

Abstract
Cellular microarrays have become extremely useful in expediting the investigation of large libraries of (bio)materials for both in vitro and in vivo biomedical applications. We have developed an exceedingly simple strategy for the fabrication of non-cell-adhesive substrates supporting the immobilization of diverse (bio)material features, including both monomeric and polymeric adhesion molecules (e.g. RGD and polylysine), hydrogels, and polymers.
Keywords: biomedical applications, polymeric materials, hydrogels, medical applications, surface modification
Progress in fundamental cell biology, diagnostics, and regenerative medicine is oftentimes brought about by advances in materials science and engineering through the development of next generation (bio)materials. High-throughput microarrays are increasingly employed for rapid screening of interactions between proteins or cells with large libraries of small molecules, proteins, peptides, and/or polymeric biomaterials,1-4 leading to new materials of interest, new in vitro and in vivo biomedical applications, or increased fundamental biological understanding. In the case of proteins, such arrays can be used to detect interactions leading to new therapeutics.4-6 With cell microarrays, specific interactions contributing to adhesion, proliferation, expansion, differentiation, or expression of a desirable phenotype (e.g. Oct4, SSEA and/or NANOG in pluripotent stem cells) can be rapidly evaluated.7-10 Indeed, the natural extracellular matrix (ECM) contains numerous insoluble proteins and by extracting signaling motifs and printing them on inert substrates in a high-throughput, parallel fashion, it is possible to build robust comprehension of important biological interactions. Moreover, microarrays provide the opportunity to identify novel materials for cell culture by using combinatorial mixing of polymers, allowing investigation of thousands of potential (bio)materials on a single substrate, a tactic that yielded the first defined surface supporting clonal expansion of pluripotent stem cells.1
Cell microarray manufacture can be performed by ink-jet or contact printing of solutions containing compounds of interest onto a non-fouling substrate. The surface coating must provide a low-fouling background resistant to protein adsorption and cell attachment, while at the same time facilitating straightforward and reliable attachment of printed features.11-13 Cell attachment between features complicates the observation of cellular responses and often leads to undesired cell-cell signaling. Thus, a key challenge for maximizing the application of microarrays and gaining control over the interaction between cells and printed features is suppression of cellular adhesion.14-19 Alternatively, substrates must also employ robust, efficient and orthogonal chemistries with expanded functional group compatibility allowing covalent immobilization of diverse molecules to develop modular bioassays. It is therefore of interest to develop substrates compatible with diverse printing tools and capable of selectively attaching compounds of interest while suppressing non-specific protein/cell attachment during cell culture.
Current techniques for producing cell microarrays of- ten include multiple steps and lengthy reactions to achieve conjugation and maintain specificity. Silanization is often used to introduce functionality such as carboxylic acids, succinimidyl esters, epoxides, or maleimide groups onto glass slides.12, 20-23 Biomaterials printed onto these substrates can be covalently conjugated via amine or thiol functional groups. Subsequent backfilling around the printed bio-material features with non-cell-adhesive materials, such as bovine serum albumin (BSA), poly(ethylene glycol) (PEG), or poly(hydroxyethymethacrylate) (PHEMA), reduces undesirable cell attachment between printed features.22, 24 However, passivation techniques such as these can significantly add to fabrication time and are often not completely cell resistant.25 Moreover, silanization with (meth)acrylate, thiol or alkene functionality has also been widely exploited in fabricating glass slides for solid support of hydrogels for use as artificial ECM materials to investigate cell behavior.26-30 In studies in 3D encapsulation of cells, however, the hydrophobic silane-based coating on the glass slides often leads to undesirable interactions with the motile encapsulated cells, complicating the observation of cellular responses.
