

Abstract
Mitochondrial disorders are defined as defects that affect the oxidative phosphorylation system (OXPHOS). They are characterized by a heterogeneous array of clinical presentations due in part to a wide variety of factors required for proper function of the components of the OXPHOS system. There is no cure for these disorders owing our poor knowledge of the pathogenic mechanisms of disease. To understand the mechanisms of human disease numerous mouse models have been developed in recent years. Here we summarize the features of several mouse models of mitochondrial diseases directly related to those factors affecting mtDNA maintenance, replication, transcription, translation as well to other proteins that are involved in mitochondrial dynamics and quality control which affect mitochondrial OXPHOS function without been intrinsic components of the system. We discuss how these models have contributed to our understanding of mitochondrial diseases and their pathogenic mechanisms.
Keywords: Mitochondrial DNA, mitochondrial transcription, mouse models, mitochondrial dynamics, quality control, mitochondrial diseases
1. Introduction
Mammalian mitochondrial DNA (mtDNA) is a double-stranded, circular molecule of about 16,5 Kb encoding for 37 genes: 13 genes for oxidative phosphorylation (OXPHOS) complexes subunits, 22 tRNAs and 2 rRNAs necessary for mitochondrial translation (Anderson, 1981). Mitochondrial DNA follows peculiar non-Mendelian genetic rules, including matrilinear transmission, polyploidy, threshold effect and mitotic segregation. Mitochondrial replication, transcription and translation occur in a semi-autonomous fashion, since all the proteins necessary for these processes are encoded by nuclear DNA and imported from the cytoplasm (Falkenberg, 2007). The intricate molecular machinery involved in the maintenance, transcription and translation of mtDNA includes several proteins found mutated in human neurological disorders that share a common feature the presence of mtDNA deletions and/or depletion (Koopman, 2012; Spinazzola, 2011). These proteins are either essential for mtDNA replication (POLγ and TWINKLE), transcription/translation (TFAM, TFB1M, mTERFs, POLRMT, LRPPRC and DARS2), for the correct maintenance of the mitochondrial dNTPs pool (TP, TK, ANT1, and RRM2B). In addition, proteins involved in mitochondrial dynamics (MFN1, MFN2, OPA1, and DRP1) and in protein quality control (CLPP, AFG3L2, PARAPLEGIN, OMA1, PHBs, OMI and PARL) are also essential for proper mitochondrial function. Defects in the factors aforementioned can cause mitochondrial diseases. We have described mouse models of mitochondrial diseases that are directly related to components of the OXPHOS system in Part I of this review miniseries. In this chapter, Part II of the review miniseries, we summarize the mouse models that recapitulate defects in mtDNA instability, transcription, translation, mitochondrial dynamics, quality control and others. We discuss how thesemousemodels have contributed to our knowledge of the specific function of these factors and their role in the pathogenic mechanisms of mitochondrial disorders. A summary of these mousemodels is shown in Fig. 1. Part III of the review miniseries discusses the different therapeutic strategies that have been tested in the mouse models described in Parts I and II.
Figure 1. Mouse models of mitochondrial diseases.
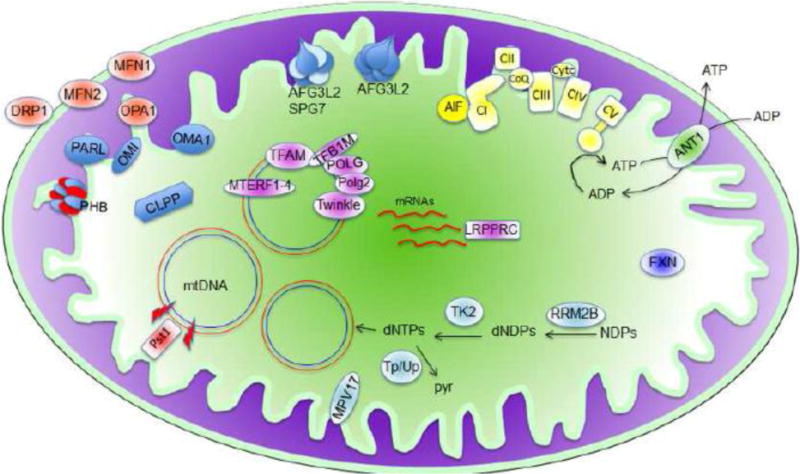
Mouse models that affect the oxidative phosphorylation system which is represented in light yellow in the mitochondrial inner membrane. Mouse models affecting mitochondrial dynamics are represented in light orange (MFN1, MFN2, OPA1 and DRP1). Mouse models of proteins involved in mtDNA replication, transcription and translation are represented in light pink (POLG, POLG2, TFAM, TFB1, TWINKLE, MTERFs and LRPPRC). Mouse models of proteins required for dNTPs synthesis, and mtDNA synthesis and stability are represented in light blue (RRM2B, TK2, TP, UP and MPV17). Mouse model for mtDNA deletion/depletion caused by double-strand brakes (red bolt) is represented in red (mito-Pst1). Mouse models of protein quality control are represented in blue (CLPP, PHB, PARL, OMI, OMA1, AFG3L2+SPG7, AFG3L2). Other mouse models (AIF1, ANT1 and FXN). NDPs: nucleotide diphosphates; dNDPs: deoxy-NDPs; dNTPs: deoxy nucleotide triphosphates; Pyr: pyrimidines; mtDNA: mitochondrial DNA.
1. Mouse models of proteins involved in mtDNA stability, replication, transcription and translation
The mammalian mtDNA genome must be replicated, transcribed and translated to assure a proper mitochondrial function. Regulation of these processes is very important for modulating the OXPHOS capacity in response to metabolic requirements or to pathological/stress conditions. All the components needed for replication and transcription of the mtDNA are encoded in the nuclear genome. For the translation of the mtDNA, 22 tRNAs and 2 rRNAs encoded in the mitochondrial DNA are required in addition to numerous proteins that are imported from the cytoplasm. The mtDNA replication seems to relay in a single DNA polymerase, polymeraseγ. The core machinery required for basal mtDNA transcription in mammals (mtRNA-polymerase, TFAM, mtTFB2) and a few proteins that regulate mtDNA transcription has been described (mtTERFS, LRPPRC). Although the regulation of the mitochondrial transcription is a not completely understood process, the creation of knockout mouse models have clearly contributed to the understanding of mitochondrial transcription in mammals (reviewed in {Peralta, 2012 #591}). Mouse models of proteins involved in the maintenance of mitochondrial DNA, replication, transcription and translation are summarized in Table I.
Table I.
Mouse models of proteins involved in mtDNA replication/transcription and translation
| Gene | Mitochondrial function | Genetic Manipulation | Phenotype | References |
|---|---|---|---|---|
| Polg | mtDNA replication (catalytic subunit of Polγ) | Knockout: -Whole body (Homologous recombination) | Embryonic lethal (E7.5–8.5) | (Hance, 2005) |
| Overexpression of human POLGY955C (associated with PEO) | Early death, cardiomegaly | (Lewis, 2007) | ||
| Overexpression of PolgD181A (lacking proof reading activity) in: -Heart -Pancreatic β-cells -Neurons |
Cardiomegaly Early onset diabetes Neuronal dysfunction after 5m of age |
(Zhang, 2000) (Bensch, 2009) (Kasahara, 2006) (Kong, 2011) |
||
| Knock-in: -PolgD257A (lacking proof reading activity) | Reduced lifespan and premature aging | (Kujoth, 2005; Trifunovic, 2004) | ||
| Polg2 | mtDNA replication (accesory subunit of Polγ) | Knockout: -Whole body (Homologous recombination) | Embryonic lethal (E8.0–8.5) | (Humble, 2013) |
| Twinkle | mtDNA replication (Helicase) | Knockout: -Whole body (Homologous recombination) | Embryonic lethal | (Milenkovic, 2013) |
| Conditional knockout: -Cardiac/skeletal muscle (Mck-Cre) | Progressive cardiomyopathy and early death | (Milenkovic, 2013) | ||
| Overexpression of TwinkleA360T (Ubiquitous) | Late-onset myopathy | (Tyynismaa, 2005) | ||
| Overexpression of Twinkle dup353–365 (Ubiquitous) | ||||
| Overexpression of Twinkledup353–365 in dopaminergic neurons (TH-promoter) | No overt phenotype. Dopaminergic neurodegeneration. | (Song, 2012) | ||
| Tfam | mtDNA maintenance and mtDNA transcription | Knockout: -Whole body (Homologous recombination) | Embryonic lethal (E8.5–10.5) | (Larsson, 1998) |
| Conditional knockout: -Skeletal muscle (Mlc-1f-Cre) | Myopathy | (Wredenberg, 2002) | ||
| -Cardiac muscle (Mck-Cre) | Cardiomyopathy. Early death (10–14 weeks) | (Freyer, 2010; Hansson, 2004; Wang, 1999a) (Li, 2000) | ||
| -Pancreatic β-cells (Rip-Cre) | Diabetes | (Silva, 2000) | ||
| -Forebrain neurons (CaMKIIa-Cre) | Late-onset neurodegeneration | (Dufour, 2008; Sorensen, 2001) | ||
| -Dopaminergic neurons (Dat-Cre) | Progressive parkinsonism | (Ekstrand, 2007) | ||
| -Adipose tissue (aP2-Cre) | Protection against obesity and insulin resistance | (Vernochet, 2012) | ||
| Tfam /Twinkle | mtDNA maintenance and replication | Overexpresion of: wt mouse Twinkle and wt human Tfam | Partial OXPHOS deficiency heart and skeletal muscle | (Ylikallio, 2010) |
| Tfb1m | Mitochondrial transcription and translation | Knockout: Full body (β-actin) | Embryonic lethal (E8.5) | (Metodiev, 2009) |
| Conditional knockout: -Heart and skeletal muscle (Mck-Cre) | Cardiomyopathy Mitochondrial protein synthesis defect No phenotype in skeletal muscle | (Metodiev, 2009) | ||
| -Pancreatic β-cells (Rip-Cre) | Development of diabetes with progressive islet dysfunction | (Sharoyko, 2014) | ||
| Mterf1 | Transcription initiation at the light-strand promoter of the mtDNA | Knockout: -Whole body (Homologous recombination) | Viable, with normal oxidative phosphorylation capacity | (Terzioglu, 2013) |
| Mterf2 | mtDNA Transcription (positive regulator) | Knockout: -Whole body (Homologous recombination) | Viable. Mild myopathy and memory defects. Decreased levels mitochondrial transcripts. | (Wenz, 2009) |
| Mterf3 | Transcription terminator of the mtDNA | Knockout: -Whole body (Homologous recombination) -Heart and skeletal muscle (Mck-Cre) |
Embryonic lethal (E8.5–E10.5) Severe cardiomyopathy and premature death |
(Park, 2007) (Park, 2007) (Andersson, 2011) |
| Mterf4 | Mitochondrial ribosomal biogenesis | Knockout: -Whole body (Homologous recombination) -Heart and skeletal muscle (Mck-Cre) |
Embryonic lethal (Before E8.5) Severe cardiomyopathy and premature death |
(Cámara, 2011) (Cámara, 2011) |
| Polrmt | Mitochondrial RNA polymerase | -Heart and skeletal muscle (Mck-Cre) | Not described | (Kuhl, 2014). |
| Lrpprc | Translation/stability of the mitochondrial mRNAs | Knockout: -Whole body (Gene trap) | Embryonic lethal (Before E12.5) | (Xu, 2012) |
| Heart and skeletal muscle (Mck-Cre) | Cardiomyopathy and reduced life span | (Ruzzenente, 2012) | ||
| Dars2 | Mitochondrial aspartyl-tRNA synthetase | Knockout: Whole body (Gene trap) | Embryonic lethal (E8.5) | (Dogan, 2014) |
| Heart and skeletal muscle (Mck-Cre) | Cardiomyopathy and premature death by 6–7 weeks of age. |
1.1. Polg
Mammalian polymerase γ (POLγ) is a heterotrimer composed by a catalytic subunit, encoded by the POLG gene, with DNA-polymerase and 3′-5′ exonuclease activities, and two identical accessory subunits, encoded by the POLG2 gene, conferring high processivity to the catalytic subunit by increasing the affinity of the heterotrimer for template DNA.
Mutations in human POLG gene have been associated with different clinical presentations, ranging from diverse infantile encephalopathies to late-onset myopathies, but sharing mtDNA instability as a common feature (for a recent review see (Milone and Massie, 2010)). Close to 250 pathogenic mutations have been described for the POLG gene (http://tools.niehs.nih.gov/polg/index.cfm) making it one of the most common causes of mitochondrial diseases (for review see (Copeland and Longley, 2014). Several mouse models have been generated modifying the murine Polg gene; however they do not fully resemble the clinical spectrum of patients. A summary of all Polg mouse models is available in Table I. Homozygous Polg knockout mice died in utero at E7.5–8.5 due to an almost complete lack of mtDNA, indicating that DNA-polymerase activity is essential for mtDNA replication during embryonic development (Hance, 2005).
One of the most common mutations of the human mitochondrial polymerase associated with Progressive External Ophthalmoplegia (PEO) is the Y955C mutation. This mutation is known to affect the DNA-polymerase activity by promoting enzyme stalling and errors in replication. Transgenic mice created by expressing the human POLGY955C mutant form died prematurely and presented with massive cardiomegaly with bilateral atrial enlargement (Lewis, 2007). Tissue analyses revealed the presence of mtDNA depletion and mtDNA damage, most likely caused by increased oxidative stress. Additionally, proliferation of mitochondrial showing cristae disorganization was observed presumably as a compensatory mechanism (Lewis, 2007).
Another mutation in Polg that causes impairment of the 3′-5′ exonuclease activity was modeled by the transgenic overexpression of the mutant PolgD181A in heart, pancreatic β-cells and neurons. The mutant PolgD181A invariably induced an accumulation of mtDNA point mutations and large-scale deletions (Bensch, 2009; Kasahara, 2006; Zhang, 2000). When the transgene was expressed in the heart, mice developed a severe progressive cardiomegaly associated with a profound alteration of transcriptional profile of genes involved in cardiac remodeling; heart and muscle contractility; energy and lipid metabolisms; and stress response and apoptosis (Zhang, 2005). When targeted to neurons, PolgD181A overexpression induced mtDNA instability in multiple brain regions, such as cortex, basal ganglia, hippocampus and retinal cells (Kasahara, 2006; Kong, 2011). These transgenic mice were perfectly normal in terms of general health, longevity and fertility; without any gross alteration in brain or retinal structure; or functional motor or coordination impairment. However, after 20 weeks of age, mice started to show some signs of neuronal dysfunction, including distorted circadian rhythms, enhanced startle response, and altered secretion pattern of monoamines (i.e. serotonin, dopamine and noradrenaline), mirroring some of the mood disorder symptoms found in PEO patients (Kasahara, 2006; Lamperti and Zeviani, 2009; Mancuso, 2004). The accumulation of mtDNA point mutations and deletions in the PolgD181A mice caused a mild mitochondrial dysfunction in the retina, with altered electroretinogram patterns but no loss of retinal ganglion cells (RGCs) or retinal thinning were found (Kong, 2011). The same transgene was targeted to pancreatic islets in a mouse model of early-onset diabetes, characterized by increased β-cells apoptosis and insulin insufficiency, supporting the mitochondrial dysfunction theory for the etiology of age-dependent diabetes (Bensch, 2009).
Two knock-in mutants of Polg have been generated independently by inducing the amino acid substitution D257A in the proofreading 3′-5′ exonuclease domain two (Table 1) (Kujoth, 2005; Trifunovic, 2004). The lack of proofreading activity of the so-called “Mutator mice” caused the accumulation of mtDNA point mutations and non-canonical multiple deletions in different tissues, leading to a progressive OXPHOS deficiency (Edgar, 2009; Trifunovic, 2004). Despite the different age of onset, these mice showed a reduced lifespan and developed a premature aging phenotype (Fox, 2012; Kujoth, 2005; Trifunovic, 2004). Unexpectedly, the accumulation of mtDNA mutations did not induce a massive chronic oxidative stress and the progeroid phenotype was mainly caused by increased apoptosis (Kujoth, 2005; Trifunovic, 2005). Although it is well established that the Mutator mouse is characterized by having mtDNA instability, whether the premature aging phenotype is caused by the accumulation of mtDNA mutations or by the multiple deletions is still a matter of discussion (Bailey, 2009; Edgar, 2009; Vermulst, 2007; Vermulst, 2008; Williams, 2010). Nonetheless, the peculiar progeroid phenotype makes the Mutator mouse a unique model for the study of the mitochondrial contribution to aging.
Dopaminergic neurons of the Mutator mouse mirrored the accumulation of mtDNA deletions found in patients with Parkinson’s disease and aged individuals (Perier, 2013). However, despite the occurrence of mtDNA deletions, the mitochondrial function of the Mutator mouse was generally comparable to controls, despite the presence of few cytochrome c oxidase (COX) negative neurons. Interestingly, the dopaminergic neurons displayed higher levels of mtDNA-deleted copies. In addition, a global compensatory response was activated in these neurons, inducing higher dopamine levels and resistance to neurotoxins (Perier, 2013). As the Mutator mouse aged (12 to 14 months), it displayed a dopaminergic dysfunction that was associated with impaired locomotor activity and rotarod performance, reduced body mass and metabolic alterations (Dai, 2013).
The decline of mitochondrial function in skeletal muscle of the Mutator mouse was associated with a profound sarcopenia and exercise intolerance, mainly due to the loss of type I muscle fibers (Hiona, 2010; Safdar, 2011). Muscle from older Mutator mice displayed higher mitochondrial fission and autophagy levels, which most likely contributed to the sarcopenic phenotype observed in premature aging and differed from the response observed in normally-aged muscle (Joseph, 2013). Moreover, mtDNA instability induced a significant remodeling of gastrocnemius transcriptional profile, with a striking activation of several genes involved in RNA synthesis and ribosomal maturation, whereas a down-regulation of genes involved in energy metabolism was found (Hiona, 2010).
The Mutator mouse also developed age-dependent macrocytic anemia that invariantly led to premature death (Chen, 2009; Trifunovic, 2004). Abnormal erythroid development and defective lymphopoiesis found in the Mutator mouse were transferred to wild type littermates by bone marrow transplantation, suggesting a possible involvement of mitochondrial defects in the pathogenesis of some age-related myelodysplastic cases of anemia (Chen, 2009). This peculiar phenotype seems to be related to the presence of dysfunctional hematopoietic and neural somatic stem cell progenitors, already from embryogenesis (Ahlqvist, 2012).
Recent work demonstrated that the Mutator mouse also showed profound changes in intestinal absorption of nutrients even at 3 to 7 months of age, with restricted absorption of dietary lipids due to a severe impairment of stem/progenitor cells (Fox, 2012).
1.2. Polg2
Mutations in POLG2, which are not as predominant as mutations in POLG, have been described in patients with early- and late-onset autosomal dominant progressive external ophthalmoplegia (adPEO) (Longley, 2006; Walter, 2010; Young, 2011). Presumably, defective POLG2 with impaired processivity activity causes POLγ to stall leading to accumulation of mutations during mtDNA replication. To better understand the role of this accessory subunit and how it causes pathogenesis, a mouse model carrying the ablation of Polg2 has been developed (Table I) (Humble, 2013). Heterozygous Polg2+/− mice were undistinguishable from controls in terms of development, lifespan and mitochondrial function. On the contrary, homozygous deletion of Polg2 resulted in embryonic lethality at E8.0–8.5 with loss of mtDNA and mtDNA-encoded proteins. Lipid accumulation, severe ultra-structural defects and cristae disorganization were detected in the mitochondria of the Polg2−/− embryos, indicating that POLG2 function is critical for mtDNA replication and mammalian embryogenesis (Humble, 2013).
