

Abstract
Objective
The Alzheimer disease (AD) APOEε4 risk allele associates with an earlier age of onset and increased amyloid-β deposition, whereas the protective APOEε2 allele delays the onset and appears to prevent amyloid-β deposition. Yet the clinical and pathological effects of APOEε2 remain uncertain because of its relative rarity. We investigated the effects of APOE ε2 and ε4 alleles on AD pathology and cognition in a large US dataset of well characterized AD patients.
Methods
We studied individuals from the National Alzheimer's Coordinating Center (NACC) autopsy cohort across the entire clinico-pathological continuum of AD. Multivariable models were built to examine the associations between APOE alleles and AD neuropathological changes, using the APOEε3/ε3 group as comparator. Mediation analysis was used to estimate the direct and indirect effects of APOE alleles on AD pathology and cognition (CDR-SOB and MMSE).
Results
Compared to APOEε3/ε3, APOEε2 is independently associated with lower Braak NFT stages and, possibly, fewer neuritic plaques, but has no direct effect on CAA severity, whereas APOEε4 is associated with more neuritic plaques and CAA, but has no independent effect on Braak NFT stage. Unadjusted analyses showed marked differences among APOE genotypes with respect to cognitive performance (ε2>ε3>ε4). Mediation analysis suggests that this is largely explained through effects on pathology.
Interpretation
Even when adjusted for age of onset, symptom duration and other demographic variables, APOEε2 is associated with milder AD pathology and less severe antemortem cognitive impairment compared to APOE ε3 and ε4 alleles, suggesting a relative neuroprotective effect of APOEε2 in AD.
Keywords: Alzheimer disease, amyloid plaques, apolipoprotein E, cerebral amyloid angiopathy, neurofibrillary tangles
Introduction
The association of the apolipoprotein E (APOE) gene with sporadic Alzheimer disease (AD) is the strongest of any known risk gene. Compared with the major ε3 allele, the APOEε4 allele increases the risk of AD in a dose-dependent but not proportional fashion: carrying one copy of the ε4 allele confers a 3-4 times increased life-risk of developing AD, whereas carrying two copies increases this risk 8-12 times1–4. By contrast, compared to the major APOEε3 allele, the APOEε2 allele reduces the risk of developing AD by almost half5–9.
A number of postmortem quantitative studies have established that the APOEε4 allele correlates with increased amyloid plaque deposition in the brain3,10–20 and with increased vascular amyloid deposition in the form of cerebral amyloid angiopathy (CAA)10,20–26. These findings have been more recently corroborated by in vivo amyloid PET imaging not only in AD patients, but also in cognitively intact elderly people27–31. While APOEε3/ε3 carriers can still develop AD, taken together, this evidence emphasizes the role of APOEε4 as an initiator of the amyloid cascade that ultimately leads to fully established AD 32,33. Conversely, although some postmortem studies14,16,17,19,34 have ascribed increased numbers of neurofibrillary tangles (NFTs) —the other pathological hallmark of the disease— to the APOEε4 allele, others have not found such association11,20, including those controlling for plaque burden in multivariable models35,36.
In contrast to APOEε4 carriers, APOEε2 carriers are thought to bear fewer amyloid plaques14,17,19,34,37,38 and possibly also fewer NFTs14,34. However, in part because of the relative rarity of this allele, sufficiently powered and detailed postmortem studies examining the effects of the APOEε2 allele on the pathobiology of AD are still lacking.
We examined the association between the APOE alleles and the AD pathological hallmarks and cognitive outcomes using the National Alzheimer's Coordinating Center (NACC) autopsy cohort, a large multicenter longitudinal cohort study of aging involving the 34 past and present National Institute on Aging-funded Alzheimer Disease Centers (NIA-funded ADCs) across the United States. We hypothesized that the large size of this multi-center intensively studied cohort would enable us to find novel pathophysiological relationships that may have remained undetectable in prior studies due to lack of statistical power. Because concurrent Lewy bodies and cerebrovascular pathologies have been reported to contribute to cognitive impairment in prior population-based clinico-pathological studies, in order to examine the impact of APOE alleles on AD-related pathological and cognitive outcomes, we selected individuals with no primary neuropathological diagnosis other than AD neuropathological changes. Specifically, we aimed to evaluate the effects of the ε2 and ε4 alleles on the main pathological hallmarks of AD [neuritic plaques, neurofibrillary tangles (NFTs), and cerebral amyloid angiopathy (CAA)]; and assess if there is a direct or indirect effect of these APOE alleles on the severity of cognitive impairment using mediation analysis.
Subjects & Methods
Inclusion and exclusion criteria
Subjects were participants in a longitudinal cohort study of aging in any of the past and present 34 NIA-funded ADCs. This multicenter study has been described in detail elsewhere39–41. Briefly, subjects undergo a baseline visit and annual follow-up visits in which a Uniform Data Set (UDS) is completed, including a minimum subject demographics data set, and standard motor, behavioral, functional, and neuropsychological assessments. Subjects were eligible for this study if they met the following inclusion criteria: 1) no primary neuropathological diagnosis other than AD neuropathological changes was found at autopsy; 2) final clinical evaluation within 2 years of death; 3) age of death 50 years old or older; and 4) APOE genotype available. Exclusion criteria were aimed at minimizing statistical noise in genotype-phenotype correlations and included: 1) a primary neuropathological diagnosis other than AD neuropathological changes (i.e., frontotemporal lobar degeneration, dementia with Lewy bodies, hippocampal sclerosis, vascular dementia, prion disease, Parkinson disease, Huntington disease, hypoxia, ischemia, necrosis, hemorrhage, other non-neurodegenerative diagnosis); 2) cognitive impairment attributable to alcohol use, depression, medication use, or medical illness; 3) carrying an APOEε2/ε4 genotype (since we aimed at investigating the separate effects of APOEε2 and APOEε4 on AD pathological lesions and cognitive outcomes), and 4) being cognitively intact (CDR-SOB=0) at last clinical evaluation (in order to prevent artificially significant results due to the “anchoring” effect of cognitively intact subjects, who typically have low levels of AD pathology and are less likely to carry the APOEε4 allele).
Data collection
Demographic and clinical data used in this study included sex, years of education, age of death, age of onset, symptom duration (age of death – age of onset), APOE genotype, last clinical dementia rating sum of boxes (CDR-SOB)42 score, and last Mini Mental State Examination (MMSE)43 score. The education level was categorized in 4-year intervals roughly corresponding to the education stages of high school, undergraduate college, and post-college education. Since we excluded subjects with CDR-SOB=0, values of CDR-SOB in this study range from 0.5 to 18, with higher values indicating worse cognitive/functional status. Supplemental tables 1 and 2 provide information about cognitively intact (CDR-SOB=0) subjects for comparison purposes.
Neuropathological variables included the Braak stage of NFTs (0: none; I-II: entorhinal; III-IV: limbic, and V-VI: isocortical)44,45, the CERAD score of neuritic plaques (none/sparse, moderate, or frequent)46, the presence of incidental Lewy bodies in any region, and the extent of vascular pathology (CAA, small and large vessel disease, and hippocampal sclerosis). Although the Thal phases of amyloid deposition47 have been recently implemented in NACC neuropathological assessment, they were not available for most subjects in this study, therefore we could not use the ABC score of AD neuropathological changes48,49 for clinico-pathologic correlations. While more objective and quantitative methods of assessment are under development, NACC neuropathology guidelines recommend the use of a qualitative and subjective grading system of the overall severity (rather than an individual vessel) to assess arteriosclerosis, atherosclerosis, and CAA (none, mild, moderate, or severe). Arteriosclerosis refers to the hyalinosis of the media and adventitia (arteriolosclerosis) of small parenchymal and/or leptomeningeal vessels, whereas atherosclerosis refers to the presence of intimal and medial fibro-fatty atheromatous plaques in large arteries at the base of the brain (i.e., circle of Willis). Hippocampal sclerosis was defined as the presence of selective neuronal loss and gliosis (“sclerosis”) limited to CA1 and subiculum, with variable additional involvement of endplate, CA2, entorhinal cortex, and amygdala.