Other techniques have relied on physical retention of bio- materials of interest on non-cell-adhesive substrates,1, 31, 32 resolving issues with difficult and tedious covalent conjugation reactions, however, leaching or detachment of the bio-material features from the surface hampers long-term cell culture studies. Moreover, extensive work has produced a variety of non-fouling polymer brushes.33-35 Recently, polyglycerol brushes have exhibited suppression of cell attachment while allowing for array fabrication by co-printing of amine-functional RGD adhesion peptides with a strong oxidant,36 but again, these suffer from limitations in the diversity of chemical reactions useful for covalent conjugation. Furthermore, thiol-ene chemistry has been exploited to fabricate PEG-based hydrogels, allowing for either direct or orthogonal post-fabrication functionalization with arrayed biomaterial features displaying assorted chemical signals on the non-fouling substrates.11 These materials, however, have not yet been shown to allow for printing of polymeric microarrays, which have proven to be useful for identifying new materials for expansion of clinically relevant cell types1, 2, 10 or new materials resistant to bacterial attachment.8, 9
To overcome these various issues, we sought to develop PEG-based non-cell-adhesive substrates with broad utility for the development of printed microarrays. These substrates offer a wealth of advantages, most importantly being the ability to covalently immobilize diverse chemical functionality from cell adhesion peptides (e.g. RGD peptides) or natural biopolymers (e.g. poly(lysine)), to hydrogels or synthetic polymeric biomaterials. The synthesis of these substrates is facile, inexpensive, rapid, and scalable.
Methoxy-terminated PEG (MeO-PEG-OH) of two molecular weights (PEG500, Mn = 500 Da; PEG2k, Mn = 2,000 Da) and a commercially available, methacrylate-functional PEG derivative (MA-PEG-OH; PEG-MA, Mn = 525 Da) were chosen as the primary polymeric materials for preparation of non-cell-adhesive substrates on account of their favorable solubility, previously demonstrated efficacy in low/non-fouling surfaces, and hydroxyl/methacrylate functionality (Figure 1). These PEG precursors were mixed (10% PEG-MA w/w) and readily functionalized using commercially available isocyanatopropyl triethoxysilane in a one-step reaction performed at ambient temperature in dichloromethane using dibutyltin dilaurate (TDL) as a catalyst (Figure 1).37 Without purification, the reaction mixture could be diluted with anhydrous toluene and added directly to UV/ozone-treated glass slides, yielding a robust PEG-coating overnight. The slide surface was purified of unreacted PEG-silane by washing with ethanol and drying under vacuum. These synthetic protocols are facile, rapid and easily scaled.
Figure 1.
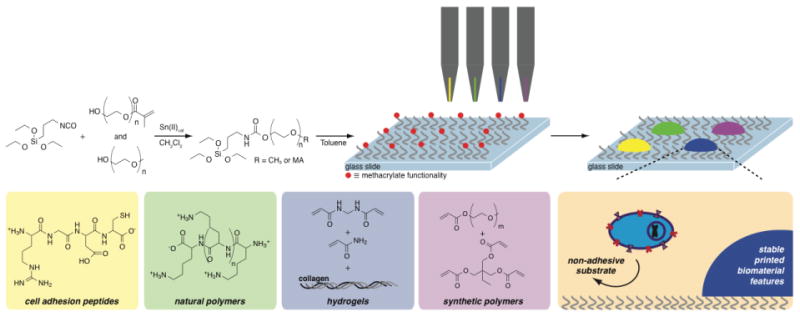
Synthesis of non-cell-adhesive substrates by a simple two-step, one-pot functionalization of glass surfaces with a mixed self-assembled mono-layer of oligomeric poly(ethylene glycol) (PEG). By incorporating a small proportion of methacrylate-functional PEG moieties into the formulation of the silane, catalyzed by dibutyltin dilaurate (TDL), followed by surface functionalization, non-cell-adhesive substrates are prepared allowing for diverse bio-functionalization by covalent attachment of cell adhesion peptides (e.g. RGD peptides), natural polymers (e.g. poly(d-lysine)), hydrogels, and/or synthetic polymeric biomaterials.
