

Abstract
The successful implementation of tyrosine kinase inhibitors (TKIs) for the treatment of chronic myeloid leukemia (CML) remains a flagship for molecularly targeted therapy in cancer. This focused review highlights critical elements of the underlying biology of CML and provides a summary of the molecular mechanisms that lead to TKI resistance: BCR-ABL1 mutation-based resistance and therapy escape through alternative pathway activation despite inhibition of BCR-ABL1 tyrosine kinase activity. We direct attention to the most current manifestations of these issues, including emergence of pan-TKI-resistant BCR-ABL1 compound mutants, new strategies for identification and therapeutic targeting of alternative pathways, and the exciting, controversial topic of cessation of TKI therapy leading to durable treatment-free remissions for a subset of patients. Further gains in our understanding of the biology of Philadelphia chromosome-positive (Ph-positive) leukemia and mechanisms of resistance to BCR-ABL1 TKIs will benefit patients and also provide a blueprint for similar discovery in other cancers.
Keywords: Chronic myeloid leukemia (CML), tyrosine kinase inhibitor (TKI), BCR-ABL1 compound mutation, treatment-free remission (TFR), BCR-ABL1-independent resistance, imatinib
Introduction – Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia
The paradigm of tyrosine kinase inhibitors (TKIs) for the treatment of patients with chronic myeloid leukemia (CML) is a success story of the highest order in molecularly targeted therapy [1, 2]. The majority of chronic phase CML patients treated with first-line imatinib demonstrate rapid achievement of durable remission, and five-year overall and progression-free survival rates approach 90%. For those patients who fail therapy due to resistance or intolerance, a quiver of additional FDA-approved second-generation TKIs are available for deployment. In total, five TKIs are approved for first-line and/or salvage treatment of CML in the U.S.: imatinib, dasatinib, nilotinib, bosutinib, and ponatinib (Table 1). The recent approval of nilotinib [3] and dasatinib [4] for first-line CML therapy, along with the ongoing clinical evaluation of bosutinib for first-line and salvage use [5–7] and the approval of the pan-BCR-ABL1 inhibitor ponatinib for refractory CML [8], has also broadened prospects for minimizing resistance and maximizing long-term disease control.
Table 1.
FDA-approved and investigational BCR-ABL1 inhibitors targeting the kinase domain or the myristate pocket.
| Inhibitor | Chemical structure | Binding site/ Inhibitor type |
Regulatory status/ approval |
|---|---|---|---|
| imatinib (Gleevec) |
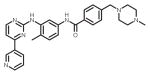
|
ATP-binding site/ATP-competitive | FDA approved/Frontline therapy |
| nilotinib (Tasigna) |
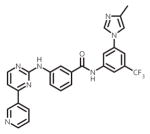
|
ATP-binding site/ATP-competitive | FDA approved/Frontline therapy |
| dasatinib (Sprycel) |
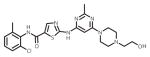
|
ATP-binding site/ATP-competitive | FDA approved/Frontline therapy |
| bosutinib (Bosulif) |
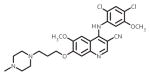
|
ATP-binding site/ATP-competitive | FDA approved/2nd-line therapy |
| ponatinib (Iclusig) |
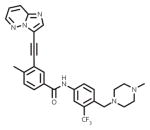
|
ATP-binding site/ATP-competitive | FDA approved/2nd-line therapy |
| ABL001 | (Currently proprietary) | Myristate pocket/Allosteric | Phase I/2nd-line therapy |
These clinical advances have largely capitalized on the molecular biology of CML, wherein the BCR-ABL1 kinase encoded by the t(9;22) chromosomal translocation is importantly present in all CML cells but not normal cells and required for disease transformation [9–11]. The fusion of BCR and ABL1 facilitates dimerization of BCR-ABL1 proteins via the coiled-coil domain of BCR. Mutual transphosphorylation of the juxtaposed ABL1 kinase domains results in constitutively active tyrosine kinase activity. Activated BCR-ABL1 drives a variety of downstream pro-survival, growth, and anti-apoptosis signaling pathways including JAK/STAT, RAS/RAF/MEK/ERK, PI3K/AKT, and BAD/BCL-XL. Furthermore, expression of BCR-ABL1 in murine bone marrow transplantation models results in an aggressive myeloid leukemia phenotype that remains sensitive to treatment with ABL1 TKIs [12].
