

Abstract
Emerging data suggest that megakaryocytes (MKs) play a significant role in skeletal homeostasis. Indeed, osteosclerosis observed in several MK-related disorders may be a result of increased numbers of MKs. In support of this idea, we have previously demonstrated that MKs increase osteoblast (OB) proliferation by a direct cell-cell contact mechanism and that MKs also inhibit osteoclast (OC) formation. As MKs and OCs are derived from the same hematopoietic precursor, in these osteoclastogenesis studies we examined the role of the main MK growth factor, thrombopoietin (TPO) on OC formation and bone resorption. Here we show that TPO directly increases OC formation and differentiation in vitro. Specifically, we demonstrate the TPO receptor (c-mpl or CD110) is expressed on cells of the OC lineage, c-mpl is required for TPO to enhance OC formation in vitro, and TPO activates the MAPK, JAK/STAT, and NFκB signaling pathways, but does not activate the PI3K/AKT pathway. Further, we found TPO enhances OC resorption in CD14+CD110+ human OC progenitors derived from peripheral blood mononuclear cells (PBMCs), and further separating OC progenitors based on CD110 expression enriches for mature OC development. The regulation of OCs by TPO highlights a novel therapeutic target for bone loss diseases and may be important to consider in the numerous hematologic disorders associated with alterations in TPO/c-mpl signaling as well as in patients suffering from bone disorders.
Keywords: Osteoclasts, Thrombopoietin, c-mpl, Megakaryocytes, Bone Resorption, Growth Factors, Cytokines
INTRODUCTION
TPO, the main MK growth factor, is critical for normal MK proliferation and differentiation (Deng, et al., 1998; Broudy, et al., 1995; Bartley, et al., 1994; Kaushansky, et al., 1995; de Sauvage, et al., 1994; Wendling, et al., 1994; Zeigler, et al., 1994), and is a key initiator of thrombocytosis in many diseases. As we have previously reviewed (Kacena, et al., 2006a; Kacena and Horowitz, 2006), MKs and/or TPO can play a role in skeletal homeostasis. In brief, MKs have been shown to: 1) Express and/or secrete several bone-related proteins (Thiede, et al., 1994; Kelm, et al., 1992; Breton-Gorius, et al., 1992; Chenu and Delmas, 1992; Frank, et al., 1993; Sipe, et al., 2004; Bord, et al., 2005; Pearse, et al., 2001; Chagraoui, et al., 2003a); 2) Stimulate OB proliferation (Kacena, et al., 2004; Ciovacco, et al., 2009; Lemieux, et al., 2010; Ciovacco, et al., 2010; Kacena, et al., 2012; Cheng, et al., 2013; Miao, et al., 2004); 3) Alter OB differentiation (Bord, et al., 2005; Ciovacco, et al., 2009); and 4) Inhibit OC formation (Beeton, et al., 2006; Kacena, et al., 2006b). Further, in humans, myeloproliferative diseases in which increases in MKs is accompanied by osteosclerosis have been reported (Thiele, et al., 1999; Lennert, et al., 1975; Chagraoui, et al., 2006), and at least 4 mouse models have been described in which MK number is significantly elevated and these mice also exhibit an increased bone phenotype (Yan, et al., 1995; Yan, et al., 1996; Villeval, et al., 1997; Frey, et al., 1998a; Frey, et al., 1998b; Kacena, et al., 2004; Kacena, et al., 2005; Suva, et al., 2008). With respect to mouse models, mice overexpressing TPO have approximately a 4-fold increase in MK number and have an osteosclerotic bone phenotype (Villeval, et al., 1997; Yan, et al., 1996). While some researchers (Chagraoui, et al., 2003b; Kakumitsu, et al., 2005) have implicated the upregulation of osteoprotegerin (OPG), which inhibits OC development, as being responsible for the high bone mass in TPO overexpressing mice, others have implicated TPO itself (Wakikawa, et al., 1997). To test whether TPO inhibited OC development Wakikawa et al. (1997) performed a series of in vitro studies which demonstrated that TPO dose-dependently reduced OC number in bone marrow (BM) cultures. Importantly, TPO treatment also increased MK number in cultures. Thus, the inhibition of OC formation seen by Wakikawa et al. (1997) is most likely the result of increased number of MKs from TPO stimulation inhibiting OC number rather than TPO directly inhibiting OC number. Indeed, studies from our laboratory (Kacena, et al., 2006b; Ciovacco, et al., 2010) and others (Beeton, et al., 2006) have shown that MKs cultured in the absence of TPO, dose-dependently inhibit OC formation. In addition, TPO-free, MK conditioned medium also dose-dependently inhibited OC formation (Kacena, et al., 2006b). Further, MKs derived from OPG-deficient mice also inhibited OC development (Kacena, et al., 2006b). The combination of these data suggests the following. First, MK-secreted OPG, alone, is not responsible for MK-mediated inhibition of osteoclastogenesis. Second, OC inhibition by TPO likely has an indirect effect on OC formation by directly stimulating MKs and the MKs in turn inhibit OC formation. Thus, in this study we examined whether TPO and/or its receptor, c-mpl, also play a direct role in osteoclastogenesis.
MATERIALS AND METHODS
Mice
For these studies c-mpl−/− and wild-type C57BL/6 mice were utilized. c-mpl−/− mice were kindly provided by Genentech. Generation and breeding of c-mpl−/− mice was previously described (Tong and Lodish, 2004; de Sauvage, et al., 1994). c-mpl−/− mice were maintained on the C57BL/6 background. C57BL/6 mice were obtained from Jackson Laboratories. All procedures (detailed below) were approved by the Indiana University Institutional Animal Care and Use Committee (IACUC) and followed NIH guidelines as well as the Guide for the Care and Use of Laboratory Animals.
Preparation of Murine Fetal Liver Derived MKs
Murine MKs were prepared as previously described (Kacena, et al., 2004, Kacena, et al., 2006b). Briefly, fetal livers were obtained from C57BL/6 mice at E13–15 and single cell suspensions generated. Cells were washed twice and cultured in DMEM + 10% FCS + 1% murine TPO (Villeval, et al., 1997). After 5 days, MKs were obtained by separating them from the lymphocytes and other cells using a one-step albumin gradient to obtain a 90–95% pure MK population (Drachman, et al., 1997).
Preparation of Murine Bone Marrow (BM) Cells
Long bones were dissected from 6–10 week old mice, the epiphyses removed, and the marrow flushed with ice cold α-MEM with 10% FCS. A single cell suspension was prepared and the cells were washed twice prior to use.
Preparation of Murine Bone Marrow Macrophages (BMMs)
BM cells were prepared as above. 5×107 cells/ml were seeded into 100 mm tissue culture dishes, in α-MEM supplemented with 10% FCS and 20 ng/ml of M-CSF (R&D Systems). After 3 days in culture, adherent cells were lifted with trypsin, and seeded at 100,000 cells/ml for OC generation as described below.
Generation of Peripheral Blood Mononuclear Cells (PBMCs)
Following Indiana University Institutional Review Board approval in compliance with the World Medical Association Declaration of Helsinki, peripheral blood was collected in heparin-sulfate tubes from healthy adult volunteers or obtained as buffy coat preparations from the Indiana Blood Center. Blood was diluted 1:4 with HBSS and centrifuged over Ficoll/Hypaque (GE Healthcare Biosciences), and interphase low density cells were collected and washed twice prior to seeding as detailed below.