PEG-coated substrates were characterized for hydrophobicity with water-contact angle (WCA) measurements and for surface roughness with atomic force microscopy (AFM). For comparison, PHEMA-functional substrates were also prepared, according to published procedures,1 by dip-coating glass slides in ethanolic PHEMA solutions and allowing to dry. All characterization was performed to compare tissue-culture polystyrene (TCPS), standard glass slides (SiO2), and PHEMA, PEG500, and PEG2k -coated glass slides. WCA measurements (Figure 2a), performed with deionized water, revealed that both PEG derivatives and PHEMA impart roughly identical hydrophobicity (∼ 47°) to the substrates, which is much higher than clean SiO2 (∼ 26°), and much lower than TCPS (∼ 86°). These measurements reveal that the synthetic protocols significantly alter the surface chemistry of the glass slides. Moreover, AFM measurements indicated a low root mean squared (RMS) roughness for PEG-functional substrates, suggesting that the silanization procedure allows for formation of low-defect monolayers of PEG on the surface. Indeed, the PEG-silanization of the glass substrates was directly verified using Fourier transform infrared spectroscopy (FTIR; Figure 2c), which demonstrated the presence of carbonyl stretching at the surface of the substrates arising from both the carbamate linkage (generated by the isocyanate coupling reaction) and the methacrylate functionality.
Figure 2.
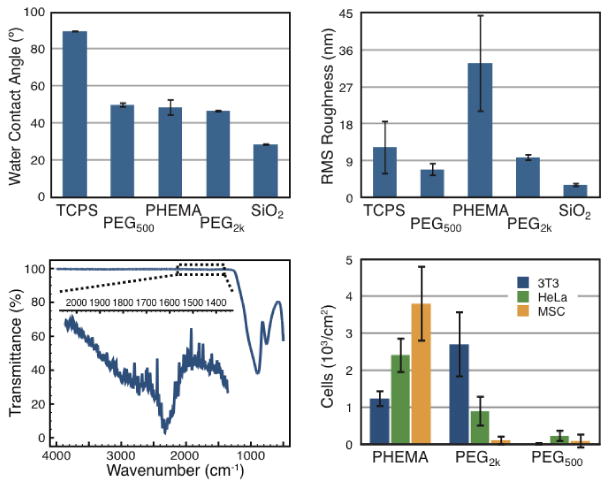
(a) Water contact angle (n=5) determined with PBS (pH=7.4). (b) Root mean squared (RMS) surface roughness (n=3) determined using atomic force microscopy (AFM). (c) FTIR characterization of PEG500-coated substrates demonstrating carbonyl stretching from the carbamate functionality arising from the isocyanate coupling and the methacrylate functionality. (d) Cell adhesion assays with NIH 3T3 fibroblast, HeLa, and mesenchymal stem (MSCs) cells demonstrating negligible adhesion of cells to PEG550-coated substrates (3T3 = (0.01 ± 0.02) ×103 cells/cm2, HeLA = (0.22 ± 0.2) ×103 cells/cm2, MSCs = (0.08 ± 0.2) ×103 cells/cm2).
Adhesion of cells to the functionalized slides was assessed using three cell types: 3T3 fibroblasts, HeLa, and mesenchymal stem cells (MSCs). Cells were seeded at a density of 5,000 cells/cm2 and were incubated with the substrates for 24h before fixing with paraformaldehyde (3.4%) and staining with Hoechst (nucleus) for quantification of adhered cells. The results of these assays are reported in Figure 2d and clearly indicate the extremely low adhesion of cells to the PEG-based substrates. PEG500-coated substrates in particular were capable of inhibiting cellular adhesion and proliferation across all of the cell types investigated. For reference, TCPS was found to strongly support the growth and proliferation of each of these cell types (7-10 × 103 cells/cm2). In contrast, PEG2k and PHEMA-coated substrates exhibited some cell-specific adhesion, where PEG2k-coated substrates were able to inhibit attachment and proliferation of MSCs, but not 3T3 fibroblasts. On the other hand, PHEMA-coated substrates inhibited 3T3 fibroblast attachment, but not attachment of HeLa cells or MSCs.