1.3. Mitochondrial helicase: Twinkle
TWINKLE is a mitochondrial hexameric helicase belonging to the RecA superfamily that forms, along with POLγ and the single-stranded mtDNA binding protein (mtSSB), the minimal mtDNA replisome (Korhonen, 2004; Spelbrink, 2001; Tyynismaa, 2004). Mutations in Twinkle have been associated with adPEO (Spelbrink, 2001), with recessively inherited infantile onset spinocerebellar ataxia (IOSCA) (Nikali, 2005) and with mtDNA depletion syndrome (MDS) (Hakonen, 2007; Sarzi, 2007). The common feature of all these diseases is the presence of mtDNA instability (depletion/deletions) caused by an alteration of the replication machinery. Homozygous ablation of Twinkle was embryonic lethal demonstrating that this protein is essential for mouse embryonic development (Milenkovic, 2013). Tissue-specific (heart and skeletal muscle) ablation of Twinkle resulted in premature death at the age of 19 weeks. The mice developed a progressive cardiomyopathy, with a significant reduction in mtDNA levels and OXPHOS dysfunction in heart and skeletal muscle (Milenkovic, 2013) (Table I).
Two transgenic mice overexpressing different mutant forms of Twinkle (TwinkleA360T and the Twinkledup353–365) have been developed. These mice are commonly named the “Deletor mice” (Tyynismaa, 2005). Both mutant mice developed the same pathological phenotype on their adulthood. Skeletal muscle from 1-year-old animals revealed signs of mitochondrial myopathy, including COX negative fibers, mitochondrial proliferation and abnormal fiber size, caused by the accumulation of deleted mtDNA molecules. Multiple deletions, mtDNA depletion and COX negative cells were also evident in brain of 18 month old animals (Tyynismaa, 2005). The study of the replication pattern of various tissues of the Deletor mouse provided molecular details on the generation and accumulation of mtDNA multiple deletions. The authors proposed that the pausing of the mutant TWINKLE in the replication fork induced the accumulation of mtDNA replication intermediates and transcriptional defects. The replication fork stalling may enhance the generation of mtDNA deleted molecules by activating a mtDNA repair system or by inducing a local depletion followed by an inappropriate repopulation (Goffart, 2009). The accumulation of mtDNA multiple deletions in the Deletor mouse was closely linked to a decline of OXPHOS function and metabolic alterations as demonstrated by the accumulation of COX negative cells, abnormal levels of phospholipids, ketone bodies and free amino acids in plasma and increased phosphatidylcholine and sphingomyelin in skeletal muscle (Ahola-Erkkilä, 2010). Gene expression profile of the Deletor mouse in skeletal muscle revealed a marked up-regulation of the amino acid starvation response pathway (Tyynismaa, 2010). In particular, the metabolic hormone fibroblast growth factor 21 (FGF21), secreted by liver and adipose tissue upon prolonged fasting, was upregulated in the Deletor mouse and it has been also found to be a reliable predictive biomarker for human mitochondrial myopathies (Suomalainen, 2011; Tyynismaa, 2010). By mimicking the phenotype of late-onset progressive mitochondrial myopathies, the Deletor mouse represents a valuable tool for the prognostic and development of therapeutic strategies and provided valuable insights in the understanding of metabolic adaptive responses.
A transgenic mouse overexpressing the mutant Twinkledup353–365 in dopaminergic neurons was developed to investigate the effects of accumulation of mtDNA deletions in Parkinson’s disease (Table I) (Song, 2012). Transgenic mice were not significantly different from controls in terms of longevity and body weight. No overt signs of movement disorders were found, however, Twinkle mutants displayed increased levels of age-related mtDNA deletions, reduced number of dopaminergic neurons and abnormalities in rotarod performance. A mild reduction of mitochondrial basal respiration was found in association with reduced levels of Parkin and increased mitophagy, suggesting a possible involvement of Twinkle in the regulation of mitochondrial quality control in dopaminergic neurons.
1.4. Mitochondrial transcription factor Tfam
The mitochondrial transcription factor A or TFAM protein plays an essential dual function in the mtDNA transcription (Fisher and Clayton, 1988) and mtDNA maintenance (Ekstrand, 2004). TFAM belongs to the high-mobility-group (HMG) box domain protein family (Parisi and Clayton, 1991), which can bind, unwind and bend DNA without sequence specificity. TFAM is the main protein component of the nucleoids and is involved in mtDNA packaging (Bonawitz, 2006; Cámara, 2011). Whole body ablation of the Tfam gene in mice (Tfam−/−) causes severe mtDNA depletion, leading to a severe respiratory chain deficiency and embryonic lethality between E8.5 and E10.5 (Larsson, 1998). To overcome the embryonic lethality, several Tfam conditional KO mouse models have been generated (Table I).
Specific inactivation of Tfam in skeletal muscle (Mlc-1f-Cre/LoxP system) resulted in myopathy with ragged-red fibers (RRFs), and progressive decline of OXPHOS function (Wredenberg, 2002). As a consequence, a marked compensatory mitochondrial proliferation was evident in skeletal muscle.
Disruption of Tfam in mouse cardiomyocytes was generated by expressing Cre recombinase under the muscle creatine kinase promoter (Mck-Cre). This conditional KO animal showed progressive dilated cardiomyopathy with heart conduction block, observed at 2 weeks of age. Animals worsened with time and died prematurely at 10 to 14 weeks of age (Wang, 1999a). Contrary to what was observed in the Mlc-1-f-Tfam KO mice, in the Mck-Tfam KO mitochondrial proliferation was evident only at later stages of disease progression (8 weeks) and it was not sufficient to sustain ATP deficiency.
Deletion of Tfam in pancreatic β-cells (Rip-Cre) led to diabetes at 5 weeks and severe mtDNA depletion and OXPHOS deficiency at 7 to 9 weeks of age leading to decreased insulin release and reduced β -cell mass (Silva, 2000). This mouse model nicely reproduced the β-cell pathology of human mitochondrial diabetes form, that constitutes about 0.5–1% of all types of diabetes mellitus and clearly undelines the crucial role of the OXPHOS system in insulin secretion.
Ablation of Tfam specifically in forebrain neurons by the CaMKIIa-Cre caused mtDNA depletion starting at 2 months of age followed by reduced levels of mitochondrial transcripts and OXPHOS deficiency at 4 months of age (Sorensen, 2001). Postnatal disruption of the OXPHOS system in forebrain neurons induced a Mitochondrial Late-Onset Neurodegeneration process (MILON mice). The onset of the neurodegenerative disease was observed at 5 months of age and animals died thereafter due to massive neuronal death and gliosis in neocortex and hippocampus (Sorensen, 2001).
Loss of TFAM protein in the dopaminergic neurons using Dat-Cre led to a parkinsonism phenotype with adult onset, progressive motor impairment, presence of intraneuronal inclusions and dopaminergic neuronal death (Ekstrand, 2007).
Specific inactivation of Tfam in both white and brown adipose tissue (using the aP2 promoter for Cre expression) results in decreased complex I and IV enzymatic activities (Vernochet, 2012). Complex II dependent respiratory function, citrate synthase activity and basal respiratory capacity were markedly increased presumably as compensatory mechanism for the bioenergetic defect. Interestingly, Tfam deletion in adipose tissue resulted in increased mitochondrial capacity and mice were protected against obesity and insulin resistance after ingestion of a high fat diet (Vernochet, 2012).
To understand how mtDNA copy number is regulated in vivo, Suomalainen’s group developed a double transgenic mouse ubiquitously overexpressing the mouse Twinkle and the human TFAM (hTFAM) genes (Ylikallio, 2010). Double transgenic mice had increased COX negative fibers in heart and skeletal muscle and accumulated enlarged and abnormal mitochondria in muscle. Molecular analysis revealed significantly higher mtDNA copy number in the double transgenic than in mice expressing single transgene particularly in muscle and heart. Twinkle appeared to regulate de novo mtDNA synthesis. Interestingly, the high mtDNA copy number in the Twinkle/hTFAM mouse was associated with an increase in size of the nucleoids, which led to defects in transcription, increased mtDNA deletions and subsequent defects of the respiratory chain (Ylikallio, 2010).
1.5. Mitochondrial transcription factor B1
Mitochondrial transcription factors B1 and B2 (TFB1M and TFB2M) regulate mtDNA transcription by forming heterodimers with the mitochondrial RNA polymerase (Falkenberg, 2002) and mitochondrial translation by exerting their rRNA methyltransferase activity (Cotney and Shadel, 2006). Null mice for Tfb1m were not viable and died at about E8.5 showing severe developmental delay whereas the conditional disruption of Tfb1m in heart and skeletal muscle produced severe cardiomyopathy and premature death by 24 weeks of age (Metodiev, 2009). Interestingly, no mitochondrial abnormalities were found in the skeletal muscle of the KO mice, whereas the heart of the Tfb1m KO displayed impaired OXPHOS function and increased mitochondrial biogenesis most likely to compensate for the bioenergetic defect. Mitochondrial protein translation was abolished in the KO mice as a consequence of impaired ribosomal assembly due to the low levels 12S rRNA and the lack of adenine dimethylation of the rRNA (Metodiev, 2009). This mouse model confirmed the function of TFB1M and its importance on ribosomal assembly and mitochondrial protein translation (Table I).
A gene variant of Tfb1m has been identified as one of the risk genes for development of Type II diabetes in humans (Koeck, 2011). To investigate the role of TFB1M in diabetes, a conditional mouse model harbouring the ablation of Tfb1m in pancreatic β-cells was created. Ablation of this gene in β-cells resulted in a progressive dysfunction of islets and development of diabetes resulting from the impaired mitochondrial function, which led to perturbations in insulin secretion. Among the pathological phenotypes are increased cell death of β-cells, inflammation and oxidative stress (Sharoyko, 2014). This mouse model underscores the importance of Tfb1m as a risk factor for diabetes.
1.6. Mitochondrial terminator factors-Mterf
The mitochondrial termination factor (MTERF) protein family is composed by 5 transcription termination factors, named MTERF1–5, which are implicated in the regulation of mitochondrial transcription and protein synthesis and in the coordination between transcription and replication (Bruni, 2012; Linder, 2005; Park, 2007; Roberti, 2009).
Park and collaborators generated the first KO mouse by inactivating of Mterf3 in the germ line (Table I) (Park, 2007). Homozygous embryos presented an improper development and died between E8.5 and E10.5. The specific inactivation of Mterf3 in heart and skeletal muscle produced a severe mitochondrial cardiomyopathy leading to premature death (Park, 2007). Shortly before death, these mice developed progressive bradycardia that later developed into an atrioventricular block (Andersson, 2011). Accumulation of aberrant mitochondria in the heart was associated with a severe OXPHOS impairment, characterized by the progressive disassembly of respiratory complexes with the exception of complex II in the KO mice. The loss of MTERF3 did not affect mtDNA abundance, but increased transcription initiation on both mtDNA strands, leading to decreased expression of critical promoter-distal tRNA genes, which may possibly explain the bioenergetic dysfunction (Park, 2007).
Moraes group also observed an imbalance of mitochondrial tRNA levels in Mterf2−/− mice, although the loss of this factor caused an opposite effect on the steady-state levels of mitochondrial mRNAs by decreasing them (Wenz, 2009). Mterf2−/− mice were viable and presented normal basal metabolism and heat production, with only a modest tendency to weight and adipose tissue loss, which was more evident in females. These mice did not display an overt phenotype, but only a mild exercise intolerance and muscle weakness that worsened when feeding mice with a ketogenic diet. Under such metabolic stress conditions, Mterf2−/− mice developed memory and cognitive impairment concomitant with an imbalance of steady-state levels mitochondrial tRNAs levels, multiple OXPHOS deficiencies and lower ATP production in multiple tissues despite compensatory mitochondrial biogenesis (Wenz, 2009).
Ubiquitous inactivation of Mterf4 is embryonic lethal (E8.5), whereas heterozygous mice were indistinguishable from wild type littermates. When Mterf4 is conditionally deleted in heart and skeletal muscle, mice manifested weight loss and mitochondrial cardiomyopathy that led to a premature death (maximum lifespan 21 weeks) (Table I) (Cámara, 2011). The heart of the Mterf4−/− mice showed a drastic reduction of fully assembled OXPHOS complexes, bioenergetic defects and an activation of compensatory mitochondrial biogenesis. Furthermore, a massive increase in mitochondrial transcripts including several tRNAs was evident. Inactivation of Mterf4 in murine heart revealed its involvement in the regulation of mitochondrial translation, instead of mitochondrial transcription as previously proposed (Cámara, 2011). In fact, despite the abundance of mitochondrial ribosomal proteins and 12S and 16S rRNA, the in vitro mitochondrial translation was severely impaired and mitoribosomes were not completely assembled due to the lack of the MTERF4/NSUN4 complex. MTERF4 forms a complex with the ribosomal RNA methyltransferase NSUN4 to control mitochondrial ribosomal biogenesis and mitochondrial translation (Cámara, 2011).
Unexpectedly and in contrast to Mterf3 and Mterf4 KO mice, Mterf1 KO animals were viable and had no OXPHOS defects (Terzioglu, 2013). Mitochondrial DNA replication, transcription and translation were not affected by the Mterf1 ablation since mitochondrial rRNA, 12S rRNA, 16S rRNA, and various mRNAs levels were not altered. However, the Mterf1 KO mice showed increased levels of rRNA antisense transcripts and decreased abundance of the light-strand promoter (LSP) proximal 7S transcript leading to the hypothesis that MTERF1 terminates the L-strand transcription at its binding site in tRNALeucine and thereby prevents the elongation of the L-strand transcription complexes by interfering with transcription initiation at the LSP. Thus, in contrast to previous in vitro results that suggested that MTERF1 regulated heavy-strand transcription levels, loss of Mterf1 in vivo strongly suggests that this protein influences transcription initiation at the LSP (Terzioglu, 2013).
1.7. Mitochondrial DNA-directed RNA polymerase, Polrmt
POLRMT is a nuclear encoded RNA polymerase that is located in mitochondria and helps in the transcription of mitochondrial genes. It has been reported that a splice variant of POLRMT, spRNAP-IV, acts inside the nucleus by transcribing a subset of genes. To shed light on the function of POLRMT, a conditional KO mouse has been generated by targeting the deletion of exon 3 that disrupts the expression of both the mitochondrial and the splice variant (Kuhl, 2014). Ablation of the floxed Polrmt in skeletal muscle and heart using the creatine kinase Cre caused a drastic reduction of de novo synthesis of mitochondrial proteins. However, transcripts of nuclear genes that are targets of spRNAP-IV, did not show any differences between Polrmt KO mice and controls. Accordingly, no nuclear expression of spRNAP-IV or other isoforms was detected in several tissues demonstrating that Polrmt has exclusively a mitochondrial localization and function (Kuhl, 2014). Unfortunately, the phenotype of the Polrmt KO was not described in this brief communication.
1.8. Leucine-rich pentatricopeptide repeat-containing protein, Lrpprc
LRPPRC encodes for a mitochondrial protein involved in the translation and/or stability of the mtDNA-encoded transcripts (Harmel, 2013; Ruzzenente, 2012; Xu, 2004). LRPPRC belongs to the pentatricopeptide repeat (PPR) protein family, whose members are characterized by a repeated motif of 35 amino acids and implicated in RNA binding (Small and Peeters, 2000). Several independent studies demonstrated that LRPPRC is mainly localized into the mitochondrial matrix (Sasarman, 2010; Sterky, 2010; Xu, 2004), however it has also been reported that it is able to interact with the PGC-1α suggesting a possible nuclear function (Cooper, 2006). The founder recessive c.1119C>T mutation in the LRPPRC gene causes the French-Canadian variant of Leigh Syndrome (FCLS), a severe unusual form of infantile LS characterized by a profound complex IV defect in brain and liver (Debray, 2011; Merante, 1993; Mootha, 2003). Clinical presentations of this form include mild regression of psychomotor skills and fatal lactic acidosis leading to death between 3 and 10 years of age. The mechanism by which LRPPRC regulates the mtDNA expression is still controversial, however, it has been demonstrated that this protein forms a complex with the stem-loop interacting RNA binding protein (SLIRP), a protein involved in the maintenance of mtRNAs (Baughman, 2009).
The generation of KO mice has partially elucidated the protein function in vivo, providing multiple hints for the identification of the molecular mechanisms underlying the pathogenesis of FCLS. Constitutive ablation of Lrpprc in homozygosis induced a developmental delay in murine embryos starting from E8.5–10.5 and a reduced CIV activity that rapidly led to death before E12.5 (Ruzzenente, 2012; Xu, 2012). On the other hand, conditional KO of Lrpprc in heart and skeletal muscle induced a dramatic reduction in mice lifespan and a progressive mitochondrial cardiomyopathy with CIV deficiency and compensatory mitochondrial proliferation (Ruzzenente, 2012)(Table I). The selectivity of CIV deficiency found in both mutant mice and FCLS patients has been partially explained by the binding of LRPPRC to a specific segment of Cox1 mRNA (Xu, 2012) and by a reduction on the translation of mtDNA-encoded CIV genes due to the destabilization of a RNA-dependent complex involved in the regulation of mitochondrial mRNAs stability (Ruzzenente, 2012; Xu, 2012).
Recent studies on the heart specific Lrpprc KO mouse revealed that the bioenergetic dysfunction observed was due to a defect on the assembly of the ATP synthase (CV) rather than to the CIV deficiency initially described. The assembly defect in CV led to altered mitochondrial cristae morphology, increased ROS production and increased mitochondrial membrane potential (Mourier, 2014).
1.9. Mitochondrial aspartyl-tRNA synthetase
The mitochondrial aspartyl-tRNA synthetase catalyzes the aminoacylation of its cognate tRNA. The aspartyl-tRNA synthetase is encoded by the DARS2 gene and mutations on this gene are associated with leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation (LBSL) (Scheper, 2007). Patients with LBSL develop spasticity, progressive cerebellar ataxia, spinal cord dysfunction and cognitive deficit. Unexpectedly, fibroblasts and lymphoblast derived from LBSL patients did not uncover any OXPHOS defect (Scheper, 2007). It is possible that the mutant protein still retains residual activity and is able to sustain normal mitochondrial protein synthesis or perhaps this protein has other unknown functions or tissue specific effects. Ablation of Dars2 in mouse caused embryonic lethality at E8.5 whereas heterozygous mice did not showed any phenotype even until 2 years of age, indicating that one copy of the gene is sufficient to maintain normal mitochondrial protein synthesis (Dogan, 2014). Conditional ablation of floxed Dars2 in heart and skeletal muscle achieved by the creatine kinase Cre caused cardiomyopathy and premature death by 6–7 weeks of age (Table I). Both heart and muscle Dars2 KO tissues showed impaired mitochondrial protein synthesis leading to OXPHOS deficiency. Interestingly, different adaptive responses to the genetic defect were observed in heart and muscle tissues independently of the OXPHOS defect. In heart, the ablation of Dars2 increased mitochondrial biogenesis, UPRmt (mitochondrial unfolded protein response), downregulation of autophagy and upregulation of the cytokine Fgf21. In contrast, muscle tissue seemed to have a higher capacity for perturbed protein homeostasis and did not elicit the compensatory mechanisms observed in heart (Dogan, 2014). Although the human disorder caused by mutations in DARS2 does not have a muscular or cardiac component in the pathology and progression of the disease, the heart and muscle conditional Dars2 KO mouse is a good animal model to illustrate how different tissues react and adapt to the OXPHOS deficiency and to give insights into the pathological mechanisms of mitochondrial diseases.