In Vitro OC-like Cell Formation Models
OC-like cells were generated as follows. Murine: 100,000 murine BMMs were cultured in α-MEM supplemented with 10% FCS and 30 ng/ml of M-CSF (R&D Systems) and 50 ng/ml RANKL (R&D Systems). Media was changed every third day for 6–8 days (until OCs formed). TPO was kindly provided by Genentech and was titrated (0–1000ng/ml) into cultures. Human: PBMCs were seeded into 96-, 48- and 24-well culture dishes at an initial density of 2.1×105 cells/mm2 and cultured in alpha-MEM (Invitrogen) supplemented with 10% FBS (Hyclone) and 20 ng/ml of recombinant human M-CSF (Peprotech) for 2 days and then supplemented with 20 ng/ml of recombinant human M-CSF and 80 ng/ml of recombinant human RANKL (Peprotech) for the remaining duration of the experiment. Cells were cultured in the absence or presence of TPO (100 ng/ml). The cell culture medium was changed every third day until OCs were visible. For both mouse and human cells, once OCs had formed, the cells were fixed with either 2.5% glutaraldehyde or 3.7% formaldehyde in phosphate buffered saline for 30 minutes at room temperature, stained for tartrate resistant acid phosphatase (TRAP, Sigma-Aldrich), and TRAP+, multinucleated (≥3) OC cells were counted.
The resorption activity of OCs cultured in the absence or presence of TPO (100 ng/ml) was evaluated using a standard pit assay (Tanaka, et al., 1996). PBMCs were isolated as above and plated into 6-well culture dishes at 2×106 cells/well. Cells were incubated in alpha-MEM containing 10% FBS and 20 ng/ml M-CSF for 2 days. The media was removed and replaced with fresh media containing 20 ng/ml M-CSF and 80 ng/ml RANKL until mature OCs were observed. Mature OCs were detached by trypsinization, washed once, re-plated onto dentin slices (Immunodiagnostics Systems Inc, Fountain Hills, AZ) and cultured for an additional 48 hrs in media containing 20 ng/ml M-CSF and 80 ng/ml RANKL (± 100 ng/ml TPO). Dentin slices were washed, incubated in 6% NaOCl for 5 min, and sonicated for 20 s to remove cells. Resorption pits were stained with a solution containing 1% toluidine blue and 1% sodium borate for 1 min, washed with water and air-dried. Pit surface area was quantified using the ImagePro 7.0 on a Leica DMI4000 with a 10X objective. Results were normalized for TRAP+ OC number, as detailed above. Experiments were performed in triplicate and results represent average pit area per dentin slice/OC number.
RNA Extraction and Real-Time PCR
Cells were washed 2 times with PBS. RNA was isolated from the cells using a NucleoSpin II RNA Purification kit (BD Biosciences) incorporating an on-column DNase treatment to remove contaminating genomic DNA. For real-time PCR, cDNA was prepared from 5μg of total RNA using Sprint PowerScript Reverse Transcriptase (BD Biosciences) and oligo(dT)12–18 primers. The cDNA was purified using an Amicon YM30 filter device (Millipore). Quantitative real-time PCR was performed on a Cepheid Smart Cycler using Platinum Taq polymerase (Invitrogen) and Sybr Green I (Molecular Probes) incorporation.
The quantitative comparison between samples was calculated using the comparative CT method. The following primer sequences were used:
c-mpl forward primer: 5′ TCACCTTGGTGACTGCTCTG
c-mpl reverse primer: 5′ GGACTTAGGGCTGCAGTGTC
GAPDH forward primer: 5′ CGTGGGGCTGCCCAGAACAT
GPADH reverse primer: 5′ TCTCCAGGCGGCACGTCAGA
Western Blotting
Seventy-five percent confluent cultures of C57BL/6 BMM, RAW 264.7, C57BL/6 MKs, or Ba/F3 cells were placed in reduced serum media (0.5%) for 16–18 hours. The cells were stimulated with recombinant human (rh) TPO (100 ng/ml) for 0, 1, 3, 5, 10, and 30 minutes in the presence of 100 μM sodium orthovanadate. In studies using pharmacological inhibitors BMMs were treated for 30 minutes with 100 ng/ml TPO and pharmacological inhibitors (Calbiochem) of the JAK-STAT (1,2,3,4,5,6-Hexa-bromocyclohexane), MAPK (PD98059), or NF-κB (Wedelolactone) pathways and cell lysates collected in the presence of inhibitors (Halt Protease Inhibitor Cocktail and Halt Phosphatase Cocktail, Pierce). For immunoprecipitation, cell lysates (200–500 μg) were precleared with Protein A-Sepharose for 1 hour at 4°C. Protein A-Sepharose was removed and cell supernatants were incubated in the appropriate antibody (e.g. αP-Tyr) for 2–16 hours at 4°C. STAT3, p-AKT, αP-Tyr, and IgG antibodies were purchased from Millipore, and ERK1/2 (MAPK) was purchased from Chemicon International. The antigen-antibody complexes were recovered during a 30–60 minute incubation using rabbit anti-mouse immunoglobulin and protein A-Sepharose or protein A-Sepharose alone. The immune complexes were washed 3–5 times with lysis buffer and the immunoprecipitated proteins were eluted into SDS-PAGE sample buffer (3% SDS, 60mM Tris, pH 6.9, 2 mM EDTA, 4% glycerol) by heating the samples to 100°C for 5 minutes.
Immunoprecipitated material (or aliquots of the total cell lysates, 25–30 μg), was analyzed by 10% SDS-PAGE under reducing conditions. Proteins were transferred to nitrocellulose via electrophoresis. Western blotting procedures were performed essentially as outlined in the ECL system (Amersham). To reprobe blots, nitrocellulose filters were stripped of antibodies in a solution containing 0.1M glycine pH2.8, at 55°C for 30 minutes.
Electrophoretic Mobility Shift Assay (EMSA)
Seventy-five percent confluent cultures of C57BL/6 BMM cells were placed in reduced serum media (0.5%) for 16–18 hours. The cells were stimulated with human recombinant TPO (100 ng/ml) for 30 minutes and nuclear proteins extracted as described by Andrews et al. (1991) EMSA was performed on 7μg of nuclear protein using a commercially available EMSA kit and NF-kB probe according to the manufacturer’s instructions (Panomics).
Flow Cytometric Sorting
PBMCs were isolated as detailed above. Cells were washed 2X with HBSS containing 2% FBS. Staining was performed in HBSS with 2% FBS. The following antibodies were purchased from PharMingen: CD110 (c-mpl), CD11b, and CD14. Light scatter and fluorescence of individual cells was measured by a FACS ARIA flow cytometer (BD), and cells were sorted based on their antigen expression.
Statistics
Unless otherwise stated, all data are presented as the Mean ± SD and a Student’s t-test was used to determine significant differences, with p<0.05 (Systat 6.0 for Microsoft Windows, SPSS Inc., Chicago). Within individual experiments, data points are based on a minimum of triplicate representative samples and experiments were repeated at least twice.