Statistical analyses
The association of the presence of APOE alleles with ordered categories of AD pathological hallmarks (neuritic plaques, NFTs, or CAA) in the cognitively impaired (CDR-SOB>0) group was examined with adjacent-categories logit models50, which allow a covariate to have a constant effect across some or all categories, using the “VGAM” package in R software51. This flexibility of the adjacent-categories logit model enabled us to fit a parsimonious regression model for ordered categories of AD pathological hallmarks without strong parametric assumptions on the relationship among ordered categories. For each covariate, the assumption that it has the same effect for each category was checked using likelihood ratio tests of nested models. Whenever the assumption was violated, nested sequences of weaker assumptions permitting separate effects of a covariate for some logits were considered and were checked using likelihood ratio tests, until the most parsimonious assumption allowing different effects for different logits was not rejected. We have previously used this approach to examine the correlations between neuropathology and cognition (CDR-SOB) across the AD continuum52.
To assess the possible pathobiological pathways linking APOE alleles with AD pathological hallmarks and whether there is a direct effect of each of these alleles on cognitive performance and/or an indirect effect through any of the AD pathological hallmarks, mediation analysis was conducted using the counterfactual framework53. To adjust for multiple mediators simultaneously, the methods proposed by Imai and Yamamoto54 were adopted. Because neuritic plaques and NFTs from different APOE genotypes are biochemically and immunohistochemically similar, we assumed that no interaction exists between APOE genotype and the AD pathological hallmarks with respect to cognitive performance; in other words, that the effect of the pathological mediators on cognition does not differ across APOE genotypes for a given severity score or stage of pathology (i.e., Braak NFT stage VI). We made this same assumption of no interaction between APOE genotype and each AD pathological hallmark with respect to the other pathological hallmarks. The “mediation” package in R software55 was used to perform the mediation analysis with multiple mediators. The estimation algorithms involve fitting varying coefficient linear structural equations models: one model has the outcome of interest (e.g., CDR-SOB) as the dependent variable and the indicator of APOE allele, the multiple mediators, and the confounders as covariates whereas, for each mediator, there is another model having this mediator variable (e.g., Braak NFT stage) as the dependent variable and the indicator of APOE allele, the confounders, and all the other mediators as covariates. A Monte Carlo simulation-based method is then used to estimate the average direct and indirect effects53. Because the mediation analysis software that is available does not handle ordinal outcome data, which includes CERAD neuritic plaque scores, Braak NFT stages, extent of CAA, CDR-SOB and MMSE, we treated them as continuous measures. This artificially inflated the power of these analyses (Tables 4 and 5), as it assumes a linear relationship among the ordinal categories of the variables; however, it is likely not problematic for the cognitive outcomes, as they have many ordinal categories that may behave piecewise linearly. For this reason, and because mediation analyses were our primary interest, we did not undertake the ordinal regression analysis for CDR-SOB or MMSE. We treated plaques and NFTs as binary mediator variables in the CDR-SOB and MMSE analyses (cutoff values are presented in footnote of Table 4). In addition, the mediation analyses for the APOE alleles ε2 and ε4 were conducted separately, that is, subjects with the ε4 allele were excluded from the ε2 allele mediation analysis, and vice versa.
Table 4. Mediation analyses of the possible pathobiological pathways linking APOE alleles with AD pathological hallmarks.
| Outcome | Presence of APOEε2 allele | Presence of APOEε4 allele | ||
|---|---|---|---|---|
| Neuritic plaques | Estimate (95% CI) | P value | Estimate (95% CI) | P value |
| Total effect | -0.85 (-1.25, -0.46) | <0.001 | 0.33 (0.18, 0.48) | <0.001 |
| Direct effect | -0.42 (-0.77, -0.09) | 0.02 | 0.23 (0.11, 0.35) | <0.001 |
| Indirect effects: | ||||
| through NFTs | -0.42 (-0.60, -0.25) | <0.001 | 0.10 (0.02, 0.17) | 0.008 |
| through CAA | -0.01 (-0.04, 0.03) | 0.56 | -0.001 (-0.03, 0.02) | 0.96 |
| Neurofibrillary tangles | Estimate (95% CI) | P value | Estimate (95% CI) | P value |
| Total effect | -1.33 (-1.92, -0.71) | <0.001 | 0.30 (0.06, 0.54) | 0.02 |
| Direct effect | -0.60 (-1.16, -0.06) | 0.03 | 0.06 (-0.12, 0.26) | 0.51 |
| Indirect effects: | ||||
| through plaques | -0.72 (-1.14, -0.31) | <0.001 | 0.19 (0.04, 0.35) | 0.006 |
| through CAA | -0.01 (-0.09, 0.06) | 0.69 | 0.05 (0.01, 0.10) | 0.01 |
| Cerebral amyloid angiopathy | Estimate (95% CI) | P value | Estimate (95% CI) | P value |
| Total effect | -0.14 (-0.53, 0.26) | 0.41 | 0.53 (0.36, 0.69) | <0.001 |
| Direct effect | 0.19 (-0.18, 0.54) | 0.31 | 0.45 (0.30, 0.61) | <0.001 |
| Indirect effects: | ||||
| through plaques | -0.15 (-0.29, -0.03) | 0.006 | 0.04 (0.004, 0.08) | <0.001 |
| through NFTs | -0.19 (-0.33, -0.05) | <0.001 | 0.04 (0.003, 0.08) | 0.01 |
Effect estimates represent coefficients from linear models. Cutoffs for mediators were selected based on our prior findings of pathological correlates of cognition in NACC autopsy cohort52, as follows: for neuritic plaques, none/sparse versus moderate/frequent; for Braak NFT stage, 0 to IV versus V/VI, and for CAA, none/mild versus moderate/severe. For the APOEε2 model, mediators include two of the three AD pathological hallmarks other than the one as dependent variable; additionally, the model is adjusted for age of death, gender, and education as confounders. APOEε4 analyses are similar to APOEε2, but additionally adjusted for symptom duration, presence of hippocampal sclerosis, and severity of arteriosclerosis (ischemic small vessel disease).
Presence of APOEε2 allele: APOEε2/ε2 + APOEε2/ε3 (but not APOEε2/ε4) versus APOEε3/ε3 (reference); presence of APOEε4 allele: APOEε3/ε4 + APOEε4/ε4 (but not APOEε2/ε4) versus APOEε3/ε3 (reference); CAA = cerebral amyloid angiopathy; NFT = neurofibrillary tangles.