1.10. Adenine nucleotide translocator isoform1 (Ant1)
The constant exchange between mitochondrial matrix generated ATP and cytosolic ADP, required for a correct energetic balance, is provided by the adenine nucleotide translocators (ANTs), a family of transmembrane proteins embedded in the inner mitochondrial membrane. Four different ANT isoforms are differentially expressed in human tissues. In particular, ANT1 is mainly expressed in high-energy demand tissues, such as heart, skeletal muscle and brain, and ANT3 is ubiquitously expressed (Li, 1989; Stepien, 1992). Mutations in ANT1 have been found in patients affected by severe mitochondrial diseases associated with mtDNA instability, such as adPEO and autosomal recessive mitochondrial myopathy and cardiomyopathy (Korhonen, 2004; Napoli, 2001; Palmieri, 2005). The essential role played by Ant1 in energy homeostasis has been demonstrated by its inactivation in a mouse model (Graham, 1997). Ant1−/− mice developed a myopathy and hypertrophic cardiomyopathy at 8 months. Interestingly there were no neurological defects in retina and brain in these animals (Lee, 2009; Phillips, 2010) (Table II). Skeletal muscle and heart from Ant1−/− mice showed typical traits of mitochondrial myopathy, characterized by mitochondrial proliferation, RRFs, mosaicism of COX negative fibers and aberrant mitochondria. Similarly to many patients with severe mitochondrial diseases, these mice manifested metabolic alterations, such as accumulation of lactate, Krebs cycle intermediates and alanine in serum (Graham, 1997). The muscle and heart mitochondria of the Ant1−/− mouse also exhibited increased ROS production associated to the upregulation of cellular antioxidant machinery, suggesting chronic oxidative stress (Esposito, 1999). However, the most striking feature of Ant1−/− heart and muscle was the presence of mtDNA large-scale rearrangements, in particular the accumulation of multiple deletions in the mtDNA. Thus, the dNTP pool imbalance caused by Ant1 dysfunction in combination with ROS-mediated DNA damage are likely to be responsible for the generation of deleted mtDNA species, which, in turn, could induce a severe decrease in the cellular respiratory capacity (Kaukonen, 2000; Spinazzola and Zeviani, 2005). Mutations in the Ant1 gene cause PEO in humans, where the typical hallmark is ptosis, due to dysfunction of the extra ocular muscles (EOMs). However in the Ant1−/− mouse model no ocular motor defects or specific EOMs fatigability were observed. Interestingly, the levels of the isoform Ant2 were increased in the EOMs of Ant1−/− mouse, suggesting that it may partially compensate for the loss of Ant1 and be responsible for the lack of phenotype in this particular muscle (Yin, 2005). It remains to be clarified whether the lack of Ant1 in the brain could be compensated by the overexpression of other isoforms and be the reason for the lack of neurological phenotype in the Ant1−/− model.
Table II.
Mouse models of proteins involved in the maintenance of mitochondrial dNTPs pool.
| Gene | Mitochondrial function | Genetic Manipulation | Phenotype | References |
|---|---|---|---|---|
| Ant1 | Adenine nucleotide translocase | Knockout: -Whole body (Homologous recombination) | Late-onset myopathy and hypertrophic cardiomyopathy | (Graham, 1997) |
| Tk2 | Synthesis of deoxypyrimidine nucleoside triphosphates | Knockout: -Whole body (Homologous recombination) Knock-in: Tk2378–379CG>AA (pathogenic form) |
Encephalopathy and premature death Encephalopathy and premature death |
(Zhou, 2008) (Akman, 2008) |
| Tp/Up | Metabolism of nucleotides | Knockout: -Whole body (Homologous recombination) | Leuko-encephalopahty | (Haraguchi, 2002) (López, 2009) |
| Rrm2b | Synthesis of dNTPs | Knockout: -Whole body (Homologous recombination) | Premature death, muscle and renal atrophy | (Kimura, 2003) |
| Mpv17 | Unknown function | Knockout: -Whole body (Homologous recombination) | Renal degeneration, hearing loss Mild hepatopathy and myopathy | (Clozel, 1999; Viscomi, 2009). |
1.11. Thymidine Kinase 2 (Tk2)
The correct maintenance of mtDNA depends not only on the normal activity of the enzymes composing the mitochondrial replisome, but also on a balanced pool of dNTPs. Depending on the proliferative status of the cell, dNTPs may be either imported into the mitochondria from the cytosol, where de novo synthesis occurs, or synthesized from intermediates in the degradative pathway for nucleotides in the mitochondrial matrix (the salvage pathway). The first step for the salvage pathway synthesis of deoxypyrimidine nucleoside triphosphates is provided by the constitutively expressed mitochondrial thymidine kinase (TK2) (Wang and Eriksson, 2000; Wang, 1999b). TK2 activity is more relevant in post-mitotic tissues, whereas de novo synthesis and Tk1-mediated cytosolic salvage pathways are crucial in proliferating cells (Coppock and Pardee, 1987; Ferraro, 2005; Reichard, 1988). Mutations in TK2 gene cause mitochondrial DNA depletion syndrome (MDS) and autosomic recessive Progressive External Ophthalmoplegia (arPEO) in humans (Götz, 2008; Mancuso, 2003; Oskoui, 2006; Saada, 2001; Tyynismaa, 2012; Wang, 2005). The clinical spectrum of TK2 mutations is broad, ranging from severe fatal infantile myopathy with motor regression, spinal muscular atrophy, rigid spine syndrome to mild mitochondrial myopathy (Oskoui, 2006).
Two different Tk2 deficient mice were developed (Akman, 2008; Zhou, 2008). One is a Tk2 KO mouse generated by deleting exon 4 and part of exon 5, encoding for the substrate binding domain of the enzyme active site (Zhou, 2008). The other is a knock-in mouse harboring the 378–379CG>AA mutation that induces the amino acid substitution H126N, corresponding to the human pathogenic mutation H121N (Table II) (Akman, 2008; Saada, 2001). Despite the genetic difference and the presence of some residual TK2 activity in the knock-in animals, these two Tk2 deficient mice shared traits of isolated encephalopathy without major skeletal muscle alterations. Heterozygous mice were comparable to their littermate controls, whereas homozygous mice displayed a growth delay starting from P7–P10 and died prematurely. The most relevant symptoms were ataxic gait, coarse tremors, impaired motor coordination, abnormal limb clasping and generalized weakness (Akman, 2008; Bartesaghi, 2010; Zhou, 2008). Progressive mtDNA depletion was detected in multiple tissues and organs although to different degrees (Akman, 2008; Zhou, 2008). In brain, the most affected tissue, the mtDNA depletion was associated with an unbalanced pool of dNTPs and severe mitochondrial bioenergetic defect, characterized by loss of mtDNA-encoded OXPHOS subunits, CI and CIV deficiency and ATP reduction. Tk2−/− mice were also characterized by a severe and progressive hypothermia, due to the nearly complete absence of hypodermal fat layer (Zhou, 2008). Brown adipose tissue (BAT) from both mice presented several abnormalities, including morphological alterations, accumulation of lipid vesicles, mtDNA depletion and moderate bioenergetic dysfunction, whereas white adipose tissue (WAT) was only modestly affected. Adipokines and genes involved in the regulation of thermogenesis, such as Ucp-1 and Pgc-1α, were down-regulated, suggesting an alteration in the endocrine properties of adipose tissues (Villarroya, 2011). In the liver of the Tk2−/− mice, mtDNA depletion induced mitochondrial proliferation, accumulation of fat vesicles and altered mitochondrial β-oxidation (Zhou, 2013). Increased levels of cholesterol, non-esterified fatty acids and long chain acylcarnitines were found in plasma of the Tk2−/− mice (Zhou, 2013). The main differences between patients and these animal models reside in the severe neurological symptoms and the lack of myopathic phenotype in mice. This can be partially explained by the fact that null mutations have never been found in patients, suggesting a possible incompatibility with human development (Bartesaghi, 2010). Moreover, since mouse brain mainly relies on Tk2 activity for dNTP synthesis; it would be more severely affected by mtDNA depletion than any other tissue. However, a recent study identified a mild myopathic phenotype in Tk2−/− mice skeletal muscles and heart, most likely attributable to an altered pool of myogenic progenitor cells (Paredes, 2013), indicating that the Tk2−/− mice could represent a valuable tool for the study of MDS.
1.12. Thymidine Phosphorylase (Tp)
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal recessive syndromic disease clinically defined by the occurrence of gastrointestinal dysmotility with intestinal pseudo-obstruction, peripheral neuropathy, ptosis, ophthalmoparesis and hearing loss (Lara, 2007; Nishino, 1999). Mutations in the TYMP or TP gene account for the vast majority of MNGIE cases and produce a severe defect in thymidine phosphorylase (TP) catalytic activity, causing a marked increase of thymidine and deoxyuridine in plasma and tissues of affected patients (Nishigaki, 2003; Nishino, 1999; Papadimitriou, 1998; Valentino, 2007). TP is a key enzyme of the nucleotide metabolism that in combination with uridine phosphorylase (UP) provides the reversible conversion of pyrimidine deoxyriboside to pyrimidine base. Thus, TP regulates intracellular and plasma thymidine levels. In humans, these two enzymes do not share their substrates whereas mouse UP cleaves both thymidine and uridine (el Kouni, 1993).
Double knockout of Tp−/−Up−/− mice were viable with similar growth rate and life span than the control animals although they showed hyperintense brain lesions in Magnetic Resonance Imaging and axonal swelling suggestive of a mitochondrial leukoencephalopathy (Table II) (Haraguchi, 2002). Ablation of Tp and Up genes produced increased levels of thymidine, uridine and deoxyuridine in plasma and in several tissues including brain, heart, kidney and skeletal muscle (López, 2009). The thymidine and uridine imbalance has been pointed responsible for the progressive mtDNA depletion and the mitochondrial defects found in the Tp−/−Up−/− brain, partially resembling the molecular hallmark of MNGIE patients (López, 2009). Interestingly, the fact that muscle, liver and kidney did not show mtDNA depletion, having similar thymidine and uridine imbalance levels than brain raised the question of why is this phenotype is brain-specific (López, 2009). It is possible that either the short life span of mice when compared to humans do not allow for the accumulation of significant amounts of somatic mtDNA alterations in tissues other than brain or that post mitotic neurons are more dependent on the mitochondrial pyrimidine salvage pathway than mitotically active tissues.
1.13. Ribonucleotide reductase subunit 2B (Rrm2b)
Ribonucleotide reductase is a heterotetrameric enzyme composed of two large (R1) and two small (R2) subunits and is essential for dNTPs synthesis during the S phase of the cell cycle, providing the de novo conversion of ribonucleoside 5′-diphosphates into deoxyribonucleoside 5′-diphosphates (Parker, 1994). Bourdon and collaborators identified several mutations in the RRM2B gene in seven children affected by trunk hypotonia, lactic acidosis and renal tubulopathy, caused by severe mtDNA depletion (Bourdon, 2007). A growing number of RRM2B mutations have been found as causative of different mitochondrial disorders such as other cases of mtDNA depletion syndrome and renal tubolopathy (Acham-Roschitz, 2009; Bornstein, 2008; Kollberg, 2009), PEO (Fratter, 2011; Takata, 2011; Tyynismaa, 2009) and MNGIE (Shaibani, 2009), highlighting the importance of this gene in mtDNA maintenance. Rrm2b−/− mice displayed growth retardation and muscle atrophy and prematurely died of severe renal failure by 14 weeks (Bourdon, 2007; Kimura, 2003). Despite the lack of an overt OXPHOS deficiency, severe mtDNA depletion was found in skeletal muscle, kidney and liver, most likely caused by an alteration of the dNTP pool (Table II) (Bourdon, 2007; Kimura, 2003). The phenotype of the Rrm2b KO mouse resembled the one found in patients.
1.14. Mpv17
MPV17 gene encodes for a mitochondrial inner membrane protein involved mtDNA maintenance although its precise function remains unknown (Bottani, 2014; Spinazzola, 2006; Trott and Morano, 2004). Mutations in human MPV17 gene cause a peculiar form of infantile hepatocerebral mtDNA depletion syndrome, characterized by severe hypoglycemic crises and progressive impairment of hepatic function that ultimately leads to liver cirrhosis (Spinazzola, 2006). A specific founder mutation in the MPV17 gene (R50Q) is the cause of the Navajo neurohepatopathy, an autosomal recessive syndrome prevalent in the Navajo population (Karadimas, 2006). Besides liver failure, the clinical presentation of this syndrome includes severe sensory and motor neuropathy, corneal ulcerations, leukoencephalopathy, and recurrent metabolic acidosis (Karadimas, 2006; Vu, 2001).
Knockout Mpv17−/− mouse developed a progressive kidney dysfunction leading to systemic hypertension at 2–3 months after birth (Table II) (Clozel, 1999; Weiher, 1990). Intriguingly, this renal phenotype apparently disappeared after few generations, and subsequent studies reported only a severe sensorineural hearing loss (Meyer zum Gottesberge and Felix, 2005; Meyer zum Gottesberge, 2012; Meyer zum Gottesberge, 1996). A reduction of mtDNA levels was found in multiple tissues, including skeletal muscle, kidney and brain. The most affected tissue was liver, in which the mtDNA depletion was associated with CI and CIV deficiency (Spinazzola, 2006). Interestingly, the Mpv17 KO mice did not show an overt liver dysfunction but only subclinical mitochondrial hepatopathy and myopathy. Elevated AST, ALT, CK enzyme levels and slight lactic acidosis in blood and the presence of some COX negative fibers in skeletal muscle and hepatocytes were observed in KO mice. The administration of a ketogenic diet led to severe liver failure and cirrhosis in the Mpv17−/− mice (Bottani, 2014; Viscomi, 2009). Similar to what was found in other mouse models, the severe mtDNA depletion induced by Mpv17 ablation triggered a functional compensation that involves an increase of mtDNA transcriptional events. This compensatory mechanism would account for decreased and unevenly distributed COX staining in the analyzed tissues (Viscomi, 2009 #228). Glomerular mtDNA depletion and increased mitochondrial ROS production were also detected in Mpv17−/− mice (Casalena, 2014; Viscomi, 2009).
2. Mouse models of proteins involved in mitochondrial dynamics
Mitochondria are dynamic organelles that are constantly undergoing fusion and fission events as part of their normal function to adapt to the cellular energetic demands and maintain the integrity of the mitochondrial network. In mammals, a series of proteins belonging to the dynamin related protein family are involved in mitochondrial fission and fusion (for review see (Hoppins, 2014)). Mitochondrial dynamics, when altered, can cause OXPHOS defects due to inability of the mitochondrial network to respond to the physiological requirements. Defects in mitochondrial dynamics are associated with neurodegenerative diseases. Mutations in MFN2 and OPA1 genes are responsible for type 2A Charcot-Marie-Tooth disease (CMT2A) and autosomal dominant optic atrophy (adOA) respectively (Alexander, 2000; Delettre, 2000; Züchner, 2004). Nonetheless, missense mutations in the GTPase domain of OPA1 have been associated with syndromic forms of adOA, characterized by the accumulation of mtDNA multiple deletions in skeletal muscle, unveiling a role of OPA1 in the correct maintenance of mtDNA (Amati-Bonneau, 2008; Hudson, 2008). It has been also reported a case of fatal infantile encephalopathy and lactic acidosis due to DRP1 c.1184C>A mutation that causes a defective mitochondrial and peroxisomal fission (Waterham, 2007). Mouse models of proteins involved in mitochondrial network dynamics are summarized in Table III.
Table III.
Mouse models of proteins involved in mitochondrial dynamics.
| Gene | Mitochondrial function | Genetic Manipulation | Phenotype | References |
|---|---|---|---|---|
| Mnf1 | Mitochondrial fusion | Knockout: -Whole body (Homologous recombination) | Embryonic lethal (E10.5–E11.5) | (Chen, 2003) |
| Conditional Knockout: -Skeltal muscle (Mlc-Cre) -Heart (Myh6-Cre) |
No overt phenotype No overt phenotype |
(Chen, 2010) (Chen, 2012b) |
||
| Mnf2 | Mitochondrial fusion | Knockout: -Whole body (Homologous recombination) | Embryonic lethal (E10.5–E11.5) | (Chen, 2003) |
| Conditional Knockout: -Whole body except placenta (Meox2-Cre) | Cerebellum atrophy Lethal at P1-P17 | (Chen, 2007) | ||
| -Developing cerebellar primordia (Wnt1-Cre and En1-Cre) | Cerebellum atrophy Lethal at P1–P17 | (Chen, 2007) | ||
| - Adult cerebellum Purkinje cells (L7-Cre) | Motor impairment, Purkinje cells degeneration | (Chen, 2007) | ||
| -Skeltal muscle (Mlc-Cre) | No overt phenotype | (Chen, 2010) | ||
| -Heart (α-Mhc-Cre and Myh6-Cre) | Mild cardiac hypertrophy | (Papanicolaou, 2011) (Chen, 2012b) | ||
| -Kidney (Pax-Cre) | Increased apoptotic nephrons | (Gall, 2012) | ||
| -liver and skeletal muscle (Alb-Cre, Mef2c-Cre) | Altered insulin signaling in liver and muscle | (Sebastian, 2012) | ||
| Expression of: -Mfn2T105M in motor neurons (associated with CMT2A) | Atrophy in the anterior muscles of the hind limbs | (Detmer, 2008) | ||
| Expression of: -Mfn2R94Q in neurons (associated with CMT2A) | Locomotor impairment and gait defects | (Cartoni, 2010) | ||
| Mnf1/Mnf2 | Mitochondrial fusion | Conditional Knockout: -Skeltal muscle (Mlc-Cre) | Muscle atrophy, premature dead (6–8 weeks) | (Chen, 2010) |
| Mnf1/Polg | Mitochondrial fusion/mtDNA replication | Knockout/knock-in -Mnf1−/−- PolgD257A (Whole body) | Embryonic lethal | (Chen, 2010) |
| Opa1 | Mitochondrial fusion |
Opa1Q285STOP (ENU-mutagenesis) -Homozygous -Heterozygous Opa1Q285STOP/ |
Embryonic lethal (E13.5) Visual loss Degeneration of the optic nerve, without loss of RGC |
(Davies, 2007) |
|
Opa1329–355del (Deletion ENU- mutagenesis) -Homozygous -Heterozygous |
Embryonic lethal (E8.5) Visual loss Degeneration of the optic nerve and loss of RGC. Tremor and abnormal clutching reflex |
(Alavi, 2007) (Alavi, 2009) |
||
| Opa1W616C+insLR (ENU-mutagenesis) -Homozygous | Embryonic lethal (E9.5–11.5) | (Moore, 2010) | ||
| Knock-in: Opa1delTTAG -Homozygous -Heterozygous |
Embryonic lethal (E10.5) Visual loss. Axonal and RGC degeneration. Deafness Cardiomyopathy |
(Sarzi, 2012) | ||
| Drp1 | Mitochondrial fission, mitophagy, apoptosis regulation | Whole body: exon 2 ablation | Embryonic lethal | (Ishihara, 2009) |
| Conditional KO: – Brain (Nestin-Cre) | Premature death, brain development defects, white matter hypoplasia | (Ishihara, 2009) | ||
| Tamoxifen-inducible conditional knock down: – Heart (αMHC-MerCreMer) | Premature death, left ventricular hypertrophy, cardiac dysfunction, increased sensitivity to ischemia/reperfusion, OXPHOS defects, block in mitophagy | (Ikeda, 2014) |
2.1. Mitofusins (Mfn1 and Mfn2)
Mitofusins are GTPases located in the outer mitochondrial membrane that are essential for mitochondrial fusion. Inactivation of both Mfn1 and Mfn2 in mouse resulted in embryonic lethality between E10.5–11.5, due to a severe developmental delay and placental defects (Chen, 2003; Chen, 2007). No overt phenotype was displayed when Mfn1 or Mfn2 were ablated specifically in skeletal muscle (Chen, 2010). However, double-KO mice were severely shrunken and died prematurely at 6–8 weeks of age. They displayed low blood glucose levels, reduced body temperature and lactic acidosis (Chen, 2010). The weight loss was mostly due to the presence of smaller skeletal muscles with subsarcolemmal accumulation of aberrant and fragmented mitochondria (Chen, 2010). Progressive mtDNA depletion, accumulation of point mutations and multiple deletions were found not only in young double KO mice, but also in aged Mfn1−/−/Mfn2+/− animals (Chen, 2010; Vermulst, 2008). Interestingly, the lack of Mfn1 is not compatible with the presence of an error-prone polymerase γ, since Mfn1−/−/PolgD257A/D257A double mutant mice died in utero (Chen, 2010). These data support the idea of a major involvement of mitochondrial fusion machinery in the maintenance of mtDNA integrity and stability.
Complete Mfn2 KO, produced by using Meox2-Cre that ubiquitously expresses Cre except in placenta, were born at expected Mendelian frequency, but 1/3 of them die at post-natal day P1 (P1). The Mfn2 KO survivors showed severe defects in movement and balance, suggesting a major role of Mfn2 in cerebellum development and function (Chen, 2007). The postnatal ablation of Mfn2 specifically in cerebellum produced a fast and progressive reduction of cerebellar volume due to a specific degeneration of Purkinje cells (PC). When Mfn2 was specifically disrupted in PCs after P7, mice displayed a pathological phenotype with an onset at 2 months of age characterized by shaky gait and poor rotarod performance due to progressive PC degeneration and axonal alterations (Chen, 2007).