RESULTS
C-mpl Expression in OC Progenitors
Previously, it was believed that the TPO receptor, the proto-oncogene c-mpl, was primarily restricted to cells of the MK and hematopoietic stem cell lineages (Methia, et al., 1993; Gurney, et al., 1994; Bartley, et al., 1994; de Sauvage, et al., 1994; Solar, et al., 1998). Our real-time PCR data demonstrate the novel finding that purified OC progenitors express mRNA for c-mpl. Specifically, the comparative CT method was used to compare c-mpl expression in different types of cells. MKs served as a positive control and had the greatest expression (normalized minimum threshold cycle, ΔCT = 25.0±1.1). All OC progenitors tested expressed c-mpl (all above acceptable threshold levels), with C57BL/6 bone marrow macrophages (BMM, 29.5±1.7) > RAW264.7 cells (33.08±0.04) > Pax5−/− spleen cell line (OC progenitor cell line, 33.2±2.2). Of importance, although these data demonstrate that c-mpl is expressed in BMMs, they do not confirm that these mRNAs are translated into proteins. However, we subsequently demonstrated by both Western blot analysis and immunocytochemistry techniques that c-mpl protein is indeed expressed in OC progenitors (Fig. 1A–C). Importantly, we also examined c-mpl expression by real-time PCR, Western blot, and immunocytochemistry and found that mature, multinucleated, murine, TRAP+ OC cells (developed in the presence of M-CSF and RANKL) did not express c-mpl (data not shown). The fact that c-mpl is expressed in OC progenitors but not in mature OCs suggests that TPO/c-mpl exerts its effect during the early stages of OC proliferation/differentiation.
Figure 1. OC progenitors express c-mpl protein and TPO stimulation increases OC formation.

A) Western blot analysis of protein lysates from Baf3 cells (transfected with c-mpl), four separate BMM samples, RAW 264.7 cells, and MKs probed with antibodies raised against c-mpl. B&C) BMMs stained via standard immunocytochemistry with a c-mpl antibody (green) and counter stained with DAPI (blue). C) c-mpl antibody control. D&E) Micrographs of BMMs cultured in the absence (D) and presence (E) of 100ng/ml of rhTPO. F) Quantitation of mature OC number when C57BL/6 BMMs were stimulated with rhTPO (0.1–100 ng/ml). TPO enhances mature C57BL/6 OC number by >2-fold. G) Quantitation of mature OC number when C57BL/6 and c-mpl−/− BMMs were cultured in the absence and presence of rhTPO (100 ng/ml). TPO significantly enhanced OC number in cells generated from C57BL/6 mice. OC number was unchanged with TPO treatment in c-mpl −/− cultures (G). Significantly fewer OCs were generated from c-mpl −/− mice as compared to C57BL/6 mice (G). Data are presented as the mean±SD. *Indicates statistically significant differences (p<0.05) compared to C57BL/6 BMMs cultured without TPO. It should be noted that we normalized OC cell count to 100 in the control (0 ng/ml TPO) for ease of comparison between data in Figures 1F and 1G.
TPO Enhances OC Formation In Vitro
While our data show that c-mpl is expressed in OC progenitor cells, it is critical to determine whether this expression results in a functional change in OC proliferation/differentiation. Here we show that stimulation of OC progenitor cells with TPO enhances OC formation in vitro.
OCs were generated using C57BL/6 BMMs as a source of OC precursors. BMMs were selected for these studies to minimize possible contamination of cultures with MKs so that the effects of TPO directly on OC progenitors could be assessed. rhTPO was titrated into the OC-like cell formation models at day 0 and remained in the cultures for the growth duration. Fig. 1D&E are representative micrographs showing the TPO-mediated enhancement in OC formation. As Fig. 1F illustrates, when C57BL/6 BMMs were cultured in the presence of M-CSF and RANKL, stimulation with TPO resulted in a dose-dependent increase in OC formation. Indeed, stimulation with 100 ng/ml of TPO results in up to a 6-fold increase in OC number.
Importantly, we have used recombinant human TPO from 3 separate vendors (R&D Systems, Peprotech, and Genentech) and all perform similarly and report that endotoxin levels are less than 1.0EU/μg of protein, suggesting that lipopolysaccharide (LPS) activity or other contaminants are not responsible for the increase in osteoclastogenesis observed (data not shown). We have also found that several different OC progenitor cell lines (including RAW 264.7 cells) which do not contain MKs/MK progenitors, express c-mpl, and have observed similar increases in OC number with TPO stimulation (Figure 1A and data not shown).
Requirement of c-mpl for TPO to enhance OC formation in vitro
To demonstrate that binding of TPO to c-mpl is required for TPO to enhance OC formation we cultured BMMs from C57BL/6 control and c-mpl−/− mice. BMMs were cultured in the presence of M-CSF and RANKL and with or without 100 ng/ml of TPO. Once OCs were formed, cells were fixed, stained for TRAP, and TRAP+ multinucleated cells (≥3 nuclei) were counted. As shown in Figure 1G, when wild-type control BMMs were treated with TPO, OC number was significantly elevated. However, when c-mpl−/− BMMs were treated with TPO, there was no difference in the number of OCs generated. It is important to note that significantly fewer OCs were formed when OC progenitors were generated from c-mpl−/− mice as compared to those generated from control mice, likely owing to the disruption in myeloid lineage cells previously documented in c-mpl−/− mice (Carver-Moore, et al., 1996).
TPO Signaling Pathways Augment Osteoclastogenesis
To determine whether TPO alone could stimulate OC formation we cultured BMMs with or without 100 ng/ml of TPO. In addition to cultures containing 0 or 100 ng/ml of TPO alone, other cultures contained 0 or 100 ng/ml of TPO in addition to the following supplementation: both M-CSF and RANKL, M-CSF alone, and RANKL alone. Importantly, only those cultures containing both M-CSF and RANKL generated TRAP+ OC-like cells (data not shown).
Since these data support the idea that TPO acts to augment M-CSF and RANKL mediated OC formation, we then determined which signaling pathways were activated by TPO stimulation of OC progenitors. To do this BMMs were serum starved overnight and then stimulated with 100 ng/ml of rhTPO for 0, 1, 3, 5, 10, or 30 minutes. As a positive control, primary MKs derived from the fetal livers of E13–15 C57BL/6 mice or BaF3 (c-mpl transfected cells) were stimulated with TPO for 0 or 10 minutes. Lysates were collected and immunoblotted with appropriate anti-phosphotyrosine antibodies for JAK/STAT, MAPK, and PI3K/AKT pathways. The JAK/STAT and MAPK pathways were examined because they are major TPO/c-mpl mediated signaling pathways in MKs (see Supplemental Fig. 1). The PI3K/AKT pathway was examined because it is a nodal point whereby c-mpl and c-Fms signaling may converge (see Supplemental Fig. 1). As seen in Figure 2A&B, Western blotting demonstrated that treating BMMs with 100 ng/ml of TPO activates a variety of intracellular signaling mechanisms, including phosphorylating members of the MAPK and STAT signaling pathways (ERK1/2 and STAT3, respectively). Western blotting also illustrated that AKT is not phosphorylated in BMMs treated with TPO (data not shown). We then performed similar studies using pharmacological inhibitors which target the JAK/STAT pathway (1,2,3,4,5,6-Hexa-bromocyclohexane) or the MAPK pathway (PD98059). These studies confirmed that TPO stimulation specifically activates both of these pathways (Fig. 2A&B). We next examined the importance of the NF-κB signaling pathway in OC progenitors stimulated with TPO as the NF-κB signaling pathway is of critical importance in RANKL mediated OC differentiation (see Supplemental Fig. 1). For NF-κB studies, following stimulation with or without TPO, nuclear extracts were prepared and electrophoretic mobility shift assays (EMSAs) were performed. Fig. 2C demonstrates that TPO induces NF-κB activation in OC precursors. Similar to the studies outlined above, we repeated the NF-κB gel mobility shift assays using a specific pharmacological inhibitor which targets the NF-κB pathway (Wedelolactone). Again, these studies confirmed that TPO stimulation specifically activates the NF-κB pathway (Fig. 2C).