With a non-cell-adhesive substrate in hand, we sought to investigate its ability to support printing and attachment of arrayed biomaterials. First, synthetic polymeric dot features composed of poly(ethylene glycol) acrylate (PEGA; Mn= 480 Da; 30%), trimethylolpropane triacrylate (TMPTA; 70%), and 2,2-dimethoxy-2-phenylacetophenone (DMPA; 1%) were printed with an AFM printer (Figure 3a) in 10 × 10 arrays in triplicate on PEG500-coated substrates in order to asses the stability of printed features in various solvents. This particular copolymer has been demonstrated to sup- port the clonal growth and expansion of human embryonic and induced pluripotent stem cells while retaining pluripotency.1 Polymer dots were polymerized by exposure to UV irradiation (λ = 365 nm) for 5 min and contain rhodamine-functional acrylate comonomer imparting strong red fluorescence for easy visualization during the stability assays. For comparison, arrays were also printed onto PHEMA-coated substrates and their stability assessed.
Figure 3.
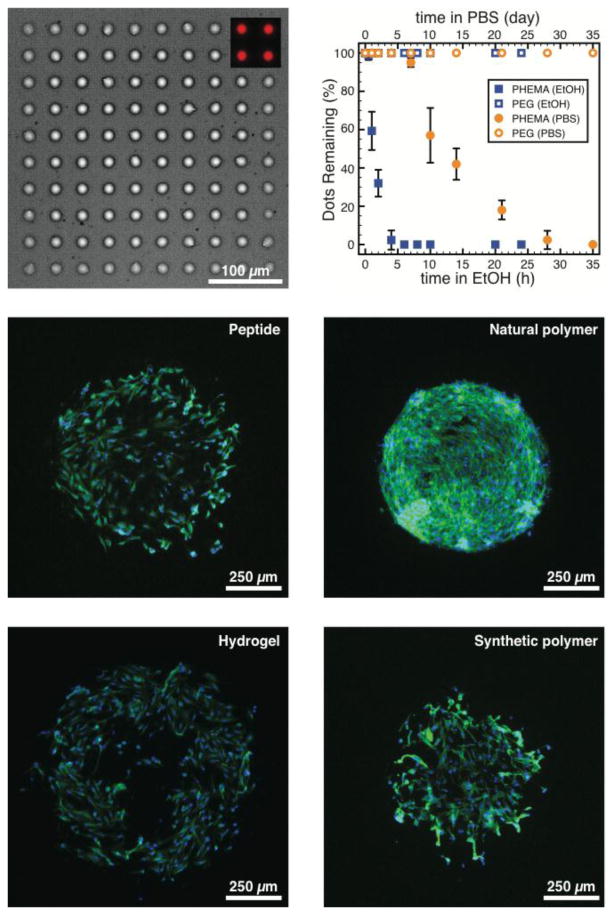
(a) Microscopy images (bright-field) of large (∼ 25μm) polymer dots composed of PEG acrylate (PEGA; 30%; Mn = 480 Da) and trimethylolpropane triacrylate (TMPTA; 70%) printed in 10 × 10 arrays in triplicate on PEG500-coated substrates. Polymer dots contain rhodamine-functional acrylate comonomer imparting strong red fluorescence (inset). (b) Stability of the printed polymer dots was investigated over time in ethanol (20°C, lower axis) and PBS (pH=7.4, 37°C, upper axis). Representative fluorescence microscopy images of 3T3 fibroblast cells grown on functional spots (d ∼ 500 μm; n=20) composed of (c) RGDC peptides (printed at 100 μg/mL), (d) poly(d-lysine) (printed at 100 μg/mL), (e) polyacrylamide hydrogel containing collagen I (0.05%), and (f) PEGA-co-TMPTA (as in part a and b), printed on PEG500-coated substrates.