The inactivation of Mfn2 but not of Mfn1 in cardiac muscle caused a hypertrophic cardiomyopathy with mitochondrial enlargement and increased left ventricle mass (Chen, 2012b; Papanicolaou, 2011). The lack of this phenotype in the heart specific Mfn1 KO indicates that MFN1 and MFN2 have different functions, as there was not a compensatory overexpression of the non-targeted protein in either of the KO mice. Studies in Ca2+ handling revealed that MFN2, but not MFN1, plays an important role in the interaction between the mitochondria and the endoplasmic reticulum beside its role in mitochondrial fusion (Chen, 2012b).
The specific Mfn2 disruption in kidney did not affect renal morphology, structure and tubular function, although a slight increase of apoptotic nephrons was observed (Gall, 2012). Primary cultures of Mfn2 deficient proximal tubule epithelial cells displayed mitochondrial fragmentation and were more prone to apoptosis after ATP deprivation (Gall, 2012).
Liver-specific ablation of Mfn2 led to glucose intolerance, enhanced hepatic gluconeogenesis and impaired response to insulin caused by an altered insulin-signaling pathway in both liver and muscle (Sebastian, 2012). OXPHOS deficiency, increased oxidative stress and endoplasmic reticulum stress were also identified in KO mice, indicating that Mfn2 plays a role in coordinating insulin signaling and mitochondria/endoplasmic reticulum crosstalk which appears to be essential for glucose homeostasis (Sebastian, 2012). Likewise, Mfn2 specific deletion in skeletal muscle also resulted in alterations on the glucose homeostasis of this tissue (Sebastian, 2012).
The mouse models described above represent useful tools for a deeper understanding of the role that Mitofusins play in cell and tissue physiology but disappointingly, the Mfn2 KO phenotype did not recapitulate the clinical features of CMT2A patients. To address this issue, two transgenic mice expressing mutant alleles of Mfn2, have been generated. The first one expressed the Mfn2T105M dominant mutant allele specifically in peripheral motor neurons (transgene expression driven by the HB9 promoter) (Detmer, 2008). Homozygous Mfn2T105M mice displayed a severe phenotype, mostly affecting hind limbs function. Interestingly, the forelimbs and the posterior musculature of the hind limbs were not affected, only the anterior musculature of the hind limbs became atrophic, causing muscle weakness similar to what was found in CMT2A patients (Detmer, 2008). As expected, sensory roots were normal, whereas motor neuron roots displayed a significant loss of axons (Detmer, 2008). The phenotype of the transgenic mouse presented incomplete penetrance, as is seen in CMT2A and other mitochondrial disorders.
The other transgenic Mfn2 mouse, called MitoCharc, was obtained by expressing a human mutant Mfn2R94Q selectively in neurons (Cartoni, 2010). Two mutant strains have been characterized; one was heterozygous and expressed the mutant Mfn2 mainly in dorsal root ganglia, whereas the other strain was homozygous and showed a preferential expression of the mutant allele in the spinal cord. Despite their different neurological phenotypes, these mice displayed gait defects and poor rotarod performance at 5 months of age. Homozygous animals presented a variable phenotype, with only some mice severely affected, suggesting a possible incomplete penetrance (Cartoni, 2010). Interestingly, no ataxic gait was found in these mice and their cerebellum development and layer organization were normal. Electron microscopy of motor neurons revealed an accumulation of enlarged mitochondria in small and medium myelinated axons. A combined CII plus CV deficiency accompanied by a severe decrease in ATP synthesis was detected in brain (Guillet, 2011). Overall, this age-dependent phenotype was compatible with a CMT2A late onset neuropathy (Cartoni, 2010).
2.2. Opa1
OPA1 is a GTPase required for mitochondrial inner membrane fusion. Mutations in Opa1 produce autosomal dominant optic atrophy (adOA) in humans which is characterized by a progressive loss of vision and blindness with an onset in childhood (Carelli, 2004). To model the adOA phenotype, two different animal models of Opa1 haploinsufficiency were generated. One mouse harbored the 1051C>T mutation which induced a Q285STOP in exon 8 (Opa1Q285STOP); the other mouse harbored the 1065+5G>A which produced a mutant form of Opa1 lacking exon 10 (Opa1329–355del) (Alavi, 2007; Davies, 2007). These mice were generated after screening an N-ethyl-N-nitrosourea (ENU)-mutagenized murine DNA library for mutants harboring Opa1 mutations and displayed some peculiar symptoms of human DOA, such visual loss associated to optic nerve fibers demyelinization (for a review see (Williams, 2011)). In homozygosis, expression of both mutations in mice resulted in embryonic lethality with a block in development between E8.5 and E13.5. Heterozygous mice for both mutant forms showed a reduction of about 50% of the levels of OPA1 in several tissues in agreement with gene dosage. Both transgenic mice developed visual loss and other neurological symptoms that progressively worsened at about 12 to 13 months of age (Alavi, 2007; Davies, 2007; Yu-Wai-Man, 2009). The major differences between these two Opa1 mutant mice were the severe loss of retinal ganglion cells (RGC) and thinning of the retinal nerve fiber layer that occurred in the Opa1329–355del but not in the Opa1Q285STOP model in which retinal architecture and morphology were normal (Alavi, 2007; Davies, 2007). In addition, the Opa1329–355del showed gliosis, reduced number of large axons in the optic nerve, swollen or disorganized axons and the loss of myelin sheets, associated with activation of astroglia and microglia (Alavi, 2007; Nguyen, 2011). Conversely, the Opa1Q285STOP mouse showed age-dependent axon demyelination, dendritic abnormalities and decreased glutamatergic synaptic density, suggesting that a functional impairment rather than a degeneration of RGCs was responsible for the vision loss (Williams, 2012).
In terms of extra-ocular phenotype, Opa1Q285STOP mice presented subtle neuromuscular abnormalities, whereas Opa1329–355del mice showed a more severe syndrome with abnormal clutching reflex, resting tremor of the upper limbs and decreased rotarod performance (Alavi, 2009). However, skeletal muscle histology was normal and no mtDNA alterations (multiple deletions, depletion, proliferation) were found (Alavi, 2007; Davies, 2007). Abnormalities in the cardiac function were also described in the Opa1329–355del mice (Piquereau, 2012) and in heterozygous Opa1+/− mice (Chen, 2012a).
Another Opa1 deficient mouse harboring an A>G transition in intron 19 that creates a cryptic splice acceptor site was described (Moore, 2010). This 6bp insertion is predicted to produce a mutant form of Opa1 with the W616C amino acid substitution as well as the addition of amino acids leucine and arginine due to the translation of the intronic sequence (Opa1W616C+insLR) (Moore, 2010). Homozygote animals died between E9.5 and E11.5. MEFs derived from homozygous embryos showed a fragmented mitochondrial network probably due to the cytosolic mislocalization of mutated OPA1 (Moore, 2010). Unfortunately, the phenotype of adult heterozygous animal carrying this mutation has not been reported.
Opa1 transgenic mouse carrying the recurrent Opa1delTTAG micro-deletion, commonly found in adOA patients, was created. A multi-systemic disorder was displayed in heterozygous mice whereas homozygotes died in utero at E10.5 (Sarzi, 2012). In particular, these mice experienced visual failure, as documented by altered visual evoked potentials and loss of RGCs. The retinal phenotype was accompanied by deafness, encephalomyopathy, peripheral neuropathy, ataxia and cardiomyopathy. Additionally, premature axonal degeneration and demyelination were observed. These phenotypes were likely due to the OXPHOS defects, in particular CIV deficiency and supercomplex instability found in multiple tissues that may lead to increased levels of autophagy and mitophagy (Sarzi, 2012).
2.3. Drp1
Mitochondrial fission is regulated by a set of nuclear encoded proteins and it has been involved in the modulation of apoptosis, mitochondria quality control and embryogenesis (reviewed in (Sesaki, 2014). Mitochondrial fission is mainly controlled by Dynamin-related protein 1 (Drp1), a cytosolic soluble protein which is recruited on the mitochondrial outer membrane through the interaction with the mitochondrial fission factor (Mff) and Fis1. Interestingly, a peculiar case of a newborn girl with a dominant-negative allele of DRP1 has been described (Waterham, 2007). This patient died at 37 days of age and showed a complex neurological syndrome characterized by microcephaly, abnormal brain development, optic atrophy and severe lactic acidosis.
The complete loss of Drp1 in mice is embryonic lethal between E10.5 and E12.5, revealing the essential role of this protein during development (Ishihara, 2009). The mutant embryos were significantly smaller, showed defects in heart, liver and brain organogenesis and presented enlarged mitochondria, which were positive for COX staining. Because apoptotic cells were detected in the neuroepithelium of Drp1−/− embryos, a conditional KO for brain was created to investigate the role of Drp1 in neurons (Ishihara, 2009). Conditional KO mice were born at expected Mendelian frequency but died within a day. Analysis of the KO mice showed white matter hypoplasia in forebrain, brainstem and cerebellum. Moreover, apoptotic events and uneven mitochondria distribution in mature neurons indicated that Drp1-dependent mitochondrial fission is essential for brain development, in particular in the formation of a proper neuronal network and synapsis function (Ishihara, 2009). In contrast, heterozygous Drp1+/− mice were undistinguishable from controls in terms of lifespan, fertility, viability and brain development (Manczak, 2012).
The specific ablation of Drp1 in heart resulted in embryonic lethality prompting the development of an inducible cardiac-specific conditional Drp1 KO mice, in which Drp1 expression was modulated by tamoxifen (Ikeda, 2014). After the tamoxifen treatment, Drp1 resulted downregulated and mice developed a progressive left ventricle hypertrophy with myocardial fibrosis and cardiac dysfunction, which led to premature death (13 weeks after tamoxifen injection). In Drp1 defective heart, a block in mitophagy induced an increase in mitochondrial mass and mtDNA copy number, which was associated with the appearance of multiple OXPHOS defects and increased ROS production. Such mitochondrial and autophagy defects promoted apoptosis and necrosis in cardiomyocyte leading to the cardiac pathological phenotype. Interestingly, heterozygous KO mice did not show any overt phenotype under basal conditions but when fasting, the left ventricle function decreased more severely than in control mice. Heterozygous KO mice were more sensitive to ischemia/reperfusion injury, indicating that Drp1 and mitochondrial function are critical for cardiac response to stress (Ikeda, 2014).
3. Mouse models of proteins involved in mitochondrial protein quality control
Mitochondria count with a surveillance system to remove damage proteins, which is composed by several proteases that are located in different mitochondrial compartments. In the matrix, damaged/misfolded proteins are degraded by the ClpXP complex or by the Lon protease and proteins containing oxidized methionine can be repaired by the MsrA/MsrB system. In the mitochondrial inner membrane, the ATP-dependent metalloproteases belonging to the AAA+ (ATPase associated with various cellular activities) superfamily, m-AAA and i-AAA regulate the degradation of most of the OXPHOS subunits and the maturation of a ribosomal protein and the PARL protease regulates the degradation of OPA1. In the intermembrane space, the HtrA2 protease also regulates the degradation of OPA1. In the mitochondrial outer membrane, damaged proteins are degraded by the ubiquitin/26S -proteasome system. [reviewed in (Fischer, 2012)]. Although defects in the mitochondrial surveillance and quality control system are not considered bona fide mitochondrial diseases, they are associated with numeorus neurological disorders that are known to have impaired mitochondrial function [reviewed in (Martinelli and Rugarli, 2010; Rugarli and Langer, 2012)]. Therefore, we had included mice models with defects in the quality control machinary in this review and are summarized in Table IV.
Table IV.
Mouse models of proteins involved in mitochondrial quality control.
| Gene | Mitochondrial function | Genetic Manipulation | Phenotype | References |
|---|---|---|---|---|
| Clpp | Mitochondrial matrix protease | Knockout: -Whole body (Homologous recombination) | Growth delay, weight loss, decreased spontaneous activity, hearing deficit and decreased pre- and postnatal survival. Infertility | (Gispert, 2013) |
| Agf3l2 | m-AAA+ protease, assembly of CI, CIII and CIV, mitochondrial protein synthesis, mitochondrial morphology and network maintenance | Paralyze Spontaneous mutation causing amino acid change R389G | Severe progressive paralysis with premature death by P15. Impaired axonal development and delayed myelination | (Maltecca, 2008) |
| provirus Emv66 insertion | ||||
| Conditional KO: -Purkinje cells (L7-Cre) | Gait disturbances, PC degeneration, mitochondrial fragmentation, impaired mitochondrial protein synthesis. OXPHOS defect (CI, CIII and CIV) | (Almajan, 2012) | ||
| Conditional KO: -Forebrain (CaMKIIa-Cre) | Neurodegeneration, tau hyperphosphorylation | (Kondadi, 2014) | ||
| Spg7 | Mitochondrial inner membrane metalloprotease | Knockout: -Whole body (Homologous recombination) -Whole body (Homologous recombination) and Afg3l2+/Emv66 |
Axonal degeneration at 3 months and impaired rotarod performance at 4 months | (Ferreirinha, 2004). |
| Axonal degeneration, cerebellar and hippocampal degeneration accompanied by gliosis. Premature death by 13–20 weeks of age. | (Martinelli, 2009). | |||
| Oma1 | Mitochondrial inner membrane protease involved in Opa1 processing | Knockout: -Whole body (Homologous recombination) | Viable. Diet-induced obesity and impaired thermogenesis. Increased tubular mitochondria in liver and brown adipose tissue. | (Quiros, 2012) |
| Phb1 | Mitochondrial Chaperone, mtDNA maintenance, Opa1 processing | Knockout: -Whole body (gene-trap): exon 7 ablation | Embryonic lethal | (He, 2008) |
| Conditional KO: -Liver (Albumin-Cre) | Liver injury, fibrosis, and hepatocellular carcinoma | (Ko, 2010) | ||
| Phb2 | Mitochondrial Chaperone, mtDNA maintenance, Opa1 processing | Knockout: -Whole body (gene-replacement) | Embryonic lethal | (Merkwirth, 2008) |
| Conditional KO: -Forebrain (CaMKIIa-Cre) | Progressive behavioral and cognitive impairment, neurodegeneration, mitochondrial deficiency | (Merkwirth, 2008) | ||
| Conditional KO: -Pancreas β-cells (Ins2-Cre) | Diabetes, altered insulin secretion, glucose homeostasis, mitochondrial deficiency. | (Supale, 2013) | ||
| HtrA2/Omi | Mitochondrial intermembrane space Serine protease | Homozygous mnd2 Spontaneous missense mutation Ser276Cys | Parkinson like phenotype: degeneration of striatal neurons, microglia activation and muscle wasting. Premature death by P30–P40 | (Jones, 2003) |
| Knockout: -Whole body (Homologous recombination) Knockout HtrA2 mice expressing Human HTRA2 in CNS (enolase-promoter) |
Parkinson like phenotype: Neurodegeneration with loss of neurons in the striatum. Spleen and thymus involution. Premature death by P30. Rescue of the neurodeneration and premature death. Accelerated aging phenotypes, such as heart enlargement, and death by 12–17 months of age. Elevated levels of clonally expanded mtDNA deletions in tissues. |
(Martins, 2004) (Kang, 2013) |
||
| Parl | Mitochondrial inner membrane protease involved in Opa1 processing | Knockout: -Whole body (PGK-Cre) | Severe weight loss, cachexia, atrophy of thymus and spleen. Premature death by 8–12 weeks of age. Neuronal death in thalamus and striatum. | (Cipolat, 2006). |
3.1. Clpp protease
CLPP or caseinolytic peptidase P is a serine proteases found in the mitochondrial matrix. CLPP associates with CLPX, a chaperone with ATPase activity, to form an active ClpXP proteolytic complex. This complex has a barrel shape proteolytic chamber, formed by hexameric rings of CLPX binding to two heptameric rings of CLPP, where unfolded substrates are degraded to small peptides which are then further processed by other exopeptidases (Kim, 2001). Mutations in CLPP have been associated with Perrault syndrome’s ovarian failure and sensorineural deafness (Jenkinson, 2013).
Disruption of the Clpp gene in mouse using the genetrap methodology caused growth delay, weight loss, decreased spontaneous activity, hearing deficit and decreased pre- and postnatal survival (Gispert, 2013). However, no overt mitochondrial dysfunction, oxidative stress or alterations in mitochondrial dynamics were observed in most tissues. Instead, increased levels of mtDNA were observed in tissues analyzed perhaps accounting for the lack of defective mitochondrial bioenergetics. Interestingly, the gonads were vulnerable to the ablation of Clpp and both male and female KO mice were infertile. In testis, spermatids and spermatozoa were missing in the seminiferous tubes. In ovaries, the folicullar layer was significantly reduced by increased cell loss (Gispert, 2013). The phenotype of the Clpp−/− mouse mimics the human hallmarks of ovarian failure and deafness in Perrault syndrome.
3.2. m-AAA protease
The m-AAA protease is oriented to the matrix side of the mitochondrial inner membrane and is involved in the degradation of damaged membrane proteins or non-assembled subunits of the respiratory chain. In mammals, the m-AAA protease is composed of paraplegin and AFG3L2, which can associate to form different isozymes: either a homo-oligomeric complex composed by AFG3L2 alone or a hetero-oligomeric complex containing paraplegin and AFG3L2.
3.2.1. Afg3l2
Mutations in AFG3L2 have been associated with spinocerebelar ataxia type 28 (SCA28) and with spastic ataxia (SPAX5). SCA28 is a dominant form of spinocerebral ataxias whereas SPAX5 is a recessive early onset and rapid neurodegenerative form of spastic ataxia (Di Bella, 2010; Pierson, 2011). The first two mouse models with defects in the Afg3l2 gene where characterized by Maltecca and collaborators (Maltecca, 2008). The first mouse model was the Afg3l2par/par, in which the authors discovered that the spontaneous mutation paralysé (par) that occurred in the C57BL/6 colony at the Pasteur Institute (Duchen, 1983) corresponded to a missense mutation in the Afg3l2 gene. This missense mutation led to the amino acid change R389G in the highly conserved AAA+ domain but did not affected the levels of expression of the protease (Maltecca, 2008). The second mouse model was the Afg3l2Emv66/Emv66, that was created by the Emv66 provirus insertion within intron 14 of the Afg3l2 gene (Maltecca, 2008). The viral insertion created a 4 base pair duplication that resulted in creation of a stop codon and the formation of a truncated protein. Neither the truncated nor the full length protease were detected in the Afg3l2Emv66/Emv66 mouse. Interestingly, both Afg3l2par/par and Afg3l2Emv66/Emv66 displayed the same phenotype characterized by a severe progressive paralysis starting at P7 with the hind limbs and by P12–P14 all limbs were completely immobile. Premature death occurred by P15. Both mouse models had impaired axonal development and delayed myelination, however, no neuronal loss was observed. Analysis of liver and muscle did not showed major abnormalities except for decreased fiber size, which was attributed to the axonal impairment. Decreased ATP levels and defects in OXPHOS were observed due to decreased assembly of complexes I and III in brain tissue of both Afg3l2par/par and Afg3l2Emv66/Emv66 mice (Maltecca, 2008). Studies on mitochondrial protein synthesis rate in organello, using isolated brain mitochondria, revealed that in the Afg3l2Emv66/Emv66 KO mouse the mitochondrial translation was severely impaired owing to defects in mitoribosome assembly (Almajan, 2012). Whether these protease is involved in mitoribosome assembly or stability remains to be determined.
Further analysis of cortical neurons in the Afg3l2Emv66/Emv66 revealed the presence of hyperphosphorylated tau, which is associated with microtubule disorganization leading to the impaired axonal development found in this KO mouse (Kondadi, 2014). Heterozygous Afg3l2+/Emv66 displayed a slow progressive decline in balance and motor coordination due to the degeneration of Purkinje cells containing dendritic hypertrophy and loss of cerebellar granular cells. Increased gliosis and oxidative stress were observed along with decreased ATP levels and defects in mitochondrial morphology and function. The mouse phenotype recapitulates pathology observed in SCA28 patients (Maltecca, 2009). The specific ablation of the floxed Afg3l2 in PC cells using the L7-Cre transgenic mouse resulted in gait disturbances noticeable at 8 weeks of age. Analysis of 4 week old KO mice showed neuronal normal appearance, however there were signs of inflammation indicated by reactive microglia and astrocytes. As the Afg3l2+/Emv66 KO mice aged, severe neurodegeneration of PC cells was observed and by 12 weeks of age all these neurons had disappeared. Mitochondrial morphology was altered and mitochondrial network appeared fragmented and about 10% of PC displayed severe COX deficiency (Maltecca, 2009).