Figure 2. TPO treatment activates the JAK/STAT, MAPK, and NF-κB signaling pathways in OC progenitors.

A&B) Western blot analysis of BMMs treated for 30 minutes with 100 ng/ml TPO and pharmacological inhibitors of the JAK/STAT (1,2,3,4,5,6-Hexa-bromocyclohexane) or the MAPK (PD98059) pathways probed with antibodies against phosphorylated and unphosphorylated STAT3 (A) and phosphorylated and unphosphorylated MAPK (B). C) EMSA of BMMs treated for 30 minutes with 100 ng/ml of TPO and pharmacological inhibitor of the NF-κB pathway (Wedelolactone) probed for NF-κB.
Effects of TPO Stimulation and c-mpl Expression in Human OC Progenitors
As all of the previous studies were conducted using murine cells, we next determined whether these findings could be replicated using human derived OC progenitors. PBMCs from normal healthy donors were cultured with M-CSF (20 ng/ml), RANKL (80 ng/ml), and TPO (0 or 100 ng/ml) as detailed in the Methods section. As depicted in Figure 3A, culture of PBMCs with M-CSF and RANKL was able to induce OC formation in all 8 specimens tested, although a large variability in the total numbers of OCs was observed. However, treatment with TPO did not alter the number of OCs formed (Fig. 3A). This may suggest that the heterogeneous population of cells contained within PBMCs as a group are not responsive to TPO, but does not address whether human OC progenitors alone are responsive to TPO. Thus, we next attempted to address this question by examining phenotypically defined OC progenitors isolated flow cytometrically based on the expression of CD11b or CD14 which are both commonly used to separate OC progenitors and we added to these well characterized OC cell surface markers, c-mpl (CD110). Specifically, in our design, CD11b and CD14 subpopulations were further separated based on the expression of CD110. As would be expected, compared to unsorted cells seeded at the same density, enriching for OC progenitors resulted in higher production of more mature OCs/cells seeded as long as the OC progenitors were CD110+ (Fig. 3B). However, few TRAP+ OCs were observed when CD11b+CD110− or CD14+CD110− populations were used as the OC progenitor populations. On the other hand, numerous TRAP+ OCs were observed when CD11b+CD110+ or CD14+CD110+ cells were used as OC progenitors and TPO further enhanced OC number in these subpopulations. Next we examined the dentin-resorbing activity of the OCs generated from the unsorted population as well as the subpopulations of OC progenitors (Fig. 3C). Mature, multinucleated OCs were re-plated on dentin slices for 48 hrs and the area resorbed (defined as the total resorption pit area on the dentin slice/TRAP+ OC) was quantified and normalized for TRAP+ OCs. When normalized for the number of TRAP+ OCs, the activity of the OCs was highest in the unsorted PBMC population. Activity was significantly lower in all of the sorted populations examined. Due to the limited number of TRAP+ OCs observed in cultures generated from CD11b+CD110− and CD14+CD110− cells (panel B) we were unable to obtain activity data for this population. Overall, these activity data suggest that accessory cells in total PBMCs cultures augment osteoclastic resorption. Of interest, while TPO treatment only modestly enhanced the bone resorbing activity of CD11b+CD110+ cells, it robustly enhanced the activity in CD14+CD110+ cells when compared to any of the other populations tested. Finally, as detailed in Fig. 3D, c-mpl expression on CD14+ cells resulted in an expression-dependent increase in mature OC formation. Specifically, we found that CD14+ cells could be divided into 3 subpopulations based on CD110 expression (CD110+ or high expression, CD110dim or low expression, and CD110− or no detectable expression) and the numbers of OCs generated from these populations positively correlated with the level of c-mpl expression. It should be noted that we did not assess c-mpl expression profile in CD11b+ cells due to the expected difficulty in obtaining sufficient numbers of cells from cell sorting.
Figure 3. Effects of TPO stimulation and c-mpl expression on OC formation and resorption.

A) PBMCs, from healthy volunteers, were isolated and cultured with M-CSF (20 ng/ml), RANKL (80 ng/ml), and TPO (0 or 100 ng/ml). Although OCs formed in all specimens tested, treatment with TPO did not alter OC number. B &C) PBMCs were separated into subpopulations based on the expression of either CD11b or CD14, cell surface markers for OC progenitors, and then further separated based on c-mpl (CD110) expression. Representative data from one of the healthy volunteers is shown. B) Few TRAP+ OCs were generated from CD11b+CD110− or CD14+CD110− populations. Numerous TRAP+ OCs were generated from CD11b+CD110+ or CD14+CD110+ populations. C) The activity of the OCs generated from these subpopulations was also assessed. When normalized for the number of TRAP+ OCs, the activity of the OCs (defined as the total resorption pit area on the dentin slice/TRAP+ OCs) was highest in the unsorted PBMC population. Activity was significantly lower in all of the sorted populations examined. Of interest, TPO treatment significantly enhanced the activity, in CD14+CD110+ cells. Due to the limited number of TRAP+ OCs generated from CD11b+CD110− and CD14+CD110− cells (panel B) we were unable to obtain activity data for these populations. D) In another study CD14+ cells were further separated into CD110+, CD110dim, and CD110− populations, cultured as above, and OC number determined. A CD110 expression-dependent increase in OC number was observed.
Expression Schema Enriching for OC Progenitors
Next, as both CD11b+CD110+ and CD14+CD110+ populations generated large numbers of mature OCs, we determined whether the cells giving rise to mature OCs were an overlapping cell population (e.g. CD14+CD11b+CD110+). Fig. 4A shows the cell sorting strategy used to obtain the cell populations of interest. As detailed in Fig. 4B, cells positive for all three surface proteins (CD14+CD11b+CD110+ cells) gave rise to 320±87 mature OCs whereas all of the other populations tested (CD14−CD11b+CD110+, CD14+CD11b−CD110+, CD14−CD11b−CD110+, and all of the CD110− cells which were either CD14+ and/or CD11b+) gave rise to less than 18±10 OCs, with no visible OCs in either of the CD14− populations tested. Finally, it should be noted that we attempted to confirm these human PBMC data using bone marrow from C57BL/6 mice, but were unable to conduct these studies as none of the commercially available anti-mouse CD110 antibodies were suitable for multicolor flow cytometric assessment and few displayed unmanageable non-specific binding. Nevertheless, our data demonstrates that, in humans, an enriched population of OC progenitors can be obtained using the following cell sorting schema CD14+CD11b+CD110+.
Figure 4. Cell sorting schema to obtain an enriched population of OC progenitors.

A) Histogram and dot plots showing the fluorescence activated cells sorting strategy used to obtain the populations of cells tested. The numbered populations in panel A correspond to the populations in panels B. CD14+CD11b+CD110+ cells give rise to more mature OCs than do any other population of cells tested.