Stability assays were performed with ethanol (20°C) and PBS (37°C) and performance assessed by the proportion of polymer dots remaining over time (Figure 3b). Intuitively, printed dots were rapidly released from PHEMA-coated substrates in ethanol (likely due to the solubility of the PHEMA in the solvent). Additionally, as observed previously,1 printed dots began detaching from PHEMA-coated substrates after 5 days in PBS, making these unsuitable for longer term cell culture studies. In contrast, dots printed on PEG500-coated substrates were found to be completely stable in either solvent, likely on account of the covalent attachment of the printed features to the substrates.
We then investigated the printing of various biomaterials onto PEG500-coated slides. We investigated four classes of materials: (a) monovalent (RGDC) and (b) polymeric (poly(d-lysine)) cell adhesion molecules, (c) hydrogels (polyacrylamide), and (d) synthetic polymers (PEGA/TMPTA). RGDC moieties were chosen as they contain a pendant thiol on the c-terminal cysteine residue, which has been utilized previously for Michael addition to methacrylate functionality using amine catalysts.38 RGDC solutions were prepared in DMSO (100 μg/mL) containing diisopropylethylamine (100 μg/mL). Poly(d-lysine) (Mw = 100 kDa) possess many pendant primary amine groups also capable of Michael addition to methacrylates, however, this reaction is slow at room temperature and physiological pH. Thus, poly(d-lysine) solutions were prepared in a high pH borate buffer (pH=10; 100 μg/mL) in order to promote the Michael addition reaction over a favorable timeframe. Hydrogel precursor solutions contained acrylamide (Am; 10% w/w), methylenebisacrylamide (MBAm; 0.2% w/w), a natural ECM component, collagen I (ColI; 0.05% w/w), and Irgacure 2529 (0.05% w/w) as a photo-initiator. Monomer solutions were prepared, as above, composed of PEGA (30%), TMPTA (70%), and DMPA (1% w/w) as a photo-initiator.
These solutions were printed onto PEG500-coated slides in a 4 × 20 (d ∼ 500 μm) array using metal pins (946MP3B, Arrayit). Following printing, the slides were subjected to UV light (λ = 365 nm) for 5 min in order to polymerize both the hydrogels and the polyacrylates, and were then kept at room temperature overnight to allow the Michael addition reactions to proceed. Once the microarrays were washed and sterilized with UV light, 3T3 fibroblast cells were seeded onto them at a density of 5000 cells/cm2. After incubation for 24h, the cells were fixed with paraformaldehyde (3.4%) and stained with Hoechst (nucleus) and Phalloidin (actin filaments) and representative images are demonstrated in Figure 3c-f. All four of the materials yielded stable circular printed features on the PEG500-coated slides and clearly supported robust cell attachment and proliferation. The 3T3 cells exhibited a preference for the poly(d-lysine) features in the parallel cell culture assay, yet nonetheless were observed to adhere and spread on all feature types. Moreover, no cells were found to adhere to the PEG500-coated slides between the printed features, corroborating observations made above. The chemistry of the substrates, therefore, allows for the covalent attachment of diverse printed features that are stabile under cell culture conditions, while suppressing undesirable non-specific cell adhesion.
In summary, we have developed an exceedingly simple strategy for the fabrication of non-cell-adhesive substrates supporting the immobilization of diverse (bio)materials. The chemistry of the substrates not only prevents adhesion of several cell types, including fibroblasts, HeLa, and mesenchymal stem cells, but also allows for the covalent attachment of printed features of diverse chemistries, including both monomeric and polymeric adhesion molecules (e.g. RGD and polylysine), hydrogels, and polymers for cell culture. This new substrate functionalization strategy has the potential for widespread application both in cellular microarrays and in hydrogels to be used as artificial extracellular matrix materials.
Supplementary Material
Acknowledgments
E. A. A. is grateful for financial support through a Wellcome Trust-MIT Postdoctoral Fellowship. This work is supported in part by NIH-R01 DE016516.
Footnotes
Supporting Information: Supporting Information is available from the Wiley Online Library or from the author.
