

Abstract
As part of the dynamic interactions between leukemic cells and cells of the bone marrow microenvironment, specific niches provide a sanctuary where subpopulations of leukemic cells evade chemotherapy-induced death and acquire a drug-resistant phenotype. This review focuses on the cellular and molecular biology of the leukemia stem cell (LSC) niche and of microenvironment/leukemia interactions. Key emerging therapeutic targets include chemokine receptors, adhesion molecules, the sympathetic nervous system, and hypoxia-related proteins, as well as the genetic and epigenetic abnormalities of the leukemia-associated stroma. The complex interplay between LSCs and microenvironment components provides a rationale for appropriately tailored molecular therapies designed to improve outcomes in leukemia. Further understanding of the contribution of the bone marrow niche to the process of leukemogenesis may provide new targets that allow destruction of LSCs without adversely affecting normal stem cell self-renewal.
Keywords: Bone marrow microenvironment, acute myeloid leukemia (AML), leukemia stem cell (LSC), hematopoietic stem cell (HSC), endosteal niche, vascular niche
Introduction
The chemotherapy-based and targeted therapies have driven significant progress in human acute myeloid leukemia (AML) over the past decade. However, relapse after an initial response remains common, which indicates the persistence of chemoresistant leukemia stem cells (LSCs) in specific niches in the bone marrow (BM) microenvironment. More than 30 years ago, a specialized regulatory BM microenvironmental “niche” was proposed, where stem cells reside, receive appropriate support for maintaining self-renewal and multi-lineage differentiation capacity, and are protected from environmental stress (1). These niches are part of a complex of BM cells, including bone-lining cells (osteoblasts and osteoclasts), mesenchymal stem cells (MSCs), sinusoidal endothelium and perivascular stromal cells, nonmyelinating Schwann cells, and immune cells, that play different roles in hematopoietic regulation (2, 3). The BM niche maintains normal hematopoietic stem cells (HSCs) primarily in a quiescent state by providing signals that inhibit cell proliferation, and only upon receipt of a stimulating signal does the stem cell become activated to divide and proliferate. Expansion of the leukemic clone is known to associate with impairment of normal hematopoiesis, resulting in severe anemia, thrombocytopenia, and immunodeficiency (4). Understanding the mechanisms underlying the regulation of leukemia-initiating LSCs by the BM niche and the alteration of the BM microenvironment by LSCs is crucial to eradicating AML (5).
BM niches fuel the growth of leukemia cells and contribute to the therapy resistance and metastatic potential of leukemia cells by shielding LSCs (6). It has become increasingly evident that myeloproliferative neoplasia and myelodysplastic syndrome (MDS) cells as well as AML cells remodel the BM niche into a malignant niche that contributes to their development through cellular, structural, and functional changes. Not only a “microenvironment-induced oncogenesis” but also a “malignancy-induced microenvironment” has been proposed (7). This review describes the key components and regulation, via cytokines, chemokines, adhesion molecules, the sympathetic nervous system, and hypoxic conditions, of the BM endosteal and vascular niches in healthy and in leukemic BM. The genetic abnormalities of leukemia-associated stroma, which are currently a subject of intense investigation to understand their contribution to LSC survival and expansion, will be also discussed.
Components of microenvironmental niches for normal hematopoietic stem cells
Historically, two distinct anatomical niches for HSCs have been described: the endosteal and vascular niches (8-11). The endosteal and vascular niches are closely related to distinct vascular structures, arterioles and sinusoids (12, 13), respectively, and work in concert (14, 15). Recent advances in microscopy, flow cytometry, and transgenic animal models have revolutionized the understanding of these niches (16) and the intrinsic and extrinsic cues that regulate the behavior of HSCs.
Arterioles run in proximity to the endosteal surface, accompanied by sympathetic nerve fibers ensheathed by nonmyelinating Schwann cells, and quiescent HSCs associate with periarteriolar niches found within the endosteal BM (12). In turn, sinusoids, fenestrated and lined by reticular-shaped sinusoidal cells, are associated with less-quiescent HSCs re-localizing to perisinusoidal niches (12, 13).