Conditional deletion of Afg3l2 in neurons under the CaMKII promoter led to neuronal loss by 8 weeks of age in both cortex and hippocampus. No evidence of oxidative stress (protein carbonyls or increased SOD2 levels) was found in the 8 week neuron specific Afg3l2 KO, however tau hyperphosphorylation was observed indicating increased ROS production that activates stress kinases PKA and ERK leading to tau phosphorylation (Kondadi, 2014). Unfortunately the full description of the phenotype of this neuron-specific Afg3l2 KO mouse was not mentioned in the manuscript.
3.2.2. Paraplegin
Mutations in the SPG7 gene, which encodes for paraplegin, have been associated with autosomal recessive form of hereditary spastic paraplegia (HSP) characterized by the retrograde degeneration of cortical motor neuron axons leading to a progressive weakness of the lower limbs (Harding, 1993). Paraplegin is located in the mitochondrial inner membrane, however, an alternative splicing form of SPG7 creates paraplegin-2 which is located in the endoplasmic reticulum (Mancuso, 2012).
A mitochondrial specific paraplegin Spg7−/− mouse was created by the targeted deletion of exons 1 and 2 of Spg7 by homologous recombination leading to the absence of the mRNA and paraplegin protein (Ferreirinha, 2004) The KO mouse appeared normal, however showed a progressive decrease in rotarod perfomance starting at 4 months of age. A late onset axonal swelling was observed at 1 year or age and axonal degeneration was evident 3 months later. The severity of the neuropatology in the Spg7−/− mouse was found from distal to proximal along the axons (retrograde degeneration). Mitochondrial morphological abnormalities were also observed along with the accumulation of neurofilaments and orgenelles in the axonal swollen regions suggesting an impairement of the retrograde axonal transport (Ferreirinha, 2004). This mouse model recapitulates nicely the axonal degeneration found in HSP patients.
Interestingly, the axonal degeneration of the Spg7−/− mouse was acelerated in the presence of the Afg3l2/Emv66 allele (Martinelli, 2009). The Spg7−/−Afg3l2+/Emv66 mouse were less active and had imparied gait coordination particularly in the hind limbs at about 6 weeks of age. The phenotype worsened rapidly and by 9 weeks of age the Spg7−/−Afg3l2+/Emv66 mice had ataxic movements, kyphosis and severe weight loss eventually leading to premature death by 13−20 weeks of age. In addition to the axonal degeneration, there was also a cerebellar and hippocampal degeneration accompanied by gliosis. Mitochondrial abnormalities were also observed with OXPHOs defects and decreased mtDNA levels in the tissues affected (Martinelli, 2009).
3.3. Oma1
OMA1 is a zinc metalloprotease located in the inner mitochondrial membrane that was first identified in yeast as a protease with overlapping activities with the m-AAA proteases (Kaser, 2003) OMA1 cleaves OPA1 at S1 site leading to the loss of the long isoforms of OPA1 (L-OPA1) and resulting in the inactivation of OPA1 and cessation of inner membrane fusion (Ehses, 2009; Head, 2009).
Mice deficient in OMA1 (Oma1−/−) developed normally, were fertile, life span was normal and no neuropathological abnormalities were observed. The KO miced did not have severe abnormalities except for increased body weight suggesting defects in metabolic homeostasis (Quiros, 2012). When challenged with a high fat diet, Oma1−/− animals showed diet-induced obesity. Significant alterations in lipid metabolism such as increased levels of triglycerides and hepatic steatosis was observed in the KO mice. Moreover, Oma1−/− mice exhibited impaired thermogenesis after cold stress. In contrast to control mice, Oma1−/− mice were unable to deplete lipid droplets from brown fat adipocytes after cold stress (Quiros, 2012). Analysis of OPA1 processing revealed that it was clearly impaired in the Oma1−/− mice and this defect induced mitochondrial morphology alterations in MEFs, liver and brown adipose tissue upon stress. These results indicate that although OMA1 mediates the in vivo cleavage of OPA1, it is not essential for embryonic developmental and suggest an important and unexpected role of OMA1 in stress-induced OPA1 processing that is required to maintain metabolic homeostasis. Interestingly, OXPHOS function was not altered in Oma1−/− MEFs and tissues (Quiros, 2012). Because mitochondrial fragmentation has been associated with acute kidney injury, Oma1−/− mice has been used in an experimental model of acute kidney injury in order to assess whether OMA1-dependent OPA1 proteolysis plays a pathological role in vivo (Xiao, 2014). When mice were subjected to renal ischemia-reperfusion, several features observed in control animals after the injury, such as mitochondrial fragmentation, tubular damage and apoptosis were attenuated in the Oma1−/− mice. The OPA1 long isoform (L2) disappeared during renal ischemia-reperfusion in controls mice but was preserved in Oma1−/− mice. These results indicate that OMA1-mediated OPA1 proteolysis plays an important role in the disruption of mitochondrial dynamic in ischemic acute kidney injury (Xiao, 2014)
In addition to OMA1, the ATP dependent protease YME1L cleaves OPA1 at position S2, to balance mitochondrial fusion and fission (Griparic, 2007). Yme1L deficient MEFs showed fragmented mitochondria whereas double knockout for Oma1 and Yme1L are able to maintain tubular mitochondria in the absence of the short OPA1 isoforms (Anand, 2014). These results indicate that OPA1 processing is dispensable for mitochondrial fusion and suggest a stimulatory role of small-OPA1 isoforms for mitochondrial fission. Unfortunatedly, no information of the phenotype of the Yme1L or the double KO mice was provided in the manuscript.
3.4. Prohibitins
Mitochondrial quality control involves also the prohibitin (PHB) complex, a ubiquitous protein complex located in the inner mitochondrial membrane, which is comprised by the two PHB family members, namely PHB1 and PHB2. Mammalian PHB1 has been identified as a potential tumor suppressor (McClung, 1989), although it has been subsequently found that such anti-proliferative activity was mediated by the 3′-UTR and not by the protein itself. PHB1 and PHB2 heterodimers form a ring-like structure within the mitochondrial inner membrane, which has been shown to regulate membrane protein degradation by the mitochondrial m-AAA protease (Steglich, 1999), to function as a chaperone (Nijtmans, 2000), and it is also involved in mtDNA maintenance (Kasashima, 2008) and mitochondrial dynamics (Merkwirth, 2008). Moreover, it has been recently demonstrated that PHB2 forms a complex with DNAJC19, which regulates mitochondrial cristae morphogenesis and cardiolipin acylation, similar to what occur in Tafazzin deficient models (Richter-Dennerlein, 2014). Interestingly, DNAJC19 and Tafazzin have been found mutated in two previously unlinked mitochondrial disorders, namely dilated cardiomyopathy with ataxia and Barth syndrome. Beside their role in controlling mitochondrial functions, prohibitins have been also linked to cell cycle progression, transcriptional regulation, apoptosis and cell signaling (Merkwirth and Langer, 2009).
Mouse models of PHB have been developed in the past years. Loss of prohibitin genes in C. elegans and mice results embryonic lethal, highlighting their essential functions during early phases of development (Artal-Sanz, 2003; He, 2008; Merkwirth, 2008).
The specific ablation of Phb1 in liver induced a severe liver injury with signs of necrosis and inflammation starting at 3 weeks of age (Ko, 2010). Pre-neoplastic markers resulted highly expressed at 3 weeks and hepatic nodules were evident from 14 weeks and worsened to multifocal hepatocellular carcinoma, indicating that Phb exert a tumor suppressor function in hepatocytes. Ultra structural analysis of liver of the Phb1 KO showed swollen mitochondria with aberrant morphology and lack of cristae organization (Ko, 2010).
Neuronal specific conditional KO of Phb2 mice were born at expected Mendelian frequency and were indistinguishable from littermate controls until week 12 of age, when they started to show weight loss, cachexia and kyphosis (Merkwirth, 2012). Moreover, the Phb2 KO mice displayed a progressive behavioral and cognitive impairment, due to a severe progressive neurodegeneration leading to premature death by 22 weeks of age. The neuron specific Phb2 KO mice also displayed a late onset mitochondrial deficiency, characterized by altered mitochondrial morphology and Opa1 processing, mtDNA depletion and reduced oxygen consumption. Moreover, loss of Phb2 induced a massive activation MAP kinases leading to tau hyperphosphorylation and accumulation of aberrant filamentous structures in neurons (Merkwirth, 2012).
Prohibitins have been shown to be also essential for pancreatic function and survival, as conditional ablation of Phb2 in β-cells induced alteration of insulin secretion and glucose homeostasis, loss of β-cells and severe diabetes (Supale, 2013). Similar to the other Phb defective models, mitochondrial fragmentation, aberrant OPA1 processing, mtDNA depletion and altered OXPHOS complexes activity were also evident in the β-cells specific KO mice (Supale, 2013).
To date, PHBs have never been found mutated in human pathologies, but these studies on mouse models clearly indicate their crucial function in cell proliferation, cell survival and energy metabolism, pointing these genes as possible candidates for diverse human diseases.
3.5. HtrA2/Omi
HTRA2, also known as OMI, is an ATP-independent serine protease that is located in the intermembrane space of the mitochondrion and is involved in the clearance of unfolded proteins. Mice with a defective enzymatic activity of the HtrA2, due to a missense mutation (mnd2 mice) or to the targeted deletion (HtrA2 KO mice) of the gene encoding for the HtrA2 protease, exhibited Parkinson’s disease-like phenotype characterized by muscle wasting, neurodegeneration, involution of spleen and thymus and premature death by P30–P40 (Jones, 2003; Martins, 2004). The mnd2 mice were rescued by the transgenic expressing the human gene, HTRA2, specifically in the brain under the control of the neuron-specific enolase promoter (Kang, 2013) (Kang, 2007). The rescued mice did not show early lethality and no sign of neurodegeneration or any involvement of the spleen and thymus were found at 2 months of age. Interestingly, the rescued mice displayed peculiar signs of premature aging as osteoporosis, lordokyphosis, weight loss, hair loss, muscle atrophy with and onset at about 21 weeks and death occurring at around 67 weeks. As the rescued mnd2 mice got older (60 weeks), it showed additional signs of aging as cardiomyopathy, splenomegaly, thymus atrophy and increased extramedullary hematopoiesis. Moreover, heart, skeletal muscle and duodenum contained several COX negative fibers and large number of mtDNA deletions (Kang, 2013). The accumulation of mtDNA deletions is considered to be a major contributor in premature aging (Kujoth, 2005). Autophagy was also increased in all tissues with the exception of the brain in 5 week old rescued mice, probably as a compensatory quality control mechanism. The reason why different tissues lead to such different phenotypes is not clear but shows a differential susceptibility to impaired mitochondrial quality control. Brain was the most vulnerable tissue to the absence of HrtA2 and the accumulation of damaged/unfolded proteins.
3.6. PARL
PARL (presenilin-ssociated rhomboid-like) is a rhomboid protease located in the mitochondrial inner membrane and appears to regulate the processing of OPA1. A Parl−/− mouse was created by the ubiquitous deletion of the floxed gene using the phosphoglycerate kinase promoter, PGK-Cre (Cipolat, 2006). The Parl KO mouse developed severe weight loss, cachexia, atrophy of thymus and spleen that resulted in premature death by 8–12 weeks of age. No abnormalities were found in the spinal motoneurons to explain the muscular degeneration however neuronal cell loss was observed int thalamus and striatum. Interestingly, ATP levels and OXPHOS function were not altered in muscle but increased apoptosis was observed in spleen and thymus (Cipolat, 2006).
4. Other models of OXPHOS deficiencies
4.1. Aif
The apoptosis-inducing factor (AIF) is a FAD-dependent mitochondrial protein that is not a subunit of complex I but it has been indirectly linked to its maintenance/assembly. Aif is drastically reduced in the Harlequin mice due to a mutation caused by a retroviral insertion (Klein, 2002) (Table V). The Harlequin mice displayed a dramatic reduction in CI activity and levels in cerebellum and retina (the most affected tissues), underscoring a possible involvement of this loss in the neurodegeneration observed in these mice (Vahsen, 2004). More direct evidence of the pathogenic role of AIF and its relationship with CI failure came by conditionally deleting the gene in the skeletal muscle and heart (Joza, 2005). The ablation of Aif led to progressive skeletal muscle atrophy and dilated cardiomyopathy around 9 weeks of age. CI activity was reduced about 80% although the biochemical defect was of late onset (5 months of age). Lactate levels were also increased in the heart, consistent with mitochondrial defects causing cardiomyopathies (Echaniz-Laguna, 2012; Wortmann, 2012). The Aif KO tissues displayed increased oxidative stress as confirmed by the increased amounts of lipid peroxidation markers.
Table V.
Other mouse models of OXPHOS deficiency.
| Gene | Mitochondrial function | Genetic Manipulation | Phenotype | References |
|---|---|---|---|---|
| Aif | Unknown function | Knockout: -Whole body (Retroviral insertion) | Neuronal degeneration, Reduction in CI activity | (Klein, 2002; Vahsen, 2004) |
| Conditional Knockout: - Skeletal muscle and heart (Mck-Cre) |
Skeletal muscle atrophy and dilated cardiomyopathy, oxidative stress, reduction in CI activity | (Joza, 2005) | ||
| - Telencephalon (Foxg1-Cre) | Embrionic lethal (E14) | (Cheung, 2006) | ||
| -Muscle (Mck-Cre) | Combined OXPHOS deficiency, insulin resistance | (Pospisilik, 2007) | ||
| -Liver (Alb-Cre) | Combined OXPHOS deficiency, insulin resistance | (Pospisilik, 2007) | ||
| PstI | Mitochondria-targeted restriction endonuclease | Overexpression of mito-PstI -Skeletal muscle (Actin-mito-PstI) |
mtDNA depletion and deletions, COX deficiency and RRG | (Srivastava and Moraes, 2005) |
| Overexpression of Pst1 -Forebrain neurons (Inducible CaMKIIa-tTA) |
mtDNA depletion and deletions, limb ‘clasping’ neurological phenotype | (Fukui and Moraes, 2009) | ||
| Overexpression of Pst1 -Forebrain neurons (Inducible CaMKIIa-tTA) |
mtDNA depletion and OXPHOS deficiency | (Pickrell, 2011a) | ||
| Overexpression of Pst1 -Dopaminergic neurons (DAT-tTA) |
Neurodegeneration of the dopaminergic neurons, and striatal DA depletion. | (Pickrell, 2011b) | ||
| Overexpression of Pst1 -Whole body (Rosa26-rtTA) |
Muscle wasting, decreased locomotor activity and mtDNA depletion | (Wang, 2013) | ||
| Fxn | Fe/S cluster biosynthesis | Knockout: -Whole body (Homologous recombination) | Embryonic lethal (E6.5) | (Cossee, 2000) |
| Conditional Knockout: - Skeletal muscle and heart (Mck-Cre) |
Cardiac hypertrophy Iron accumulation | (Puccio, 2001) | ||
| - Neuron specific (Nse-Cre) | Progressive ataxia, cardiac hypertrophy | (Puccio, 2001) | ||
| -Pancreatic β-cells (Ins2-Cre) | Diabetes and loss of β-cells mass | (Ristow, 2003) | ||
| - DRG, spinal cord and cerebellum (Prp-Cre) | Cerebellar and sensory ataxia, loss of propioception | (Simon, 2004) | ||
| -Liver (Alb-Cre) | Reduced survival, liver regeneration | (Martelli, 2012) | ||
| Knock-in/Knockout: -Fxn−/(GAA)230 (KIKO) | No overt phenotype, mild heart fibrosis | (Miranda, 2002) | ||
| Overexpression in knockout Fxn mice of: -Human Fxn carrying GAA repeats (YG8R and YG22R) | Mild progressive coordination phenotype, iron deposits. Vacuoles in large sensory neurons. | (Al-Mahdawi, 2004; Al-Mahdawi, 2006) | ||
| Ethe1 | Mitochondrial sulfur dioxygenase (SDO) | Knockout: -Whole body (Homologous recombination) | EE syndrome Growth arrest at postnatal day 15. Lethal at 5–6 weeks after birth. Reduced motor activity. | (Tiranti, 2009) |
| Conditional Knockout: - Liver (Alb-Cre) |
No overt phenotype | (Di Meo, 2011) | ||
| - Muscle (MyoD-Cre) | No overt phenotype Isolated COX deficiency in muscle | |||
| -Brain (Nestin-Cre) | No overt phenotype Isolated COX deficiency in brain |
Abbreviations: adOA: Autosomal dominant optic atrophy; CMT2A: Charcot-Marie-Tooth type 2;CI: Complex I; DA: Dopamine; DRG: Dorsal root ganglia; EE: Ethylmalonic Encephalopathy FRDA: Friedreich ataxia; MCK: Muscle creatine kinase; NSE: Neuron specific enolase; OXPHOS: Oxidative phosphorylation system; PEO: Progressive External Ophthalmoplegia; RRF: Ragged red fibers; RGC: Retinal Ganglion Cells; Polγ: mtDNA polymerase
Conditional deletion of Aif in the telencephalon resulted in embryonic lethality at E14 and mice showed reduced cortical thickness due to neuronal death. Morphological analysis of mitochondria, in cultured KO neurons, showed aberrant mitochondria with disorganized cristae that may explain the respiratory chain deficiency observed in Aif KO mice (Cheung, 2006). Recent observations have shown that a decreased OXPHOS capacity in muscle may lead to insulin resistance (Rabøl, 2006). Conditional ablation of Aif in muscle and liver provided an in vivo model to study this phenomenon. Authors found that Aif inactivation resulted in a down regulation of OXPHOS gene expression without major changes in mitochondrial morphology but leading to a combined respiratory chain complex deficiency (Pospisilik, 2007). Metabolic analysis in muscle and liver of KO mice showed increased glucose uptake, increased glycolysis and reduced lipid storage, even when they were fed with a high-fat diet. The same phenotype was observed in the systemic Aif KO mice.
Although Aif mouse models have offered the opportunity to study pathological consequences of CI defects in vivo, recent studies of patients carrying mutations in Aif did not revealed defects in CI (Ghezzi, 2010), questioning the link between Aif and CI deficiencies.
4.2. Mito-Pst1
A mouse model of mtDNA deletions/depletion was ingeniously created by producing mtDNA double strand breaks via the expression and target the restriction endonuclease PstI to the mitochondria (mito-PstI) (Table V). In the first study, mito-PstI was expressed in the skeletal muscle (Srivastava and Moraes, 2005). This transgenic mice developed COX deficiency and ragged red fibers due to a muscle specific mtDNA depletion and deletions (Srivastava and Moraes, 2005). Subsequently, an inducible mito-PstI transgene was engineered and expressed in the adult neurons using the tetracycline-regulated expression system (Fukui and Moraes, 2009). Animals were shortly induced for 3 days at 4 weeks of age and analyzed 10 weeks later. This transient expression of mito-PstI was shown to induce mtDNA deletions and depletions (Fukui and Moraes, 2009). Interestingly, mtDNA with large deletions accumulate faster than those with smaller deletions, implying a replicative advantage of smaller mtDNAs. Therefore, this study revealed that double strand breaks, DNA repair systems and replicative advantage are likely mechanisms underlying the generation of age-associated accumulation of mtDNA deletions in mammalian neurons. Long-term expression of PstI in the adult neurons led to an OXPHOS deficiency, mostly due to mtDNA depletion associated with a progressive neurodegenerative phenotype particularly affecting the striatum. (Pickrell, 2011a).
Expression of mito-PstI in dopaminergic neurons (DA) has provided a novel model to study the role of mitochondrial defects in the pathophysiology of Parkinson’s disease. The DA-mito-PstI mice recapitulated most of the major features of PD and showed mtDNA depletion in the striatum (Pickrell, 2011b).
Transient systemic overexpression of mito-Pst1 led to mtDNA depletion, muscle wasting and a decline in locomotor activity later in life (Wang, 2013). Mice showed a significant decline in muscle satellite cells suggesting that the systemic mitochondrial dysfunction plays an important role in age-related muscle wasting by preferentially affecting the myosatellite cell pool. Collectively, these studies indicate that the mito-Pst1 mouse is a suitable model for the study of mitochondrial depletion syndromes.
