographic data of inhibitor 1 in Plm IV (PDB ID, 2ANL) was used to generate the initial docking conformation of 49 in Plm II. According to the binding mode of 1, Asp214 was intentionally protonated. After several energy minimization processes with an MMFF94x force field including a 20 Å of water soak around the inhibitor, a molecular dynamics (MD) simulation was performed (Fig. 3). The binding direction of 49 was same with that of 1 but opposite to that of pepstatin A (Fig. 3A). The hydrogen bond interactions of HMC with the two catalytic Asps were deliberately kept in a similar configuration to those observed in HIV protease complexes, that is, the hydroxyl group of HMC interacted with the carboxylate anion of one of the Asps, and the carbonyl interacted with the carboxylic acid proton of the other Asp.40,41 However, these interactions were collapsed in the MD simulation, and the both Asp residues tended to interact only with the hydroxyl group of HMC. Hydrogen bond interactions except HMC were similar to our previous study of tripeptidic inhibitors34 and those suggested by Åqvist’s group.37 The difference was found at the phenoxyacetyl moiety, that is, the p-position of 2,6-dimethylphenoxyacetyl group, was oriented to water outside; therefore, further extension of the pyridine-4-yl-methyl moiety from the p-amino group was favored to be surrounded mostly by water molecules (Fig. 3B and C). This is consistent with Plm II inhibitory activity of 49, suggesting that it is easier for the nitrogen of the 4-pyridine-ring to contact the water than 2- and 3-pyridine. A significant movement was observed in the phenoxyacetyl moiety during the MD simulation with RMSD 1.341 Å ± 0.366, suggesting a loss of contact with the enzyme, resulting in moderate reduction in the activity compared to that of 2. The moderate reduction of 42 with only a hydroxyl group different from 2 may be explained by the same reasoning.
Figure 3.

MD simulated pose of compound 49 bound to Plm II. Asp214 was protonated. (A) Superimposition of pepstatin A (cyan), 1 (yellow) and 49 (red). (B) Pyridinylmethyl group of 49 (green stick) was surrounded by water molecules (lines, shown only 5 Å from the ligand), not in the Plm II binding sites (red surface). (C) Schematic representation of 49. Figures A and B were generated using MacPyMOL (DeLano Scientific LLC, CA, USA).42
Antimalarial activities of pyridinylmethyl derivatives 47–49 were maintained at the sub-micromolar level. The EC50 value of 0.3 μM of 47 is the best among tested derivatives, about a 20-fold improvement over 2. The cytotoxicity of these pyridinemethyl analogues was in the acceptable range for clinical inhibitors. These results suggest that the pyridinemethyl analogues are promising for the development of new antimalarial agents. Interestingly, all of the tested monoalkylamino derivatives exhibited sub-micromolar potency against parasites even though the activities are not proportional to Plm inhibitory activities. This indicates the strong dependence on other factors for the compounds to reach the target Plm. Similar attempts using basic nitrogen were reported by Hallberg’s group.43,44 However, the effect of this basic group could be caused by a different inhibition mechanism without Plm inhibition.45 Further investigations to clarify the effect of compound basicity for delivery of the drug to the acidic food vacuole are required.
4. Conclusions
We synthesized a series of derivatives of 2 with specific substitutions on the phenoxyacetyl moiety, which contain the Apns-Dmt scaffold to exhibit potent inhibitory activity against the Plms. We observed the enhancement of antimalarial activity in cases of compounds modified by attaching both lipophilic and hydrophilic moieties, suggesting an importance of the balance of inhibitor characteristics. In particular, attachment of basic moieties such as a monoalkylamino group contributed to enhance antimalarial activity up to the submicromolar level. In this study, we identified some alkylamino compounds with promising antimalarial activity, maintaining their potent Plm II inhibitory activity and low cytotoxicity. Evaluations of these analogues using malaria parasite-infected mouse models are underway to develop potential antimalarial drugs.
5. Experimental
5.1. Materials
Reagents and solvents were purchased from Wako Pure Chemical Ind., Ltd (Osaka, Japan), Nacalai Tesque (Kyoto, Japan), Aldrich Chemical Co. Inc. (Milwaukee, WI, USA), and Tokyo Kasei Kogyo Co., Ltd (Tokyo, Japan) and used without further purification. TLC was performed using Merck Silica gel 60 F254 precoated plates. Column chromatography was performed on Merck 107734 silica gel 60 (70–230 mesh). Melting points were measured on a Yanagimoto micro melting apparatus. Preparative HPLC was carried out on a C18 reverse phase column (20 × 250 mm; YMC Pack ODS SH343-5) with a binary solvent system: a linear gradient of CH3CN in 0.1% aqueous trifluoroacetic acid (TFA) with a flow rate of 5.0 mL/min and detection at 230 nm. 1H NMR spectra were obtained on a JEOL AL300 (300 MHz) spectrometer with TMS as an internal standard. Mass spectra (electrospray ionization, methanol as the mobile phase) were analysed with SHIMADZU LCMS-2010 spectrometer. MALDI-TOF MS was performed on a Voyager-DE RP spectrometer (PerSeptive Biosystems, Inc.). High resolution FAB-MS was performed on a JEOL JMS-SX102A spectrometer equipped with the JMA-DA7000 data system. Analytical HPLC was performed using a C18 reverse phase column (4.6 × 150 mm; YMC Pack ODS AM-302) with a binary solvent system: linear gradient of CH3CN 20–80% in 0.1% aqueous TFA in 30 min at a flow rate of 0.9 mL/min, detected at 230 nm. The purity of the desired compounds was greater than 95%.
5.2. Chemistry
5.2.1. Ethyl 4-(tert-butyloxycarbonylamino)-2,6-dimethylphenoxyacetate (7)
To a mixture of 2,6-dimethyl-4-nitrophenol (10.0 g, 60 mmol), K2CO3 (10.0 g, 72 mmol) in DMF (100 mL) was added ethyl bromoacetate (9.27 mL, 84 mmol) in an ice-water bath, and the mixture was stirred overnight at room temperature. After removal of the solvent in vacuo, the residue was dissolved in EtOAc, filtered insoluble solid, washed sequentially with 10% citric acid, 5% NaHCO3, and saturated NaCl, dried over MgSO4, and concentrated in vacuo. A mixture of the residue and 10% Pd–C in MeOH (100 mL) was vigorously stirred with a H2 balloon. After 4 h stirring, the reaction mixture was filtered, concentrated in vacuo to give amino intermediate 6; yield 96%. MS (ES+) m/z: 224 for [M+H]+.
To a solution of the residue and triethylamine (12.1 mL) in THF–H2O (1:1, 100 mL) was added (Boc)2O (14.0 g, 64 mmol) and stirred overnight at room temperature. The reaction mixture was concentrated in vacuo, diluted with EtOAc, washed with acid and base mentioned above, dried over MgSO4, and concentrated in vacuo. Precipitation with n-hexane gave 18.0 g of the titled compound as white solid; yield 92%; mp 106–108 °C. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 9.10 (s, 1H), 7.09 (s, 2H), 4.38 (s, 2H), 4.18 (q, J = 7.1 Hz, 2 H), 2.16 (s, 6 H), 1.46 (s, 9 H) 4.18 (t, J = 7.1 Hz, 3H); MS (ES+) m/z: 346 for [M+Na]+.
5.2.2. 4-(N-tert-Butyloxycarbonyl-N′-methyl)amino-2,6-dimethylphenoxyacetic acid (11)
Compound 11 was prepared from intermediate 7 in a manner similar to that described for compound 15; yield 74%; mp 97–99 °C. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.8 (br, 1H), 6.91 (s, 2H), 4.35 (s, 2H), 3.11 (s, 3H), 2.21 (s, 6H), 1.38 (s, 9H); MS (ES+) m/z: 308 for [M–H]−.