Hypoxia promotes maintenance of HSCs in a quiescent and pluripotent state(17), while HSCs that reside in close proximity to the vascular niche are actively cycling and are responsible for replenishing circulating cells (18). Hypoxia-Inducible Factor α (HIF-1α), the master transcriptional regulator of hypoxia response, maintains HSCs in a quiescent state by promoting expression of various target genes, including vascular endothelial growth factor (VEGF) and C-X-C chemokine receptor type 4 (CXCR4) (3, 19, 20). On the other hand, the high abundance of capillaries in cancellous bone is incompatible with the hypoxia in BM niches, and histologic images of HSCs indicate that HSCs reside in proximity to the proliferative progenitor cells throughout the BM (9). A recent quantitative imaging study of HSCs further demonstrated that, despite their preferential endosteal localization, HSCs closely interact with BM microvessels and yet exhibit a hypoxic profile (21). These novel observations suggest that regulation of the hypoxic HSC state is mediated through a cell-intrinsic system rather than lack of blood supply.
The endosteal niche
Osteoblasts
The surface of the endosteum is lined by osteoblasts and osteoclasts. Osteoblasts are progenitor bone-forming cells derived from pluripotent MSCs; they work in tandem with osteoclasts in the process of osteogenesis (22). Osteopontin (OPN) and N-cadherin, extracellular matrix (ECM) proteins of endosteal osteolineage cells, mediate HSC localization (23). Signaling through Jagged-1 (Jag-1) on osteoblasts and its receptor Notch on HSCs is involved in the expansion of the HSC pool (8). On the other hand, interaction of angiopoietin-1 (Angpt1) in osteoblasts with its receptor Tie-2 on HSCs results in activation of β1-integrin and N-cadherin and enhanced adhesion between the niche cells and the HSCs, which contributes to the maintenance of stem cell quiescence (24). The C-X-C motif chemokine 12 (CXCL12) produced by osteoblasts is the major chemoattractant for HSCs (25).
Osteoclasts
Bone-resorbing osteoclasts participate in the initial formation as well as the maintenance of the cavities that constitute the endosteal niche (22, 26). Osteoclastic bone resorption produces abundant active transforming growth factor beta (TGF-β) from bone, the largest latent reservoir of TGF-β (27). Osteoclast inhibition by bisphosphonates causes severe depression of HSC formation and delay of hematopoietic recovery (28). The relevance of osteoclasts in retaining high calcium concentration, which is critical for localization of calcium-sensing receptor-expressing HSCs on the endosteal surface, has been demonstrated in a mouse model (10).
Sympathetic nervous system
The sympathetic nervous system is responsible, via norepinephrine signaling, for regulation of HSCs residing in the periarteriolar position (29). HSCs residing in contact with nonmyelinating Schwann cells, ensheathed autonomic nerves, are reported to be highly quiescent. Mechanistic analysis showed that nonmyelinating Schwann cells are responsible for activation of latent TGF-β produced by a variety of BM cells, including osteoblasts and MSCs. TGF-β/Smad signaling plays a critical role in HSC maintenance, and autonomic denervation reduced the number of active TGF-β-producing cells, resulting in rapid loss of HSCs from the BM (30). Consistent with these findings, chemotherapy-induced nerve injury impaired hematopoietic regeneration in a murine model (31).
Mature hematopoietic cells and niche regulation
T-regulatory (Treg) cells provide a relative immune sanctuary for HSC on the endosteal surface (13, 32), in which co-localized accumulation of HSCs with Treg cells has been shown to be lost after depletion of Treg cells (32). Other multiple adjunct cell types, including macrophages and megakaryocytes, participate in HSC localization and regulation of HSC quiescence (13).
The vascular niche
The perivascular niches are the important resident area of HSCs (1). The candidate niche cells include CXCL12-abundant reticular (CAR) cells (33), Nestin-positive (Nestin+) MSCs (34), and leptin receptor-positive (LepR+) MSCs (11), which exhibit significant overlap and express multiple soluble and membrane-bound factors that regulate HSC self-renewal and retention (35).