Supporting Information: Title ((no stars))
Author(s), and Corresponding Author(s)* ((write out full first and last names))
((Please insert your Supporting Information text/figures here. Please note: Supporting Display items, should be referred to as Figure S1, Equation S2, etc., in the main text…)
References
- 1.Mei Y, Saha K, Bogatyrev SR, Yang J, Hook AL, Kalcioglu ZI, Cho SW, Mitalipova M, Pyzocha N, Rojas F, Van Vliet KJ, Davies MC, Alexander MR, Langer R, Jaenisch R, Anderson DG. Nature Mater. 2010;9:768. doi: 10.1038/nmat2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson DG, Levenberg S, Langer R. Nature Biotechnol. 2004;22:863. doi: 10.1038/nbt981. [DOI] [PubMed] [Google Scholar]
- 3.Anderson DG, Putnam D, Lavik EB, Mahmood TA, Langer R. Biomaterials. 2005;26:4892. doi: 10.1016/j.biomaterials.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 4.Vegas AJ, Fuller JH, Koehler AN. Chem Soc Rev. 2006;37:1385. doi: 10.1039/b703568n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macbeath G, Koehler AN, Schreiber SL. J Am Chem Soc. 1999;121:7967. [Google Scholar]
- 6.Bradner JE, McPherson OM, Koehler AN. Nature Protocols. 2006;1:2344. doi: 10.1038/nprot.2006.282. [DOI] [PubMed] [Google Scholar]
- 7.Hook AL, Anderson DG, Langer R, Williams P, Davies MC, Alexander MR. Biomaterials. 2012;31:187. doi: 10.1016/j.biomaterials.2009.09.037. [DOI] [PubMed] [Google Scholar]
- 8.Hook AL, Chien-Yi C, Yang J, Luckett J, Cockayne A, Atkin- son S, Mei Y, Bayston R, Irvine DJ, Langer R, Anderson DG, Williams P, Davies MC, Alexander MR. Nature Biotechnol. 2012;30:868. doi: 10.1038/nbt.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hook AL, Chang CY, Yang J, Atkinson S, Langer R, Anderson DG, Davies MC, Williams P, Alexander MR. Adv Mater. 2013;25:2542. doi: 10.1002/adma.201204936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Celiz AD, Smith JGW, Patel AK, Langer R, Ander- son DG, Barrett DA, Young LE, Davies MC, Denning C, Alexander MR. Biomaterials Sci. 2014;2:1604. doi: 10.1039/c4bm00054d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta N, Lin BF, Campos LM, Dimitriou MD, Hikita ST, Treat ND, Tirrell MV, Clegg DO, Kramer EJ, Hawker CJ. Nature Chem. 2009;2:138. doi: 10.1038/nchem.478. [DOI] [PubMed] [Google Scholar]
- 12.Kurkuri MD, Driever C, Johnson G, McFarland G, Thissen H, Voelcker NH. Biomacromolecules. 2009;10:1163. doi: 10.1021/bm801417s. [DOI] [PubMed] [Google Scholar]
- 13.Hook AL, Thissen H, Voelcker NH. Biomacromolecules. 2009;10:573. doi: 10.1021/bm801217n. [DOI] [PubMed] [Google Scholar]
- 14.Hucknall A, Rangarajan S, Chiltoki A. Adv Mater. 2009;21:2441. doi: 10.1002/adma.200803125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang S, Cao Z. Adv Mater. 2010;22:920. doi: 10.1002/adma.200901407. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez-Emmenegger C, Brynda E, Riedel T, Sedlakova Z, Houska M, Alles AB. Langmuir. 2009;25:6328. doi: 10.1021/la900083s. [DOI] [PubMed] [Google Scholar]
- 17.Kostina NY, Rodriguez-Emmenegger C, Houska M, Brynda E, Michalek J. Biomacromolecules. 2012;13:4164. doi: 10.1021/bm301441x. [DOI] [PubMed] [Google Scholar]