5.2.3. 4-(N-tert-Butyloxycarbonyl-N′-ethyl)amino-2,6-dimethylphenoxyacetic acid (12)
Compound 12 was prepared from intermediate 7 in a manner similar to that described for compound 15; yield 97%; mp 121–123 °C. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 6.80 (s, 2H), 3.90 (s, 2H), 3.51 (q, J = 6.9 Hz, 2H), 2.18 (s, 6H), 1.36 (s, 9H), 1.03 (t, J = 6.9 Hz, 3H); MS (ES+) m/z: 322 for [M–H]−.
5.2.4. 4-(N-tert-Butyloxycarbonyl-N′-pyridin-2-yl)amino-2,6-dimethylphenoxyacetic acid (13)
Compound 13 was prepared from intermediate 7 in a manner similar to that described for compound 15; yield 90%; mp 156–158 °C. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.50–8.46 (m, 1H), 7.80–7.73 (m, 1H), 7.30 (d, J = 8.1 Hz, 1H), 7.26–7.22 (m, 1H), 6.97 (s, 2H), 4.80 (s, 2H), 4.32 (s, 2H), 2.16 (s, 6H), 1.29 (s, 9H); MS (ES+) m/z: 385 for [M–H]−.
5.2.5. 4-(N-tert-Butyloxycarbonyl-N′-pyridin-3-yl)amino-2,6-dimethylphenoxyacetic acid (14)
Compound 14 was prepared from intermediate 7 in a manner similar to that described for compound 15; yield 95% as oil. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.44 (dd, J = 4.5 Hz, 1.5 Hz, 1H), 8.40 (d, J = 2.1 Hz, 1H), 7.60 (d, J = 8.1 Hz, 1H), 7.34 (dd, J = 7.8 Hz, 4.8 Hz, 1H), 6.87 (s, 2H), 4.78 (s, 2H), 4.33 (s, 2H), 2.17 (s, 6H), 1.35 (s, 9H); MS (ES+) m/z: 385 for [M–H]−.
5.2.6. 4-(N-tert-Butyloxycarbonyl-N′-pyridin-4-yl)amino-2,6-dimethylphenoxyacetic acid (15)
To a solution of compound 7 (100 mg, 0.31 mmol) in dry THF (5 mL) was added sodium hydride (74 mg, 3.1 mmol) and stirred for 1 h under ice-water cooling and Ar atmosphere, then 4-(bromomethyl) pyridine hydrobromide (235 mg, 0.93 mmol) was added and stirred overnight at room temperature. Additional sodium hydride (74 mg, 3.1 mmol) was added and stirred overnight at room temperature. The reaction mixture was diluted with ether, extracted with 5% NaHCO3. The aqueous layer was neutralized with citric acid, saturated with NaCl, extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. Precipitation with n-hexane gave 102 mg of the titled compound as a brownish solid; yield 86%; mp 188–190 °C. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.50 (d, J = 4.5 Hz, 2H), 7.22 (d, J = 4.5 Hz, 2H), 6.92 (s, 2H), 4.78 (s, 2H), 4.33 (s, 2H), 2.17 (s, 6H), 1.33 (s, 9H); MS (ES+) m/z: 385 for [M–H]−.
5.2.7. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide hydrochloride salt (18)
To a solution of Boc-Dmt-OH (16, 8.47 g, 32.4 mmol) in DMF (80 mL) were added triethylamine (6.79 mL, 48.6 mmol), (1 S,2R)-1-amino-2-indanol (5.33 g, 35.7 mmol) and BOP (15.74 g, 35.6 mmol) in an ice-water bath, and the mixture was stirred overnight at room temperature. After removal of the solvent in vacuo, the residue was mixed with EtOAc, washed sequentially with 10% citric acid, 5% NaHCO3, and saturated NaCl, dried over MgSO4, and concentrated in vacuo to give 13.6 g of 17 as a white solid (MS (ES+) m/z: 393 for [M+H]+).
A mixture of 19, anisole (7.04 mL, 64.8 mmol), and 4 M HCl in dioxane (100 mL) was stirred at room temperature for 1.5 h. After removal of the solvent in vacuo, the residue was precipitated from ether and filtered to give the hydrochloride salt. To a solution of the salt in DMF (80 mL) were added HOBt·H2O (5.45 g, 35.6 mmol), Boc-Apns-OH (10.51 g, 35.6 mmol), and EDC·HCl (6.82 g, 35.6 mmol). Under stirring in an ice-water bath, triethylamine (4.53 mL, 32.4 mmol) was slowly added for 20 min. The mixture was stirred overnight at room temperature. After removal of the solvent in vacuo, the residue was mixed with EtOAc, washed sequentially with 10% citric acid, 5% NaHCO3, and saturated NaCl, dried over MgSO4, and concentrated in vacuo. Purification of the product by silica gel column chromatography (n-hexane–EtOAc) gave 14.85 g of white solid; yield 80% (MS (ES+) m/z: 593 for [M+Na]+). A mixture of the intermediate, anisole (5.67 mL, 52.2 mmol), and 4 M HCl in dioxane (85 mL) was stirred at room temperature for 1.5 h. After removal of the solvent in vacuo, the residue was precipitated from ether to give 12.34 g of the titled compound; yield 75% from 18; mp 141–143 °C. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.19 (d, J = 9.0 Hz, 1H), 8.02 (br, 3H), 7.41–7.12 (m, 9H), 6.22 (br, 1H), 5.25 (dd, J = 8.6 Hz, 5.0 Hz, 1H), 5.09 (br, 1H), 4.91 (d, J = 8.7 Hz, 1H), 4.71–4.68 (m, 2H), 4.56–4.51 (m, 1H), 4.44–4.38 (m, 1H), 3.58–3.49 (m, 1H, over-lapped with H2O), 3.04 (dd, J = 16.5 Hz, 4.5 Hz, 1H), 2.96–2.78 (m, 3H), 1.55 (s, 3H), 1.46 (s, 3H); MS (ES+) m/z: 470 for [M+H]+.
5.2.8. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2-methylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (19)
To a solution of 18 (100 mg, 0.20 mmol) in DMF (3 mL) were added triethylamine (55.2 μL, 0.40 mmol), 2-methylphenoxyacetic acid (36.1 mg, 0.22 mmol) and BOP (96.1 mg, 0.22 mmol) in an ice-water bath and the mixture was stirred overnight at room temperature. After removal of the solvent in vacuo, the residue was dissolved in EtOAc, washed sequentially with 10% citric acid, 5% NaHCO3, and saturated NaCl, dried over MgSO4, and concentrated in vacuo. The residue was precipitated from hexane to give the crude product. Purification of the product by preparative HPLC gave the titled compound as a white foam; yield 72%; 1H NMR (DMSO-d6) δ (ppm): 8.23 (d, J = 9.0 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.36–6.99 (m, 11H), 6.85–6.81 (m, 1H), 6.59 (d, J = 7.8 Hz, 1H), 5.43 (d, J = 7.2 Hz, 1H), 5.29 (dd, J = 8.6 Hz, 5.0 Hz, 1H), 5.03 (d, J = 4.5 Hz, 1H), 4.94 (s, 2H), 4.74 (s, 1H), 4.49–4.25 (m, 4H), 3.09–3.01 (m, 1H), 2.84–2.68 (m, 3H), 2.20 (m, 3H), 1.56 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 619 [M+H]+; HRMS (FAB+) calcd for C34H39N3O6SNa [M+Na]+ 640.2457, found m/z 640.2452.
5.2.9. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(3-methylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (20)
Compound 20 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.26 (d, J = 9.0 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.34–7.06 (m, 10H), 6.75 (d, J = 7.8 Hz, 1H), 6.67 (s, 1H), 6.57 (d, J = 8.7 Hz, 1H), 5.40 (d, J = 6.9 Hz, 1H), 5.31–5.24 (m, 1H), 5.04 (d, J = 3.9 Hz, 1H), 4.95 (s, 2H), 4.75 (s, 1H), 4.49–4.21 (m, 5H), 3.07–3.02 (m, 1H), 2.85–2.68 (m, 3H), 2.24 (m, 3H), 1.55 (s, 3H), 1.46 (d, J = 5.7 Hz, 3H); MS (TOF) m/z: 619 [M+H]+; HRMS (FAB+) calcd for C34H39N3O6SNa [M+Na]+ 640.2457, found m/z 640.2454.