CXCL12-abundant reticular cells
Components of the BM niches, including osteoblasts, CAR cells, and Nestin+ MSCs, are known to be CXCL12-secreting cells. In the sinusoidal areas of the BM where HSCs predominantly reside, the HSCs are in direct contact with CAR cells, which secrete higher levels of CXCL12 than osteoblasts. Most CAR cell populations express PPARγ, Runx2, and Osterix in the BM, and short-term ablation of CAR cells in vivo severely impaired the adipogenic and osteogenic differentiation potential of BM cells, indicating that CAR cells are adipo-osteogenic bipotential progenitors (36).
CXCL12 attracts HSCs expressing its cognate receptor CXCR4. CXCL12-CXCR4 signaling is involved in homing of HSCs into BM and in adhesion through activation of several integrins, and supports survival of HSCs (33). Downregulation of CXCL12 or depletion of CXCR4 leads to mobilization of HSCs into the peripheral blood and reduction of the HSC population (33), suggesting that CXCL12-CXCR4 chemokine signaling plays an essential role in maintaining the HSC pool.
Nestin-positive MSCs and leptin receptor-positive MSCs
MSCs are identifiable by expression of indicator genes from promoters for Nestin, LepR, Prx-1, or Mx-1, with overlaps between the populations not well defined (13). Specific Nestin+ MSCs, which co-localize with HSCs and adrenergic nerve fibers (34), have been reported to participate in the regulation of BM niches (7). Depletion of Nestin+ MSCs significantly reduced BM homing of hematopoietic progenitors and HSC content in the BM in an in vivo model (34). The conditional deletion of stem cell factor (SCF) from LepR+ perivascular stromal cells, including Nestin+ MSCs and CAR cells, significantly reduced HSC number (11), whereas SCF deletion from osteoblasts did not affect HSC frequency and function (11).
Bone marrow niche for leukemic stem cells
LSC behaviour, like that of HSCs, is modulated by interactions and signals received within their BM niches (8, 24, 37). Although LSCs share the molecules with HSCs to mediate the interaction with the BM niches, recent studies indicate that LSCs create their “foster home,” inducing reversible changes in niche cell function or composition that contribute to LSC engraftment into the niches and subsequent leukemia development, survival, and drug resistance (7, 38). Suppression of normal hematopoiesis in leukemia patients with relatively low tumor burden may reflect disruption of normal HSC BM niches and creation of leukemia niches by leukemic cells (39).
Homing to the BM niche: CXCL12-CXCR4 signaling
Interaction of LSCs and BM niches is recognized as the major cause of AML relapse. Many in vitro and in vivo studies demonstrated that inhibition of CXCL12-CXCR4 interactions results in abolishment of CXCL12-induced Chemotaxis, inactivation of prosurvival signaling pathways, and decreases in stromal protective effects on chemotherapy-induced apoptosis in AML cells (40-42). CXCR4 expression has been reported to be higher in Flt3/internal tandem duplication AML than in Flt3/wild-type AML (43), and CXCR4 inhibition increased sensitivity of FLT3-mutated leukemic cells to the FLT3 inhibitor sorafenib under stromal co-culture conditions (44). Similarly, treatment with CXCR4 inhibitor plerixafor combined with TGF-β-neutralizing antibody and cytarabine decreased leukemia burden and prolonged survival in a leukemia mouse model, proving that TGF-β and CXCL12, produced abundantly in the BM niche, play a role in AML chemoresistance (45). In AML cells, chemotherapy-induced upregulation of surface CXCR4 has been demonstrated, which caused stromal protection from additional chemotherapy-induced apoptosis (46). These results suggest that CXCL12-CXCR4 interactions in the BM microenvironment contribute to the chemoresistance of leukemic cells and that disruption of these interactions by CXCR4 inhibitors represents a rational strategy for blocking LSC homing to a BM niche and/or sensitizing AML cells to chemotherapy or kinase inhibitors.