If left untreated clinically, CML progresses from a chronic phase (CP-CML) featuring excessive proliferation of the full lineage of myeloid cells in the bone marrow through an accelerated phase to an unstable and aggressive blast crisis (which may present in myeloid or lymphoid form) characterized by excessive marrow and peripheral blood blasts and a block in differentiation. Additionally, approximately 20–30% of adult cases and 3–5% of pediatric cases of Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph-positive ALL) harbor the t(9;22) translocation. TKIs targeting BCR-ABL1 are often also part of the treatment approach for these patients, but in this setting and in the advanced phases of CML, responses are almost inevitably transient and the prognosis remains poor compared to CP-CML.
Given the significant milestones achieved in understanding the molecular pathogenesis of CML and the successful implementation of TKIs in its treatment to date, there may be a perception that “CML is done” as far as the need for additional research. While we wish this were true, several challenging hurdles remain to be cleared. Here, we survey and discuss progress on three of the most pressing current problems pertaining to the treatment and management of in Ph-positive leukemia.
Problem 1: Emergence of TKI-resistant versions of BCR-ABL1
In the field of TKI resistance, the oldest trick in the book is acquisition of a mutation in the gene encoding the target enzyme that results in decreased or eliminated inhibitor effectiveness without undue compromise in catalytic function. In the case of CML, approximately 20–30% of patients initially treated with imatinib will develop resistance to therapy, most commonly through gain of point mutations within the kinase domain of BCR-ABL1 that directly interfere with or indirectly disfavor imatinib binding. To date, over 100 different BCR-ABL1 kinase domain mutations have been reported in resistance to imatinib and to a lesser extent, second-generation TKIs, primarily centered around or within the phosphate binding loop (P-loop), ATP binding site, and activation loop [2, 13, 14].
With respect to single mutations in BCR-ABL1, this clinical challenge has been met with a succession of second-generation TKIs (Table 1). In principle, there is now a therapeutic TKI option for every clinically reported BCR-ABL1 point mutant linked to TKI resistance [2]. In the case of the gatekeeper T315I mutant, the only TKI approved for clinical use is ponatinib [15]. Despite the clinical availability of five TKIs, when one considers the limited options for certain mutations, most notably BCR-ABL1T315I, as well as patient-to-patient variability in pharmacokinetic and TKI tolerability profiles, it is our opinion that there is still a small, unmet need in the area of TKIs for BCR-ABL1 point mutations.
A so-far rare but much more concerning unmet need is a strategy to deal with BCR-ABL1 compound mutants, in which the identities of two or more amino acid residues are changed in the same BCR-ABL1 molecule (Fig. 1) [16]. Generally thought to emerge under selective pressure associated with sequential TKI treatment, over 60 different BCR-ABL1 compound mutations have been reported to date in connection with TKI resistance [16–21]. Evidence that BCR-ABL1 kinase has limited tolerance to accrue successive missense mutations indicates that TKIs with activity against compound mutants may block mutational escape [17, 22]. Drug sensitivity profiling of a broad panel of two-component compound mutants indicates that T315I-inclusive compound mutants in particular confer a high degree of resistance to all currently available BCR-ABL1 TKIs, suggesting that patients who harbor such a mutation may have very limited therapeutic options and underscoring the need to distinguish polyclonal mutations from true compound mutations [16–18]. Furthermore, many patients who start on ponatinib therapy harbor a T315I mutation at baseline and T315I-inclusive compound mutations have been confirmed in a subset of clinical failures of ponatinib [8, 18]. This warrants close monitoring of such patients for evidence of potential emergence of compound mutants. By contrast, BCR-ABL1 compound mutants that do not include a T315I component show variable sensitivity to the clinically available TKIs, such that one or more TKI may represent a rational treatment option [18]. Additionally, we reported that a small group of key positions is highly represented in clinically observed two-component compound mutants, with select positions (e.g. 315) pairing with nearly all other key positions and some positions (e.g. 252) only pairing with one or two others [18]. Although early detection of extremely low-level BCR-ABL1 compound mutations may eventually prove to be of diagnostic value, technical limitations dating back to the first reports on BCR-ABL1 compound mutations remain to be overcome [16, 17, 23].
Figure 1.

Resistance to BCR-ABL1 tyrosine kinase inhibitors may take the form of (left) point or compound mutation-based, BCR-ABL1-dependent resistance or (middle) recruitment of alternative pathway signaling upon effective inhibition of BCR-ABL1. (right) A subset of patients achieving deep remissions on TKI therapy who elect to stop therapy subsequently demonstrate apparently durable treatment-free remission (TFR).