Table 5. Impact of APOE alleles on cognition (CDR-SOB and MMSE) and mediators of their impact.
| CDR-SOB | ||||
|---|---|---|---|---|
| Presence of APOE ε2 allele | Presence of APOEε4 allele | |||
| Estimate (95% CI) | P value | Estimate (95% CI) | P value | |
| Total effect | -2.89 (-4.77, -0.93) | 0.004 | 0.62 (-0.17, 1.35) | 0.10 |
| Direct effect | -0.76 (-2.56, 0.99) | 0.42 | -0.29 (-0.98, 0.32) | 0.36 |
| Indirect effects: | ||||
| through neuritic plaques | -0.82 (-1.56, -0.09) | 0.004 | 0.24 (0.08, 0.41) | <0.001 |
| through NFTs | -1.65 (-2.50, -0.79) | <0.001 | 0.34 (0.12, 0.57) | <0.001 |
| through CAA | NA | NA | 0.16 (0.00, 0.33) | 0.05 |
| through age of onset | -0.32 (-0.88, 0.25) | 0.24 | 0.19 (0.03, 0.35) | 0.01 |
| MMSE | ||||
| Presence of APOE ε2 allele | Presence of APOEε4 allele | |||
| Estimate (95% CI) | P value | Estimate (95% CI) | P value | |
| Total effect | 3.49 (-0.13, 6.47) | 0.07 | -3.10 (-4.35, -1.80) | 0.002 |
| Direct effect | -0.33 (-3.15, 2.60) | 0.83 | -0.98 (-2.13, 0.18) | 0.13 |
| Indirect effects: | ||||
| through neuritic plaques | 1.17 (0.11, 2.24) | 0.01 | -0.33 (-0.59, -0.07) | 0.002 |
| through NFTs | 2.42 (1.10, 3.74) | <0.001 | -0.66 (-1.09, -0.22) | 0.004 |
| through CAA | NA | NA | -0.56 (-0.93, -0.19) | <0.001 |
| through age of onset | 0.33 (-0.63, 1.30) | 0.46 | -0.62 (-1.00, -0.24) | <0.001 |
Effect estimates represent coefficients from linear models. For the APOEε2 model, mediators include two AD pathological hallmarks (neuritic plaques and NFTs) and age of onset, whereas confounders include gender, education, symptom duration, and CAA. We could not include CAA as a mediator for APOEε2 due to the small sample size of this group. For the APOEε4 model, mediators include the three AD pathological hallmarks (neuritic plaques, NFTs, and CAA) and age of onset, whereas confounders include gender, education, and symptom duration.
Presence of APOEε2 allele: APOEε2/ε2 + APOEε2/ε3 (but not APOEε2/ε4) versus APOEε3/ε3 (reference); presence of APOEε4 allele: APOEε3/ε4 + APOEε4/ε4 (but not APOEε2/ε4) versus APOEε3/ε3 (reference); CAA = cerebral amyloid angiopathy; CDR-SOB = clinical dementia rating scale sum of boxes; MMSE = Mini Mental State Examination; NFT = neurofibrillary tangles.
In this study we did not adjust for potential selection bias associated with the decision to undergo autopsy as we found similar results with and without this adjustment, performed with inverse probability weighting, in our prior study on the NACC autopsy cohort52.
Results
Description of the sample
The flow-chart in Figure 1 depicts the selection procedure and the number of subjects excluded by each exclusion criterion. As of March 2014, the 2005-2014 NACC autopsy cohort consisted of 2987 subjects, of whom 793 subjects met all the eligibility criteria and did not meet any of the exclusion criteria. The distribution of APOE genotypes in this final sample was the following: ε2/ε2=1 (0.1%), ε2/ε3=40 (5.0%), ε3/ε3=339 (42.7%), ε3/ε4=320 (40.3%), and ε4/ε4=93 (11.7%). Allelic frequencies in this AD enriched autopsy cohort were distributed as follows: ε2=2.6%, ε3=65.4%, and ε4=31.9%.
Figure 1.
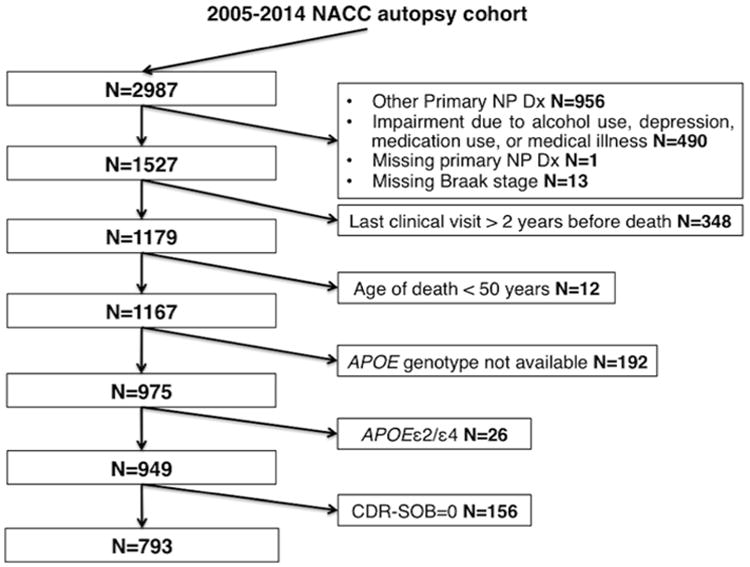
Flow-chart of the selection process from March 2014 data freeze of the National Alzheimer's Coordinating Center (NACC) autopsy cohort. Other primary NP Dx: other primary neuropathological diagnoses refer to conditions other from Alzheimer disease and includes frontotemporal lobar degeneration, progressive supranuclear palsy, corticobasal degeneration, dementia with Lewy bodies, Parkinson disease, hypoxia, hemorrhage/hematoma, necrosis, vascular dementia, hippocampal sclerosis, and prion-associated diseases.
Table 1 shows the demographic characteristics of the study subjects by APOE genotype. For the purpose of comparison, Supplemental Table 1 describes the subjects with CDR-SOB=0 who were excluded from the study. Sex and education were not significantly different across genotypes, but there were major differences in AD-related clinical phenotypes across the different genotypes. Besides the well-established association between genotype and age of symptom onset and death (ε4<ε3<ε2), these unadjusted analyses also revealed a strong correlation between genotype and antemortem degree of cognitive impairment (ε4>ε3>ε2). The latter differences were striking despite excluding cognitively intact subjects (CDR-SOB=0): the average MMSE score of APOEε2 carriers was 20.5, whereas the average MMSE score of APOEε4 carriers was only 11.9 for one copy and 9.7 for two copies of the allele.
Table 1. Demographic and clinical characteristics of the cognitively impaired subjects (CDR-SOB>0) by APOE genotype.
| ε2/ε2+ε2/ε3 | ε3/ε3 | ε3/ε4 | ε4/ε4 | P value | |
|---|---|---|---|---|---|
| Number of subjects, n | 41 | 339 | 320 | 93 | NA |
| Sex, n female (%) | 21 (51.2) | 163 (48.1) | 128 (40.0) | 40 (43.0) | 0.155 |
| Education (y) | 14.5±3.5 | 14.9±3.3 | 14.9±3.2 | 15.1±3.2 | 0.955 |
| Age of death (y) | 84.4±12.1 | 82.9±10.6 | 81.3±9.1 | 78.6±8.0 | <0.0001hj |
| Age of onset (y) | 77.6±14.0 | 73.4±12.6 | 71.5±10.5 | 68.7±8.7 | <0.0001bhj |
| Symptom duration (y) | 6.8±4.5 | 9.2±6.3 | 9.7±6.5 | 9.7±4.4 | 0.012 bc |
| CDR-SOB | 6.0 (2.0-12.0) | 13.0 (6.0-18.0) | 16.0 (9.5-18.0) | 17.0 (12.0-18.0) | <0.0001eklm |
| MMSE | 20.5±9.1 | 15.3±9.6 | 11.9±8.8 | 9.7±8.3 | <0.0001ailmo |
Values represent mean±SD except for CDR-SOB that is expressed as median (interquartile range), and sex that is indicated as number (%) of females. Comparisons were performed with Kruskal-Wallis ANOVA with Dunn's multiple comparison test, except for sex, that was compared using Chi-square with Fisher´s exact test.
p<0.05 ε2/ε2+ε2/ε3 vs ε3/ε3;
p<0.05 ε2/ε2+ε2/ε3 vs ε3/ε4;
p<0.05 ε2/ε2+ε2/ε3 vs ε4/ε4;
p<0.05 ε3/ε3 vs ε3/ε4;
p <0.05 ε3/ε3 vs ε4/ε4;
p<0.01 ε2/ε2+ε2/ε3 vs ε3/ε3;
p<0.01 ε2/ε2+ε2/ε3 vs ε3/ε4;
p<0.01 ε2/ε2+ε2/ε3 vs ε4/ε4;
p<0.01 ε3/ε3 vs ε3/ε4;
p<0.01 ε3/ε3 vs ε4/ε4;
p<0.0001 ε2/ε2+ε2/ε3 vs ε3/ε3;
p <0.0001 ε2/ε2+ε2/ε3 vs ε3/ε4;
p <0.0001 ε2/ε2+ε2/ε3 vs ε4/ε4;
p <0.0001 ε3/ε3 vs ε3/ε4;
p<0.0001 ε3/ε3 vs ε4/ε4.