DISCUSSION
In 1997, Wakikawa et al. published a manuscript entitled “Thrombopoietin inhibits in vitro osteoclastogenesis from murine bone marrow cells”. In this study they demonstrated that in vitro treatment of BM cells with TPO inhibited OC formation. As would be expected, BM cultures treated with TPO also contained high numbers of MKs. Of importance, when OC progenitors are cultured with MKs, or MK conditioned medium, OC development is inhibited by up to 10-fold (Beeton, et al., 2006; Kacena, et al., 2006b). Further, OPG expression alone was not responsible for this inhibition as MKs derived from OPG deficient mice were similarly able to inhibit OC formation (Kacena, et al., 2006b). Interestingly, in the study by Wakikawa and colleagues (1997) it is the indirect effect of TPO which mediates an increase in MK numbers that is responsible for the inhibition of OC formation in vitro, rather than the direct action of TPO on OC progenitor cells. Thus, if whole bone marrow is used, as used by Wakikawa et al. (1997), which contains mature MKs or MK progenitors, the inhibitory effects of MKs on OC formation appear to outweigh the positive effects of TPO on OC formation, resulting in a net inhibition of OC formation.
Therefore TPO, with both indirect and direct effects on osteoclastogenesis, has a somewhat paradoxical effect on OC number. This may in part be due to the fact that MKs, which express increased levels of c-mpl, are competing with OC progenitors for the TPO present within the bone marrow cavity (Sungaran, et al., 1997). Importantly, in normal bone homeostasis, the bone marrow cavity contains all four of these elements: TPO, MKs, OBs, and OCs. Thus, elucidation of the interactions between these elements will allow a better understanding of normal and pathological skeletal regulation.
Fig. 5 describes our current working model of the complex interactions between OBs, OCs, MKs, and TPO. Specifically, MKs induce OB activation in vitro via mechanisms requiring direct physical contact between the two cell types (Kacena, et al., 2005; Kacena, et al., 2004; Kacena et al., 2012; Cheng et al., 2013). MKs also inhibit OC development in vitro via the secretion of as-yet unidentified soluble factor(s) (Beeton, et al., 2006; Kacena, et al., 2006b). The net result, as demonstrated in vivo, is an increase in MK numbers that can lead to concomitant increases in bone mass (Kacena, et al., 2005; Kacena, et al., 2004; Cheng et al., 2013).
Figure 5. Working model showing how TPO directly stimulates OC progenitor cells while indirectly stimulating OB proliferation through MKs.
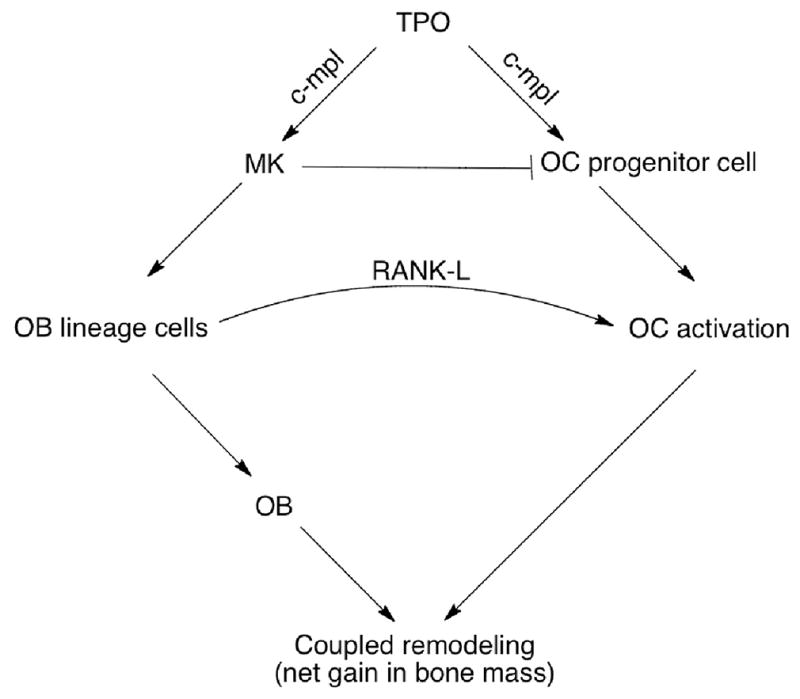
MKs also elaborate unidentified soluble factor(s) which are likely to inhibit OC formation. The enhancement in OB lineage cell number by MKs also increases overall RANKL expression and subsequent activation of OCs.
However, a study by Perry et al. (2007) examined the phenotype of c-mpl deficient mice. In these mice there is a significant reduction in MK number (~90%) and bone volume was predicted to decrease as a result of the decrease in MK number. Yet, they found no change in bone volume. Our data may explain this observation. Here we show TPO activation of multiple signaling cascades in OC progenitors which results in increases in OC number. However, in c-mpl deficient mice (global knockout) OC progenitors would not express c-mpl, and therefore TPO cannot directly enhance OC formation. Thus, compared to normal mice in which BM TPO can directly increase OC number, in c-mpl deficient mice OC number in response to TPO is unchanged. Therefore, in c-mpl deficient mice there may be a relative “reduction” in OC number which could result in a decrease in bone resorption. This relative “reduction” in OC number could lead to a net increase in bone volume as compared to what would be predicted if c-mpl were functional in OC progenitors (or if osteoclastogenesis was not enhanced with TPO stimulation) but not in MKs. In their manuscript, Perry et al. (2007) did not report on the in vitro or in vivo OB or OC populations. This analysis is of critical importance to better understand the potential role of MK-mediated skeletal effects. This is particularly true in light of our data showing that OC progenitors express c-mpl and that in vitro stimulation of these cells with TPO enhances OC development; and in light of previous studies by de Sauvage and colleagues (Carver-Moore, et al., 1996) demonstrating that c-mpl deficient mice have fewer myeloid lineage cells as well as our data presented here showing that fewer OCs are generated from c-mpl deficient mice as compared to that observed from wild-type mice. In other studies we are working to better characterize the bone phenotype of the c-mpl deficient mice in terms of OB- and OC-specific parameters.
Since our data support the idea that TPO acts to augment M-CSF and RANKL mediated OC formation, we began determining which signaling pathways were activated by TPO stimulation of OC progenitors. Our data suggest that TPO can activate the JAK/STAT, MAPK, and NF-κB signaling pathways in OC progenitors, but not the PI3K/AKT pathway (data not shown). Of particular importance to osteoclastogenesis, the main signaling pathway activated by RANKL, the NF-κB pathway, was activated in response to TPO stimulation. Interestingly, the NF-κB pathway has also been reported to be activated in MKs and hepatoblastoma cells in response to TPO stimulation. One study demonstrated that TPO treatment of a megakaryocytic cell line resulted in the immediate increase in Iκκ activity (Zhang, et al., 2002). Another study showed that incubation of hepatoblastoma cells with TPO resulted in a marked increase in IκBα phosphorylation which was similar to the observed appearance of nuclear NF-κB (Romanelli, et al., 2006). Taken together, these observations suggest that stimulation of OC progenitors with TPO results in enhanced mature OC formation through augmented activation of the NF-κB signaling pathway.
Next, using human peripheral blood cells we were able to assay more purified populations of cells. TPO treatment of whole human PBMC cultures did not alter OC number. However, when we enriched for OC progenitors based on the expression of CD11b+ or CD14+ in conjunction with c-mpl expression (CD110+), we found that TPO treatment enhanced OC number, especially in CD14+ cells. These data also confirm that human OC progenitors express c-mpl. The fact that few, if any, mature OCs were formed from CD110−, CD11b−, and CD14− cell populations suggests that CD14+CD11b+CD110+ cells are an enriched population of OC progenitors.