5.2.10. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(4-methylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (21)
Compound 21 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.26 (d, J = 8.4 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.34–6.99 (m, 11H), 6.68 (d, J = 8.7 Hz, 2H), 5.40 (d, J = 6.9 Hz, 1H), 5.31–5.26 (m, 1H), 5.04 (d, J = 4.5 Hz, 1H), 4.95 (s, 2H), 4.75 (s, 1H), 4.49–4.19 (m, 5H), 3.08–3.02 (m, 1H), 2.85–2.68 (m, 3H), 2.20 (s, 3H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 619 [M+H]+; HRMS (FAB+) calcd for C34H39N3O6SNa [M+Na]+ 640.2457, found m/z 640.2451.
5.2.11. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2-methoxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (22)
Compound 22 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.18 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.29–7.05 (m, 9H), 7.00–6.90 (m, 2H), 6.79–6.69 (m, 2H), 5.39 (br, 1H), 5.28 (dd, J = 8.4 Hz, 5.1 Hz, 1H), 5.04 (br, 1H), 4.96 (s, 2H), 4.75 (s, 1H), 4.50 (s, 1H), 4.39–4.22 (m, 4H), 3.78 (s, 3H), 3.08–3.02 (m, 1H), 2.87–2.79 (m, 2H), 2.71–2.62 (m, 1H), 1.56 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 635 [M+H]+; HRMS (FAB+) calcd for C34H39N3O7SNa [M+Na]+ 656.2406, found m/z 656.2399.
5.2.12. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(3-methoxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (23)
Compound 23 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.26 (d, J = 9.0 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.34–7.29 (m, 2H), 7.23–7.08 (m, 8H), 6.54–6.50 (m, 1H), 6.45 (s, 1H), 6.37–6.34 (m, 1H), 5.40 (br, 1H), 5.28 (dd, J = 8.7 Hz, 5.1 Hz, 1H), 5.03 (br, 1 H), 4.95 (s, 2H), 4.75 (s, 1H), 4.48–4.24 (m, 5H), 3.69 (s, 3H), 3.08–3.02 (m, 1H), 2.85–2.73 (m, 3H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 635 [M+H]+; HRMS (FAB+) calcd for C34H39N3O7SNa [M+Na]+ 656.2406, found m/z 656.2415.
5.2.13. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(4-methoxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (24)
Compound 24 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.24 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 9.3 Hz, 1H), 7.35–7.29 (m, 3H), 7.22–7.08 (m, 9H), 6.86–6.82 (m, 4H), 5.41 (d, J = 7.2 Hz, 1H), 5.28 (dd, J = 8.9 Hz, 5.0 Hz, 1H), 5.04 (d, J = 4.5 Hz, 1H), 4.95 (s, 2H), 4.75 (s, 1H), 4.49–4.41 (m, 2H), 4.33–4.24 (m, 3H), 3.68 (s, 3H), 3.09–3.02 (m, 1H), 2.85–2.73 (m, 3H), 1.56 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 635 [M+H]+; HRMS (FAB+) calcd for C34H39N3O7SNa [M+Na]+ 656.2406, found m/z 656.2404.
5.2.14. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2-hydroxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (25)
Compound 25 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 9.19 (s, 1H), 8.45 (d, J = 9.0 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.43–7.28 (m, 3H), 7.22–7.05 (m, 6H), 6.82–6.77 (m, 3H), 6.70–6.44 (m, 1H), 5.28 (dd, J = 8.4 Hz, 5.1 Hz, 1H), 5.05 (d, J = 9.0 Hz, 1H), 4.97 (d, J = 9.3 Hz, 1H), 4.76 (s, 1H), 4.55 (d, J = 3.0 Hz, 1H), 4.43–4.26 (m, 4H), 3.06 (dd, J = 6.6 Hz, 4.7 Hz, 1H), 2.90–2.71 (m, 3 H), 1.56 (d, J = 15.3 Hz, 3H), 1.47 (s, 3H); MS (TOF) m/z: 621 [M+H]+; HRMS (FAB+) calcd for C33H37N3O7SNa [M+Na]+ 642.2250, found m/z 642.2256.
5.2.15. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(3-hydroxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (26)
Compound 26 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 9.39 (s, 1H), 8.17 (d, J = 8.7 Hz, 1H), 8.07 (d, J = 9.0 Hz, 1H), 7.31 (t, J = 6.2 Hz, 1H), 7.25–7.06 (m, 6H), 6.98 (t, J = 8.1 Hz, 1H), 6.38–6.35 (m, 1H), 6.30 (d, J = 2.3 Hz, 1H), 6.22 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 5.31–5.26 (m, 1H), 4.95 (s, 1H), 4.74 (s, 1H), 4.48 (d, J = 3.0 Hz, 1H), 4.44–4.40 (m, 1H), 4.35–4.22 (m, 3H), 3.05 (dd, J = 16.2 Hz, 4.5 Hz, 1H), 2.89–2.71 (m, 3H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 621 [M+H]+; HRMS (FAB+) calcd for C33H37N3O7SNa [M+Na]+ 642.2250, found m/z 642.2245.
5.2.16. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(4-hydroxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (27)
Compound 27 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.92 (br, 1H), 8.13 (d, J = 8.7 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.22–7.11 (m, 5H), 7.06 (t, J = 7.2 Hz, 1H), 6.64–6.57 (m, 4H), 5.26 (dd, J = 8.6 Hz, 4.8 Hz, 1H), 4.93 (s, 2H), 4.72 (s, 1H), 4.45 (d, J = 3.3 Hz, 1H), 4.42–4.38 (m, 1H), 4.33–4.22 (m, 3H), 3.03 (dd, J = 15.9 Hz, 4.5 Hz, 1H), 2.87–2.66 (m, 3H), 1.54 (s, 3H), 1.45 (s, 3H); MS (TOF) m/z: 621 [M+H]+; HRMS (FAB+) calcd for C33H37N3O7SNa [M+Na]+ 642.2250, found m/z 642.2257.
5.2.17. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2-hydroxymethylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (28)
Compound 28 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.28 (d, J = 8.7 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 7.41–7.28 (m, 4H), 7.22–7.08 (m, 7H), 6.98–6.91 (m, 1H), 6.64 (d, J = 7.8 Hz, 1H), 5.35–5.23 (m, 2H), 5.17–5.09 (m, 1H), 5.04–4.93 (m, 3H), 4.75 (s, 1H), 4.56–4.41 (m, 7H), 4.33–4.21 (m, 1H), 3.07–3.02 (m, 1H), 2.84–2.65 (m, 3H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 657 [M+Na]+; HRMS (FAB+) calcd for C34H39N3O7SNa [M+Na]+ 656.2406, found m/z 656.2411.
5.2.18. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(3-hydroxymethylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (29)
Compound 29 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.27 (d, J = 8.4 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.35–7.08 (m, 10H), 6.90–6.83 (m, 2H), 6.63 (d, J = 8.1 Hz, 1H), 5.40 (d, J = 6.9 Hz, 1H), 5.31–5.15 (m, 2H), 5.04 (d, J = 4.5 Hz, 1H), 4.96 (s, 2H), 4.75 (s, 1H), 4.78–4.19 (m, 7H), 3.07–3.03 (m, 1H), 2.84–2.68 (m, 3H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 635 [M+H]+; HRMS (FAB+) calcd for C34H39N3O7SNa [M+Na]+ 656.2406, found m/z 656.2402.