An intriguing possibility for sidestepping the problem of BCR-ABL1 kinase domain mutation-mediated TKI resistance in CML is to disable BCR-ABL1 using an allosteric inhibitor directed to a site other than the ATP pocket. A series of studies with the related compounds, GNF-2 and GNF-5, validated this concept. Somewhat surprisingly, perhaps, is the reported observation that engagement of these allosteric inhibitors by BCR-ABL1 restored the efficacy of BCR-ABL1 TKIs, including nilotinib against the T315I mutant [24–27]. More recently, Novartis has initiated a phase 1, multicenter, open-label study of oral ABL001, a BCR-ABL1 myristate site binder that enforces an autoinhibited conformation of the enzyme, in patients with refractory CML or Ph-positive ALL (http://clinicaltrials.gov/show/NCT02081378; Table 1). Capacity of such inhibitors to block compound mutations as well as the possibility of acquired resistance mutations within the allosteric binding site emerging clinically will require additional investigation.
While a subset of BCR-ABL1 compound mutations represent a formidable clinical challenge at present, longer clinical experience and establishment of a universal, gold standard method that can accurately detect low level compound mutations may permit determination of whether a limited set of compound mutants account for most instances of therapy escape via this route. Efforts to understand the scope of this problem and to develop a structural rationale for targeting compound mutants are ongoing.
Problem 2: Some patients experience treatment failure despite effective BCR-ABL1 inhibition
From the earliest reports of clinical resistance to TKI therapy in CML, it has been clear that BCR-ABL1 mutations are not always the explanation for relapse. Now that the field has become reasonably proficient at controlling BCR-ABL1 mutation-based resistance, the quest to understand and intercept other escape routes has finally begun to get the attention it requires. In BCR-ABL1-independent resistance, TKIs successfully suppress BCR-ABL1 kinase activity, but alternative signaling through pathways such as SRC [28, 29], PI3K [30], KRAS [31] and JAK2 [32] compensate for this loss (Fig. 1B). However, no unifying concept of BCR-ABL1-independent resistance has emerged and clinical management relies on cytotoxic agents or transplant.
The inability to identify and target BCR-ABL1-independent alternative pathways has important clinical consequences. Interim results from a phase 1 study of ponatinib in patients with refractory Ph-positive leukemia in which 94% of enrolled patients had failed ≥2 prior TKIs demonstrated that responses occurred irrespective of BCR-ABL1 mutation status and were mostly durable in CP-CML, but major molecular response rates were lower in patients without evidence of a BCR-ABL1 mutation at baseline, suggesting involvement of BCR-ABL1-independent mechanisms in ponatinib resistance [33]. Further analysis from the large, ongoing phase 2 study (PACE trial) suggests that at least half of the occurrences of clinical ponatinib resistance cannot be explained by BCR-ABL1 single or compound mutations, similarly implicating activation of alternative co-critical pathways. In addition, given that responses to any of the clinically available BCR-ABL1 TKIs are generally poorer and transient rather than durable in patients with advanced CML or Ph-positive ALL, and it is unlikely that any single agent TKI, including ponatinib, will change this. To contend with the range of resistance mechanisms, we need TKIs that target compound mutants and we also need to clinically implement TKI/second inhibitor combinations that induce synthetic lethality.
Recent studies on NFAT [34] and MEK [35–37] feedback signaling circuits, for example, reveal new targets for synthetic lethality approaches in therapy-resistant CML and for targeting CML stem cells. Ma and colleagues recently employed a large-scale shRNA-based screen to identify a subset of genes whose down-regulation resulted in decreased imatinib sensitivity in CML cells [37]. While the individual genes implicated were quite varied, nearly all conditions exhibited persistent RAF/MEK/ERK signaling activity attributed to increased PRKCH expression despite inhibition of BCR-ABL1 kinase activity by TKIs. Intriguingly, the combination of BCR-ABL1 TKIs with the MEK inhibitor trametinib resulted in synergistic kill of these cells and prolonged survival in a CML mouse model of BCR-ABL1-independent resistance. Notably, the concept of synthetic lethality opportunity between BCR-ABL1 and MEK targets is also in line with previous studies describing paradoxical RAS-dependent activation of RAF/MEK/ERK in nilotinib-treated CML cells harboring drug-resistant BCR-ABL1 mutations [35] and the role of high levels of MEK-dependent negative feedback in BCR-ABL1-mediated oncogene addiction [36].