Adhesion to the BMniche
Adhesion to the BM niche directly protects LSCs from damage by chemotherapy or kinase inhibitors, and cell adhesion receptor integrins are required for lodging of LSCs in the BM niche (47). Integrins are known to link to CD44 through their interactions with selectins. The transmembrane glycoprotein CD44, existing as a standard isoform (CD44s) and a range of variant isoforms (CD44v), modulates interactions of LSCs with ECM components hyaluronan and a range of growth factor ligands (41, 48). Jin et al. demonstrated that administration of CD44 antibody markedly reduced leukemic repopulation in human AML-transplanted mice (48). As an elemental mechanism in promoting leukemic progression and chemoresistance, CD44 signaling modulates microRNA expression, which regulates promoter methylation status and gene expression (49), and induces leukemia cell reprogramming to exhibit a more stem cell-like LSC phenotype (49).
Hypoxia/HIF-1α signaling
The overexpression of oxygen-regulated component HIF-1α has been shown in clusters of leukemic cells in BM specimens (50). One of the accepted functions of hypoxia and HIF-1α is upregulation of growth factor VEGF and stimulation of angiogenesis. The microvasculature is an active component of the BM microenvironment, supplying appropriate oxygen and nutrients. VEGF secreted by leukemic cells activates receptors on both leukemic and endothelial cells and plays a vital role in the growth of leukemia cells (51). The direct HIF-1α inhibitor PX-478 decreased hypoxia-mediated VEGF expression in tumor xenografts, resulting in antitumor activity (52). Furthermore, HIF-1α has been demonstrated to regulate CXCL12 gene expression in endothelial cells, which increased migration and homing of circulating CXCR4-positive progenitor cells into the ischemic tissue (53), and hypoxia upregulates CXCR4 expression in AML cells (54).
Sympathetic nervous system
Leukemic cells have been shown to alter the niche in a manner that affects normal HSC migration, pool size, and differentiation (39). Schepers et al. demonstrated that the malignant myeloid cells reprogrammed the endosteal BM osteoblasts into the pro-inflammatory BM niche that supports LSCs while creating an inhospitable environment for normal HSCs (55).
Induction of neoplastic niche alteration by AML cells through sympathetic neuropathy has been demonstrated recently (56). Hanoun et al. showed that MLL-AF9 AML cells rapidly transform the HSC niche, reducing the numbers of arteriole-associated niche cells and the density of their sympathetic nerve network, which is critical for MSC quiescence. HSCs isolated from AML mice had decreased BM repopulation capacity, with a greater extent in the peripheral blood and spleen, indicating that AML colonization of the BM significantly alters the HSC niche (23).
A similar role for sympathetic neuropathy has been demonstrated in malignant cells during disease progression in myeloproliferative neoplasia (MPN) (57). The destruction of sympathetic nerve fibers in the perivascular niche by MPN cells induces Nestin+ MSC apoptosis, HSC niche alteration, and MPN pathogenesis. Interleukin-lb produced by MPN cells destroy Schwann cells, which support the sympathetic nerve fibers, followed by the apoptotic loss of Nestin+ cells and reduction of HSC maintenance factors in the microenvironment, such as CXCL12, resulting in peripheral mobilization of HSCs and acceleration of MPN cell expansion in the BM.
Both studies provide new insight into the critical role of the sympathetic nervous system in regulating the HSC niche and in malignant niche transformation during disease progression in AML and MPN.