CDR-SOB = clinical dementia rating scale sum of boxes; MMSE = Mini Mental State Examination; NA = not applicable.
Table 2 depicts the scores of each AD pathological hallmark as recorded within the NACC database (CERAD score of neuritic plaques, Braak NFT stage, and CAA severity) by APOE genotype. Hippocampal sclerosis, arteriosclerosis, atherosclerosis, and Lewy bodies are also presented. The subjects with CDR-SOB=0 excluded from the analyses below are described in Supplemental Table 2 for the purpose of comparison. Major differences between genotypes can be observed in the summary data of Table 2: among APOEε2 carriers with impaired cognition at death, just fewer than half (18 out of 41, ∼44%) had sparse or no neuritic plaques whereas this number is almost 10-fold lower for APOEε4 carriers (21 out of 413, ∼5%). Similarly, only ∼27% of APOEε2 carriers (11 out of 41) were Braak V/VI, the most severe NFT stages, whereas almost 90% of APOEε4/4 carriers (81 out of 93) had this widespread distribution of NFTs. Last, moderate or severe CAA was present in only ∼27% of APOEε2 carriers (10 out of 41), as opposed to ∼72% of APOEε4/ε4 carriers (67 out of 93).
Table 2. Pathological characteristics of the cognitively impaired subjects (CDR-SOB>0) by APOE genotype.
| ε2/ε2+ε2/ε3 | ε3/ε3 | ε3/ε4 | ε4/ε4 | P value | |
|---|---|---|---|---|---|
| Number of subjects, n | 41 | 339 | 320 | 93 | NA |
| Neuritic plaques, n (%) | <0.0001 | ||||
| None/sparse | 18 (43.9) | 49 (14.5) | 20 (6.3) | 1 (1.1) | |
| Moderate | 12 (29.3) | 97 (28.6) | 47 (14.7) | 13 (14.0) | |
| Frequent | 11 (26.8) | 193 (56.9) | 253 (79.1) | 79 (85.0) | |
| Braak NFT stage, n (%) | <0.0001 | ||||
| 0/I/II | 13 (31.7) | 35 (10.3) | 15 (4.7) | 0 (0.0) | |
| III/IV | 17 (41.5) | 82 (24.2) | 56 (17.5) | 12 (12.9) | |
| V | 4 (9.8) | 84 (24.8) | 91 (28.4) | 26 (28.0) | |
| VI | 7 (17.1) | 138 (40.7) | 158 (49.4) | 55 (59.1) | |
| CAA, n (%) | <0.0001 | ||||
| None | 20 (48.8) | 133 (39.2) | 53 (16.6) | 12 (12.9) | |
| Mild | 9 (22.0) | 101 (29.8) | 104 (32.5) | 14 (15.1) | |
| Moderate | 6 (14.6) | 64 (18.9) | 100 (31.3) | 32 (34.4) | |
| Severe | 5 (12.2) | 35 (10.3) | 54 (16.9) | 35 (37.6) | |
| Not assessed/unknown | 1 (2.4) | 6 (1.8) | 9 (2.8) | 0 (0.0) | |
| Arteriosclerosis, n (%) | 0.887 | ||||
| None | 8 (19.5) | 66 (19.5) | 46 (14.4) | 15 (16.1) | |
| Mild | 11 (26.8) | 96 (28.3) | 88 (27.5) | 26 (28.0) | |
| Moderate | 11 (26.8) | 74 (21.8) | 76 (23.8) | 19 (20.4) | |
| Severe | 3 (7.3) | 26 (7.7) | 27 (8.4) | 10 (10.8) | |
| Not assessed/unknown | 8 (19.5) | 77 (22.7) | 83 (25.9) | 23 (24.7) | |
| Atherosclerosis, n (%) | 0.829 | ||||
| Yes | 35 (85.4) | 281 (82.9) | 261 (81.6) | 79 (85.0) | |
| No | 6 (14.6) | 58 (17.1) | 59 (18.4) | 14 (15.0) | |
| Hippocampal sclerosis, n (%) | 0.560 | ||||
| Yes | 2 (4.9) | 26 (7.7) | 30 (9.4) | 7 (7.5) | |
| No | 37 (90.2) | 298 (87.9) | 286 (89.4) | 83 (89.3) | |
| Not assessed/unknown | 2 (4.9) | 15 (4.4) | 4 (1.3) | 3 (3.2) | |
| Lewy bodies, n (%) | 0.121 | ||||
| Yes | 9 (22.0) | 87 (25.7) | 112 (35.0) | 29 (31.2) | |
| No | 32 (78.0) | 250 (73.8) | 208 (65.0) | 64 (68.8) | |
| Not assessed/unknown | 0 (0.0) | 2 (0.6) | 0 (0.0) | 0 (0.0) |
Numbers in parenthesis represent column percents, that is, percent of subjects of each APOE genotype with a given neuropathologic feature. CAA = cerebral amyloid angiopathy. NA = not applicable. NFT = neurofibrillary tangles. P value is for comparison of the distribution of each neuropathological variable among APOEε2 carriers (ε2/ε2+ε2/ε3) and APOEε4 carriers (ε3/ε4+ε4/ε4).
Next we examined the complex relationships between APOE alleles, AD neuropathology, and cognitive performance prior to death.
Effects of APOE alleles on AD pathological hallmarks
To investigate the independent effects of the APOEε2 and the APOEε4 alleles on the presence and severity of each of these AD pathological hallmarks, we constructed multivariable models including all three pathological lesions and all demographic and clinical variables (age of death, sex, education, and symptom duration), using the APOEε3/ε3 group as reference group (Table 3). While individuals with a primary neuropathological diagnosis of Lewy body disease, hippocampal sclerosis, and cerebrovascular disease were excluded from the study, the “incidental” finding of these lesions was allowed. However, univariate analyses in this group of cognitively impaired (CDR-SOB>0) individuals revealed no correlation of Lewy bodies, hippocampal sclerosis, arteriosclerosis (parenchymal small vessel disease), or atherosclerosis of large arteries with carrying either of the APOE alleles. Therefore these neuropathological variables were not included in the multivariable models.
Table 3. Associations between the APOE alleles and AD pathological lesions.
| Outcome | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Braak stage of NFTa | CERAD score of neuritic plaquesb | CAAc | ||||||||
| Mild versus none | Moderate versus mild | Severe versusmoderate | ||||||||
| Log [OR (95%CI)] | P value | Log[OR (95%CI)] | P value | Log[OR (95%CI)] | P value | Log[OR (95%CI)] | P value | Log[OR (95%CI)] | P value | |
| Presence of APOEε2 allele | -0.25 (-0.49, -0.01) | 0.04 | -0.19 (-0.48, 0.09) | 0.178 | 0.09 (-0.10, 0.28) | 0.33 | 0.09 (-0.10, 0.28) | 0.33 | 0.09 (-0.10, 0.28) | 0.33 |
| Presence of APOEε4 allele | -0.08 (-0.18, 0.02) | 0.14 | 0.33 (0.17, 0.48) | <0.001 | 0.38 (0.19, 0.57) | <0.001 | 0.20 (0.01, 0.39) | 0.038 | 0.04 (-0.18, 0.26) | 0.71 |
| Presence of 1 APOEε4 allele | -0.08 (-0.18, 0.03) | 0.15 | 0.31 (0.15, 0.48) | <0.001 | 0.44 (0.25, 0.64) | <0.001 | 0.05 (-0.06, 0.17) | 0.36 | 0.05 (-0.06, 0.17) | 0.36 |
| Presence of 2 APOEε4 alleles | -0.08 (-0.24, 0.09) | 0.38 | 0.39 (0.09, 0.69) | <0.001 | 0.17 (-0.21, 0.56) | 0.38 | 0.43 (0.11, 0.75) | 0.008 | 0.36 (0.08, 0.63) | 0.010 |
Effect estimates represent the logarithm of odd ratios. The logarithm of odd ratios was calculated to facilitate comparisons with the estimates resulting from mediation analysis in table 4.