Another strategy that has begun to be explored rather than trying to identify and inhibit individual, proximal signaling pathways is to target TKI-induced feedback activation of STAT3 [38, 39]. STAT3 is a mediator of extrinsic TKI resistance conferred on CML cells by bone marrow-derived factors [40, 41]. STAT3Y705 is phosphorylated in TKI-resistant primary CML cells in a cell-autonomous (intrinsic) fashion, suggesting that pSTAT3Y705 integrates intrinsic and extrinsic resistance pathways. As leukemia cells have limited ways to compensate for loss of BCR-ABL1 signaling, a signal integrator such as STAT3 is perhaps an ideal therapeutic target. Targeting transcription factors is notoriously difficult, and development of BP-5-087, a novel and potent mechanism-based STAT3 inhibitor, required a choreographed combination of synthetic chemistry, sensitive in vitro reporter assays and dynamic computational modeling. Additional structure-activity relationship and absorption, distribution, metabolism, excretion studies are ongoing, with the goal of bringing clinical STAT3 inhibitors within reach for the first time [38]. As the field delves further into exploitable resistance mechanisms for novel therapeutic intervention, it is our viewpoint that findings with respect to effective synthetic lethality approaches in Ph-positive leukemia will inform similar strategies in other malignancies such as acute myeloid leukemia (AML).
Along with more thoroughly defining mechanisms of resistance, considerable progress has been made in understanding the additional complexities that define advanced CML and Ph-positive ALL as compared to CP-CML. For example, Beer and colleagues reported that protein levels of the tumor suppressor IKAROS are barely detectable or absent in bone marrow blasts in the majority of CML patients with advanced myeloid disease, compared with substantial levels in CP-CML cells [42]. Forced expression of IK6, a dominant negative isoform of IKAROS, in CD34+ CP-CML cells in vitro conferred features of accelerated phase CML. Deletion of IKAROS has also been previously reported in Ph-positive ALL [43]. These findings link loss or reduction of IKAROS to advanced as compared to chronic phase disease, providing a potential biomarker for impending disease progression.
Problem 3: TKI therapy is not curative; most patients require lifelong TKI therapy
Even at the level of phase 1 clinical trials, imatinib demonstrated astounding efficacy. In the ensuing 15 years, the practice of TKI-based disease management has been continuously improved. One point, however, has always been taken as gospel: TKIs enforce maximum disease control but do not target stem cells and are not curative. As such, any patient discontinuing TKI therapy would be expected to be at risk of immediate or eventual relapse, and there is substantial anecdotal clinical evidence and underlying CML stem cell biology supporting this assumption [44, 45]. This of course also has very significant implications for the financial burden of the treatment of the disease for patients.
The impetus to characterize and effectively target CML at its hematopoietic roots has been a long fought battle. CML originates in the hematopoietic stem cell compartment, and is renewed by poorly defined leukemic stem cells (LSCs). As best we can experimentally determine, LSCs are BCR-ABL1-positive, though whether they express high or low levels of BCR-ABL1 is controversial [46]. Our working hypothesis is that TKIs are, in principle, capable of reaching LSCs and blocking their BCR-ABL1 activity but that this intervention is insufficient to eliminate LSCs [47, 48]. In other words, LSCs are not solely or strictly dependent on BCR-ABL1 kinase activity for survival. There is also evidence that the bone marrow niche is a hypoxic microenvironment that may act to promote LSC maintenance independent of BCR-ABL1 kinase activity, suggesting that combining BCR-ABL1 TKIs with inhibitors of hypoxia-inducible factor 1 α signaling may be a feasible strategy for LSC eradication [49]. In recent reports, several factors have been convincingly implicated in CML LSC maintenance, survival, and resistance to TKI therapy, including arachidonate 15-lipoxygenase [50], the IL-2/CD25 signaling axis [51], and the Wnt/β-catenin axis as influenced by N-cadherin [52]. Furthermore, given that in vitro studies do not reflect all of the barriers inherent in the bone marrow microenvironment [53], recent evidence suggests blockade of adhesion molecule-ligand interactions that are more important for homing and engraftment of LSCs than normal HSCs is a strategy for improving accessibility of TKIs to these cells [54, 55].
Despite this somewhat engrained dogma of CML LSC persistence and inevitable relapse upon stopping treatment, however, some patients who achieved and sustained deep molecular responses for years on TKI therapy accepted the terms of a carefully conceived and controlled clinical trial to see if treatment-free remission (TFR) is possible, and the findings continue to be both intriguing and incompletely understood [56]. The first large-scale trial exploring this question was called STop IMatinib (STIM) (Fig. 1) [57]. A different set of investigators carried out the similarly designed TWISTER study, which utilized a slightly different trigger point for restarting TKI therapy [58]. The interim results for the two trials are remarkably congruent, with in the neighborhood of 40% of patients maintaining TFR at two years, and the vast majority of patients experiencing molecular recurrence doing so within the first seven months after treatment cessation.