Modulation of LSC niche
LSCs that receive the support of a BM niche for their survival may in turn contribute to deregulation of the BM niche by their dominant proliferation-promoting signals. Recently, MSCs have been recognized as participating in the malignant process. MSCs in the BM constitute a heterogeneous population (58), and heterotypic signaling from diseased “reprogrammed” MSCs may affect other cells in the BM. MDS is characterized by BM failure and predisposition for evolution into AML (58). Medyouf et al. demonstrated that MSCs are implicated in MDS initiation and progression (59). CD34+ hematopoietic cells derived from MDS patients, co-injected with their corresponding MSCs into the BM cavity of mice, demonstrated greater reconstitution ability than CD34+ cells alone or co-injected with normal MSCs. Transcriptional profiling revealed the aberrant gene expression implicated in intercellular crosstalk, osteo/adipogenesis, inflammation, and fibrosis in MDS MSCs, indicating that MSCs are required to drive disease initiation and progression (58). Similarly, deficiency of phosphatase and tensin homolog (PTEN), a tumor suppressor and antagonist of the PI3K pathway, in both hematopoietic cells and the microenvironment resulted in myeloproliferation that progressed to overt leukemia/lymphoma (60). Notably, PTEN deletion in hematopoietic cells in the presence of a wild-type BM microenvironment promoted HSC depletion without evidence of myeloproliferation or leukemic development (60).
These findings indicate the importance of interactions between hematopoietic cells and the BM niche/microenvironment and suggest that additional genetic mutations within the BM microenvironment may be necessary for leukemic transformation. Raaijmakers et al. demonstrated that conditional knockout of DICER1, a gene that regulates microRNA processing, in osteoblastic precursors results in BM failure and leukemia predisposition.
DICERl deletion caused reduced expression of SBDS, the gene mutated in Schwachman-Bodian-Diamond syndrome. Deletion of SBDS in mouse osteoprogenitors induced myelodysplasia and the development of AML (7). Other genetic changes in the BM microenvironment also have been demonstrated to contribute to or be required for leukemogenesis. Walkley et al. reported that dysfunction of the retinoblastoma protein, a central regulator of the cell cycle and a tumor suppressor, or of retinoic acid receptor y in the BM microenvironment contributes to development of preleukemic myeloproliferative disease (61, 62). These findings highlight the suggestion that primary stromal dysfunction can result in secondary neoplastic disease, supporting the concept of niche-induced oncogenesis.
It has been shown that β-catenin signaling, NF-κB activation, and Jagged/Notch activation play critical roles in the development of AML LSCs. Wang et al. showed that the Wnt/β-catenin signaling pathway was required for self-renewal of murine LSCs derived from MLL-AF9-induced AML (63). Very recently, Kode et al. demonstrated that activation of β-catenin in mature osteoblastic cells induces AML via Notch activation (64). The activation of NF-κB or the absence of its inhibitor IκBα in myelopoietic cells changed the nonhematopoietic compartment, resulting in increased numbers of dysplastic hematopoietic cells with progression into secondary AML via upregulated perinatal expression of Jag-1 in IκBαΔΔ hepatocytes and activation of Notch 1 in neutrophils (65). Very recently, the interaction between vascular cell adhesion molecule 1 (VCAM-1) and very late antigen-4 (VLA-4) has been shown to play an integral role in the activation of NF-κB in the stromal and leukemia cell compartments (66).
Intriguingly, recent studies have indicated the link between microenvironment and AML metabolism. In an in vitro screening of 14,718 compounds in LSC-enriched AML cells, the cholesterol-lowering drug lovastatin induced cell-autonomous inhibition of LSCs in a co-culture system with MSCs but did not inhibit AML cells cultured alone. Lovastatin pretreatment of LSC-stromal co-cultures also prolonged the survival of mice injected with these cells (67, 68). In turn, AML cells alter the immune microenvironment via release of high concentrations of arginase II, which suppresses T cell proliferation, polarizes surrounding monocytes into a suppressive M2-like phenotype, and finally inhibits proliferation and differentiation of murine granulocyte-monocyte progenitors and human CD34+ progenitors (69). These findings imply that metabolic features supporting the AML BM niche may yield novel therapeutic targets.
Conclusion
Recent and current studies are elucidating the role of the BM microenvironment in the pathogenesis of hematologic tumors. Identification of the precise mechanisms involved in leukemia-host interactions that contribute to drug resistance will provide important insights for development of novel therapies targeting both leukemic cells and the cells in their surrounding microenvironment. Although the impacts of the leukemic microenvironment on therapeutic outcome or the potential targets of the leukemic microenvironment have not been well characterized, the increasing insights into LSC development in their specific BM microenvironment will ultimately result in novel therapeutic strategies in a framework for targeting niche cells to attenuate leukemic progression or targeting LSCs without adversely affecting normal stem cell self-renewal.