With respect to osteoclastic bone resorption two intriguing findings were noted. First, while OC progenitor cultures enriched for CD11b+ or CD14+ cells generated higher numbers of mature OCs, the resulting OCs were less active than their unsorted counterparts. This is likely due to the secretion of additional growth factors/cytokines by accessory cells contained in the unsorted population, which positively impacted osteoclastic resorption. Second, while TPO treatment modestly enhanced CD11b+CD110+ activity, it robustly enhanced the activity in CD14+CD110+ cells.
Based on the enhancements observed in CD14+ cell populations as compared to CD11b+ cell populations, we then sought to determine whether an overlapping population of cells was responsible for the promotion of osteoclastogenesis and bone resorption in these sorted populations. The data in Figure 4B demonstrate significant overlap in the populations and that CD14+CD11b+CD110+ cells give rise to more mature OCs than do any of the other populations tested. This suggests that cells with the phenotype CD11b+CD14+CD110+ are the major contributors to the osteoclastogenesis. The fact that only ~24% of CD11b+ cells are CD110+ whereas a much higher percentage of CD14+ cells are also CD110+ (~89%) may explain why CD14+ cells better promote osteoclastogenesis than do their CD11b+ counterparts.
In closing, our research has demonstrated a novel role for TPO as a stimulatory cytokine promoting osteoclastogenesis. Moreover, we show not only is c-mpl expressed on OC progenitor cells, but it is also a functional receptor, likely responding to activation by TPO. Increasing evidence implicates a role for MKs in skeletal homeostasis (Lennert et al., 1975; Thiele et al., 1999; Chagraoui et al., 2006). However, until now, the role of TPO has not been studied independently of MKs. Here we show that TPO potentially plays a direct role in skeletal homeostasis by enhancing OC development. Better understanding the interactions between bone cells and MKs and TPO may reveal mechanisms underlying skeletal homeostasis and may provide insight regarding the development of novel therapeutic treatments for bone loss diseases such as osteoporosis. As there are numerous hematologic disorders with alterations in TPO concentrations or mutations in c-mpl and or c-mpl signaling (Moliterno et al., 1998; Pardanani et al., 2006; Khan and Mikhael, 2010), knowledge that c-mpl is expressed and functioning in OC progenitors may allow physicians to better treat these patients in terms of potential secondary bone-related complications such as osteosclerosis and/or bone loss. Finally, as about half of RA patients have thrombocytosis with elevated TPO levels (Ertenli et al., 1998; Farr et al., 1983), and these patients have more severe disease and more osteoclastic joint destruction, it is possible that addition of a thrombocyte-targeted therapy (e.g. therapy targeted to TPO or c-mpl) may serve to increase the effectiveness of treatments for these patients.
Supplementary Material
Acknowledgments
Contract grant sponsor: NIH; Contract grant number: R01 AR060332
Contract grant sponsor: NIH; Contract grant number: R01 AR060863
Contract grant sponsor: NIH; Contract grant number: T32 HL007910
Contract grant sponsor: NIH; Contract grant number: P30 DK090948
Contract grant sponsor: NIH; Contract grant number: P30 CA082709
Contract grant sponsor: NIH; Contract grant number: UL1TR001108
This work was sponsored in part by the Department of Orthopaedics and Rehabilitation at Yale University School of Medicine (MAK), the Department of Orthopaedic Surgery at Indiana University School of Medicine (MAK), the Department of Oral Biology at Indiana University School of Dentistry (AB), a Yale University School of Medicine Medical Student Research Fellowship (CLTB), a Biomedical Research Grant and Pilot Funding for Research Use of Core Facilities Award both from Indiana University School of Medicine (MAK), by a Research Support Funds Grant from Indiana University Purdue University Indianapolis (MAK), by a grant from the Ralph W. and Grace M. Showalter Research Trust Fund (MAK), by the Indiana - Clinical and Translational Sciences Institute funded in part by NIH grants UL1TR001108 (MAK), the Indiana University Center of Excellence in Molecular Hematology funded by NIH P30 DK090948 (MAK, EFS), and by NIH grants R01 AR060332 (MAK, AB), R01 AR060863 (MAK), T32 HL007910 (MB). We would like to thank Genentech and Drs. Fredrick de Sauvage and Wei Tong for providing the c-mpl−/− mice. We would also like to thank Genentech for providing us with the human recombinant TPO. Finally, we would like to thank the operators of the Indiana University Melvin and Bren Simon Cancer Center Flow Cytometry Resource Facility (FCRF) for their technical help and support. The FCRF is partially funded by P30 CA082709.
ABBREVIATIONS
- BMM
bone marrow macrophage
- DAPI
4′,6-diamidino-2-phenylindole
- EMSA
electrophoretic mobility shift assay
- JAK
Janus kinase
- rh
recombinant human
- TPO
thrombopoietin
- MAPK
mitogen-activated protein kinases
- M-CSF
macrophage colony-stimulating factor
- MK
megakaryocyte
- ml
milliliter
- NF-κB
nuclear factor-kappaB
- ng
nanogram
- OB
osteoblast
- OC
osteoclast
- PBMC
peripheral blood mononuclear cell
- RANKL
receptor activator of nuclear factor kappa-B ligand
- STAT
signal transducer and activator of transcription
- TRAP
tartrate resistant acid phosphatase
Footnotes
The authors have no conflicts of interest.