Of note, this is ~40% of an already very select population characterized by deep, durable molecular response to TKIs as assessed by quantitative RT-PCR of BCR-ABL1 transcript levels indexed to an international scale [59]. All in all, only ~5% of patients are likely to be eligible for TKI cessation. Current efforts and trial designs are geared toward determining whether use of second-generation TKIs such as dasatinib [4] or nilotinib increase the rate of TFRs, either in the first-line setting or after suboptimal response on imatinib [60, 61]. There is also emphasis on defining the best threshold for trial enrollment and for mandating re-start of TKI therapy [62]. It is becoming clear that these values will need to be tailored to specific situations, as exemplified by the nilotinib-based ENESTcmr trial [61] and follow-up suite of TFR studies (ENESTfreedom, ENESTop, ENESTgoal, ENESTpath).
For the time being, the exciting and somewhat daring prospect of stopping TKI therapy and monitoring for TFR is panning out spectacularly for a small minority of patients [56], but we are not sure how to prospectively identify these patients [62]. One certainty is that any plan to test the waters of TFR at this time should be done only in the setting of a clinical trial. Extensive effort into determining TFR-specific signatures is of great interest and warrants the attention of the field.
Closing Thoughts and Outlook
Many of us will face cancer in our lifetime, and certainly none of us will view it as good news. For those who receive a diagnosis of CML, the availability of TKIs that target the enzymatic activity of the causative BCR-ABL1 fusion tyrosine kinase provides an effective treatment strategy but generally not a cure. Beginning with the regulatory approval of imatinib in May of 2001, the use of TKIs in CML has been honed to a fine art, much to patients’ benefit.
Key current issues include the need for design and clinical implementation of TKIs that inhibit BCR-ABL1 compound mutants and development of inhibitor combinations targeting BCR-ABL1 and alternative pathways. TKI resistance in several other cancers also involves either compound mutations or alternative pathway activation, suggesting a general principle in kinase-targeted therapy. For example, FLT3 ITD-positive AML patients resistant to quizartinib (AC220) exhibit secondary mutations in the kinase activation loop, a subset of which are ponatinib-sensitive [63–65]. Many gastrointestinal stromal tumor (GIST) patients with resistance to imatinib and sunitinib exhibit compound mutations including the KIT gatekeeper residue; overexpression of AXL or focal adhesion kinase is implicated in some cases without secondary KIT mutations [66, 67]. The recent literature is replete with innovative strategies to identify alternative pathway inhibitors that cause cell death when combined with BCR-ABL1 TKIs. For example, our recent report on the role of STAT3 as a signaling node central to TKI resistance and the use of optimized STAT3 inhibitors with ex vivo activity in cells from patients with treatment-refractory CML may eventually impact other cancers lacking effective treatments [39].
The overriding primary goal in treating Ph-positive leukemia is to stay on the chronic phase side of the chronic phase/advanced disease border. Accelerated and especially blastic phase CML as well as Ph-positive ALL take on the problems and limited therapeutic options associated with more deadly diseases such as AML. The second goal is to minimize disease burden and establish durable, event-free remissions, the epitome of which is sustained, deep molecular response. If the quantitative aspects of such a remission are aligned with attempting a carefully monitored TFR in a clinical trial setting, that once seemingly impossible horizon of ‘operational cure’ may be reached by a few. We view the criteria for ‘operational cure’, coined by the late CML pioneer John Goldman, as: a quality of life and life expectancy unaffected by CML and the absence of any intervention other than periodic monitoring of BCR-ABL1 transcript levels as specified by one’s physician.
There is work to be done in Ph-positive leukemia research, most notably continuing to contend with resistance to TKIs and striving toward a cure either through eradication of CML LSCs or by better understanding and exploiting the rules for achieving operational cure with TKIs.