5.2.19. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(4-hydroxymethylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (30)
1H NMR (DMSO-d6) δ (ppm): 8.28 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.35–7.06 (m, 11H), 6.73 (d, J = 8.7 Hz, 2H), 5.40 (d, J = 6.9 Hz, 1H), 5.31–5.26 (m, 1H), 5.07–5.03 (m, 2H), 4.95 (s, 2H), 4.75 (s, 1H), 4.49–4.22 (m, 7H), 3.07–3.03 (m, 1H), 2.84–2.68 (m, 3H), 1.55 (s, 3H), 1.46 (s, 3H); MS (TOF) m/z: 635 [M+H]+; HRMS (FAB+) calcd for C34H39N3O7SNa [M+Na]+ 656.2406, found m/z 656.2400.
5.2.20. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2-aminophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (31)
Compound 31 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.16 (d, J = 9.0 Hz, 1H), 7.30–7.12 (m, 10H), 6.95–6.87 (m, 4H), 5.42 (br, 1H), 5.23–5.18 (m, 1H), 5.05 (br, 1H), 4.93 (s, 2H), 4.74 (d, J = 6.6 Hz, 1H), 4.58 (s, 1H), 4.55 (s, 2H), 4.42–4.35 (m, 1H), 4.14 (d, J = 6.9 Hz, 1H), 3.07–2.98 (m, 3H), 2.85–2.72 (m, 1H), 1.56 (s, 3H), 1.45 (s, 3H); MS (TOF) m/z: 620 [M+H]+; HRMS (FAB+) calcd for C33H38N4O6SNa [M+Na]+ 641.2410, found m/z 641.2415.
5.2.21. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(3-aminophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (32)
Compound 32 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.37 (d, J = 8.7 Hz, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.35–7.08 (m, 10H), 6.95–6.81 (m, 2H), 6.64 (d, J = 8.1 Hz, 1H), 5.29–5.25 (m, 1H), 4.97 (d, J = 3.6 Hz, 2H), 4.75 (s 1H), 4.51 (d, J = 3.0 Hz, 3H), 4.46–4.37 (m, 3H), 4.33–4.25 (m, 2H), 3.05–3.01 (m, 1H), 2.85–2.73 (m, 3H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 620 [M+H]+; HRMS (FAB+) calcd for C33H38N4O6SNa [M+Na]+ 641.2410, found m/z 641.2415.
5.2.22. (R)-N-[(1S,2R)-2-Hhydroxyindan-1-yl]-3-[(2S,3S)-3-(4-aminophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (33)
Compound 33 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.58 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.35–7.08 (m, 11H), 6.86–6.80 (m, 2H), 5.30–5.25 (m, 1H), 4.96 (d, J = 3.0 Hz, 2H), 4.76 (s, 1H), 4.49–4.41 (m, 4H), 4.34–4.24 (m, 1H), 3.05 (d, J = 11.4 Hz, 1H), 2.85–2.72 (m, 3H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 620 [M+H]+; HRMS (FAB+) calcd for C33H38N4O6SNa [M+Na]+ 641.2410, found m/z 641.2403.
5.2.23. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2-nitrophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (34)
Compound 34 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.18 (d, J = 8.7 Hz, 1H), 8.08 (d, J = 8.7 Hz, 1H), 7.88 (dd, J = 8.1 Hz, 1.8 Hz, 1H), 7.33–7.27 (m, 3H), 7.27–7.06 (m, 7H), 6.86 (d, J = 8.4 Hz, 1H), 5.35 (br, 1H), 5.27 8.18 (dd, J = 8.9 Hz, 5.0 Hz, 1H), 5.15 (br, 1H), 4.96 (s, 2H), 4.75 (s, 1H), 4.64 (s, 2H), 4.54–4.49 (m, 1H), 4.43–4.38 (m, 1H), 4.32–4.23 (m, 1H), 3.04 (dd, J = 15.9 Hz, 4.8 Hz, 1H), 2.90–2.78 (m, 2H), 2.75–2.63 (m, 1H), 1.55 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 650 [M+H]+; HRMS (FAB+) calcd for C33H36N4O8SNa [M+Na]+ 671.2152, found m/z 671.2155.
5.2.24. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(3-nitrophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (35)
Compound 35 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.34 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.7 Hz, 1H), 7.83–7.78 (m, 1H), 7.65 (t, J = 2.3 Hz, 1H), 7.50 (t, J = 8.3 Hz, 1H), 7.31–7.04 (m, 10H), 5.30–5.24 (m, 1H), 4.96 (d, J = 3.3 Hz, 2H), 4.75 (s, 1H), 4.57 (s, 2H), 4.50 (d, J = 3.3 Hz, 1H), 4.43–4.39 (m, 1H), 4.33–4.22 (m, 1H), 3.08–3.00 (m, 1H), 2.88–2.68 (m, 3H), 1.55 (s, 3H), 1.46 (s, 3H); MS (TOF) m/z: 650 [M+H]+; HRMS (FAB+) calcd for C33H36N4O8SNa [M+Na]+ 671.2152, found m/z 671.2159.
5.2.25. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(4-nitrophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (36)
Compound 36 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.40 (d, J = 9.0 Hz, 1H), 8.14–8.06 (m, 3H), 7.35–7.04 (m, 9H), 6.92 (d, J = 9.0 Hz, 2H), 5.27 (dd, J = 8.7 Hz, 5.1 Hz, 1H), 4.96 (d, J = 2.4 Hz, 2H), 4.75 (s, 1H), 4.63 (s, 2H), 4.50 (d, J = 3.3 Hz, 1H), 4.43–4.39 (m, 1H), 4.32–4.22 (m, 1H), 3.04 (dd, J = 16.1 Hz, 4.7 Hz, 2H), 2.89–2.68 (m, 3H), 1.55 (s, 3H), 1.46 (s, 3H); MS (TOF) m/z: 650 [M+H]+; HRMS (FAB+) calcd for C33H36N4O8SNa [M+Na]+ 671.2152, found m/z 671.2148.
5.2.26. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-difluorophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (37)
Compound 37 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.17 (d, J = 8.4 Hz, 1H), 8.07 (d, J = 9.0 Hz, 1H), 7.33–7.27 (m, 3H), 7.22–7.04 (m, 9H), 5.28 (dd, J = 9.0 Hz, 4.8 Hz, 1H), 4.94 (s, 2H), 4.74 (s, 1H), 4.51–4.47 (m, 3H), 4.43–4.39 (m, 1H), 4.33–4.24 (m, 1H), 3.08–3.01 (m, 1H), 2.87–2.78 (m, 2H), 2.75–2.66 (m, 1H), 1.56 (s, 3H), 1.46 (s, 3H); MS (TOF) m/z: 641 [M+H]+; HRMS (FAB+) calcd for C33H35F2N3O6SNa [M+Na]+ 662.2112, found m/z 662.2108.
5.2.27. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dichlorophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (38)
Compound 38 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.17 (d, J = 9.0 Hz, 1H), 8.08 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 4.1 Hz, 2H), 7.36 (d, J = 7.2 Hz, 2H), 7.29 (d, J = 7.5 Hz, 1H), 7.24–7.11 (m, 6H), 7.04 (t, J = 7.1 Hz, 1H), 5.42 (br, 1H), 5.31–5.26 (m, 1H), 5.17 (br, 1H), 4.96 (d, J = 5.1 Hz, 2H), 4.75 (s, 1H), 4.54–4.50 (m, 1H), 4.44–4.36 (m, 3H), 4.25 (d, J = 13.8 Hz, 1H), 3.05 (dd, J = 16.1 Hz, 5.0 Hz, 1H), 2.91–2.71 (m, 3H), 1.56 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 673 [M+H]+; HRMS (FAB+) calcd for C33H35Cl2N3O6SNa [M+Na]+ 694.1521, found m/z 694.1526.