For Braak stage of NFTs, estimates refer to the following comparisons: VI versus V, V versus III/IV, and III/IV versus 0/I/II.
For CERAD score of neuritic plaques, estimates refer to the following comparisons: frequent versus moderate and moderate versus none/sparse.
For CAA, estimates refer to the comparisons indicated in the table. With each one of the three pathological lesions as dependent variable, the model adjusted for the other two of the three pathological lesions, and all demographic and clinical variables (age of death, sex, education, and symptom duration), using the APOEε3/ε3 group as reference group.
Presence of APOEε2 allele: APOEε2/ε2 + APOEε2/ε3 (but not APOEε2/ε4) versus APOEε3/ε3 (reference); presence of APOEε4 allele: APOEε3/ε4 + APOEε4/ε4 (but not APOEε2/ε4) versus APOEε3/ε3 (reference); CAA = cerebral amyloid angiopathy; NFT = neurofibrillary tangles.
Effect of APOE ε2 and ε4 alleles on neuritic plaques
After controlling for age of death, sex, education, symptom duration, Braak stage of NFTs, and severity of CAA, there was a significant positive association between the presence of the APOEε4 allele and the density of neuritic plaques as assessed with CERAD scores compared to the APOEε3/ε3 genotype, with a higher density in APOEε4 carriers. By contrast, compared to the APOEε3/ε3 group, the presence of the APOEε2 allele was not significantly associated with a lower density of neuritic plaques (although see further analysis and discussion below).
Effect of APOE ε2 and ε4 alleles on cerebral amyloid angiopathy
Similarly, after controlling for age of death, sex, education, symptom duration, Braak stage of NFTs, and CERAD score of neuritic plaques, there was a significant positive association between the presence of the APOEε4 allele and the risk of CAA, with a higher risk of having mild versus none CAA and moderate versus mild CAA, but not severe versus moderate CAA, in APOEε4 carriers compared to the APOEε3/ε3 group. Split analyses by number of APOEε4 alleles revealed a clear dose-dependent association. Carrying one APOEε4 allele was associated with a higher risk of mild but not moderate or severe CAA, whereas carrying two APOEε4 alleles was associated with a higher risk of having moderate and severe but not mild CAA. By contrast, compared to the APOEε3/ε3 genotype, there was no significant association between the presence of the APOEε2 allele and the risk of CAA.
Effect of APOE ε2 and ε4 alleles on neurofibrillary tangles
Neither the presence of the APOEε4 allele nor the number of APOEε4 alleles was significantly associated with a higher risk of having more advanced Braak stages than the APOEε3/ε3 group, when controlling for age of death, sex, education, symptom duration, CERAD score of neuritic plaques, and CAA severity. By contrast, carrying an APOEε2 allele was significantly associated with a lower risk of having a higher Braak stage of NFTs, compared to the APOEε3/ε3 genotype.
Pathobiological pathways linking APOE alleles with AD pathological hallmarks
The above results indicate that, compared to the most common genotype APOEε3/ε3, the APOEε2 allele is independently associated with a lower Braak NFT stage, whereas the APOEε4 allele is independently associated with both a higher density of neuritic plaques and a higher severity of CAA. To investigate possible pathobiological pathways leading from these APOE alleles to their pathological correlates, we performed separate mediation analyses with each of the three AD pathological hallmarks as outcome variables, and the other two as mediators (Table 4). All analyses were adjusted by age of death, sex, and education, and APOEε4 analyses were also adjusted by symptom duration, presence of hippocampal sclerosis, and severity of arteriosclerosis (ischemic small vessel disease). The APOEε2 analyses could not adjust for these additional variables due to the small sample size.
These mediation analyses revealed that the protective effect of the APOEε2 allele against NFTs can be explained by two mechanisms: a direct effect of the APOEε2 allele and an indirect pathway mediated through its effect on neuritic plaques, both with similar magnitude. Of note, these analyses also suggested a protective effect of the APOEε2 allele against neuritic plaques resulting from two effects of similar size: a direct effect against neuritic plaques and an indirect consequence of its protective effect on NFTs. Mediation analyses also revealed that the APOEε2 allele has no significant direct effect on CAA severity but does have a significant indirect protective effect mediated through its lowering of both neuritic plaques and NFTs.
The accumulation of neuritic plaques associated with the APOEε4 allele is the result of a direct effect and, to a lesser extent, of an indirect pathway involving its effect on NFTs, but not on CAA. Similarly, the increasing severity of CAA associated with the APOEε4 allele stems mainly from a direct effect, although its effects on neuritic plaques and NFTs also contribute to some extent. Last, the APOEε4 allele has no direct effect on NFTs, but may promote NFTs formation and/or spreading by indirect pathways involving its significant enhancing effects on neuritic plaques and, to a lesser extent, CAA.
Lack of independent effect of APOEε2 and ε4 alleles on cognition
We have recently reported in this same autopsy cohort that the extent of neuritic plaques (as measured by CERAD scores) and NFTs (as measured by Braak stage), severe CAA, severe parenchymal small vessel disease, and the presence of hippocampal sclerosis, all independently predict a higher degree of antemortem cognitive impairment as measured by CDR-SOB, whereas education level is an independent protective factor against cognitive impairment52. Herein, we examined whether the APOE genotype impacts cognition independently of both demographics and neuropathology. Specifically, we tested the hypothesis that possessing the APOEε2 allele directly or indirectly protects against cognitive deficits, whereas carrying the APOEε4 allele directly or indirectly contributes to cognitive impairment, as captured by CDR-SOB score or MMSE score prior to death. To distinguish between an effect mediated by neuropathological variables (indirect) or an independent (direct) effect of each allele, mediation analyses were carried out controlling for all the demographic/clinical (age of death, sex, education, age of onset, and symptom duration) and neuropathological characteristics. Although the total effect of APOEε2 on CAA was non-significant (Table 4), the effect was not negligible (0.14) and the confidence interval was wide and non-symmetric (-0.53,0.26). For these reasons, in spite of the non-significance, we considered it as a mediator of APOEε2 in the CDR-SOB model. Because the APOEε2 and ε4 alleles are known to have opposite effects on age of onset of cognitive decline —APOEε2 delays onset whereas APOEε4 anticipates it—, we also tested age of onset as a mediator of indirect effects of APOE alleles on cognition.
Neither the APOEε2 allele nor the APOEε4 allele had a significant independent (direct) impact on cognition as measured with CDR-SOB and as compared to the more common genotype APOEε3/ε3 (Table 5). Similar results were observed using the MMSE score as the cognitive outcome. However, the APOEε2 allele did exert an indirect protective effect against cognitive impairment that was mediated through its lowering effect on NFTs and, to a lesser extent, through its lowering effect on neuritic plaques. CAA could not be tested as a mediator of APOEε2 allele-induced cognitive effects due to instability of the model. The protective effect on antemortem cognition associated with the APOEε2 allele was not mediated by the delayed age of onset also associated with this allele. Conversely, the APOEε4 allele had an indirect deleterious effect on cognition mediated by all three AD neuropathological lesions. In addition, the effect of APOEε4 on the age of onset of cognitive decline significantly contributed to the severity of cognitive impairment prior to death, even after controlling for symptom duration.