Footnotes
Conflict of Interest
Dr. Christopher A. Eide declares that he has no conflict of interest. Dr. Thomas O’Hare declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Eiring AM, Deininger MW. Individualizing kinase-targeted cancer therapy: the paradigm of chronic myeloid leukemia. Genome Biol. 2014;15(9):461. doi: 10.1186/s13059-014-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12(8):513–26. doi: 10.1038/nrc3317. [DOI] [PubMed] [Google Scholar]
- 3.Hughes TP, Saglio G, Kantarjian HM, Guilhot F, Niederwieser D, Rosti G, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353–60. doi: 10.1182/blood-2013-06-510396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jabbour E, Kantarjian HM, Saglio G, Steegmann JL, Shah NP, Boque C, et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION) Blood. 2014;123(4):494–500. doi: 10.1182/blood-2013-06-511592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brummendorf TH, Cortes JE, de Souza CA, Guilhot F, Duvillie L, Pavlov D, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukaemia: results from the 24-month follow-up of the BELA trial. Br J Haematol. 2014 Sep 8; doi: 10.1111/bjh.13108. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gambacorti-Passerini C, Cortes JE, Lipton JH, Dmoszynska A, Wong RS, Rossiev V, et al. Safety of bosutinib versus imatinib in the phase 3 BELA trial in newly diagnosed chronic phase chronic myeloid leukemia. Am J Hematol. 2014;89(10):947–53. doi: 10.1002/ajh.23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantarjian HM, Cortes JE, Kim DW, Khoury HJ, Brummendorf TH, Porkka K, et al. Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood. 2014;123(9):1309–18. doi: 10.1182/blood-2013-07-513937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8••.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial3 of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–96. doi: 10.1056/NEJMoa1306494. These interim results (median follow-up: 15 months) for the multicenter PACE phase 2 trial of ponatinib in patients with CML or Ph+ ALL (initial dose: 45 mg once daily) find striking clinical benefit in heavily pre-treated CP-CML patients, irrespective of BCR-ABL1 mutation status at trial entry. Occurrences of arterial thrombosis and other severe vascular adverse events are noted, prompting dose adjustment and other proactive measures to optimize ponatinib treatment for patients lacking other therapeutic options. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. 2013;13(8):559–71. doi: 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5(3):172–83. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 11.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96(10):3343–56. [PubMed] [Google Scholar]
- 12.Li S, Ilaria RL, Jr, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189(9):1399–412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8(11):1018–29. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 14.Milojkovic D, Apperley J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin Cancer Res. 2009;15(24):7519–27. doi: 10.1158/1078-0432.CCR-09-1068. [DOI] [PubMed] [Google Scholar]
- 15.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–12. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117(9):2562–9. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khorashad JS, Kelley TW, Szankasi P, Mason CC, Soverini S, Adrian LT, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood. 2013;121(3):489–98. doi: 10.1182/blood-2012-05-431379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18••.Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26(3):428–42. doi: 10.1016/j.ccr.2014.07.006. This study investigates clinically observed compound mutations involving 12 key positions within the BCR-ABL1 kinase domain. Among patients harboring compound mutations at the end of ponatinib treatment, 15/16 cases involved two key positions. In vitro cell proliferation assays confirmed that the effectiveness of ponatinib was substantially decreased in T315I-inclusive compound mutations. In contrast, non-T315I compound mutations exhibited variable sensitivity to ponatinib and other TKIs, raising the possibility that early detection of low-level compound mutations could be combined with inhibitor sensitivity studies to select TKIs capable of suppressing resistant clones. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soverini S, De Benedittis C, Machova Polakova K, Brouckova A, Horner D, Iacono M, et al. Unraveling the complexity of tyrosine kinase inhibitor-resistant populations by ultra-deep sequencing of the BCR-ABL kinase domain. Blood. 2013;122(9):1634–48. doi: 10.1182/blood-2013-03-487728. [DOI] [PubMed] [Google Scholar]
- 20.Gibbons DL, Pricl S, Posocco P, Laurini E, Fermeglia M, Sun H, et al. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc Natl Acad Sci U S A. 2014;111(9):3550–5. doi: 10.1073/pnas.1321173111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kastner R, Zopf A, Preuner S, Proll J, Niklas N, Foskett P, et al. Rapid identification of compound mutations in patients with Philadelphia-positive leukaemias by long-range next generation sequencing. Eur J Cancer. 2014;50(4):793–800. doi: 10.1016/j.ejca.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buffa P, Romano C, Pandini A, Massimino M, Tirro E, Di Raimondo F, et al. BCR-ABL residues interacting with ponatinib are critical to preserve the tumorigenic potential of the oncoprotein. FASEB J. 2014;28(3):1221–36. doi: 10.1096/fj.13-236992. [DOI] [PubMed] [Google Scholar]