The content in this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartley TD, Bogenberger J, Hunt P, Li YS, Lu HS, Martin F, Chang MS, Samal B, Nichol JL, Swift S. Identification and cloning of a megakaryocyte growth and development factor that is a ligand for the cytokine receptor Mpl. Cell. 1994;77:1117–1124. doi: 10.1016/0092-8674(94)90450-2. [DOI] [PubMed] [Google Scholar]
- Beeton CA, Bord S, Ireland D, Compston JE. Osteoclast formation and bone resorption are inhibited by megakaryocytes. Bone. 2006;39:985–990. doi: 10.1016/j.bone.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Bord S, Frith E, Ireland DC, Scott MA, Craig JI, Compston JE. Megakaryocytes modulate osteoblast synthesis of type-l collagen, osteoprotegerin, and RANKL. Bone. 2005;36:812–819. doi: 10.1016/j.bone.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Breton-Gorius J, Clezardin P, Guichard J, Debili N, Malaval L, Vainchenker W, Cramer EM, Delmas PD. Localization of platelet osteonectin at the internal face of the alpha-granule membranes in platelets and megakaryocytes. Blood. 1992;79:936–941. [PubMed] [Google Scholar]
- Broudy VC, Lin NL, Kaushansky K. Thrombopoietin (c-mpl ligand) acts synergistically with erythropoietin, stem cell factor, and interleukin-11 to enhance murine megakaryocyte colony growth and increases megakaryocyte ploidy in vitro. Blood. 1995;85:1719–1726. [PubMed] [Google Scholar]
- Carver-Moore K, Broxmeyer HE, Luoh SM, Cooper S, Peng J, Burstein SA, Moore MW, de Sauvage FJ. Low levels of erythroid and myeloid progenitors in thrombopoietin-and c-mpl-deficient mice. Blood. 1996;88:803–808. [PubMed] [Google Scholar]
- Chagraoui H, Sabri S, Capron C, Villeval JL, Vainchenker W, Wendling F. Expression of osteoprotegerin mRNA and protein in murine megakaryocytes. Exp Hematol. 2003a;31:1081–1088. doi: 10.1016/s0301-472x(03)00233-9. [DOI] [PubMed] [Google Scholar]
- Chagraoui H, Tulliez M, Smayra T, Komura E, Giraudier S, Yun T, Lassau N, Vainchenker W, Wendling F. Stimulation of osteoprotegerin production is responsible for osteosclerosis in mice overexpressing TPO. Blood. 2003b;101:2983–2989. doi: 10.1182/blood-2002-09-2839. [DOI] [PubMed] [Google Scholar]
- Chagraoui H, Wendling F, Vainchenker W. Pathogenesis of myelofibrosis with myeloid metaplasia: Insight from mouse models. Best Pract Res Clin Haematol. 2006;19:399–412. doi: 10.1016/j.beha.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Cheng Y-H, Hooker RA, Nguyen K, Gerard-O’Riley R, Waning DL, Chitteti BR, Meijome TE, Chua HL, Plett AP, Orschell CM, Srour EF, Mayo LD, Pavalko FM, Bruzzaniti A, Kacena MA. Pyk2 regulates megakaryocyte-induced increases in osteoblast number and bone formation. J Bone Miner Res. 2013;28:1434–1445. doi: 10.1002/jbmr.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenu C, Delmas PD. Platelets contribute to circulating levels of bone sialoprotein in human. J Bone Miner Res. 1992;7:47–54. doi: 10.1002/jbmr.5650070108. [DOI] [PubMed] [Google Scholar]
- Ciovacco WA, Goldberg CG, Taylor AF, Lemieux JM, Horowitz MC, Donahue HJ, Kacena MA. The role of gap junctions in megakaryocyte-mediated osteoblast proliferation and differentiation. Bone. 2009;44:80–86. doi: 10.1016/j.bone.2008.08.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciovacco WA, Cheng YH, Horowitz MC, Kacena MA. Immature and mature megakaryocytes enhance osteoblast proliferation and inhibit osteoclast formation. J Cell Biochem. 2010;109:774–781. doi: 10.1002/jcb.22456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B, Banu N, Malloy B, Hass P, Wang JF, Cavacini L, Eaton D, Avraham H. An agonist murine monoclonal antibody to the human c-Mpl receptor stimulates megakaryocytopoiesis. Blood. 1998;92:1981–1988. [PubMed] [Google Scholar]
- de Sauvage FJ, Hass PE, Spencer SD, Malloy BE, Gurney AL, Spencer SA, Darbonne WC, Henzel WJ, Wong SC, Kuang WJ. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature. 1994;369:533–538. doi: 10.1038/369533a0. [DOI] [PubMed] [Google Scholar]
- Drachman JG, Sabath DF, Fox NE, Kaushansky K. Thrombopoietin signal transduction in purified murine megakaryocytes. Blood. 1997;89:483–492. [PubMed] [Google Scholar]
- Ertenli I, Kiraz S, Arici M, et al. P-selectin as a circulating molecular marker in rheumatoid arthritis with thrombocytosis. J Rheumatol. 1998;25:1054–1058. [PubMed] [Google Scholar]
- Farr M, Scott DL, Constable TJ, Hawker RJ, Hawkins CF, Stuart J. Thrombocytosis of active rheumatoid disease. Ann Rheum Dis. 1983;42:545–549. doi: 10.1136/ard.42.5.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank JD, Balena R, Masarachia P, Seedor JG, Cartwright ME. The effects of three different demineralization agents on osteopontin localization in adult rat bone using immunohistochemistry. Histochemistry. 1993;99:295–301. doi: 10.1007/BF00269102. [DOI] [PubMed] [Google Scholar]
- Frey BM, Rafii S, Crystal RG, Moore MA. Adenovirus long-term expression of thrombopoietin in vivo: a new model for myeloproliferative syndrome and osteomyelofibrosis. Schweiz Med Wochenschr. 1998a;128:1587–1592. [PubMed] [Google Scholar]
- Frey BM, Rafii S, Teterson M, Eaton D, Crystal RG, Moore MA. Adenovector-mediated expression of human thrombopoietin cDNA in immune-compromised mice: insights into the pathophysiology of osteomyelofibrosis. J Immunol. 1998b;160:691–699. [PubMed] [Google Scholar]
- Gurney AL, Carver-Moore K, de Sauvage FJ, Moore MW. Thrombocytopenia in c-mpl-deficient mice. Science. 1994;265:1445–1447. doi: 10.1126/science.8073287. [DOI] [PubMed] [Google Scholar]
- Kacena MA, Shivdasani RA, Wilson K, Xi Y, Troiano N, Nazarian A, Gundberg CM, Bouxsein ML, Lorenzo JA, Horowitz MC. Megakaryocyte-osteoblast interaction revealed in mice deficient in transcription factors GATA-1 and NF-E2. J Bone Miner Res. 2004;19:652–660. doi: 10.1359/JBMR.0301254. [DOI] [PubMed] [Google Scholar]
- Kacena MA, Gundberg CM, Nelson T, Horowitz MC. Loss of the transcription factor p45 NF-E2 results in a developmental arrest of megakaryocyte differentiation and the onset of a high bone mass phenotype. Bone. 2005;36:215–223. doi: 10.1016/j.bone.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Kacena MA, Horowitz MC. The role of megakaryocytes in skeletal homeostasis and rheumatoid arthritis. Curr Opin Rheumatol. 2006;18:405–410. doi: 10.1097/01.bor.0000231910.42666.31. [DOI] [PubMed] [Google Scholar]