5.2.28. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethoxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (39)
Compound 39 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.27 (d, J = 9.3 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 7.28 (d, J = 7.2 Hz, 2H), 7.23–7.03 (m, 10H), 6.73 (d, J = 8.4 Hz, 2H), 5.28 (dd, J = 8.4 Hz, 5.1 Hz, 1H), 5.00 (d, J = 9.0 Hz, 1H), 4.91 (d, J = 9.0 Hz, 1H), 4.77 (s, 1H), 4.52 (d, J = 2.7 Hz, 1H), 4.44–4.37 (m, 2H), 4.30 (d, J = 15.9 Hz, 1H), 4.17 (d, J = 15.6 Hz, 1H), 3.84 (s, 6H), 3.05 (dd, J = 16.2 Hz, 4.5 Hz, 1H), 2.92–2.60 (m, 3H), 1.56 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 665 [M+H]+; HRMS (FAB+) calcd for C33H41N3O8SNa [M+Na]+ 686.2512, found m/z 686.2505.
5.2.29. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-diisopropylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (40)
Compound 40 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.14 (d, J = 9.0 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.36 (d, J = 6.9 Hz, 2H), 7.30–7.09 (m, 9H), 7.02 (t, J = 7.5 Hz, 1H), 5.27 (dd, J = 8.4 Hz, 5.1 Hz, 1H), 4.99 (s, 2H), 4.76 (s, 1H), 4.54 (d, J = 3.3 Hz, 1H), 4.46–4.37 (m, 2H), 4.13 (d, J = 14.1 Hz, 1H), 3.96 (d, J = 14.1Hz, 1H), 3.23–3.01 (m, 3H), 2.94–2.74 (m, 3H), 1.56 (s, 3H), 1.49 (s, 3H), 1.14–1.10 (m, 12H); MS (TOF) m/z: 689 [M+H]+; HRMS (FAB+) calcd for C39H49N3O6SNa [M+Na]+ 710.3240, found m/z 710.3237.
5.2.30. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-fluorophenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (41)
Compound 41 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.20 (d, J = 9.0 Hz, 1H), 8.08 (d, J = 9.0 Hz, 1H), 7.37 (d, J = 6.9 Hz, 2H), 7.29–7.11 (m, 6H), 7.02 (d, J = 7.1 Hz, 1H), 6.86 (s, 1H), 6.83 (s, 1H), 5.27 (dd, J = 8.4 Hz, 5.1 Hz, 1H), 4.96 (s, 2H), 4.75 (s, 1H), 4.50 (d, J = 3.3 Hz, 1H), 4.44–4.29 (m, 2H), 4.18 (d, J = 14.1 Hz, 1H), 3.99 (d, J = 14.1 Hz, 1H), 3.04 (dd, J = 15.9 Hz, 5.1 Hz, 1H), 2.92–2.71 (m, 3H), 2.15 (s, 6H), 1.56 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 650 [M+H]+; HRMS (FAB+) calcd for C35H40FN3O6SNa [M+Na]+ 672.2520, found m/z 672.2516.
5.2.31. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-hydroxyphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (42)
Compound 42 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.97 (s, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.10 (d, J = 9.0 Hz, 1H), 7.37 (d, J = 7.2 Hz, 2H), 7.30–7.11 (m, 6H), 7.06–7.00 (m, 1H), 6.37 (s, 2H), 5.44 (d, J = 6.6 Hz, 1H), 5.30–5.26 (m, 1H), 5.04 (d, J = 4.2 Hz, 1H), 4.96 (s, 2H), 4.76 (s, 1H), 4.52–4.46 (m, 1H), 4.42–4.26 (m, 1H), 4.11 (d, J = 14.4 Hz, 1H), 3.89 (d, J = 14.4 Hz, 1H), 3.08–3.01 (m, 1H), 2.89–2.70 (m, 3H), 2.09–2.07 (m, 6H), 1.56 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 649 [M+H]+; HRMS (FAB+) calcd for C35H41N3O7SNa [M+Na]+ 670.2563, found m/z 670.2558.
5.2.32. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(4-amino-2,6-dimethylphenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (43)
Compound 43 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.21 (d, J = 8.8 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.37 (d, J = 7.3 Hz, 2H), 7.31–7.10 (m, 8H), 7.03 (t, J = 7.2 Hz, 1H), 6.78 (s, 2H), 5.28 (dd, J = 8.1 Hz, 4.7 Hz, 1H), 4.96 (s, 2H), 4.76 (s, 1H), 4.50 (d, J = 2.9 Hz, 1H), 4.45–4.27 (m, 2H), 4.19 (d, J = 14.3 Hz, 1H), 4.07–3.96 (m, 1H), 3.05 (dd, J = 16.0 Hz, 5.1 Hz, 1H), 2.92–2.71 (m, 3H), 2.14 (s, 6H), 1.56 (s, 3H), 1.48 (s, 3H); HRMS (FAB+) calcd for C35H42N4O6SNa [M+Na]+ 669.2723, found m/z 669.2728.
5.2.33. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-methylamino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (44)
Compound 44 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.14 (d, J = 9.0 Hz, 1H), 8.07 (d, J = 9.0 Hz, 1H), 7.37 (d, J = 6.9 Hz, 2H), 7.30–7.11 (m, 6H), 7.03 (t, J = 7.2 Hz, 1H), 6.54 (s, 2H), 5.28 (dd, J = 8.6 Hz, 5.0 Hz, 1H), 4.96 (d, J = 2.7 Hz, 2H), 4.75 (s, 1H), 4.50 (d, J = 3.3 Hz, 1H), 4.44–4.34 (m, 2H), 4.15 (d, J = 14.4 Hz, 1H), 3.94 (d, J = 14.1 Hz, 1H), 3.05 (dd, J = 16.1v, 5.3 Hz, 1H), 2.91–2.77 (m, 3H), 2.73 (s, 3H), 2.11 (s, 6H), 1.56 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 662 [M+H]+; HRMS (FAB+) calcd for C36H44N4O6SNa [M+Na]+ 683.2879, found m/z 683.2876.
5.2.34. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-dimethylamino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (45)
Compound 45 was prepared from intermediate 18 in a manner similar to that described for compound 19. 1H NMR (DMSO-d6) δ (ppm): 8.15 (d, J = 8.7 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.37 (d, J = 6.9 Hz, 2H), 7.30–7.11 (m, 6H), 7.03 (t, J = 6.9 Hz, 1H), 6.66 (s, 2H), 5.28 (dd, J = 8.6 Hz, 5.0 Hz, 1H), 4.96 (d, J = 2.7 Hz, 2H), 4.75 (s, 1H), 4.50 (d, J = 3.3 Hz, 1H), 4.44–4.34 (m, 2H), 4.15 (d, J = 14.4 Hz, 1H), 3.94 (d, J = 14.1 Hz, 1H), 3.04 (dd, J = 16.1 Hz, 5.3 Hz, 1H), 2.91 (s, 6H), 2.88–2.75 (m, 3H), 2.14 (s, 6H), 1.56 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 676 [M+H]+; HRMS (FAB+) calcd for C37H46N4O6SNa [M+Na]+ 697.3036, found m/z 697.3042.
5.2.35. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-ethylamino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (46)
Compound 46 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.17 (d, J = 8.7 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.37 (d, J = 6.9 Hz, 2H), 7.31–7.10 (m, 6H), 7.03 (t, J = 6.9 Hz, 1H), 6.59 (s, 2H), 5.28 (dd, J = 8.1 Hz, 5.1 Hz, 1H), 4.96 (d, J = 2.4 Hz, 2H), 4.75 (s, 1H), 4.50 (d, J = 3.3 Hz, 1H), 4.44–4.34 (m, 2H), 4.16 (d, J = 13.8 Hz, 1H), 3.95 (d, J = 13.8 Hz, 1H), 3.12–3.00 (m, 3H), 2.90–2.72 (m, 3H), 2.11 (s, 6H), 1.56 (s, 3H), 1.48 (s, 3H), 1.15 (t, J = 7.1 Hz, 3H); MS (TOF) m/z: 676 [M+H]+; HRMS (FAB+) calcd for C37H46N4O6SNa [M+Na]+ 697.3036, found m/z 697.3030.