Figure 1. Key components of the leukemic bone marrow microenvironment.
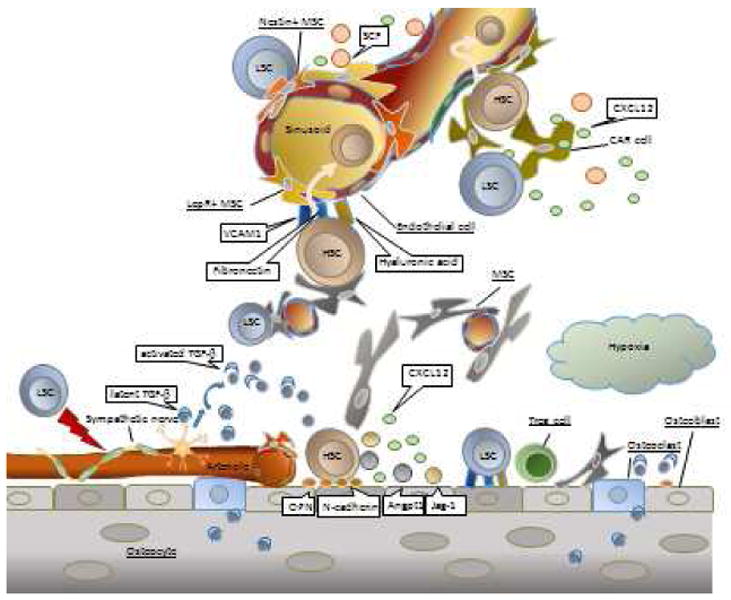
Normal hematopoietic stem cell (HSC) niches consist of multiple cell types, including osteoblasts, CXCL12-abundant reticular (CAR) cells, Nestin-positive mesenchymal stem cells (Nestin+ MSCs), leptin receptor-expressing MSCs (LepR+ MSCs), endothelial cells, and Schwann cells wrapping sympathetic nerve fibers. Leukemia stem cells (LSCs) hijack HSC bone marrow (BM) spaces, including perivascular and endosteal niches, partially by sympathetic nerve system denervation. The BM stromal cells and osteoblasts produce complex extracellular matrix, comprising molecules such as vascular cell-adhesion molecule-1 (VCAM1), fibronectin, and hyaluronic acid, which facilitates engraftment and adhesion of LSCs. Osteoblasts within endosteal niches generate osteopontin (OPN), N-cadherin, angiopoietin-1 (Angpt1), and Jagged-1 (Jag-1), which in turn promote leukemia cell dormancy and decrease their chemosensitivity. Osteoclastic bone resorption preserves high calcium concentration as well as transforming growth factor beta (TGF-β) from bone, which are critical for localization of HSC. T-regulatory (Treg) cells provide an immune sanctuary for HSCs on the endosteal surface. Latent TGF-β is activated by the sympathetic nervous system, and autonomic denervation by LSCs induces rapid loss of HSCs from the BM. CAR cells, Nestin+ MSCs, LepR+ MSCs, and endothelial cells may play roles in leukemia cells migration to the perivascular microenvironment via interaction with cytokines, chemokines, and adhesion molecules.
Acknowledgments
Supported in part by NIH/NCI R01 CA155056-04 (MK), Leukemia and Lymphoma Society (MK) and NIH/NCI P30 CA016672 (MK)
Footnotes
Compliance with Ethics Guidelines: Conflict of Interest: Dr. Yoko Tabe and Dr. Marina Konopleva each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
Yoko Tabe, Email: tabe@juntendo.ac.jp.
Marina Konopleva, Email: mkonople@mdanderson.org.