Discussion
The examination of raw data and unadjusted statistical analyses describing this large cognitively impaired autopsy cohort (Tables 1 and 2) suggested that, compared to the most common APOEε3/ε3, carrying the APOEε4 allele may be associated with both a more severe clinical expression (i.e., younger age of onset and poorer cognition prior to death) and a more advanced pathological phenotype (i.e., higher scores for neuritic plaques and CAA) of AD, whereas carrying the APOEε2 allele may imply developing a clinically and pathologically milder form. This would be in agreement with the known opposite effects of these APOE alleles on the risk of developing AD. However, the relationships between genotype and phenotype can be so complex that robust statistical methods and multivariable analyses are necessary to account for confounding and mediator factors. The results of these analyses support the above interpretation and can be summarized as follows: 1) Compared to APOEε3/ε3, APOEε2 is independently associated with both lower Braak stages of NFTs and fewer neuritic plaques, but has no independent effect on CAA severity; 2) compared to APOEε3/ε3, APOEε4 is associated with higher numbers of neuritic plaques and higher severity of CAA, but has no independent effect on NFTs; 3) besides these direct effects of APOE ε2 and ε4 alleles on each AD lesion, additional indirect effects mediated through the other two AD pathological hallmarks were identified; 4) neither APOEε2 nor APOEε4 has an independent impact on cognition prior to death, but each indirectly affects cognition through its effects on AD neuropathological changes.
The main novel finding of this study is a significant negative direct effect of the APOEε2 allele on the Braak stage of NFT, suggesting a protective effect of the apolipoprotein E2 against NFT formation and spreading, whereas APOEε4 does not exhibit the opposite effect. Mediation analyses indicated that two effects equally contributed to this negative association between APOEε2 and NFTs: a direct effect of APOEε2 and an indirect mechanism through its plaque-lowering effect. Several proposed pathophysiological mechanisms could underlie these findings. First, apolipoprotein E (apoE) may directly interact with tau protein, the main constituent of NFTs, and apoE2 may interact with tau to a different extent than the other apoE isoforms56–60. Second, APOEε2 may indirectly contribute to restrict NFT spreading to cortical areas and determine a lower Braak stage by protecting against the accumulation of soluble Aβ oligomeric species —probably not well captured by the CERAD neuritic plaque score— and plaques61 and by protecting against synaptotoxicity and neurotoxicity of existing plaques downstream Aβ accumulation61,62. By contrast, the association of APOEε4 allele and Braak stage of NFT was not significant after controlling for plaques and CAA, arguing against a significant direct effect of the APOEε4 allele on NFT formation and spreading. Indeed, in agreement with prior work36,63, our mediation analyses suggested that the positive association between the APOEε4 allele and the Braak NFT stage was largely explained by the APOEε4 allele effect on plaques.
The mediation analys also demonstrate that the augmentation of neuritic plaques and CAA induced by the APOEε4 allele was primarily a direct effect, supporting its role as initiator of amyloid deposition both at the brain parenchyma and at the blood vessel wall in the form of CAA. This result is in agreement with numerous postmortem quantitative and amyloid PET studies that have established that the increased risk of AD induced by APOEε4 is associated with a higher amyloid burden. Recent experimental data indicate that the apoE4 increases the amount of synaptotoxic soluble Aβ oligomeric species61,64,65 and impairs Aβ clearance61,66.
While the ordinal regression model in Table 3 detected a protective effect of the APOEε2 allele against neuritic plaques when compared to the APOEε3/ε3, this association was not found to be statistically significant likely due to lack of power, as suggested by the upper limit of the 95% confidence interval for the log odds ratio just above zero. Mediation analyses did yield a statistically significant result, although this statistical significance was likely contributed by the greater power afforded by the mediation analysis due to its modeling of CERAD neuritic plaque score as continuous, rather than ordinal. In addition, due to the small sample size, the APOEε2 mediation analysis was not able to control for symptom duration, which may be a confounder also contributing to this statistical significance given its significant association with a higher Braak stage (p<0.0001) and marginally significant association with a higher CERAD score (p=0.07) in the ordinal regression models. In any case, the protective effect of the APOEε2 allele against neuritic plaques was equally explained by a direct effect on plaques and an indirect mechanism, mediated through its NFT-lowering effect. This latter indirect effect appears to imply a synergistic interaction between tangles and plaques that has been postulated based on experimental data67,68.
Although significant indirect protective effects of the APOEε2 allele on CAA severity were detected by mediation analysis, the total and direct effects were not significant, and the association between APOEε2 allele and CAA severity was not significant in the ordinal regression model. This neutral total effect of the APOEε2 allele on the extent of CAA is consistent with prior work suggesting that the APOEε2 allele is not associated with a higher incidence of vascular amyloid deposition26,69. The current NACC autopsy data set did not allow us to confirm the previously described association between the APOEε2 allele and a higher incidence of vasculopathic changes in amyloid-β-laden vessels leading to a higher risk of CAA-related hemorrhages.
Whether the APOE genotype independently and directly influences the rate of cognitive decline in AD has been a matter of controversy. Multiple clinical studies have attributed to the APOEε4 allele either an accelerating, a neutral70–77, or even a slowing effect78–80 on the rate of cognitive decline once AD has been diagnosed. By contrast, compared to the APOEε3/ε3 genotype, the APOEε2 allele has been associated with a reduced decline81,82. However, all these clinical studies lacked autopsy confirmation and, therefore, were not able to study the effect of the APOE genotype on cognition in the context of the neuropathological lesions. This is of critical importance because up to 30% of subjects diagnosed with probable AD in US NIA-funded ADCs have other neuropathological diagnoses causing or contributing to their cognitive deficits, or insufficient AD neuropathological changes at autopsy to fulfill current AD neuropathological diagnostic criteria83,49,48,84. In this study APOEε2 predicted substantially less severe cognitive impairment at the time of death, whereas APOEε4 was associated with a more severe cognitive impairment. However, these effects were seemingly entirely mediated by the impact of both alleles on AD neuropathological changes. Indeed, we observed no significant direct effect (i.e., independent of pathological lesions) of either of the APOE alleles on the CDR-SOB or MMSE score prior to death. We did, however, observe significant indirect effects of both APOE alleles on cognition mediated by their opposite effects with respect to the accumulation of AD neuropathological lesions. In addition, part of the detrimental effect of the APOEε4 allele on antemortem cognition was mediated by its effect on the age of onset of cognitive decline. The APOEε4 allele is known to anticipate the onset of cognitive decline in AD, and APOEε4 carriers exhibited a more severe cognitive impairment prior to death the younger they were at clinical onset, even after controlling for symptom duration.
In order to prevent “false” statistically significant results derived from an overrepresentation of the APOEε2 allele and an under representation of the APOEε4 allele among the cognitively intact subjects, we decided to exclude subjects with CDR-SOB=0 and focused our statistical analyses in the continuum of cognitive decline ranging from subjective cognitive complaints and mild cognitive impairment (CDR-SOB=0.5-3.0) to end-stage dementia (CDR-SOB=18). While this conservative design reinforces the validity of the statistically significant results obtained, the power to detect other statistically significant and clinically relevant associations may have been hampered by a smaller sample size, particularly for the APOEε2 allele group (n=41) and for the detection of early direct or indirect effects of APOE alleles on cognition.