- 23.Parker WT, Phillis SR, Yeung DT, Hughes TP, Scott HS, Branford S. Many BCR-ABL1 compound mutations reported in chronic myeloid leukemia patients may actually be artifacts due to PCR-mediated recombination. Blood. 2014;124(1):153–5. doi: 10.1182/blood-2014-05-573485. [DOI] [PubMed] [Google Scholar]
- 24.Khateb M, Ruimi N, Khamisie H, Najajreh Y, Mian A, Metodieva A, et al. Overcoming Bcr-Abl T315I mutation by combination of GNF-2 and ATP competitors in an Abl-independent mechanism. BMC Cancer. 2012;12:563. doi: 10.1186/1471-2407-12-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray NS, Fabbro D. Discovery of allosteric bcr-abl inhibitors from phenotypic screen to clinical candidate. Methods Enzymol. 2014;548:173–88. doi: 10.1016/B978-0-12-397918-6.00007-0. [DOI] [PubMed] [Google Scholar]
- 26.Iacob RE, Zhang J, Gray NS, Engen JR. Allosteric interactions between the myristate- and ATP-site of the Abl kinase. PLoS One. 2011;6(1):e15929. doi: 10.1371/journal.pone.0015929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463(7280):501–6. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101(2):690–8. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- 29.Gioia R, Leroy C, Drullion C, Lagarde V, Etienne G, Dulucq S, et al. Quantitative phosphoproteomics revealed interplay between Syk and Lyn in the resistance to nilotinib in chronic myeloid leukemia cells. Blood. 2011;118(8):2211–21. doi: 10.1182/blood-2010-10-313692. [DOI] [PubMed] [Google Scholar]
- 30.Burchert A, Wang Y, Cai D, von BN, Paschka P, Muller-Brusselbach S, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19(10):1774–82. doi: 10.1038/sj.leu.2403898. [DOI] [PubMed] [Google Scholar]
- 31.Agarwal A, Eide CA, Harlow A, Corbin AS, Mauro MJ, Druker BJ, et al. An activating KRAS mutation in imatinib-resistant chronic myeloid leukemia. Leukemia. 2008;22(12):2269–72. doi: 10.1038/leu.2008.124. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Cai D, Brendel C, Barett C, Erben P, Manley PW, et al. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 2007;109(5):2147–55. doi: 10.1182/blood-2006-08-040022. [DOI] [PubMed] [Google Scholar]
- 33.Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075–88. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’Hare T, et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18(1):74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Packer LM, Rana S, Hayward R, O’Hare T, Eide CA, Rebocho A, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715–27. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Asmussen J, Lasater EA, Tajon C, Oses-Prieto J, Jun YW, Taylor BS, et al. MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction. Cancer Discov. 2014;4(2):200–15. doi: 10.1158/2159-8290.CD-13-0235. Asmussen and colleagues use phosphoproteomics analysis on CML cells transiently exposed to dasatinib to show that BCR-ABL1 may rewire and suppress normal growth factor-dependent signaling. Gene expression analysis implicates a MEK-driven negative feedback circuit in this process, which remains intact upon TKI-mediated interruption of BCR-ABL1 signaling and enforces an irreversible apoptosis program in temporal advance of rescue by pro-survival growth factors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J, et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6(252):252ra121. doi: 10.1126/scitranslmed.3009073. Ma and colleagues identify a panel of different genes whose silencing by RNAi results in imatinib resistance in CML cells, nearly uniformly leading to upregulation of PRKCH. This in turn drives persistent activation of RAF/MEK/ERK signaling after imatinib treatment, and these findings are validated in primary TKI-refractory CML specimens and using in vivo mouse models. The authors also report selective persistent RAF/MEK/ERK activity in CML stem vs. progenitor cells, a difference that may potentially be exploited with BCR-ABL1/MEK inhibitor combinations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eiring AM, Page BD, Kraft IL, Mason CC, Vellore NA, Resetca D, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2014 Aug 19; doi: 10.1038/leu.2014.245. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 39.Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26(2):207–21. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 40.Traer E, Mackenzie R, Snead J, Agarwal A, Eiring AM, O’Hare T, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26:1140–43. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther. 2008;7(10):3169–75. doi: 10.1158/1535-7163.MCT-08-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beer PA, Knapp DJ, Miller PH, Kannan N, Sloma I, Heel K, et al. Disruption of IKAROS activity in primitive chronic phase CML cells mimics myeloid disease progression. Blood. 2014 Nov 4; doi: 10.1182/blood-2014-06-581173. pii: blood-2014-06-581173 Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–4. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 44.Mauro MJ, Druker BJ, Maziarz RT. Divergent clinical outcome in two CML patients who discontinued imatinib therapy after achieving a molecular remission. Leuk Res. 2004;28(Suppl 1):S71–3. doi: 10.1016/j.leukres.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Kuwabara A, Babb A, Ibrahim A, Milojkovic D, Apperley J, Bua M, et al. Poor outcome after reintroduction of imatinib in patients with chronic myeloid leukemia who interrupt therapy on account of pregnancy without having achieved an optimal response. Blood. 2010;116(6):1014–6. doi: 10.1182/blood-2010-04-280206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood. 2012;119(2):530–9. doi: 10.1182/blood-2010-08-303495. [DOI] [PubMed] [Google Scholar]