4.3. Frataxin
FRATAXIN is a mitochondrial protein that is involved in the biosynthesis of the iron-sulfur clusters and is encoded by the Fxn gene (Martelli, 2007). Expansion of the GAA triplet in the first intron of the Fnx gene leads to transcriptional silencing and is the molecular cause of Friedreich’s ataxia (FRDA), a syndrome presenting neurodegeneration, hypertrophyc cardiomyopathy and diabetes (Bayot, 2011). The three biochemical features found in patients are: (i) the presence of iron deposits in cardiomyocytes, (ii) decreased iron sulfur cluster-containing enzyme activities in heart homogenates (aconitase and respiratory chain complex I, II and CIII), and (iii) the presence of markers of oxidative damage in blood and urine (Perdomini, 2013). Several mouse models have been generated to study the pathophysiology of FRDA. Complete ablation of Fxn is not compatible with life in mouse (Cossee, 2000). Conditional knockout mouse models in heart (Puccio, 2001), neurons (Puccio, 2001; Simon, 2004), β-cells (Ristow, 2003) and liver (Martelli, 2012) reproduced most of the charateristic features of the human disease: hypertrophic cardiomyopathy, progressive spinocerebellar and sensory ataxia and progressive diabetes. However, the phenotypes in mice were more rapid and severe when compared to the FRDA patients, but have contributed to the understanding of the molecular mechanisms of disease. Interestingly, in contrast to what has been observed extensively in clinical studies, the mice did not show any oxidative damage. This difference could be explained because in mice the total deletion of Fxn happens in a certain tissue at a specific time, whereas FRDA is characterized by partial Frataxin deficiency in all cells throughout the lifetime.
A knock-in mouse carrying the GAA repeat expansion into the first intron of the mouse Fxn locus has been created to reproduce the genomic context of the disease (Miranda, 2002). Heterozygous knock-in, knock-out Fxn mice (KIKO) showed 25% residual frataxin compared to control animals, resemblesing the levels found in patients. However, no overt phenotype was detected but significant transcriptional changes were found in genes related to mitochondrial and amino acid metabolism, nucleic acid binding, signal transduction and stress response (Coppola, 2006).
Transgenic overexpression of the human Fxn containing GAA repeats in the first intron in mice reproduced a mild progressive phenotype, with locomotor defects and giant vacuoles in the large sensory neurons of the dorsal root ganglia (Al-Mahdawi, 2004; Al-Mahdawi, 2006). Contrary to the conditional KO models described above, slight signs of oxidative damage were reported in several tissues of this trangenic model (Al-Mahdawi, 2006).
4.4. Ethe1
ETHE1 encodes a ubiquitous mitochondrial sulfur dioxygenase (SDO) involved in the detoxification of hydrogen sulfide (H2S) (Tiranti, 2009), which is produced by the catabolism of sulfurated amino acids in tissues and by anaerobic bacteria in the large intestine. In mammalian tissues, H2S is a gasotransmitter acting as a signaling molecule, but at supraphysiological concentrations it is a potent inhibitor of COX (Dorman, 2002). Mutations in ETHE1 gene in humans cause ethylmalonic encephalopathy (EE), a fatal autosomal recessive disorder characterized by early-onset encephalopathy, microangiopathy, chronic diarrhea, defective COX in muscle and brain, high concentration of C4 and C5 acylcarnitines in blood and high excretion of ethylmalonic acid in urine. Constitutive ablation of Ethe1 gene in mouse caused growth arrest from P15, reduced motor activity, and early death, between 5 and 6 weeks of age (Tiranti, 2009). COX activity was severely impaired in brain and muscle in the Ethe1−/− mouse, in which very high levels of H2S were observed. Thiosulfate levels in the plasma, a biomarker reflecting the accumulation of H2S, were very elevated in Ethe1−/− mouse compared to controls, resembling the situation in patients. Also increased amounts of lactate and C4 and C5 acylcarnitines in blood, and increased levels of ethylmalonic acid in urine of were detected in the Ethe1−/− mouse. The phenotype of the Ethe1−/− mouse mimics all the hallmarks observed in the human ethylmalonic encephalopathy from the early onset and fatal course to the biochemical profile in blood and urine to the COX deficiency in brain and muscle tissue (Tiranti, 2009). Ethe1 was conditionally deleted in muscle, brain and liver (Di Meo, 2011). Besides the COX defect specifically in brain and muscle, none of the three conditional mouse models showed the clinical and biological hallmarks of EE, at least during the first 3 months of life. Urine thiosulfate levels were similar between the 3 conditional mouse models and wild-type (Di Meo, 2011). This result indicated that the main pathogenic mechanism in the EE is the accumulation of the H2S and it byproducts in the blood rather than the COX deficiency in the tissues.
Concluding remarks
Here we described those mouse models of different factors that affect the proper function of the electron transport chain. Interestingly, the majority of these models recapitulated human phenotypes associated with mitochondrial diseases and other disorders. Additionally, these mouse models represent great tools to study adaptive responses and compensatory mechanisms (mitochondrial biogenesis) to counteract the bioenergetics defect. Many of the knockout models have helped to understand the function of a particular gene. However, in many instances the creation of knock-in models may be more informative and relevant when trying to recapitulate the disease phenotype in animals. Nevertheless, all mouse models have had a great impact in our understanding of the pathological mechanisms and have advanced the field of mitochondrial diseases forward in relatively short time.
Highlights.
Mitochondrial diseases can be caused by defects in factors that do not form part of the OXPHOS system but that can affect its function.
Defects in proteins involved in mtDNA replication, transcription, translation and maintenance can cause mitochondrial diseases.
Defects in proteins involved in mitochondrial dynamics and protein quality control also contribute to the development of defects in mitochondrial function.
Mouse models of these factors have contributed to elucidate their function and the molecular bases of the disorders.
Acknowledgments
We are thankful to Nadee Nissanka for revision of this manuscript. FD work has been funded by the James and Esther King Research Program Florida Department of Health (grant 08KN-01), the National Institute of Health (grant GM101225) and the United Mitochondrial Disease Foundation (grant 14050R).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: Authors have nothing to declare.
Contributor Information
Luisa Iommarini, Email: luisa.iommarini2@unibo.it.
Susana Peralta, Email: SPeralta@med.miami.edu.
Alessandra Torraco, Email: alessandra.torraco@opbg.net.
References
- Acham-Roschitz B, Plecko B, Lindbichler F, Bittner R, Mache CJ, Sperl W, Mayr JA. A novel mutation of the RRM2B gene in an infant with early fatal encephalomyopathy, central hypomyelination, and tubulopathy. Mol Genet Metab. 2009;98:300–304. doi: 10.1016/j.ymgme.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Ahlqvist KJ, Hämäläinen RH, Yatsuga S, Uutela M, Terzioglu M, Götz A, Forsström S, Salven P, Angers-Loustau A, Kopra OH, Tyynismaa H, Larsson NG, Wartiovaara K, Prolla T, Trifunovic A, Suomalainen A. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012;15:100–109. doi: 10.1016/j.cmet.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Ahola-Erkkilä S, Carroll CJ, Peltola-Mjösund K, Tulkki V, Mattila I, Seppänen-Laakso T, Oresic M, Tyynismaa H, Suomalainen A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19:1974–1984. doi: 10.1093/hmg/ddq076. [DOI] [PubMed] [Google Scholar]
- Akman HO, Dorado B, López LC, García-Cazorla A, Vilà MR, Tanabe LM, Dauer WT, Bonilla E, Tanji K, Hirano M. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum Mol Genet. 2008;17:2433–2440. doi: 10.1093/hmg/ddn143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Ruddle P, Carroll C, Webster Z, Pook M. GAA repeat instability in Friedreich ataxia YAC transgenic mice. Genomics. 2004;84:301–310. doi: 10.1016/j.ygeno.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, Webster Z, Blake J, Cooper JM, King R, Pook MA. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, Ruttiger L, Beck SC, Tonagel F, Pichler BJ, Knipper M, Peters T, Laufs J, Wissinger B. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain. 2007;130:1029–1042. doi: 10.1093/brain/awm005. [DOI] [PubMed] [Google Scholar]
- Alavi MV, Fuhrmann N, Nguyen HP, Yu-Wai-Man P, Heiduschka P, Chinnery PF, Wissinger B. Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp Neurol. 2009;220:404–409. doi: 10.1016/j.expneurol.2009.09.026. [DOI] [PubMed] [Google Scholar]
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- Almajan ER, Richter R, Paeger L, Martinelli P, Barth E, Decker T, Larsson NG, Kloppenburg P, Langer T, Rugarli EI. AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. The Journal of clinical investigation. 2012;122:4048–4058. doi: 10.1172/JCI64604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissière A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, Labauge P, Figarella-Branger D, Marcorelles P, Furby A, Beauvais K, Letournel F, Liguori R, La Morgia C, Montagna P, Liguori M, Zanna C, Rugolo M, Cossarizza A, Wissinger B, Verny C, Schwarzenbacher R, Martín MA, Arenas J, Ayuso C, Garesse R, Lenaers G, Bonneau D, Carelli V. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. The Journal of cell biology. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Andersson DC, Fauconnier J, Park CB, Zhang SJ, Thireau J, Ivarsson N, Larsson NG, Westerblad H. Enhanced cardiomyocyte Ca(2+) cycling precedes terminal AV-block in mitochondrial cardiomyopathy Mterf3 KO mice. Antioxid Redox Signal. 2011;15:2455–2464. doi: 10.1089/ars.2011.3915. [DOI] [PubMed] [Google Scholar]
- Artal-Sanz M, Tsang WY, Willems EM, Grivell LA, Lemire BD, van der Spek H, Nijtmans LG. The mitochondrial prohibitin complex is essential for embryonic viability and germline function in Caenorhabditis elegans. J Biol Chem. 2003;278:32091–32099. doi: 10.1074/jbc.M304877200. [DOI] [PubMed] [Google Scholar]
- Bailey LJ, Cluett TJ, Reyes A, Prolla TA, Poulton J, Leeuwenburgh C, Holt IJ. Mice expressing an error-prone DNA polymerase in mitochondria display elevated replication pausing and chromosomal breakage at fragile sites of mitochondrial DNA. Nucleic Acids Res. 2009;37:2327–2335. doi: 10.1093/nar/gkp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi S, Betts-Henderson J, Cain K, Dinsdale D, Zhou X, Karlsson A, Salomoni P, Nicotera P. Loss of thymidine kinase 2 alters neuronal bioenergetics and leads to neurodegeneration. Hum Mol Genet. 2010;19:1669–1677. doi: 10.1093/hmg/ddq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Nilsson R, Gohil VM, Arlow DH, Gauhar Z, Mootha VK. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009;5:e1000590. doi: 10.1371/journal.pgen.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayot A, Santos R, Camadro JM, Rustin P. Friedreich’s ataxia: the vicious circle hypothesis revisited. BMC medicine. 2011;9:112. doi: 10.1186/1741-7015-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensch KG, Mott JL, Chang SW, Hansen PA, Moxley MA, Chambers KT, de Graaf W, Zassenhaus HP, Corbett JA. Selective mtDNA mutation accumulation results in beta-cell apoptosis and diabetes development. Am J Physiol Endocrinol Metab. 2009;296:E672–680. doi: 10.1152/ajpendo.90839.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24:813–825. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
- Bornstein B, Area E, Flanigan KM, Ganesh J, Jayakar P, Swoboda KJ, Coku J, Naini A, Shanske S, Tanji K, Hirano M, DiMauro S. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul Disord. 2008;18:453–459. doi: 10.1016/j.nmd.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottani E, Giordano C, Civiletto G, Di Meo I, Auricchio A, Ciusani E, Marchet S, Lamperti C, d’Amati G, Viscomi C, Zeviani M. AAV-mediated liver-specific MPV17 expression restores mtDNA levels and prevents diet-induced liver failure. Mol Ther. 2014;22:10–17. doi: 10.1038/mt.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chrétien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rötig A. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- Bruni F, Manzari C, Filice M, Loguercio Polosa P, Colella M, Carmone C, Hambardjieva E, Garcia-Diaz M, Cantatore P, Roberti M. D-MTERF5 is a novel factor modulating transcription in Drosophila mitochondria. Mitochondrion. 2012;12:492–499. doi: 10.1016/j.mito.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cámara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, Franz T, Erdjument-Bromage H, Tempst P, Hallberg BM, Gustafsson CM, Larsson NG. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011;13:527–539. doi: 10.1016/j.cmet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Progress in retinal and eye research. 2004;23:53–89. doi: 10.1016/j.preteyeres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Arnaud E, Médard JJ, Poirot O, Courvoisier DS, Chrast R, Martinou JC. Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain. 2010;133:1460–1469. doi: 10.1093/brain/awq082. [DOI] [PubMed] [Google Scholar]
- Casalena G, Krick S, Daehn I, Yu L, Ju W, Shi S, Tsai SY, D’Agati VD, Lindenmeyer MT, Cohen CD, Schlondorff D, Bottinger EP. Mpv17 in mitochondria protects podocytes against mitochondrial dysfunction and apoptosis in vivo and in vitro. Am J Physiol Ren Physiol. 2014;306:F1372–F1380. doi: 10.1152/ajprenal.00608.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of cell biology. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, Cortopassi G, Chiamvimonvat N, Bers DM, Votruba M, Knowlton AA. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. Journal of the American Heart Association. 2012a;1:e003012. doi: 10.1161/JAHA.112.003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Logan TD, Hochberg ML, Shelat SG, Yu X, Wilding GE, Tan W, Kujoth GC, Prolla TA, Selak MA, Kundu M, Carroll M, Thompson JE. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. Blood. 2009;114:4045–4053. doi: 10.1182/blood-2008-08-169474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW, 2nd, Maack C. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circulation research. 2012b;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Joza N, Steenaart NA, McClellan KA, Neuspiel M, McNamara S, MacLaurin JG, Rippstein P, Park DS, Shore GC, McBride HM, Penninger JM, Slack RS. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. The EMBO journal. 2006;25:4061–4073. doi: 10.1038/sj.emboj.7601276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, Dejaegere T, Pellegrini L, D’Hooge R, Scorrano L, De Strooper B. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Clozel M, Hess P, Fischli W, Löffler BM, Zwacka RM, Reuter A, Weiher H. Age-dependent hypertension in Mpv17-deficient mice, a transgenic model of glomerulosclerosis and inner ear disease. Exp Gerontol. 1999;34:1007–1015. doi: 10.1016/s0531-5565(99)00074-1. [DOI] [PubMed] [Google Scholar]
- Cooper MP, Qu L, Rohas LM, Lin J, Yang W, Erdjument-Bromage H, Tempst P, Spiegelman BM. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1alpha/LRP130 complex. Genes & development. 2006;20:2996–3009. doi: 10.1101/gad.1483906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland WC, Longley MJ. Mitochondrial genome maintenance in health and disease. DNA Rep. 2014;19:190–198. doi: 10.1016/j.dnarep.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppock DL, Pardee AB. Control of thymidine kinase mRNA during the cell cycle. Mol Cell Biol. 1987;7:2925–2932. doi: 10.1128/mcb.7.8.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Choi SH, Santos MM, Miranda CJ, Tentler D, Wexler EM, Pandolfo M, Geschwind DH. Gene expression profiling in frataxin deficient mice: microarray evidence for significant expression changes without detectable neurodegeneration. Neurobiology of disease. 2006;22:302–311. doi: 10.1016/j.nbd.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossee M, Puccio H, Gansmuller A, Koutnikova H, Dierich A, LeMeur M, Fischbeck K, Dolle P, Koenig M. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum Mol Genet. 2000;9:1219–1226. doi: 10.1093/hmg/9.8.1219. [DOI] [PubMed] [Google Scholar]
- Cotney J, Shadel GS. Evidence for an early gene duplication event in the evolution of the mitochondrial transcription factor B family and maintenance of rRNA methyltransferase activity in human mtTFB1 and mtTFB2. Journal of molecular evolution. 2006;63:707–717. doi: 10.1007/s00239-006-0075-1. [DOI] [PubMed] [Google Scholar]
- Dai Y, Kiselak T, Clark J, Clore E, Zheng K, Cheng A, Kujoth GC, Prolla TA, Maratos-Flier E, Simon DK. Behavioral and metabolic characterization of heterozygous and homozygous POLG mutator mice. Mitochondrion. 2013;13:282–291. doi: 10.1016/j.mito.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, Votruba M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. 2007;16:1307–1318. doi: 10.1093/hmg/ddm079. [DOI] [PubMed] [Google Scholar]
- Debray FG, Morin C, Janvier A, Villeneuve J, Maranda B, Laframboise R, Lacroix J, Decarie JC, Robitaille Y, Lambert M, Robinson BH, Mitchell GA. LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J Med Genet. 2011;48:183–189. doi: 10.1136/jmg.2010.081976. [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Detmer SA, Vande Velde C, Cleveland DW, Chan DC. Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A. Hum Mol Genet. 2008;17:367–375. doi: 10.1093/hmg/ddm314. [DOI] [PubMed] [Google Scholar]
- Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, Finardi A, Cagnoli C, Tempia F, Frontali M, Veneziano L, Sacco T, Boda E, Brussino A, Bonn F, Castellotti B, Baratta S, Mariotti C, Gellera C, Fracasso V, Magri S, Langer T, Plevani P, Di Donato S, Muzi-Falconi M, Taroni F. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–321. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- Di Meo I, Fagiolari G, Prelle A, Viscomi C, Zeviani M, Tiranti V. Chronic exposure to sulfide causes accelerated degradation of cytochrome c oxidase in ethylmalonic encephalopathy. Antioxid Redox Signal. 2011;15:353–362. doi: 10.1089/ars.2010.3520. [DOI] [PubMed] [Google Scholar]
- Dogan SA, Pujol C, Maiti P, Kukat A, Wang S, Hermans S, Senft K, Wibom R, Rugarli EI, Trifunovic A. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014;19:458–469. doi: 10.1016/j.cmet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Dorman DC, Moulin FJ, McManus BE, Mahle KC, James RA, Struve MF. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicological sciences: an official journal of the Society of Toxicology. 2002;65:18–25. doi: 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Gomez S, Guenet JL, Love S. Paralyze – a New Neurological Mutant Mouse with Progressive Atrophy and Loss of Motor-Nerve Terminals. J Physiol-London. 1983;345:P166–P166. [Google Scholar]
- Dufour E, Terzioglu M, Sterky FH, Sorensen L, Galter D, Olson L, Wilbertz J, Larsson NG. Age-associated mosaic respiratory chain deficiency causes trans-neuronal degeneration. Hum Mol Genet. 2008;17:1418–1426. doi: 10.1093/hmg/ddn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Chassagne M, Ceresuela J, Rouvet I, Padet S, Acquaviva C, Nataf S, Vinzio S, Bozon D, Mousson de Camaret B. Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J Med Genet. 2012;49:146–150. doi: 10.1136/jmedgenet-2011-100504. [DOI] [PubMed] [Google Scholar]
- Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009;10:131–138. doi: 10.1016/j.cmet.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. The Journal of cell biology. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Kouni MH, el Kouni MM, Naguib FN. Differences in activities and substrate specificity of human and murine pyrimidine nucleoside phosphorylases: implications for chemotherapy with 5-fluoropyrimidines. Cancer Res. 1993;53:3687–3693. [PubMed] [Google Scholar]
- Esposito LA, Melov S, Panov A, Cottrell BA, Wallace DC. Mitochondrial disease in mouse results in increased oxidative stress. Proc Natl Acad Sci U S A. 1999;96:4820–4825. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet. 2002;31:289–294. doi: 10.1038/ng909. [DOI] [PubMed] [Google Scholar]
- Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- Ferraro P, Pontarin G, Crocco L, Fabris S, Reichard P, Bianchi V. Mitochondrial deoxynucleotide pools in quiescent fibroblasts: a possible model for mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) J Biol Chem. 2005;280:24472–24480. doi: 10.1074/jbc.M502869200. [DOI] [PubMed] [Google Scholar]
- Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, Auricchio A, Piemonte F, Tozzi G, Gaeta L, Casari G, Ballabio A, Rugarli EI. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. The Journal of clinical investigation. 2004;113:231–242. doi: 10.1172/JCI20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Hamann A, Osiewacz HD. Mitochondrial quality control: an integrated network of pathways. Trends in biochemical sciences. 2012;37:284–292. doi: 10.1016/j.tibs.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Clayton DA. Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol. 1988;8:3496–3509. doi: 10.1128/mcb.8.8.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RG, Magness S, Kujoth GC, Prolla TA, Maeda N. Mitochondrial DNA polymerase editing mutation, PolgD257A, disturbs stem-progenitor cell cycling in the small intestine and restricts excess fat absorption. Am J Physiol Gastrointest Liver Physiol. 2012;302:G914–924. doi: 10.1152/ajpgi.00402.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratter C, Raman P, Alston CL, Blakely EL, Craig K, Smith C, Evans J, Seller A, Czermin B, Hanna MG, Poulton J, Brierley C, Staunton TG, Turnpenny PD, Schaefer AM, Chinnery PF, Horvath R, Turnbull DM, Gorman GS, Taylor RW. RRM2B mutations are frequent in familial PEO with multiple mtDNA deletions. Neurology. 2011;76:2032–2034. doi: 10.1212/WNL.0b013e31821e558b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyer C, Park CB, Ekstrand MI, Shi Y, Khvorostova J, Wibom R, Falkenberg M, Gustafsson CM, Larsson NG. Maintenance of respiratory chain function in mouse hearts with severely impaired mtDNA transcription. Nucleic Acids Research. 2010;38:6577–6588. doi: 10.1093/nar/gkq527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2009;18:1028–1036. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall JM, Wang Z, Liesa M, Molina A, Havasi A, Schwartz JH, Shirihai O, Borkan SC, Bonegio RG. Role of mitofusin 2 in the renal stress response. PLoS One. 2012;7:e31074. doi: 10.1371/journal.pone.0031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D’Adamo P, Novara F, Zuffardi O, Uziel G, Zeviani M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gispert S, Parganlija D, Klinkenberg M, Drose S, Wittig I, Mittelbronn M, Grzmil P, Koob S, Hamann A, Walter M, Buchel F, Adler T, Hrabe de Angelis M, Busch DH, Zell A, Reichert AS, Brandt U, Osiewacz HD, Jendrach M, Auburger G. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum Mol Genet. 2013;22:4871–4887. doi: 10.1093/hmg/ddt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffart S, Cooper HM, Tyynismaa H, Wanrooij S, Suomalainen A, Spelbrink JN. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet. 2009;18:328–340. doi: 10.1093/hmg/ddn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz A, Isohanni P, Pihko H, Paetau A, Herva R, Saarenpää-Heikkilä O, Valanne L, Marjavaara S, Suomalainen A. Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome. Brain. 2008;131:2841–2850. doi: 10.1093/brain/awn236. [DOI] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. The Journal of cell biology. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillet V, Gueguen N, Cartoni R, Chevrollier A, Desquiret V, Angebault C, Amati-Bonneau P, Procaccio V, Bonneau D, Martinou JC, Reynier P. Bioenergetic defect associated with mKATP channel opening in a mouse model carrying a mitofusin 2 mutation. FASEB J. 2011;25:1618–1627. doi: 10.1096/fj.10-173609. [DOI] [PubMed] [Google Scholar]
- Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lönnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130:3032–3040. doi: 10.1093/brain/awm242. [DOI] [PubMed] [Google Scholar]
- Hance N, Ekstrand MI, Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum Mol Genet. 2005;14:1775–1783. doi: 10.1093/hmg/ddi184. [DOI] [PubMed] [Google Scholar]
- Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci U S A. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi M, Tsujimoto H, Fukushima M, Higuchi I, Kuribayashi H, Utsumi H, Nakayama A, Hashizume Y, Hirato J, Yoshida H, Hara H, Hamano S, Kawaguchi H, Furukawa T, Miyazono K, Ishikawa F, Toyoshima H, Kaname T, Komatsu M, Chen ZS, Gotanda T, Tachiwada T, Sumizawa T, Miyadera K, Osame M, Noda T, Yamada Y, Akiyama S. Targeted deletion of both thymidine phosphorylase and uridine phosphorylase and consequent disorders in mice. Mol Cell Biol. 2002;22:5212–5221. doi: 10.1128/MCB.22.14.5212-5221.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AE. Hereditary spastic paraplegias. Seminars in neurology. 1993;13:333–336. doi: 10.1055/s-2008-1041143. [DOI] [PubMed] [Google Scholar]
- Harmel J, Ruzzenente B, Terzioglu M, Spahr H, Falkenberg M, Larsson NG. The leucine-rich pentatricopeptide repeat-containing protein (LRPPRC) does not activate transcription in mammalian mitochondria. J Biol Chem. 2013;288:15510–15519. doi: 10.1074/jbc.M113.471649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Feng Q, Mukherjee A, Lonard DM, DeMayo FJ, Katzenellenbogen BS, Lydon JP, O’Malley BW. A repressive role for prohibitin in estrogen signaling. Molecular endocrinology. 2008;22:344–360. doi: 10.1210/me.2007-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. The Journal of cell biology. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, Someya S, Miyakawa T, Nakayama C, Samhan-Arias AK, Servais S, Barger JL, Portero-Otín M, Tanokura M, Prolla TA, Leeuwenburgh C. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One. 2010;5:e11468. doi: 10.1371/journal.pone.0011468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S. The regulation of mitochondrial dynamics. Current opinion in cell biology. 2014;29C:46–52. doi: 10.1016/j.ceb.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG, Ahlqvist K, Suomalainen A, Reynier P, McFarland R, Turnbull DM, Chinnery PF, Taylor RW. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–337. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- Humble MM, Young MJ, Foley JF, Pandiri AR, Travlos GS, Copeland WC. Polg2 is essential for mammalian embryogenesis and is required for mtDNA maintenance. Hum Mol Genet. 2013;22:1017–1025. doi: 10.1093/hmg/dds506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2014;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nature cell biology. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jenkinson EM, Rehman AU, Walsh T, Clayton-Smith J, Lee K, Morell RJ, Drummond MC, Khan SN, Naeem MA, Rauf B, Billington N, Schultz JM, Urquhart JE, Lee MK, Berry A, Hanley NA, Mehta S, Cilliers D, Clayton PE, Kingston H, Smith MJ, Warner TT, University of Washington Center for Mendelian G. Black GC, Trump D, Davis JR, Ahmad W, Leal SM, Riazuddin S, King MC, Friedman TB, Newman WG. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet. 2013;92:605–613. doi: 10.1016/j.ajhg.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnoczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–727. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- Joseph AM, Adhihetty PJ, Wawrzyniak NR, Wohlgemuth SE, Picca A, Kujoth GC, Prolla TA, Leeuwenburgh C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS One. 2013;8:e69327. doi: 10.1371/journal.pone.0069327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Oudit GY, Brown D, Bénit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HC, Lee YM, Kim HD, Lee JS, Slama A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia. 2007;48:82–88. doi: 10.1111/j.1528-1167.2006.00906.x. [DOI] [PubMed] [Google Scholar]
- Kang S, Louboutin JP, Datta P, Landel CP, Martinez D, Zervos AS, Strayer DS, Fernandes-Alnemri T, Alnemri ES. Loss of HtrA2/Omi activity in non-neuronal tissues of adult mice causes premature aging. Cell death and differentiation. 2013;20:259–269. doi: 10.1038/cdd.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadimas CL, Vu TH, Holve SA, Chronopoulou P, Quinzii C, Johnsen SD, Kurth J, Eggers E, Palenzuela L, Tanji K, Bonilla E, De Vivo DC, DiMauro S, Hirano M. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544–548. doi: 10.1086/506913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Kubota M, Miyauchi T, Noda Y, Mouri A, Nabeshima T, Kato T. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol Psychiatry. 2006;11:577–593. 523. doi: 10.1038/sj.mp.4001824. [DOI] [PubMed] [Google Scholar]
- Kasashima K, Sumitani M, Satoh M, Endo H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Experimental cell research. 2008;314:988–996. doi: 10.1016/j.yexcr.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Kaser M, Kambacheld M, Kisters-Woike B, Langer T. Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J Biol Chem. 2003;278:46414–46423. doi: 10.1074/jbc.M305584200. [DOI] [PubMed] [Google Scholar]
- Kaukonen J, Juselius JK, Tiranti V, Kyttälä A, Zeviani M, Comi GP, Keränen S, Peltonen L, Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nature structural biology. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- Kimura T, Takeda S, Sagiya Y, Gotoh M, Nakamura Y, Arakawa H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat Genet. 2003;34:440–445. doi: 10.1038/ng1212. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Ko KS, Tomasi ML, Iglesias-Ara A, French BA, French SW, Ramani K, Lozano JJ, Oh P, He L, Stiles BL, Li TW, Yang H, Martinez-Chantar ML, Mato JM, Lu SC. Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology. 2010;52:2096–2108. doi: 10.1002/hep.23919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeck T, Olsson AH, Nitert MD, Sharoyko VV, Ladenvall C, Kotova O, Reiling E, Ronn T, Parikh H, Taneera J, Eriksson JG, Metodiev MD, Larsson NG, Balhuizen A, Luthman H, Stancakova A, Kuusisto J, Laakso M, Poulsen P, Vaag A, Groop L, Lyssenko V, Mulder H, Ling C. A common variant in TFB1M is associated with reduced insulin secretion and increased future risk of type 2 diabetes. Cell Metab. 2011;13:80–91. doi: 10.1016/j.cmet.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Kollberg G, Darin N, Benan K, Moslemi AR, Lindal S, Tulinius M, Oldfors A, Holme E. A novel homozygous RRM2B missense mutation in association with severe mtDNA depletion. Neuromuscul Disord. 2009;19:147–150. doi: 10.1016/j.nmd.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Kondadi AK, Wang S, Montagner S, Kladt N, Korwitz A, Martinelli P, Herholz D, Baker MJ, Schauss AC, Langer T, Rugarli EI. Loss of the m-AAA protease subunit AFG(3)L(2) causes mitochondrial transport defects and tau hyperphosphorylation. The EMBO journal. 2014;33:1011–1026. doi: 10.1002/embj.201387009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong YX, Van Bergen N, Trounce IA, Bui BV, Chrysostomou V, Waugh H, Vingrys A, Crowston JG. Increase in mitochondrial DNA mutations impairs retinal function and renders the retina vulnerable to injury. Aging Cell. 2011;10:572–583. doi: 10.1111/j.1474-9726.2011.00690.x. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. The New England journal of medicine. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- Korhonen JA, Pham XH, Pellegrini M, Falkenberg M. Reconstitution of a minimal mtDNA replisome in vitro. The EMBO journal. 2004;23:2423–2429. doi: 10.1038/sj.emboj.7600257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl I, Kukat C, Ruzzenente B, Milenkovic D, Mourier A, Miranda M, Koolmeister C, Falkenberg M, Larsson NG. POLRMT does not transcribe nuclear genes. Nature. 2014;514:E7–11. doi: 10.1038/nature13690. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lamperti C, Zeviani M. Encephalomyopathies caused by abnormal nuclear-mitochondrial intergenomic cross-talk. Acta Myol. 2009;28:2–11. [PMC free article] [PubMed] [Google Scholar]
- Lara MC, Valentino ML, Torres-Torronteras J, Hirano M, Martí R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): biochemical features and therapeutic approaches. Biosci Rep. 2007;27:151–163. doi: 10.1007/s10540-007-9043-2. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- Lee J, Schriner SE, Wallace DC. Adenine nucleotide translocator 1 deficiency increases resistance of mouse brain and neurons to excitotoxic insults. Biochim Biophys Acta. 2009;1787:364–370. doi: 10.1016/j.bbabio.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, Haase CP, Keebaugh ES, Long R, Ludaway T, Russ R, Steltzer J, Tioleco N, Santoianni R, Copeland WC. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab Invest. 2007;87:326–335. doi: 10.1038/labinvest.3700523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:3467–3472. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Warner CK, Hodge JA, Minoshima S, Kudoh J, Fukuyama R, Maekawa M, Shimizu Y, Shimizu N, Wallace DC. A human muscle adenine nucleotide translocator gene has four exons, is located on chromosome 4, and is differentially expressed. J Biol Chem. 1989;264:13998–14004. [PubMed] [Google Scholar]
- Linder T, Park CB, Asin-Cayuela J, Pellegrini M, Larsson NG, Falkenberg M, Samuelsson T, Gustafsson CM. A family of putative transcription termination factors shared amongst metazoans and plants. Curr Genet. 2005;48:265–269. doi: 10.1007/s00294-005-0022-5. [DOI] [PubMed] [Google Scholar]
- Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, Nightingale S, Turnbull DM, Copeland WC, Chinnery PF. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López LC, Akman HO, García-Cazorla A, Dorado B, Martí R, Nishino I, Tadesse S, Pizzorno G, Shungu D, Bonilla E, Tanji K, Hirano M. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum Mol Genet. 2009;18:714–722. doi: 10.1093/hmg/ddn401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltecca F, Aghaie A, Schroeder DG, Cassina L, Taylor BA, Phillips SJ, Malaguti M, Previtali S, Guenet JL, Quattrini A, Cox GA, Casari G. The mitochondrial protease AFG3L2 is essential for axonal development. J Neurosci. 2008;28:2827–2836. doi: 10.1523/JNEUROSCI.4677-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltecca F, Magnoni R, Cerri F, Cox GA, Quattrini A, Casari G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J Neurosci. 2009;29:9244–9254. doi: 10.1523/JNEUROSCI.1532-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso G, Barth E, Crivello P, Rugarli EI. Alternative splicing of Spg7, a gene involved in hereditary spastic paraplegia, encodes a variant of paraplegin targeted to the endoplasmic reticulum. PLoS One. 2012;7:e36337. doi: 10.1371/journal.pone.0036337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso M, Filosto M, Bellan M, Liguori R, Montagna P, Baruzzi A, DiMauro S, Carelli V. POLG mutations causing ophthalmoplegia, sensorimotor polyneuropathy, ataxia, and deafness. Neurology. 2004;62:316–318. doi: 10.1212/wnl.62.2.316. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Filosto M, Bonilla E, Hirano M, Shanske S, Vu TH, DiMauro S. Mitochondrial myopathy of childhood associated with mitochondrial DNA depletion and a homozygous mutation (T77M) in the TK2 gene. Arch Neurol. 2003;60:1007–1009. doi: 10.1001/archneur.60.7.1007. [DOI] [PubMed] [Google Scholar]
- Manczak M, Sesaki H, Kageyama Y, Reddy PH. Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim Biophys Acta. 2012;1822:862–874. doi: 10.1016/j.bbadis.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli A, Friedman LS, Reutenauer L, Messaddeq N, Perlman SL, Lynch DR, Fedosov K, Schulz JB, Pandolfo M, Puccio H. Clinical data and characterization of the liver conditional mouse model exclude neoplasia as a non-neurological manifestation associated with Friedreich’s ataxia. Disease models & mechanisms. 2012;5:860–869. doi: 10.1242/dmm.009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli A, Wattenhofer-Donze M, Schmucker S, Bouvet S, Reutenauer L, Puccio H. Frataxin is essential for extramitochondrial Fe-S cluster proteins in mammalian tissues. Hum Mol Genet. 2007;16:2651–2658. doi: 10.1093/hmg/ddm163. [DOI] [PubMed] [Google Scholar]
- Martinelli P, La Mattina V, Bernacchia A, Magnoni R, Cerri F, Cox G, Quattrini A, Casari G, Rugarli EI. Genetic interaction between the m-AAA protease isoenzymes reveals novel roles in cerebellar degeneration. Hum Mol Genet. 2009;18:2001–2013. doi: 10.1093/hmg/ddp124. [DOI] [PubMed] [Google Scholar]
- Martinelli P, Rugarli EI. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim Biophys Acta. 2010;1797:1–10. doi: 10.1016/j.bbabio.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung JK, Danner DB, Stewart DA, Smith JR, Schneider EL, Lumpkin CK, Dell’Orco RT, Nuell MJ. Isolation of a cDNA that hybrid selects antiproliferative mRNA from rat liver. Biochemical and biophysical research communications. 1989;164:1316–1322. doi: 10.1016/0006-291x(89)91813-5. [DOI] [PubMed] [Google Scholar]
- Merante F, Petrova-Benedict R, MacKay N, Mitchell G, Lambert M, Morin C, De Braekeleer M, Laframboise R, Gagné R, Robinson BH. A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am J Hum Genet. 1993;53:481–487. [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes & development. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, Rugarli EI, Langer T. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 2012;8:e1003021. doi: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metodiev MD, Lesko N, Park CB, Camara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 2009;9:386–397. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Meyer zum Gottesberge AM, Felix H. Abnormal basement membrane in the inner ear and the kidney of the Mpv17−/− mouse strain: ultrastructural and immunohistochemical investigations. Histochem Cell Biol. 2005;124:507–516. doi: 10.1007/s00418-005-0027-7. [DOI] [PubMed] [Google Scholar]
- Meyer zum Gottesberge AM, Massing T, Hansen S. Missing mitochondrial Mpv17 gene function induces tissue-specific cell-death pathway in the degenerating inner ear. Cell Tissue Res. 2012;347:343–356. doi: 10.1007/s00441-012-1326-7. [DOI] [PubMed] [Google Scholar]
- Meyer zum Gottesberge AM, Reuter A, Weiher H. Inner ear defect similar to Alport’s syndrome in the glomerulosclerosis mouse model Mpv17. Eur Arch Otorhinolaryngol. 1996;253:470–474. doi: 10.1007/BF00179952. [DOI] [PubMed] [Google Scholar]
- Milenkovic D, Matic S, Kuhl I, Ruzzenente B, Freyer C, Jemt E, Park CB, Falkenberg M, Larsson NG. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum Mol Genet. 2013;22:1983–1993. doi: 10.1093/hmg/ddt051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone M, Massie R. Polymerase gamma 1 mutations: clinical correlations. Neurologist. 2010;16:84–91. doi: 10.1097/NRL.0b013e3181c78a89. [DOI] [PubMed] [Google Scholar]
- Miranda CJ, Santos MM, Ohshima K, Smith J, Li L, Bunting M, Cossee M, Koenig M, Sequeiros J, Kaplan J, Pandolfo M. Frataxin knockin mouse. FEBS letters. 2002;512:291–297. doi: 10.1016/s0014-5793(02)02251-2. [DOI] [PubMed] [Google Scholar]
- Moore BA, Gonzalez Aviles GD, Larkins CE, Hillman MJ, Caspary T. Mitochondrial retention of Opa1 is required for mouse embryogenesis. Mamm Genome. 2010;21:350–360. doi: 10.1007/s00335-010-9272-8. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, Hudson TJ, Robinson B, Rioux JD, Lander ES. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier A, Ruzzenente B, Brandt T, Kuhlbrandt W, Larsson NG. Loss of LRPPRC causes ATP synthase deficiency. Hum Mol Genet. 2014;23:2580–2592. doi: 10.1093/hmg/ddt652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli L, Bordoni A, Zeviani M, Hadjigeorgiou GM, Sciacco M, Tiranti V, Terentiou A, Moggio M, Papadimitriou A, Scarlato G, Comi GP. A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology. 2001;57:2295–2298. doi: 10.1212/wnl.57.12.2295. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Alavi MV, Kim KY, Kang T, Scott RT, Noh YH, Lindsey JD, Wissinger B, Ellisman MH, Weinreb RN, Perkins GA, Ju WK. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011;2:e240. doi: 10.1038/cddis.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans LG, de Jong L, Artal Sanz M, Coates PJ, Berden JA, Back JW, Muijsers AO, van der Spek H, Grivell LA. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. The EMBO journal. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikali K, Suomalainen A, Saharinen J, Kuokkanen M, Spelbrink JN, Lönnqvist T, Peltonen L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet. 2005;14:2981–2990. doi: 10.1093/hmg/ddi328. [DOI] [PubMed] [Google Scholar]