In summary, the analysis of this large autopsy sample of cognitively impaired elderly subjects selected to examine the clinico-pathological continuum of Alzheimer disease with regression and mediation models led us to the novel finding that the APOEε2 allele is independently associated with a lower Braak NFT stage. This observation implies a protective effect of the APOEε2 allele against spreading of neurofibrillary tangle pathology to the neocortex and warrants future experimental studies modeling the interaction between apolipoprotein E and tau. We also confirmed the association of the APOEε4 allele with a higher density of neuritic plaques and a more severe degree of CAA. These data also emphasize the impact of the APOE genotype in the clinical presentation and course of AD, with the major impact of the APOEε4 allele on amyloid variables and a surprisingly strong impact of APOEε2 on the spread of NFTs to the cortex leading to clinically meaningful differences in severity of dementia at death. Thus, APOEε2 appears to lead to a milder form of AD.
Supplementary Material
Supplemental Table 1. Demographic and clinical characteristics of cognitively intact subjects (CDR-SOB=0) by APOE genotype.
Supplemental Table 2. Pathological characteristics of cognitively intact subjects (CDR-SOB=0) by APOE genotype.
Acknowledgments
This work was supported by the Massachusetts Alzheimer's Research Center (NIH grant P50 AG0001534 to BTH). The NACC database is funded by the NIH National Institute on Aging grant U01 AG016976.
Footnotes
Conflict of Interest: The authors report no conflict of interest
References
- 1.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 2.Poirier J, Davignon J, Bouthillier D, et al. Apolipoprotein E polymorphism and Alzheimer's disease. Lancet. 1993;342(8873):697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 3.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993;11(4):575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 4.Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43(8):1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 5.Benjamin R, Leake A, McArthur FK, et al. Protective effect of apoE epsilon 2 in Alzheimer's disease. Lancet. 1994;344(8920):473. doi: 10.1016/s0140-6736(94)91804-x. [DOI] [PubMed] [Google Scholar]
- 6.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7(2):180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 7.Smith AD, Johnston C, Sim E, et al. Protective effect of apo epsilon 2 in Alzheimer'sdisease. Oxford Project to Investigate Memory and Ageing (OPTIMA) Lancet. 1994;344(8920):473–474. [PubMed] [Google Scholar]
- 8.Talbot C, Lendon C, Craddock N, et al. Protection against Alzheimer's disease with apoE epsilon 2. Lancet. 1994;343(8910):1432–1433. doi: 10.1016/s0140-6736(94)92557-7. [DOI] [PubMed] [Google Scholar]
- 9.West HL, Rebeck GW, Hyman BT. Frequency of the apolipoprotein E epsilon 2 allele is diminished in sporadic Alzheimer disease. Neurosci Lett. 1994;175(1-2):46–48. doi: 10.1016/0304-3940(94)91074-x. [DOI] [PubMed] [Google Scholar]
- 10.Caselli RJ, Walker D, Sue L, et al. Amyloid load in nondemented brains correlates with APOE e4. Neurosci Lett. 2010;473(3):168–171. doi: 10.1016/j.neulet.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez-Isla T, West HL, Rebeck GW, et al. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer's disease. Ann Neurol. 1996;39(1):62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- 12.Hyman BT, West HL, Rebeck GW, et al. Quantitative analysis of senile plaques in Alzheimer disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome) Proc Natl Acad Sci U S A. 1995;92(8):3586–3590. doi: 10.1073/pnas.92.8.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McNamara MJ, Gomez-Isla T, Hyman BT. Apolipoprotein E genotype and deposits of Abeta40 and Abeta42 in Alzheimer disease. Arch Neurol. 1998;55(7):1001–1004. doi: 10.1001/archneur.55.7.1001. [DOI] [PubMed] [Google Scholar]
- 14.Morris CM, Benjamin R, Leake A, et al. Effect of apolipoprotein E genotype on Alzheimer's disease neuropathology in a cohort of elderly Norwegians. Neurosci Lett. 1995;201(1):45–47. doi: 10.1016/0304-3940(94)12126-b. [DOI] [PubMed] [Google Scholar]
- 15.Nagy Z, Esiri MM, Jobst KA, et al. The effects of additional pathology on the cognitive deficit in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56(2):165–170. doi: 10.1097/00005072-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Ohm TG, Scharnagl H, März W, Bohl J. Apolipoprotein E isoforms and the development of low and high Braak stages of Alzheimer's disease-related lesions. Acta Neuropathol (Berl) 1999;98(3):273–280. doi: 10.1007/s004010051080. [DOI] [PubMed] [Google Scholar]
- 17.Polvikoski T, Sulkava R, Haltia M, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333(19):1242–1247. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 18.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiraboschi P, Hansen LA, Masliah E, et al. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62(11):1977–1983. doi: 10.1212/01.wnl.0000128091.92139.0f. [DOI] [PubMed] [Google Scholar]
- 20.Yip AG, McKee AC, Green RC, et al. APOE, vascular pathology, and the AD brain. Neurology. 2005;65(2):259–265. doi: 10.1212/01.wnl.0000168863.49053.4d. [DOI] [PubMed] [Google Scholar]
- 21.Berg L, McKeel DW, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55(3):326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 22.Chalmers K, Wilcock GK, Love S. APOE epsilon 4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol Appl Neurobiol. 2003;29(3):231–238. doi: 10.1046/j.1365-2990.2003.00457.x. [DOI] [PubMed] [Google Scholar]
- 23.Greenberg SM, Rebeck GW, Vonsattel JP, et al. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38(2):254–259. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 24.Olichney JM, Hansen LA, Hofstetter CR, et al. Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E4. Arch Neurol. 2000;57(6):869–874. doi: 10.1001/archneur.57.6.869. [DOI] [PubMed] [Google Scholar]
- 25.Premkumar DR, Cohen DL, Hedera P, et al. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer's disease. Am J Pathol. 1996;148(6):2083–2095. [PMC free article] [PubMed] [Google Scholar]
- 26.Rannikmäe K, Kalaria RN, Greenberg SM, et al. APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J Neurol Neurosurg Psychiatry. 2014;85(3):300–305. doi: 10.1136/jnnp-2013-306485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barthel H, Gertz HJ, Dresel S, et al. Cerebral amyloid-β PET with florbetaben (18F) in patients with Alzheimer's disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol. 2011;10(5):424–435. doi: 10.1016/S1474-4422(11)70077-1. [DOI] [PubMed] [Google Scholar]
- 28.Drzezga A, Grimmer T, Henriksen G, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72(17):1487–1494. doi: 10.1212/WNL.0b013e3181a2e8d0. [DOI] [PubMed] [Google Scholar]
- 29.Johnson KA, Sperling RA, Gidicsin CM, et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer's disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement J Alzheimers Assoc. 2013;9(5 Suppl):S72–S83. doi: 10.1016/j.jalz.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67(1):122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106(16):6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63(3):287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagy Z, Esiri MM, Jobst KA, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer's disease. Neuroscience. 1995;69(3):757–761. doi: 10.1016/0306-4522(95)00331-c. [DOI] [PubMed] [Google Scholar]
- 35.Bennett DA, Schneider JA, Wilson RS, et al. Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J Neurol Neurosurg Psychiatry. 2005;76(9):1194–1199. doi: 10.1136/jnnp.2004.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mungas D, Tractenberg R, Schneider JA, et al. A 2-process model for neuropathology of Alzheimer's disease. Neurobiol Aging. 2014;35(2):301–308. doi: 10.1016/j.neurobiolaging.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berlau DJ, Corrada MM, Robinson JL, et al. Neocortical β-amyloid area is associated with dementia and APOE in the oldest-old. Alzheimers Dement J Alzheimers Assoc. 2013;9(6):699–705. doi: 10.1016/j.jalz.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lippa CF, Smith TW, Saunders AM, et al. Apolipoprotein E-epsilon 2 and Alzheimer's disease: genotype influences pathologic phenotype. Neurology. 1997;48(2):515–519. doi: 10.1212/wnl.48.2.515. [DOI] [PubMed] [Google Scholar]