5.2.36. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-(pyridine-2-ylmethyl)amino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (47)
Compound 47 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.61 (d, J = 5.4 Hz, 1H), 8.12–8.08 (m, 2H), 7.98 (t, J = 7.2 Hz, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.46 (t, J = 6.6 Hz, 1H), 7.36 (d, J = 7.2 Hz, 2H), 7.30–7.09 (m, 6H), 7.02 (t, J = 7.8 Hz, 1H), 6.26 (s, 2H), 5.30–5.26 (m, 1H), 4.95 (d, J = 3.0 Hz, 2H), 4.75 (s, 1H), 4.49 (d, J = 3.6 Hz, 1H), 4.44–4.37 (m, 4H), 4.09 (d, J = 15.0 Hz, 1H), 3.87 (d, J = 14.4 Hz, 1H), 3.04 (dd, J = 16.8 Hz, 5.4 Hz, 1H), 2.86–2.77 (m, 3H), 2.03 (s, 6H), 1.56 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 739 [M+H]+; HRMS (FAB+) calcd for C41H47N5O6SNa [M+Na]+ 760.3145, found m/z 760.3148.
5.2.37. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-(pyridine-3-ylmethyl)amino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (48)
Compound 48 was prepared from intermediate 18 in a manner similar to that described for compound 49. 1H NMR (DMSO-d6) δ (ppm): 8.66 (s, 1H), 8.57 (d, J = 3.6 Hz, 1H), 8.10 (d, J = 8.4 Hz, 2H), 8.01 (t, J = 6.9 Hz, 1H), 7.62–7.53 (m, 2H), 7.36 (d, J = 6.6 Hz, 2H), 7.30–7.12 (m, 8H), 7.01 (t, J = 3.6 Hz, 1H), 6.25 (s, 2H), 5.28 (dd, J = 8.1 Hz, 4.2 Hz, 1H), 4.96 (s, 2H), 4.76 (s, 1H), 4.49 (d, J = 3.6 Hz, 1H), 4.44–4.28 (m, 4H), 4.08 (d, J = 14.4 Hz, 1H), 3.86 (d, J = 14.4 Hz, 1H, overlapped with water), 3.04 (dd, J = 15.9 Hz, 3.9 Hz, 1H), 2.86–2.77 (m, 3H), 2.22–2.03 (m, 6H), 1.55 (s, 3H), 1.48 (s, 3H); MS (TOF) m/z: 739 [M+H]+; HRMS (FAB+) calcd for C41H47N5O6SNa [M+Na]+ 760.3145, found m/z 760.3148.
5.2.38. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-(pyridine-4-ylmethyl)amino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (49)
To a solution of 18 (50.6 mg, 0.10 mmol) in DMF (2 mL) were added triethylamine (34.9 μL, 0.25 mmol), compound 15 (42.5 mg, 0.11 mmol) and BOP (48.7 mg, 0.11 mmol) in an ice-water bath and the mixture was stirred overnight at room temperature. After removal of the solvent in vacuo, the residue was diluted with EtOAc, washed sequentially with 5% NaHCO3, and saturated NaCl, dried over MgSO4, and concentrated in vacuo. A mixture of the intermediate, thioanisole (35.2 μL, 0.3 mmol), m-cresol (31.4 μL, 0.3 mmol) and 4M HCl (1 mL) in dioxane was stirred at room temperature for 1 h. After removal of the solvent in vacuo, the residue was precipitated from ether to give the crude product. Purification of the product by preparative HPLC gave the titled compound as a yellowish white foam; yield 38%; 1H NMR (DMSO-d6) δ (ppm): 8.80–8.68 (m, 2H), 8.08 (d, J = 8.7 Hz, 1H), 7.90–7.75 (m, 2H), 7.36 (d, J = 8.7 Hz, 2H), 7.31–7.11 (m, 8H), 7.07–7.00 (m, 1H), 6.19 (s, 2H), 5.30–5.24 (m, 1H), 4.97 (s, 2H), 4.74 (s, 1H), 4.52–4.33 (m, 5H), 4.08 (d, J = 13.5 Hz, 1H, overlapped with water), 3.85 (d, J = 13.5 Hz, 1H, overlapped with water), 3.09–3.00 (m, 1H), 2.86–2.75 (m, 3H), 2.22–2.01 (m, 6H), 1.56 (s, 3H), 1.47 (s, 3H); MS (TOF) m/z: 739 [M+H]+; HRMS (FAB+) calcd for C41H47N5O6SNa [M+Na]+ 760.3145, found m/z 760.3140.
5.2.39. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-butylamino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (50)
To a solution of crude 43 (50 mg, 0.07 mmol) in MeOH (0.2 mL) were added triethylamine (12.3 μL, 0.09 mmol), butyraldehyde (6.6 μL, 0.07 mmol) and NaBH3CN (27.6 mg, 0.44 mmol) in an ice-water bath and the mixture was stirred for 6 h at room temperature. After removal of the solvent in vacuo, the residue was diluted with EtOAc, washed 5% NaHCO3, and saturated NaCl, dried over MgSO4, and concentrated in vacuo. Purification of the product by preparative HPLC gave the titled compound as a white foam; yield 20%; 1H NMR (DMSO-d6) δ (ppm): 8.18 (d, J = 9.0 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 7.37 (d, J = 7.2 Hz, 2H), 7.31–7.10 (m, 6H), 7.03 (t, J = 7.8 Hz, 1H), 6.64 (s, 2H), 5.28 (dd, J = 8.6 Hz, 4.7 Hz, 1H), 4.96 (s, 2H), 4.76 (s, 1H), 4.50 (d, J = 3.3 Hz, 1H), 4.44–4.33 (m, 2H), 4.16 (d, J = 14.1 Hz, 1H), 3.96 (d, J = 14.7 Hz, 1H, overlapped with water), 3.10–3.00 (m, 3H), 2.92–2.72 (m, 3H), 2.12 (s, 6H), 1.58–1.46 (m, 8H), 1.35 (apparent q, J = 7.8 Hz, 1H), 0.89 (t, J = 7.4 Hz, 3H); MS (TOF) m/z: 704 [M+H]+; HRMS (FAB+) calcd for C39H50N4O6SNa [M+Na]+ 725.3349, found m/z 725.3355.
5.2.40. (R)-N-[(1S,2R)-2-Hydroxyindan-1-yl]-3-[(2S,3S)-3-(2,6-dimethyl-4-pentylamino-phenoxyacetyl)amino-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide (51)
Compound 51 was prepared from intermediate 43 in a manner similar to that described for compound 50. 1H NMR (DMSO-d6) δ (ppm): 8.18 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 7.37 (d, J = 6.9 Hz, 2H), 7.31–7.10 (m, 6H), 7.03 (t, J = 7.2 Hz, 1H), 6.60 (s, 2H), 5.31–5.25 (m, 1H), 4.96 (s, 2H), 4.76 (s, 1H), 4.50 (d, J = 3.3 Hz, 1H), 4.44–4.34 (m, 2H), 4.16 (d, J = 14.1 Hz, 1H), 3.95 (d, J = 14.1 Hz, 1H), 3.09–3.00 (m, 3H), 2.87–2.77 (m, 3H), 2.11 (s, 6H), 1.58–1.46 (m, 8H), 1.34–1.27 (m, 4H), 0.87 (t, J = 6.6 Hz, 3H); MS (TOF) m/z: 718 [M+H]+; HRMS (FAB+) calcd for C40H52N4O6SNa [M+Na]+ 739.3505, found m/z 739.3511.