References
Papers of particular interest, published recently, have been highlighted as:
•• Of major importance
- 1.Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25. [PubMed] [Google Scholar]
- 2.Nwajei F, Konopleva M. The bone marrow microenvironment as niche retreats for hematopoietic and leukemic stem cells. Adv Hematol. 2013;2013:953982. doi: 10.1155/2013/953982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol. 2014;164:767–78. doi: 10.1111/bjh.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet. 2013;381:484–95. doi: 10.1016/S0140-6736(12)61727-9. [DOI] [PubMed] [Google Scholar]
- 5.Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat. 2009;12:103–13. doi: 10.1016/j.drup.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 7.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–7. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 9.Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- 10.Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439:599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- 11.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–43. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kfoury Y, Mercier F, Scadden DT. Snapshot: The hematopoietic stem cell niche. Cell. 2014;158:228, e1. doi: 10.1016/j.cell.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 14.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20:833–46. doi: 10.1038/nm.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perry JM, Li L. Disrupting the stem cell niche: good seeds in bad soil. Cell. 2007;129:1045–7. doi: 10.1016/j.cell.2007.05.053. [DOI] [PubMed] [Google Scholar]
- 16.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–34. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 18.Winkler IG, Barbier V, Wadley R, Zannettino AC, Williams S, Lévesque JP. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood. 2010;116:375–85. doi: 10.1182/blood-2009-07-233437. [DOI] [PubMed] [Google Scholar]
- 19.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 20.Simsek T, Kocabas F, Zheng J, DeBerardinis RJ, Mahmoud AI, Olson EN, et al. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell. 2010;7:380–90. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15:533–43. doi: 10.1038/ncb2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schröder HC, Wang XH, Wiens M, Diehl-Seifert B, Kropf K, Schloßmacher U, et al. Silicate modulates the cross-talk between osteoblasts (SaOS-2) and osteoclasts (RAW 264.7 cells): inhibition of osteoclast growth and differentiation. J Cell Biochem. 2012;113:3197–206. doi: 10.1002/jcb.24196. [DOI] [PubMed] [Google Scholar]
- 23.Price T, Sipkins DA. Rewiring the niche: sympathetic neuropathy drives malignant niche transformation. Cell Stem Cell. 2014;15:261–2. doi: 10.1016/j.stem.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 24.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Christopher MJ, Liu F, Hilton MJ, Long F, Link DC. Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood. 2009;114:1331–9. doi: 10.1182/blood-2008-10-184754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SE, Wakkach A, Blin-Wakkach C. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med. 2012;209:537–49. doi: 10.1084/jem.20110994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balooch G, Balooch M, Nalla RK, Schilling S, Filvaroff EH, Marshall GW, et al. TGF-beta regulates the mechanical properties and composition of bone matrix. Proc Natl Acad Sci U S A. 2005;102:18813–8. doi: 10.1073/pnas.0507417102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lymperi S, Ersek A, Ferraro F, Dazzi F, Horwood NJ. Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood. 2011;117:1540–9. doi: 10.1182/blood-2010-05-282855. [DOI] [PubMed] [Google Scholar]
- 29.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–21. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 30.Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–58. doi: 10.1016/j.cell.2011.09.053. [DOI] [PubMed] [Google Scholar]
- 31.Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med. 2013;19:695–703. doi: 10.1038/nm.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474:216–9. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 34.Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doan PL, Chute JP. The vascular niche: home for normal and malignant hematopoietic stem cells. Leukemia. 2012;26:54–62. doi: 10.1038/leu.2011.236. [DOI] [PubMed] [Google Scholar]
- 36.Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–99. doi: 10.1016/j.immuni.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood. 2005;106:1232–9. doi: 10.1182/blood-2004-11-4422. [DOI] [PubMed] [Google Scholar]
- 38.Dührsen U, Hossfeld DK. Stromal abnormalities in neoplastic bone marrow diseases. Ann Hematol. 1996;73:53–70. doi: 10.1007/s002770050203. [DOI] [PubMed] [Google Scholar]