- Kacena MA, Gundberg CM, Horowitz MC. A reciprocal regulatory interaction between megakaryocytes, bone cells, and hematopoietic stem cells. Bone. 2006a;39:978–984. doi: 10.1016/j.bone.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Kacena MA, Nelson T, Clough ME, Lee SK, Lorenzo JA, Gundberg CM, Horowitz MC. Megakaryocyte-mediated inhibition of osteoclast development. Bone. 2006b;39:991–999. doi: 10.1016/j.bone.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Kacena MA, Eleniste PP, Cheng Y-H, Huang S, Shivanna M, Meijome TE, Mayo LD, Bruzzaniti A. Megakaryocytes regulate the expression of Pyk2 isoforms and the caspase-mediated cleavage of actin in osteoblasts. J Biol Chem. 2012;287:17257–17268. doi: 10.1074/jbc.M111.309880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakumitsu H, Kamezaki K, Shimoda K, Karube K, Haro T, Numata A, Shide K, Matsuda T, Oshima K, Harada M. Transgenic mice overexpressing murine thrombopoietin develop myelofibrosis and osteosclerosis. Leuk Res. 2005;29:761–769. doi: 10.1016/j.leukres.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Kaushansky K, Broudy VC, Lin N, Jorgensen MJ, McCarty J, Fox N, Zucker-Franklin D, Lofton-Day C. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc Natl Acad Sci U S A. 1995;92:3234–3238. doi: 10.1073/pnas.92.8.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm RJ, Jr, Hair GA, Mann KG, Grant BW. Characterization of human osteoblast and megakaryocyte-derived osteonectin (SPARC) Blood. 1992;80:3112–3119. [PubMed] [Google Scholar]
- Khan M, Mikhael J. A review of immune thrombocytopenic papura: focus on the novel thrombopoietin agonists. J Blood Med. 2010;1:21–31. doi: 10.2147/JBM.S6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux JM, Horowitz MC, Kacena MA. Involvement of integrins alpha(3)beta(1) and alpha(5)beta(1) and glycoprotein IIb in megakaryocyte-induced osteoblast proliferation. J Cell Biochem. 2010;109:927–932. doi: 10.1002/jcb.22468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennert K, Nagai K, Schwarze EW. Patho-anatomical features of the bone marrow. Clin Haematol. 1975;4:331–351. [PubMed] [Google Scholar]
- Methia N, Louache F, Vainchenker W, Wendling F. Oligodeoxynucleotides antisense to the proto-oncogene c-mpl specifically inhibit in vitro megakaryocytopoiesis. Blood. 1993;82:1395–1401. [PubMed] [Google Scholar]
- Miao D, Murant S, Scutt N, Genever P, Scutt A. Megakaryocyte-bone marrow stromal cell aggregates demonstrate increased colony formation and alkaline phosphatase expression in vitro. Tissue Eng. 2004;10:807–817. doi: 10.1089/1076327041348473. [DOI] [PubMed] [Google Scholar]
- Moliterno AR, Hankins WD, Spivak JL. Impaired expression of the thrombopoietin receptor by platelets from patients with polycythemia vera. N Engl J Med. 1998;338:572–580. doi: 10.1056/NEJM199802263380903. [DOI] [PubMed] [Google Scholar]
- Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litzow MR, Gilliland DG, Tefferi A. MPL515 mutations in myeloproliferative and other myeloid disorders: as study of 1192 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- Pearse RN, Sordillo EM, Yaccoby S, Wong BR, Liau DF, Colman N, Michaeli J, Epstein J, Choi Y. Multiple myeloma disrupts the TRANCE/osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci U S A. 2001;98:11581–11586. doi: 10.1073/pnas.201394498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MJ, Redding KA, Alexander WS, Tobias JH. Mice Rendered Severely Deficient in Megakaryocytes through Targeted Gene Deletion of the Thrombopoietin Receptor c-Mpl Have a Normal Skeletal Phenotype. Calcif Tissue Int. 2007;81:224–231. doi: 10.1007/s00223-007-9051-z. [DOI] [PubMed] [Google Scholar]
- Romanelli RG, Petrai I, Robino G, Efsen E, Novo E, Bonacchi A, Pagliai G, Grossi A, Parola M, Navari N, Delogu W, Vizzutti F, Rombouts K, Gentilini P, Laffi G, Marra F. Thrombopoietin stimulates migration and activates multiple signaling pathways in hepatoblastoma cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:G120–8. doi: 10.1152/ajpgi.00350.2004. [DOI] [PubMed] [Google Scholar]
- Sipe JB, Zhang J, Waits C, Skikne B, Garimella R, Anderson HC. Localization of bone morphogenetic proteins (BMPs)-2, -4, and -6 within megakaryocytes and platelets. Bone. 2004;35:1316–1322. doi: 10.1016/j.bone.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Solar GP, Kerr WG, Zeigler FC, Hess D, Donahue C, de Sauvage FJ, Eaton DL. Role of c-mpl in early hematopoiesis. Blood. 1998;92:4–10. [PubMed] [Google Scholar]
- Sungaran R, Markovic B, Chong BH. Localization and regulation of thrombopoietin mRNa expression in human kidney, liver, bone marrow, and spleen using in situ hybridization. Blood. 1997;89:101–107. [PubMed] [Google Scholar]
- Suva LJ, Hartman E, Dilley JD, Russell S, Akel NS, Skinner RA, Hogue WR, Budde U, Varughese KI, Kanaji T, Ware J. Platelet dysfunction and a high bone mass phenotype in a murine model of platelet-type von Willebrand disease. Am J Pathol. 2008;172:430–439. doi: 10.2353/ajpath.2008.070417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Amling M, Neff L, Peyman A, Uhlmann E, Levy JB, Baron R. c-Cbl is downstream of c-Src in a signalling pathway necessary for bone resorption. Nature. 1996;383:528–531. doi: 10.1038/383528a0. [DOI] [PubMed] [Google Scholar]
- Thiede MA, Smock SL, Petersen DN, Grasser WA, Thompson DD, Nishimoto SK. Presence of messenger ribonucleic acid encoding osteocalcin, a marker of bone turnover, in bone marrow megakaryocytes and peripheral blood platelets. Endocrinology. 1994;135:929–937. doi: 10.1210/endo.135.3.8070388. [DOI] [PubMed] [Google Scholar]
- Thiele J, Kvasnicka HM, Fischer R. Histochemistry and morphometry on bone marrow biopsies in chronic myeloproliferative disorders - aids to diagnosis and classification. Ann Hematol. 1999;78:495–506. doi: 10.1007/s002770050546. [DOI] [PubMed] [Google Scholar]
- Tong W, Lodish HF. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med. 2004;200:569–580. doi: 10.1084/jem.20040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeval JL, Cohen-Solal K, Tulliez M, Giraudier S, Guichard J, Burstein SA, Cramer EM, Vainchenker W, Wendling F. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood. 1997;90:4369–4383. [PubMed] [Google Scholar]
- Wakikawa T, Shioi A, Hino M, Inaba M, Nishizawa Y, Tatsumi N, Morii H, Otani S. Thrombopoietin inhibits in vitro osteoclastogenesis from murine bone marrow cells. Endocrinology. 1997;138:4160–4166. doi: 10.1210/endo.138.10.5438. [DOI] [PubMed] [Google Scholar]
- Wendling F, Maraskovsky E, Debili N, Florindo C, Teepe M, Titeux M, Methia N, Breton-Gorius J, Cosman D, Vainchenker W. cMpl ligand is a humoral regulator of megakaryocytopoiesis. Nature. 1994;369:571–574. doi: 10.1038/369571a0. [DOI] [PubMed] [Google Scholar]
- Yan XQ, Lacey D, Fletcher F, Hartley C, McElroy P, Sun Y, Xia M, Mu S, Saris C, Hill D, Hawley RG, McNiece IK. Chronic exposure to retroviral vector encoded MGDF (mpl-ligand) induces lineage-specific growth and differentiation of megakaryocytes in mice. Blood. 1995;86:4025–4033. [PubMed] [Google Scholar]
- Yan XQ, Lacey D, Hill D, Chen Y, Fletcher F, Hawley RG, McNiece IK. A model of myelofibrosis and osteosclerosis in mice induced by overexpressing thrombopoietin (mpl ligand): reversal of disease by bone marrow transplantation. Blood. 1996;88:402–409. [PubMed] [Google Scholar]
- Zeigler FC, de Sauvage F, Widmer HR, Keller GA, Donahue C, Schreiber RD, Malloy B, Hass P, Eaton D, Matthews W. In vitro megakaryocytopoietic and thrombopoietic activity of c-mpl ligand (TPO) on purified murine hematopoietic stem cells. Blood. 1994;84:4045–4052. [PubMed] [Google Scholar]
- Zhang Y, Sun S, Wang Z, Thompson A, Kaluzhny Y, Zimmet J, Ravid K. Signaling by the Mpl receptor involves IKK and NF-kappaB. J Cell Biochem. 2002;85:523–535. doi: 10.1002/jcb.10141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.