- Nishigaki Y, Martí R, Copeland WC, Hirano M. Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. The Journal of clinical investigation. 2003;111:1913–1921. doi: 10.1172/JCI17828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- Oskoui M, Davidzon G, Pascual J, Erazo R, Gurgel-Giannetti J, Krishna S, Bonilla E, De Vivo DC, Shanske S, DiMauro S. Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch Neurol. 2006;63:1122–1126. doi: 10.1001/archneur.63.8.1122. [DOI] [PubMed] [Google Scholar]
- Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, Ferrero I, Zeviani M. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet. 2005;14:3079–3088. doi: 10.1093/hmg/ddi341. [DOI] [PubMed] [Google Scholar]
- Papadimitriou A, Comi GP, Hadjigeorgiou GM, Bordoni A, Sciacco M, Napoli L, Prelle A, Moggio M, Fagiolari G, Bresolin N, Salani S, Anastasopoulos I, Giassakis G, Divari R, Scarlato G. Partial depletion and multiple deletions of muscle mtDNA in familial MNGIE syndrome. Neurology. 1998;51:1086–1092. doi: 10.1212/wnl.51.4.1086. [DOI] [PubMed] [Google Scholar]
- Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes JA, Zhou X, Hoglund S, Karlsson A. Gene expression deregulation in postnatal skeletal muscle of TK2 deficient mice reveals a lower pool of proliferating myogenic progenitor cells. PLoS One. 2013;8:e53698. doi: 10.1371/journal.pone.0053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. 1991;252:965–969. doi: 10.1126/science.2035027. [DOI] [PubMed] [Google Scholar]
- Park CB, Asin-Cayuela J, Cámara Y, Shi Y, Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument-Bromage H, Tempst P, Falkenberg M, Gustafsson CM, Larsson NG. MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell. 2007;130:273–285. doi: 10.1016/j.cell.2007.05.046. [DOI] [PubMed] [Google Scholar]
- Parker NJ, Begley CG, Fox RM. Human R1 subunit of ribonucleotide reductase (RRM1): 5′ flanking region of the gene. Genomics. 1994;19:91–96. doi: 10.1006/geno.1994.1017. [DOI] [PubMed] [Google Scholar]
- Peralta S, Wang X, Moraes CT. Mitochondrial transcription: lessons from mouse models. Biochim Biophys Acta. 2012;1819(9-10):961–969. doi: 10.1016/j.bbagrm.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomini M, Hick A, Puccio H, Pook MA. Animal and cellular models of Friedreich ataxia. Journal of neurochemistry 126 Suppl. 2013;1:65–79. doi: 10.1111/jnc.12219. [DOI] [PubMed] [Google Scholar]
- Perier C, Bender A, Garcia-Arumi E, Melia MJ, Bove J, Laub C, Klopstock T, Elstner M, Mounsey RB, Teismann P, Prolla T, Andreu AL, Vila M. Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain. 2013;136:2369–2378. doi: 10.1093/brain/awt196. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Webb-Wood S, Faulkner AE, Jabbar SB, Biousse V, Newman NJ, Do VT, Boatright JH, Wallace DC, Pardue MT. Retinal function and structure in Ant1-deficient mice. Invest Ophthalmol Vis Sci. 2010;51:6744–6752. doi: 10.1167/iovs.10-5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Fukui H, Wang X, Pinto M, Moraes CT. The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J Neurosci. 2011a;31:9895–9904. doi: 10.1523/JNEUROSCI.6223-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Pinto M, Hida A, Moraes CT. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci. 2011b;31:17649–17658. doi: 10.1523/JNEUROSCI.4871-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson TM, Adams D, Bonn F, Martinelli P, Cherukuri PF, Teer JK, Hansen NF, Cruz P, Mullikin For The Nisc Comparative Sequencing Program JC. Blakesley RW, Golas G, Kwan J, Sandler A, Fuentes Fajardo K, Markello T, Tifft C, Blackstone C, Rugarli EI, Langer T, Gahl WA, Toro C. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7:e1002325. doi: 10.1371/journal.pgen.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, Huynh le H, Nicolas V, Alavi MV, Brenner C, Ventura-Clapier R, Veksler V, Joubert F. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovascular research. 2012;94:408–417. doi: 10.1093/cvr/cvs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- Quiros PM, Ramsay AJ, Sala D, Fernandez-Vizarra E, Rodriguez F, Peinado JR, Fernandez-Garcia MS, Vega JA, Enriquez JA, Zorzano A, Lopez-Otin C. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. The EMBO journal. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabøl R, Boushel R, Dela F. Mitochondrial oxidative function and type 2 diabetes. Appl Physiol Nutr Metab. 2006;31:675–683. doi: 10.1139/h06-071. [DOI] [PubMed] [Google Scholar]
- Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- Richter-Dennerlein R, Korwitz A, Haag M, Tatsuta T, Dargazanli S, Baker M, Decker T, Lamkemeyer T, Rugarli EI, Langer T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014;20:158–171. doi: 10.1016/j.cmet.2014.04.016. [DOI] [PubMed] [Google Scholar]
- Ristow M, Mulder H, Pomplun D, Schulz TJ, Muller-Schmehl K, Krause A, Fex M, Puccio H, Muller J, Isken F, Spranger J, Muller-Wieland D, Magnuson MA, Mohlig M, Koenig M, Pfeiffer AF. Frataxin deficiency in pancreatic islets causes diabetes due to loss of beta cell mass. The Journal of clinical investigation. 2003;112:527–534. doi: 10.1172/JCI18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberti M, Polosa PL, Bruni F, Manzari C, Deceglie S, Gadaleta MN, Cantatore P. The MTERF family proteins: mitochondrial transcription regulators and beyond. Biochim Biophys Acta. 2009;1787:303–311. doi: 10.1016/j.bbabio.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. The EMBO journal. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Cámara Y, Milenkovic D, Zickermann V, Wibom R, Hultenby K, Erdjument-Bromage H, Tempst P, Brandt U, Stewart JB, Gustafsson CM, Larsson NG. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. The EMBO journal. 2012;31:443–456. doi: 10.1038/emboj.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342–344. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarzi E, Angebault C, Seveno M, Gueguen N, Chaix B, Bielicki G, Boddaert N, Mausset Bonnefont AL, Cazevieille C, Rigau V, Renou JP, Wang J, Delettre C, Brabet P, Puel JL, Hamel CP, Reynier P, Lenaers G. The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain. 2012;135:3599–3613. doi: 10.1093/brain/aws303. [DOI] [PubMed] [Google Scholar]
- Sarzi E, Goffart S, Serre V, Chrétien D, Slama A, Munnich A, Spelbrink JN, Rötig A. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann Neurol. 2007;62:579–587. doi: 10.1002/ana.21207. [DOI] [PubMed] [Google Scholar]
- Sasarman F, Brunel-Guitton C, Antonicka H, Wai T, Shoubridge EA, Consortium L. LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol Biol Cell. 2010;21:1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, Muravina TI, Serkov SV, Uziel G, Bugiani M, Schiffmann R, Krageloh-Mann I, Smeitink JA, Florentz C, Van Coster R, Pronk JC, van der Knaap MS. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Adachi Y, Kageyama Y, Itoh K, Iijima M. In vivo functions of Drp1: lessons learned from yeast genetics and mouse knockouts. Biochim Biophys Acta. 2014;1842:1179–1185. doi: 10.1016/j.bbadis.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaibani A, Shchelochkov OA, Zhang S, Katsonis P, Lichtarge O, Wong LJ, Shinawi M. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol. 2009;66:1028–1032. doi: 10.1001/archneurol.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharoyko VV, Abels M, Sun J, Nicholas LM, Mollet IG, Stamenkovic JA, Gohring I, Malmgren S, Storm P, Fadista J, Spegel P, Metodiev MD, Larsson NG, Eliasson L, Wierup N, Mulder H. Loss of TFB1M results in mitochondrial dysfunction that leads to impaired insulin secretion and diabetes. Hum Mol Genet. 2014;23:5733–5749. doi: 10.1093/hmg/ddu288. [DOI] [PubMed] [Google Scholar]
- Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- Simon D, Seznec H, Gansmuller A, Carelle N, Weber P, Metzger D, Rustin P, Koenig M, Puccio H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J Neurosci. 2004;24:1987–1995. doi: 10.1523/JNEUROSCI.4549-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small ID, Peeters N. The PPR motif – a TPR-related motif prevalent in plant organellar proteins. Trends in biochemical sciences. 2000;25:46–47. doi: 10.1016/s0968-0004(99)01520-0. [DOI] [PubMed] [Google Scholar]
- Song L, Shan Y, Lloyd KC, Cortopassi GA. Mutant Twinkle increases dopaminergic neurodegeneration, mtDNA deletions and modulates Parkin expression. Hum Mol Genet. 2012;21:5147–5158. doi: 10.1093/hmg/dds365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, Larsson NG. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci. 2001;21:8082–8090. doi: 10.1523/JNEUROSCI.21-20-08082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Spinazzola A. Mitochondrial DNA mutations and depletion in pediatric medicine. Semin Fetal Neonatal Med. 2011;16:190–196. doi: 10.1016/j.siny.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D’Adamo P, Calvo S, Marsano RM, Donnini C, Weiher H, Strisciuglio P, Parini R, Sarzi E, Chan A, DiMauro S, Rötig A, Gasparini P, Ferrero I, Mootha VK, Tiranti V, Zeviani M. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic signaling. Gene. 2005;354:162–168. doi: 10.1016/j.gene.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Moraes CT. Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum Mol Genet. 2005;14:893–902. doi: 10.1093/hmg/ddi082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steglich G, Neupert W, Langer T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol Cell Biol. 1999;19:3435–3442. doi: 10.1128/mcb.19.5.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem. 1992;267:14592–14597. [PubMed] [Google Scholar]
- Sterky FH, Ruzzenente B, Gustafsson CM, Samuelsson T, Larsson NG. LRPPRC is a mitochondrial matrix protein that is conserved in metazoans. Biochemical and biophysical research communications. 2010;398:759–764. doi: 10.1016/j.bbrc.2010.07.019. [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Elo JM, Pietilainen KH, Hakonen AH, Sevastianova K, Korpela M, Isohanni P, Marjavaara SK, Tyni T, Kiuru-Enari S, Pihko H, Darin N, Ounap K, Kluijtmans LA, Paetau A, Buzkova J, Bindoff LA, Annunen-Rasila J, Uusimaa J, Rissanen A, Yki-Jarvinen H, Hirano M, Tulinius M, Smeitink J, Tyynismaa H. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet neurology. 2011;10:806–818. doi: 10.1016/S1474-4422(11)70155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supale S, Thorel F, Merkwirth C, Gjinovci A, Herrera PL, Scorrano L, Meda P, Langer T, Maechler P. Loss of prohibitin induces mitochondrial damages altering beta-cell function and survival and is responsible for gradual diabetes development. Diabetes. 2013;62:3488–3499. doi: 10.2337/db13-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Kato M, Nakamura M, Yoshikawa T, Kanba S, Sano A, Kato T. Exome sequencing identifies a novel missense variant in RRM2B associated with autosomal recessive progressive external ophthalmoplegia. Genome Biol. 2011;12:R92. doi: 10.1186/gb-2011-12-9-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzioglu M, Ruzzenente B, Harmel J, Mourier A, Jemt E, Lopez MD, Kukat C, Stewart JB, Wibom R, Meharg C, Habermann B, Falkenberg M, Gustafsson CM, Park CB, Larsson NG. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013;17:618–626. doi: 10.1016/j.cmet.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, Zeviani M. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Trott A, Morano KA. SYM1 is the stress-induced Saccharomyces cerevisiae ortholog of the mammalian kidney disease gene Mpv17 and is required for ethanol metabolism and tolerance during heat shock. Eukaryot Cell. 2004;3:620–631. doi: 10.1128/EC.3.3.620-631.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkilä S, Wenz T, Ruhanen H, Guse K, Hemminki A, Peltola-Mjøsund KE, Tulkki V, Oresic M, Moraes CT, Pietiläinen K, Hovatta I, Suomalainen A. Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet. 2010;19:3948–3958. doi: 10.1093/hmg/ddq310. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H, Sembongi H, Bokori-Brown M, Granycome C, Ashley N, Poulton J, Jalanko A, Spelbrink JN, Holt IJ, Suomalainen A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet. 2004;13:3219–3227. doi: 10.1093/hmg/ddh342. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Sun R, Ahola-Erkkilä S, Almusa H, Pöyhönen R, Korpela M, Honkaniemi J, Isohanni P, Paetau A, Wang L, Suomalainen A. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum Mol Genet. 2012;21:66–75. doi: 10.1093/hmg/ddr438. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Ylikallio E, Patel M, Molnar MJ, Haller RG, Suomalainen A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am J Hum Genet. 2009;85:290–295. doi: 10.1016/j.ajhg.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schägger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. The EMBO journal. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino ML, Martí R, Tadesse S, López LC, Manes JL, Lyzak J, Hahn A, Carelli V, Hirano M. Thymidine and deoxyuridine accumulate in tissues of patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) FEBS letters. 2007;581:3410–3414. doi: 10.1016/j.febslet.2007.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin MJ, An D, Lee KY, Ilkayeva OR, Zingaretti CM, Emanuelli B, Smyth G, Cinti S, Newgard CB, Gibson BW, Larsson NG, Kahn CR. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012;16:765–776. doi: 10.1016/j.cmet.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroya J, Dorado B, Vilà MR, Garcia-Arumí E, Domingo P, Giralt M, Hirano M, Villarroya F. Thymidine kinase 2 deficiency-induced mitochondrial DNA depletion causes abnormal development of adipose tissues and adipokine levels in mice. PLoS One. 2011;6:e29691. doi: 10.1371/journal.pone.0029691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C, Spinazzola A, Maggioni M, Fernandez-Vizarra E, Massa V, Pagano C, Vettor R, Mora M, Zeviani M. Early-onset liver mtDNA depletion and late-onset proteinuric nephropathy in Mpv17 knockout mice. Hum Mol Genet. 2009;18:12–26. doi: 10.1093/hmg/ddn309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TH, Tanji K, Holve SA, Bonilla E, Sokol RJ, Snyder RD, Fiore S, Deutsch GH, Dimauro S, De Vivo D. Navajo neurohepatopathy: a mitochondrial DNA depletion syndrome? Hepatology. 2001;34:116–120. doi: 10.1053/jhep.2001.25921. [DOI] [PubMed] [Google Scholar]
- Walter MC, Czermin B, Muller-Ziermann S, Bulst S, Stewart JD, Hudson G, Schneiderat P, Abicht A, Holinski-Feder E, Lochmuller H, Chinnery PF, Klopstock T, Horvath R. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. Journal of neurology. 2010;257:1517–1523. doi: 10.1007/s00415-010-5565-9. [DOI] [PubMed] [Google Scholar]
- Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999a;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- Wang L, Eriksson S. Cloning and characterization of full-length mouse thymidine kinase 2: the N-terminal sequence directs import of the precursor protein into mitochondria. Biochem J. 2000;351(Pt 2):469–476. [PMC free article] [PubMed] [Google Scholar]
- Wang L, Limongelli A, Vila MR, Carrara F, Zeviani M, Eriksson S. Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes. Mol Genet Metab. 2005;84:75–82. doi: 10.1016/j.ymgme.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Wang L, Munch-Petersen B, Herrström Sjöberg A, Hellman U, Bergman T, Jörnvall H, Eriksson S. Human thymidine kinase 2: molecular cloning and characterisation of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS letters. 1999b;443:170–174. doi: 10.1016/s0014-5793(98)01711-6. [DOI] [PubMed] [Google Scholar]
- Wang X, Pickrell AM, Rossi SG, Pinto M, Dillon LM, Hida A, Rotundo RL, Moraes CT. Transient systemic mtDNA damage leads to muscle wasting by reducing the satellite cell pool. Hum Mol Genet. 2013;22:3976–3986. doi: 10.1093/hmg/ddt251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. The New England journal of medicine. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Weiher H, Noda T, Gray DA, Sharpe AH, Jaenisch R. Transgenic mouse model of kidney disease: insertional inactivation of ubiquitously expressed gene leads to nephrotic syndrome. Cell. 1990;62:425–434. doi: 10.1016/0092-8674(90)90008-3. [DOI] [PubMed] [Google Scholar]
- Wenz T, Luca C, Torraco A, Moraes CT. mTERF2 regulates oxidative phosphorylation by modulating mtDNA transcription. Cell Metab. 2009;9:499–511. doi: 10.1016/j.cmet.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Williams PA, Morgan JE, Votruba M. Mouse models of dominant optic atrophy: what do they tell us about the pathophysiology of visual loss? Vision Res. 2011;51:229–234. doi: 10.1016/j.visres.2010.08.031. [DOI] [PubMed] [Google Scholar]
- Williams PA, Piechota M, von Ruhland C, Taylor E, Morgan JE, Votruba M. Opa1 is essential for retinal ganglion cell synaptic architecture and connectivity. Brain. 2012;135:493–505. doi: 10.1093/brain/awr330. [DOI] [PubMed] [Google Scholar]
- Williams SL, Huang J, Edwards YJ, Ulloa RH, Dillon LM, Prolla TA, Vance JM, Moraes CT, Züchner S. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 2010;12:675–682. doi: 10.1016/j.cmet.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortmann SB, Champion MP, van den Heuvel L, Barth H, Trutnau B, Craig K, Lammens M, Schreuder MF, Taylor RW, Smeitink JA, Wevers RA, Rodenburg RJ, Morava E. Mitochondrial DNA m.3242G > A mutation, an under diagnosed cause of hypertrophic cardiomyopathy and renal tubular dysfunction? Eur J Med Genet. 2012;55:552–556. doi: 10.1016/j.ejmg.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Hu Y, Quiros PM, Wei Q, Lopez-Otin C, Dong Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am J Physiol Renal Physiol. 2014;306:F1318–1326. doi: 10.1152/ajprenal.00036.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Addis JB, Cameron JM, Robinson BH. LRPPRC mutation suppresses cytochrome oxidase activity by altering mitochondrial RNA transcript stability in a mouse model. Biochem J. 2012;441:275–283. doi: 10.1042/BJ20110985. [DOI] [PubMed] [Google Scholar]
- Xu F, Morin C, Mitchell G, Ackerley C, Robinson BH. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem J. 2004;382:331–336. doi: 10.1042/BJ20040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Stahl JS, Andrade FH, McMullen CA, Webb-Wood S, Newman NJ, Biousse V, Wallace DC, Pardue MT. Eliminating the Ant1 isoform produces a mouse with CPEO pathology but normal ocular motility. Invest Ophthalmol Vis Sci. 2005;46:4555–4562. doi: 10.1167/iovs.05-0695. [DOI] [PubMed] [Google Scholar]
- Ylikallio E, Tyynismaa H, Tsutsui H, Ide T, Suomalainen A. High mitochondrial DNA copy number has detrimental effects in mice. Hum Mol Genet. 2010;19:2695–2705. doi: 10.1093/hmg/ddq163. [DOI] [PubMed] [Google Scholar]
- Young MJ, Longley MJ, Li FY, Kasiviswanathan R, Wong LJ, Copeland WC. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum Mol Genet. 2011;20:3052–3066. doi: 10.1093/hmg/ddr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Davies VJ, Piechota MJ, Cree LM, Votruba M, Chinnery PF. Secondary mtDNA defects do not cause optic nerve dysfunction in a mouse model of dominant optic atrophy. Invest Ophthalmol Vis Sci. 2009;50:4561–4566. doi: 10.1167/iovs.09-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Chang SW, Denniger G, Feng Z, Zassenhaus HP. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69:151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Chang SW, Stevens M, Mikolajczak P, Zassenhaus HP. Mitochondrial DNA mutations activate programmed cell survival in the mouse heart. Am J Physiol Heart Circ Physiol. 2005;288:H2476–2483. doi: 10.1152/ajpheart.00670.2004. [DOI] [PubMed] [Google Scholar]
- Zhou X, Kannisto K, Curbo S, von Dobeln U, Hultenby K, Isetun S, Gafvels M, Karlsson A. Thymidine kinase 2 deficiency-induced mtDNA depletion in mouse liver leads to defect beta-oxidation. PLoS One. 2013;8:e58843. doi: 10.1371/journal.pone.0058843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Solaroli N, Bjerke M, Stewart JB, Rozell B, Johansson M, Karlsson A. Progressive loss of mitochondrial DNA in thymidine kinase 2-deficient mice. Hum Mol Genet. 2008;17:2329–2335. doi: 10.1093/hmg/ddn133. [DOI] [PubMed] [Google Scholar]
- Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM, Battologlu E. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]