- 39••.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–5. doi: 10.1126/science.1164390. This study showed that leukemic cell growth disrupts normal hematopoietic progenitor cell bone marrow niches and creates abnormal microenvironments. [DOI] [PubMed] [Google Scholar]
- 40.Zeng Z, Samudio IJ, Munsell M, An J, Huang Z, Estey E, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5:3113–21. doi: 10.1158/1535-7163.MCT-06-0228. [DOI] [PubMed] [Google Scholar]
- 41.Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206–14. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19:357–66. doi: 10.1158/1078-0432.CCR-12-2333. [DOI] [PubMed] [Google Scholar]
- 43.Rombouts EJ, Pavic B, Löwenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104:550–7. doi: 10.1182/blood-2004-02-0566. [DOI] [PubMed] [Google Scholar]
- 44.Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–24. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tabe Y, Shi YX, Zeng Z, Jin L, Shikami M, Hatanaka Y, et al. TGF-β-Neutralizing Antibody 1D11 Enhances Cytarabine-lnduced Apoptosis in AML Cells in the Bone Marrow Microenvironment. PLoS One. 2013;8:e62785. doi: 10.1371/journal.pone.0062785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sison EA, Mclntyre E, Magoon D, Brown P. Dynamic chemotherapy-induced upregulation of CXCR4 expression: a mechanism of therapeutic resistance in pediatric AML. Mol Cancer Res. 2013;11:1004–16. doi: 10.1158/1541-7786.MCR-13-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redondo-Muñoz J, Ugarte-Berzal E, García-Marco JA, del Cerro MH, Van den Steen PE, Opdenakker G, et al. Alpha4beta1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood. 2008;112:169–78. doi: 10.1182/blood-2007-08-109249. [DOI] [PubMed] [Google Scholar]
- 48.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 49.Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood) 2013;238:324–38. doi: 10.1177/1535370213480714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wellmann S, Guschmann M, Griethe W, Eckert C, von Stackelberg A, Lottaz C, et al. Activation of the HIF pathway in childhood ALL, prognostic implications of VEGF. Leukemia. 2004;18:926–33. doi: 10.1038/sj.leu.2403332. [DOI] [PubMed] [Google Scholar]
- 51.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 52.Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, et al. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2008;7:90–100. doi: 10.1158/1535-7163.MCT-07-0463. [DOI] [PubMed] [Google Scholar]
- 53.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–64. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 54.Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood. 2009;113:1504–12. doi: 10.1182/blood-2008-06-161539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13:285–99. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15:365–75. doi: 10.1016/j.stem.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arranz AM, Delbroek L, Van Kolen K, Guimarães MR, Mandemakers W, Daneeis G, et al. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J Cell Sci. 2014 doi: 10.1242/jcs.158196. [DOI] [PubMed] [Google Scholar]
- 58.Raaijmakers MH. Disease progression in myelodysplastic syndromes: do mesenchymal cells pave the way? Cell Stem Cell. 2014;14:695–7. doi: 10.1016/j.stem.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 59.Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14:824–37. doi: 10.1016/j.stem.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 60.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 61.Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129:1081–95. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–3. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64••.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506:240–4. doi: 10.1038/nature12883. This study showed that an activating mutation of β-catenin alters the differentiation potential of myeloid and lymphoid progenitors leading to development of acute myeloid leukaemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rupec RA, Jundt F, Rebholz B, Eckelt B, Weindl G, Herzinger T, et al. Stroma-mediated dysregulation of myelopoiesis in mice lacking I kappa B alpha. Immunity. 2005;22:479–91. doi: 10.1016/j.immuni.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 66.Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of nf-κb mediates chemoresistance. Blood. 2014;123:2691–702. doi: 10.1182/blood-2013-06-511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seton-Rogers S. Tumour microenvironment: Destroying leukaemia stem cell habitats. Nat Rev Cancer. 2013;13:821. doi: 10.1038/nrc3639. [DOI] [PubMed] [Google Scholar]
- 68.Hartwell KA, Miller PG, Mukherjee S, Kahn AR, Stewart AL, Logan DJ, et al. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat Chem Biol. 2013;9:840–8. doi: 10.1038/nchembio.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. 2013;122:749–58. doi: 10.1182/blood-2013-01-480129. [DOI] [PMC free article] [PubMed] [Google Scholar]