- 39.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21(3):249–258. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 40.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord. 2006;20(4):210–216. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- 41.Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer's Disease Centers' Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord. 2009;23(2):91–101. doi: 10.1097/WAD.0b013e318191c7dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 43.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 44.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 45.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (Berl) 2006;112(4):389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41(4):479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 47.Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 48.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol (Berl) 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. 2012;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agresti A, Agresti A. Categorical Data Analysis. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2002. [cited 2013 May 30]. Internet. Available from: http://doi.wiley.com/10.1002/0471249688. [Google Scholar]
- 51.Yee TM. The VGAM package for categorical data analysis. J Stat Softw. 2010;32(10):1–34. [Google Scholar]
- 52.Serrano-Pozo A, Qiang J, Monsell SE, et al. Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. J Neuropathol Exp Neurol. 2013;72 doi: 10.1097/NEN.0000000000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imai K, Keele L, Tingley D. A general approach to causal mediation analysis. Psychol Methods. 2010;15(4):309–334. doi: 10.1037/a0020761. [DOI] [PubMed] [Google Scholar]
- 54.Imai K, Yamamoto T. Identification and Sensitivity Analysis for Multiple Causal Mechanisms: Revisiting Evidence from Framing Experiments. Polit Anal. 2013;21:141–171. [Google Scholar]
- 55.Tingley D, Yamamoto T, Hirose K, et al. mediation: R package for causal mediation analysis. J Stat Softw. 2014;59(5):1–38. [Google Scholar]
- 56.Brecht WJ, Harris FM, Chang S, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci Off J Soc Neurosci. 2004;24(10):2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fleming LM, Weisgraber KH, Strittmatter WJ, et al. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp Neurol. 1996;138(2):252–260. doi: 10.1006/exnr.1996.0064. [DOI] [PubMed] [Google Scholar]
- 58.Huang DY, Weisgraber KH, Goedert M, et al. ApoE3 binding to tau tandem repeat I is abolished by tau serine262 phosphorylation. Neurosci Lett. 1995;192(3):209–212. doi: 10.1016/0304-3940(95)11649-h. [DOI] [PubMed] [Google Scholar]
- 59.Huang Y, Liu XQ, Wyss-Coray T, et al. Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A. 2001;98(15):8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strittmatter WJ, Saunders AM, Goedert M, et al. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91(23):11183–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hudry E, Dashkoff J, Roe AD, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5(212):212ra161. doi: 10.1126/scitranslmed.3007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pooler AM, Polydoro M, Wegmann SK, et al. Tau-amyloid interactions in the rTgTauEC model of early Alzheimer's disease suggest amyloid-induced disruption of axonal projections and exacerbated axonal pathology. J Comp Neurol. 2013;521(18):4236–4248. doi: 10.1002/cne.23411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bennett DA, Schneider JA, Wilson RS, et al. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61(3):378–384. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- 64.Hashimoto T, Serrano-Pozo A, Hori Y, et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J Neurosci Off J Soc Neurosci. 2012;32(43):15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koffie RM, Hashimoto T, Tai HC, et al. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-β. Brain J Neurol. 2012;135(Pt 7):2155–2168. doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3(89):89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 68.Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 69.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology. 1998;50(4):961–965. doi: 10.1212/wnl.50.4.961. [DOI] [PubMed] [Google Scholar]
- 70.Bracco L, Piccini C, Baccini M, et al. Pattern and progression of cognitive decline in Alzheimer's disease: role of premorbid intelligence and ApoE genotype. Dement Geriatr Cogn Disord. 2007;24(6):483–491. doi: 10.1159/000111081. [DOI] [PubMed] [Google Scholar]
- 71.Dal Forno G, Rasmusson DX, Brandt J, et al. Apolipoprotein E genotype and rate of decline in probable Alzheimer's disease. Arch Neurol. 1996;53(4):345–350. doi: 10.1001/archneur.1996.00550040085017. [DOI] [PubMed] [Google Scholar]
- 72.Growdon JH, Locascio JJ, Corkin S, et al. Apolipoprotein E genotype does not influence rates of cognitive decline in Alzheimer's disease. Neurology. 1996;47(2):444–448. doi: 10.1212/wnl.47.2.444. [DOI] [PubMed] [Google Scholar]
- 73.Kleiman T, Zdanys K, Black B, et al. Apolipoprotein E epsilon4 allele is unrelated to cognitive or functional decline in Alzheimer's disease: retrospective and prospective analysis. Dement Geriatr Cogn Disord. 2006;22(1):73–82. doi: 10.1159/000093316. [DOI] [PubMed] [Google Scholar]
- 74.Kurz A, Egensperger R, Haupt M, et al. Apolipoprotein E epsilon 4 allele, cognitive decline, and deterioration of everyday performance in Alzheimer's disease. Neurology. 1996;47(2):440–443. doi: 10.1212/wnl.47.2.440. [DOI] [PubMed] [Google Scholar]
- 75.Murphy GM, Jr, Taylor J, Kraemer HC, et al. No association between apolipoprotein E epsilon 4 allele and rate of decline in Alzheimer's disease. Am J Psychiatry. 1997;154(5):603–608. doi: 10.1176/ajp.154.5.603. [DOI] [PubMed] [Google Scholar]
- 76.Norrman J, Brookes AJ, Yates C, St Clair D. Apolipoprotein E genotype and its effect on duration and severity of early and late onset Alzheimer's disease. Br J Psychiatry J Ment Sci. 1995;167(4):533–536. doi: 10.1192/bjp.167.4.533. [DOI] [PubMed] [Google Scholar]
- 77.Tschanz JT, Corcoran CD, Schwartz S, et al. Progression of cognitive, functional, and neuropsychiatric symptom domains in a population cohort with Alzheimer dementia: the Cache County Dementia Progression study. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 2011;19(6):532–542. doi: 10.1097/JGP.0b013e3181faec23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoyt BD, Massman PJ, Schatschneider C, et al. Individual growth curve analysis of APOE epsilon 4-associated cognitive decline in Alzheimer disease. Arch Neurol. 2005;62(3):454–459. doi: 10.1001/archneur.62.3.454. [DOI] [PubMed] [Google Scholar]
- 79.Stern Y, Brandt J, Albert M, et al. The absence of an apolipoprotein epsilon4 allele is associated with a more aggressive form of Alzheimer's disease. Ann Neurol. 1997;41(5):615–620. doi: 10.1002/ana.410410510. [DOI] [PubMed] [Google Scholar]
- 80.Van der Vlies AE, Koedam ELGE, Pijnenburg YAL, et al. Most rapid cognitive decline in APOE epsilon4 negative Alzheimer's disease with early onset. Psychol Med. 2009;39(11):1907–1911. doi: 10.1017/S0033291709005492. [DOI] [PubMed] [Google Scholar]
- 81.Bonner-Jackson A, Okonkwo O, Tremont G. Alzheimer's Disease Neuroimaging Initiative. Apolipoprotein E ε2 and functional decline in amnestic mild cognitive impairment and Alzheimer disease. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 2012;20(7):584–593. doi: 10.1097/JGP.0b013e3182203c32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wilson RS, Bienias JL, Berry-Kravis E, et al. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry. 2002;73(6):672–677. doi: 10.1136/jnnp.73.6.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005-2010. J Neuropathol Exp Neurol. 2012;71(4):266–273. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Serrano-Pozo A, Qian J, Monsell SE, et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann Neurol. 2014;75(4):597–601. doi: 10.1002/ana.24125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Demographic and clinical characteristics of cognitively intact subjects (CDR-SOB=0) by APOE genotype.
Supplemental Table 2. Pathological characteristics of cognitively intact subjects (CDR-SOB=0) by APOE genotype.