5.3. Plasmepsin II inhibitory activity
Inhibition constants (Ki) against Plm II were obtained as described previously.28 Briefly, the rate of substrate hydrolysis at 25 °C was measured using 400 nM protease in 10 mM sodium formate (pH 4.0), 163 μM chromogenic substrate [H-Ala-Leu-Glu-Arg-Thr-Phe-Phe(p-NO2)-Ser-Phe-Pro-Thr-OH] which is purchased from California Peptide Research Inc., Napa, CA and 2% DMSO with increasing amounts of inhibitor. Ki were estimated by fitting the data to standard equations for tight binding competitive inhibitors.
5.4. HIV protease inhibitory activity
Recombinant HIV-1 PR was purchased from Bachem AG, Bubendorf-Switzerland. HIV PR substrate [H-Lys-Ala-Arg-Val-Tyr-Phe(p-NO2)-Glu-Ala-Nle-NH2] was synthesized by conventional solid-phase method. In the inhibition assay, 25 μL of 200 mM 2-(N-morpholino) ethanesulfonic acid (MES)–NaOH buffer (pH 5.5), containing 2 mM dithiothreitol, 2 mM EDTA–2Na and 1 M NaCl was mixed with 5 μL of the inhibitor (500 nM) dissolved in DMSO and 10 μL of HIV-1 protease (2 μg/mL) in 50 mM AcOH (pH 5.0) containing 1 mM EDTA-2Na, 25 mM NaCl, 0.2% 2-mercaptoethanol, 0.2% Non-idet P-40, and 10% glycerol. The reaction was initiated by adding of 10 μL of a 1.0 mM substrate solution. After incubation for 15 min at 37 °C, the reaction was terminated by addition of 1M HCl, and the amount of cleaved N-terminal fragment (H-Lys-Ala-Arg-Val-Tyr-OH) was measured by reversed-phase HPLC on a C18 column (3.0 × 75 mm; YMC Pack ODS AS-3E7) with a linear gradient of CH3CN in 0.8% aqueous TFA at a flow rate of 1.0 mL/min and the quantity was determined by monitoring fluorescence intensity (Ex, 275 nm; Em, 305 nm). Percent inhibition was obtained compared to intensity without inhibitor.
5.5. Anti-malarial activity
Activity against P. falciparum was determined as described previously. 28 Briefly, chloroquine-sensitive infected cells (NF54) were maintained in a 2.4% suspension of type O+ human erythrocytes in RPMI 1640, supplemented with 25 mM HEPES, 27 mM NaHCO3, and 10% heat inactivated human type O+ serum, under 3% O2, 4% CO2, and 93% N2. DMSO solution of test compounds was serially diluted in 0.2% DMSO in medium. Parasite culture was added, incubated for 48 h prior. After addition of 0.6 μCi of [3H]hypoxanthine, the mixture was incubated for 20 h. Cells were harvested onto GF-C glass filter, washed, dried, and counted in scintillation cocktail. Drug concentration of 50% reduced the level of incorporation of [3H]hypoxanthine was determined from dose-response curves. The cytotoxicity of the inhibitors was evaluated using L6 cells (rat skeletal myoblasts) using standard procedures.46
5.6. Molecular dynamics simulation of plasmepsin II binding models
An X-ray crystal structure of Plm IV complexed with compound 1 (PDB, 2ANL) was aligned with Plm II (PDB, 1SME), and 1 was used to build initial conformation of inhibitor 49 in Plm II using the Molecular Operating Environment modeling package (MOE 2007.09, Chemical Computing Group, Inc., Montreal, Canada) with MMFF94x force field. A carboxyl group of an Asp214 close to the HMC carbonyl group was protonated. Anilinyl and pyridinyl nitrogens of 49 were protonated to simulate acidic assay condition. The binding area was immersed in a 20 Å sphere of TIP3P water from the ligand. The inhibitor, contacted residues and flap region of the enzyme, and surrounded water were energy-minimized while the other atoms were fixed. MD simulations were performed without heating at 298 K for 300 ps equilibration with 1.5 fs time step. An additional 50 ps of MD simulation was performed for data collection. Alignment with crystal data was performed using MOE Align. Pictures were generated using MacPyMOL (DeLano Scientific LLC, CA, USA).
Acknowledgments
This research was supported in part by the Frontier Research Program, the 21st Century COE Program of Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT), and grants from MEXT. E.F. and A.J.R. also thank the Johns Hopkins Malaria Institute for a Pilot Research Grant and a Graduate Student Fellowship as well as grants from the National Institutes of Health (GM57144 and GM56550) and the National Science Foundation (MCB-0641252). We gratefully acknowledge Mr. T. Hamada, Mr. H.-O. Kumada and Ms. S. Shibakawa for the determination of HIV-1 PR inhibitory activity, and Ms. Y. Iteya and Ms. Y. Kadowaki for synthetic assistance, and K. Oda for mass spectrometry. We thank the Swiss Tropical Institute (Prof. R. Brun) for performing the erythrocyte inhibition assays and the cytotoxicity assays.
References and notes
- 1.World Malaria Report 2005. WHO and UNICEF; 2005. [Google Scholar]
- 2.Olliaro PL, Bloland PB. In: Antimalarial Chemotherapy. Rosenthal PJ, editor. Humana Press; Totawa, NJ: 2001. pp. 65–83. [Google Scholar]
- 3.Ridley RG. Nature. 2002;415:686. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- 4.Benerjee R, Goldberg DE. In: Antimalarial Chemotherapy. Rosenthal PJ, editor. Humana Press; Totawa, NJ: 2001. pp. 43–63. [Google Scholar]
- 5.Cooms GH, Goldberg DE, Klemba M, Berry C, Kay J, Mottram JC. Trends Parasitol. 2001;17:532. doi: 10.1016/s1471-4922(01)02037-2. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. Proc Natl Acad Sci USA. 2002;99:990. doi: 10.1073/pnas.022630099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francis SE, Gluzman IY, Oksman A, Knickerbocker A, Mueller R, Bryant ML, Sherman DR, Russell DG, Goleberg DE. EMBO J. 1994;13:306. doi: 10.1002/j.1460-2075.1994.tb06263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dame JB, Reddy GR, Yowell CA, Dunn BM, Kay J, Berry C. Mol Biochem Parasitol. 1994;64:177. doi: 10.1016/0166-6851(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 9.Hill J, Tyas L, Phylip LH, Kay J, Dunn BM, Berry C. FEBS Lett. 1994;352:155. doi: 10.1016/0014-5793(94)00940-6. [DOI] [PubMed] [Google Scholar]
- 10.Berry C, Humphreys MJ, Matharu P, Granger R, Horrocks P, Moon RP, Certa U, Ridley RG, Bur D, Kay J. FEBS Lett. 1999;447:149. doi: 10.1016/s0014-5793(99)00276-8. [DOI] [PubMed] [Google Scholar]
- 11.Humphreys MJ, Moon RP, Klinder A, Fowler SD, Rupp K, Bur D, Ridley RG, Berry C. FEBS Lett. 1999;463:43. doi: 10.1016/s0014-5793(99)01597-5. [DOI] [PubMed] [Google Scholar]