- 18.Gautrot JE, Trappmann B, Oceguera-Yanez F, Connelly J, He X, Watt FM, Huck WTS. Biomaterials. 2010;31:5030. doi: 10.1016/j.biomaterials.2010.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautrot JE, Wang C, Liu X, Goldie SJ, Trappmann B, Huck WTS, Watt FM. Biomaterials. 2012;33:5221. doi: 10.1016/j.biomaterials.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandes TG, Kwon SJ, Lee MY, Clark DS, Cabral JMS, Dordick JS. Anal Chem. 2008;80:6633. doi: 10.1021/ac800848j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Paz JL, Noti C, Seeberger PH. J Am Chem Soc. 2006;128:2766. doi: 10.1021/ja057584v. [DOI] [PubMed] [Google Scholar]
- 22.Ghaemi SR, Harding F, Delalat B, Vasani R, Voelcker NH. Biomacromolecules. 2013;14:2675. doi: 10.1021/bm400531n. [DOI] [PubMed] [Google Scholar]
- 23.Calabretta A, Wasserberg D, Posthuma-Trumpie GA, Subra- maniam V, van Amerongen A, Corradini R, Tedeschi T, Sforza S, Reinhoudt DN, Marchelli R, Huskens J, Jonkheijm P. Langmuir. 2010;27:1536. doi: 10.1021/la102756k. [DOI] [PubMed] [Google Scholar]
- 24.MacBeath G, Schreiber SL. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 25.Templin MF, Stoll D, Schrenk M, Traub PC, Vohringer CF, Joos TO. Trends Biotechnol. 2002;20:160. doi: 10.1016/s0167-7799(01)01910-2. [DOI] [PubMed] [Google Scholar]
- 26.Trappmann B, Gautrot JE, Connelly JT, Strange DG, Li Y, Oyen ML, Cohen Stuart MA, Boehm H, Li B, Vogel V, Spatz JP, Watt FM, Huck WTS. Nature Mater. 2012;11:642. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 27.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin CC, S AK. Proc Natl Acad Sci. 2011;108:6380. [Google Scholar]
- 29.Yang C, Tibbitt MW, Basta L, Anseth KS. Nature Mater. 2014;13:645. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kloxin AM, Tibbitt MW, Anseth KS. Nature Protocols. 2010;5:1867. doi: 10.1038/nprot.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones CN, Lee JY, Zhu J, Stybayeva G, Ramanculov E, Zern MA, Revzin A. Anal Chem. 2008;80:6351. doi: 10.1021/ac8007626. [DOI] [PubMed] [Google Scholar]
- 32.Bauer M, Kim K, Qiu Y, Calpe B, Khademhosseini A, Liao R, Wheeldon I. ACS Comb Sci. 2012;14:471. doi: 10.1021/co300039w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sundberg SA, Barrett RW, Pirrung M, Lu AL, Kiangsoon- tra B, Holmes CP. J Am Chem Soc. 1995;117:12050. [Google Scholar]
- 34.Zhou F, Zheng Z, Yu B, Liu W, Huck WTS. J Am Chem Soc. 2006;128:16253. doi: 10.1021/ja0654377. [DOI] [PubMed] [Google Scholar]
- 35.Advincula RC, Brittain WJ, Caster KC, Ruhe J, editors. Polymer Brushes: Synthesis, Characterization, Applications. Wiley VCH; 2005. [Google Scholar]
- 36.Moore E, Delalat B, Vasani R, Thissen H, Voelcker NH. Biomacromolecules. 2014;6:15243. doi: 10.1021/am503570v. [DOI] [PubMed] [Google Scholar]
- 37.Biedermann F, Appel EA, del Barrio J, Gruendling T, Barner-Kowollik C, Scherman OA. Macromolecules. 2011;44:4828. [Google Scholar]
- 38.Li GZ, Randev RK, Soeriyadi AH, Rees G, Boyer C, Tong Z, Davis TP, Becer CR, Haddleton DM. Polym Chem. 2010;1:1196. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.