- 47.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–10. doi: 10.1182/blood-2010-12-326843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ng KP, Manjeri A, Lee KL, Huang W, Tan SY, Chuah CT, et al. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood. 2014;123(21):3316–26. doi: 10.1182/blood-2013-07-511907. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Peng C, Abraham SA, Shan Y, Guo Z, Desouza N, et al. Arachidonate 15-lipoxygenase is required for chronic myeloid leukemia stem cell survival. J Clin Invest. 2014;124(9):3847–62. doi: 10.1172/JCI66129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kobayashi CI, Takubo K, Kobayashi H, Nakamura-Ishizu A, Honda H, Kataoka K, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123(16):2540–9. doi: 10.1182/blood-2013-07-517847. [DOI] [PubMed] [Google Scholar]
- 52.Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood. 2013;121(10):1824–38. doi: 10.1182/blood-2012-02-412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacLean AL, Filippi S, Stumpf MP. The ecology in the hematopoietic stem cell niche determines the clinical outcome in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2014;111(10):3883–8. doi: 10.1073/pnas.1317072111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54•.Krause DS, Lazarides K, Lewis JB, von Andrian UH, Van Etten RA. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood. 2014;123(9):1361–71. doi: 10.1182/blood-2013-11-538694. Investigation of adhesion pathways that contribute to engraftment of BCR-ABL1-driven, CML-like myeloproliferative neoplasia in a mouse retroviral transduction/transplantation model reveal that BCR-ABL1 LSCs are more dependent on selectin-mediated homing and engraftment than normal hematopoietic stem cells. These observations and earlier reports from the same group suggest that blockade of selectin-ligand interactions could mitigate leukemic engraftment and relapse in autografted patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Ren X, Shi W, Wang S, Chen H, Zhang B, et al. Small molecule Me6TREN mobilizes hematopoietic stem/progenitor cells by activating MMP-9 expression and disrupting SDF-1/CXCR4 axis. Blood. 2014;123(3):428–41. doi: 10.1182/blood-2013-04-498535. [DOI] [PubMed] [Google Scholar]
- 56.Mahon FX, Etienne G. Deep molecular response in chronic myeloid leukemia: the new goal of therapy? Clin Cancer Res. 2014;20(2):310–22. doi: 10.1158/1078-0432.CCR-13-1988. [DOI] [PubMed] [Google Scholar]
- 57.Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–35. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 58.Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122(4):515–22. doi: 10.1182/blood-2013-02-483750. [DOI] [PubMed] [Google Scholar]
- 59.Cross NC, White HE, Muller MC, Saglio G, Hochhaus A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia. 2012;26(10):2172–5. doi: 10.1038/leu.2012.104. [DOI] [PubMed] [Google Scholar]
- 60.Shah NP, Guilhot F, Cortes JE, Schiffer CA, le Coutre P, Brummendorf TH, et al. Long-term outcome with dasatinib after imatinib failure in chronic-phase chronic myeloid leukemia: follow-up of a phase 3 study. Blood. 2014;123(15):2317–24. doi: 10.1182/blood-2013-10-532341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hughes TP, Lipton JH, Spector N, Cervantes F, Pasquini R, Clementino NC, et al. Deep molecular responses achieved in patients with CML-CP who are switched to nilotinib after long-term imatinib. Blood. 2014;124(5):729–36. doi: 10.1182/blood-2013-12-544015. [DOI] [PubMed] [Google Scholar]
- 62.Rousselot P, Charbonnier A, Cony-Makhoul P, Agape P, Nicolini FE, Varet B, et al. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J Clin Oncol. 2014;32(5):424–30. doi: 10.1200/JCO.2012.48.5797. [DOI] [PubMed] [Google Scholar]
- 63.Shah NP, Talpaz M, Deininger MW, Mauro MJ, Flinn IW, Bixby D, et al. Ponatinib in patients with refractory acute myeloid leukaemia: findings from a phase 1 study. Br J Haematol. 2013;162(4):548–52. doi: 10.1111/bjh.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith CC, Lasater EA, Zhu X, Lin KC, Stewart WK, Damon LE, et al. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood. 2013;121(16):3165–71. doi: 10.1182/blood-2012-07-442871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–3. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang WL, Conley A, Reynoso D, Nolden L, Lazar AJ, George S, et al. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer Chemotherapy and Pharmacology. 2011;67(Suppl 1):S15–24. doi: 10.1007/s00280-010-1513-8. [DOI] [PubMed] [Google Scholar]
- 67.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–78. doi: 10.1038/nrc3143. [DOI] [PubMed] [Google Scholar]