- 12.Omara-Opyene AL, Moura PA, Sulsona CR, Bonilla JA, Yowell CA, Fujioka H, Fidock DA, Dame JB. J Biol Chem. 2004;279:54088. doi: 10.1074/jbc.M409605200. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Gluzman IY, Drew ME, Goldberg DE. J Biol Chem. 2005;280:1432. doi: 10.1074/jbc.M409740200. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE. Proc Natl Acad Sci USA. 2006;103:8840. doi: 10.1073/pnas.0601876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonilla JA, Bonilla TD, Yowell CA, Fujioka H, Dame JB. Mol Microbiol. 2007;65:64. doi: 10.1111/j.1365-2958.2007.05768.x. [DOI] [PubMed] [Google Scholar]
- 16.Semenov A, Olsen JE, Rosenthal PJ. Antimicrob Agents Chemother. 1998;42:2254. doi: 10.1128/aac.42.9.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva AM, Lee AY, Gulnik SV, Majer P, Collins J, Bhat TN, Collins PJ, Cachau RE, Lurer KE, Gluzman IY, Francis SE, Oksman A, Goldberg DE, Erickson JW. Proc Natl Acad Sci USA. 1996;93:10034. doi: 10.1073/pnas.93.19.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll CD, Patel H, Johnson TO, Guo T, Orlowski M, He ZM, Cavallaro CL, Guo J, Oksman A, Gluzman IY, Connelly J, Chelsky D, Goldberg DE, Dolle RE. Bioorg Med Chem Lett. 1998;8:2315. doi: 10.1016/s0960-894x(98)00419-3. [DOI] [PubMed] [Google Scholar]
- 19.Haque TS, Skillman AG, Lee CE, Habashita H, Gluzman IY, Ewing TJA, Goldberg DE, Kuntz ID, Ellman JA. J Med Chem. 1999;42:1428. doi: 10.1021/jm980641t. [DOI] [PubMed] [Google Scholar]
- 20.Ersmark K, Samuelsson B, Hallberg A. Med Res Rev. 2006;26:626. doi: 10.1002/med.20082. [DOI] [PubMed] [Google Scholar]
- 21.Moon RP, Tyas L, Certa U, Rupp K, Bur D, Jacquet C, Matile H, Loetscher H, Grueninger-Leitch F, Kay J, Dunn BM, Berry C, Ridley RG. Eur J Biochem. 1997;244:552. doi: 10.1111/j.1432-1033.1997.00552.x. [DOI] [PubMed] [Google Scholar]
- 22.Corminboeuf O, Dunet G, Hafsi M, Grimont J, Grisostomi C, Meyer S, Binkert C, Bur D, Jones A, Prade L, Brun R, Boss C. Bioorg Med Chem Lett. 2006;16:6194. doi: 10.1016/j.bmcl.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 23.Dell’Agli M, Parapini S, Galli G, Vaiana N, Taramelli D, Sparatore A, Liu P, Dunn BM, Bosisio E, Romeo S. J Med Chem. 2006;49:7440. doi: 10.1021/jm061033d. [DOI] [PubMed] [Google Scholar]
- 24.Gluzman IY, Francis SE, Oksman A, Smith CE, Duffin KL, Goldberg DE. J Clin Invest. 1994;93:1602. doi: 10.1172/JCI117140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hellen CUT, Kraeusslich HG, Wimmer E. Biochemistry. 1989;28:9881. doi: 10.1021/bi00452a001. [DOI] [PubMed] [Google Scholar]
- 26.Mimoto T, Imai J, Tanaka S, Hattori N, Takahashi O, Kisanuki S, Nagano Y, Shintani M, Hayashi H, Sakikawa K, Akaji K, Kiso Y. Chem Pharm Bull. 1991;39:2465. doi: 10.1248/cpb.39.2465. [DOI] [PubMed] [Google Scholar]
- 27.Mimoto T, Imai J, Tanaka S, Hattori N, Kisanuki S, Akaji K, Kiso Y. Chem Pharm Bull. 1991;39:3088. doi: 10.1248/cpb.39.3088. [DOI] [PubMed] [Google Scholar]
- 28.Nezami A, Luque I, Kimura T, Kiso Y, Freire E. Biochemistry. 2002;41:2273. doi: 10.1021/bi0117549. [DOI] [PubMed] [Google Scholar]
- 29.Kiso Y. J Synth Org Chem, Jpn. 1998;56:896. [Google Scholar]
- 30.Mimoto T, Kato R, Takaku H, Nojima S, Terashima K, Misawa S, Fukazawa T, Ueno T, Sato H, Shintani M, Kiso Y, Hayashi H. J Med Chem. 1999;42:1789. doi: 10.1021/jm980637h. [DOI] [PubMed] [Google Scholar]
- 31.Vega S, Kang LW, Velazquez-Campoy A, Kiso Y, Amzel LM, Freire E. Proteins. 2004;55:594. doi: 10.1002/prot.20069. [DOI] [PubMed] [Google Scholar]
- 32.Nezami A, Kimura T, Hidaka K, Kiso A, Liu J, Kiso Y, Goldberg DE, Freire E. Biochemistry. 2003;42:8459. doi: 10.1021/bi034131z. [DOI] [PubMed] [Google Scholar]; Highlighted. Science. 2003;301:143. [Google Scholar]
- 33.Kiso A, Hidaka K, Kimura T, Hayashi Y, Nezami A, Freire E, Kiso Y. J Peptide Sci. 2004;10:641. doi: 10.1002/psc.609. [DOI] [PubMed] [Google Scholar]
- 34.Hidaka K, Kimura T, Tsuchiya Y, Kamiya M, Ruben AJ, Freire E, Hayashi Y, Kiso Y. Bioorg Med Chem Lett. 2007;17:3048. doi: 10.1016/j.bmcl.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 35.Kiso Y, Matsumoto H, Mizumoto S, Kimura T, Fujiwara Y, Akaji K. Biopolymer. 1999;51:59. doi: 10.1002/(SICI)1097-0282(1999)51:1<59::AID-BIP7>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 36.Clemente JC, Govindasamy L, Madabushi A, Fisher SZ, Moose RE, Yowell CA, Hidaka K, Kimura T, Hayashi Y, Kiso Y, Agbandje-McKenna M, Dame JB, Dunn BM, McKenna R. Acta Crystallogr. 2006;62:246. doi: 10.1107/S0907444905041260. [DOI] [PubMed] [Google Scholar]
- 37.Gutiérrez-de-Teran H, Nervall M, Dunn BM, Clemente JC, Åqvist J. FEBS Lett. 2006;580:5910. doi: 10.1016/j.febslet.2006.09.057. [DOI] [PubMed] [Google Scholar]
- 38.Hidaka K, Kimura T, Hayashi Y, McDaniel KF, Dekhtyar T, Colletti L, Kiso Y. Bioorg Med Chem Lett. 2003;13:93. doi: 10.1016/s0960-894x(02)00848-x. [DOI] [PubMed] [Google Scholar]
- 39.Chen X, Kempf DJ, Li L, Sham HL, Vasavanonda S, Wideburg NE, Saldivar A, Marsh KC, McDonald E, Norbeck DW. Bioorg Med Chem Lett. 2003;13:3657. doi: 10.1016/j.bmcl.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 40.Baldwin ET, Bhat TN, Gulnik S, Liu B, Topol IA, Kiso Y, Mimoto T, Mitsuya H, Erickson JW. Structure. 1995;3:581. doi: 10.1016/s0969-2126(01)00192-7. [DOI] [PubMed] [Google Scholar]
- 41.Wang YX, Freedberg DI, Yamazaki T, Wingfield PT, Stahl SJ, Kaufman JD, Kiso Y, Torchia DA. Biochemistry. 1996;35:9945. doi: 10.1021/bi961268z. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. MacPyMOL: A PyMOL-based Molecular Graphics Application for MacOS X. DeLano Scientific LLC; Palo Alto, CA, USA: 2006. Available from: http://www.pymol.org. [Google Scholar]
- 43.Nöteberg D, Hamelink E, Hulten J, Wahlgren M, Vrang L, Samuelsson B, Hallberg A. J Med Chem. 2003;46:734. doi: 10.1021/jm020951i. [DOI] [PubMed] [Google Scholar]
- 44.Muthas D, Nöteberg D, Sabnis YA, Hamelink E, Vrang L, Samuelsson B, Karlén A, Hallberg A. Bioorg Med Chem. 2005;13:5371. doi: 10.1016/j.bmc.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 45.Choi CYH, Schneider EL, Kim JM, Gluzman IY, Goldberg DE, Ellman JA, Marletta MA. Chem Biol. 2002;9:881. doi: 10.1016/s1074-5521(02)00183-7. [DOI] [PubMed] [Google Scholar]
- 46.Verotta L, Appendino G, Bombardelli E, Brun R. Bioorg Med Chem Lett. 2007;17:1544. doi: 10.1016/j.bmcl.2006.12.100. [DOI] [PubMed] [Google Scholar]