

Abstract
The effects of cannabinoids have been known for centuries and over the past several decades two G protein-coupled receptors, CB1 and CB2, that are responsible for their activity have been identified. Endogenous lipid-derived cannabinergic agents have been found, biosynthetic and catabolic machinery has been characterized, and synthetic agents have been designed to modulate these receptors. Selective agents including agonists, antagonists, inverse agonists, and novel allosteric modulators targeting either CB1 or CB2 have been developed to inhibit or augment their basal tone. As a result, the role these receptors play in human physiology and their potential therapeutic applications in disease states are being elucidated. The CB1 receptor, although ubiquitous, is densely expressed in the brain, and CB2 is largely found on cells of immune origin. This minireview highlights the role of CB1 in excitotoxic assaults in the brain and its potential to limit addiction liability. In addition, it will examine the relationship between receptor activity and stimulation of insulin release from pancreatic β-cells, insulin resistance, and feeding behavior leading toward obesity. The roles of CB2 in the neuropathology of amyotrophic lateral sclerosis and in the central manifestations of chronic HIV infection potentially converge at inflammatory cell activation, thereby providing an opportunity for intervention. Last, CB2 modulation is discussed in the context of an experimental model of postmenopausal osteoporosis. Achieving exquisite receptor selectivity and elucidating the mechanisms underlying receptor inhibition and activation will be essential for the development of the next generation of cannabinergic-based therapeutic agents.
There are nearly 400 distinct chemical entities that are isolated from the plant Cannabis sativa, and it was not until 1942 that Adams (1) reported the structures of a series of tetrahydrocannabinols—referred to as cannabinoids—and it was proposed that these were the active components of marijuana. This mixture has been resolved and contains approximately 60 separate centrally active or psychoactive components. Mechoulam and Gaoni reported in 1965 (2) that the major psychoactive component of marijuana was (−)-Δ9-6a,10a-trans-tetrahydrocannabinol (Δ9-THC) (Figure 1). In the decades since, much of what has been learned about the actions of these agents has been accomplished by exploring structure-activity relationships, which has led to extensive pharmacologic characterization of synthetic cannabinoids often designed from the scaffold of Δ9-THC. Moreover, synthetic cannabinoid agents have facilitated the identification of proteins that are involved in the biosynthesis and catabolism of endogenous cannabinoid “hormones” as well as membrane-associated receptors whose activity can be “tuned” by these agents—hence the “cannabinergic” system in human physiology.
Figure 1.

Representative ligands that modulate the CB1 and CB2 receptors, including the natural product Δ9-THC, endogenous lipid-derived agonists AEA and 2-AG, and synthetic agonists CP55940 and WIN55212-2. CB1-selective ligands include SR141716A, AM251, and AM6545 (all inverse agonists), whereas the CB2-selective molecules are represented by AM630 (inverse agonist), SR144528 (inverse agonist), AM1241 (agonist), and HU308 (agonist).
A synthetic nonclassical cannabinoid, CP55940 (Figure 1), was found to bind in a stereospecific and saturable manner to rat brain homogenates, a process that was salt sensitive and competitively displaced by cannabinoid ligands, allowing Howlett and colleagues (3) to demonstrate for the first time that there was a specific G protein–coupled receptor (GPCR) involved with cannabinoid activity. Subsequently, Matsuda and colleagues (4) reported the cloning and functional expression of a GPCR from rat brain that when stimulated with cannabinoids was associated with a reduction in cAMP accumulation. With this pivotal discovery, the receptor localized to the brain was termed the CB1 cannabinoid receptor. Subsequently a second receptor subtype, CB2, was cloned from splenic macrophages (5). Despite sharing elements of structural and functional homology, CB1 and CB2 are distributed throughout the body uniquely.
The groundbreaking discovery of specific receptors for plant-derived and synthetic cannabinoids prompted the search for endogenous “hormones” for the cannabinoid receptors (CBRs). One such molecule, identified from porcine brain extracts, was named anandamide (AEA) from the Sanskrit word “ananda,” meaning bliss (6). Biosynthesis of AEA is enzymatically catalyzed by a Ca2+-activated phospholipase D-mediated hydrolysis of N-arachidonyl phosphatidylethanolamine (7–9). Furthermore, another lipid-derived endogenous cannabinoid was identified as 2-arachidonyl glycerol (2-AG) (10, 11) and subsequently characterized as having higher potency than AEA (12, 13). 2-AG biosynthesis is accomplished via sn-1-diacylglycerol lipase. Several other lipid-derived substances have been found to bind to the CBRs (eg, arachidonyl glyceryl ether, n-arachidonoyldopamine, and virodhamine, a congener of AEA), but AEA and 2-AG are among the most well-characterized agents (12, 14). Moreover, fatty acid amidohydrolase and monoacylglycerol lipase hydrolytically terminate the activity of AEA and 2-AG, respectively (15, 16).
CB1 Receptor Structure and Function
The hallmark features of the superfamily of GPCRs are a heptahelical arrangement of membrane-spanning α-helical transmembrane domains (TMDs) that are connected by intervening loop regions with extracellular amino termini and intracellular carboxy termini. An early determination of a crystal structure of rhodopsin has allowed the generation of homology models of other GPCRs, and this has inspired drug discovery efforts (17). With the use of in silico models for CB1 (and for CB2), based on rhodopsin that also binds a lipophilic ligand (18, 19), the β2-adrenergic receptor that has additional structural similarities and has been crystalized with G protein (20), or the β1-adrenergic receptor that optimizes CB1 helix packing conformations (21), it has been possible to design cannabinergic agents that are more subtype selective and achieve greater specificity for the CBRs over other GPCRs, limiting off-target activity. Intense efforts have been directed at obtaining high-resolution crystal structures of GPCRs (22, 23), but, to date, the CBRs remain elusive to crystallization efforts owing to the challenges of getting the required high yield of such remarkably hydrophobic membrane proteins, the inherent high energy of their activated forms, and their conformational plasticity (24).
The classical cannabinoids with fused heterocyclic rings, such as Δ9-THC, became the scaffold for the design of synthetic cannabinergic agents such as the nonclassical cannabinoids best exemplified by CP55940, which deviate from the structurally constrained 3-ring heterocyclic classical cannabinoids (3, 25). In addition, the aminoalkylindole class of cannabinergic agents initially had limited CB1/CB2 selectivity but ultimately facilitated design of CB2-selective agents (26, 27). Last, the biarylpyrazoles, such as SR141716A and AM251, were discovered and have been developed as the first of a series of cannabinoid antagonists/inverse agonists for CB1 (28–30). Based on the lack of structural similarity among the cannabinergic ligands, it is easy to envision that CP55940 might not bind CBRs in a manner similar to that of the cannabinergic aminoalkylindole WIN55212-2 (Figure 1), and they do not. Collectively, the CB1 binding sites for classical/nonclassical cannabinoids agonists such as Δ9-THC and CP55940 arise from TMDs 3-6-7 (31–35), whereas the agonist WIN55212-2 has been implicated in interactions in TMDs 3-4-5-6 (32, 33). Furthermore, the biarylpyrazole receptor inverse agonist SR141716A has been found to interact with TMDs 2-3-5-7 (36). Not surprisingly, these ligands all occupy common “space” within the receptor and mutually displace each other but do not maintain identical contacts with the same amino acids.
Since the seminal work of Howlett and colleagues (3), it was demonstrated that the CB1 receptor couples primarily to inhibitory G proteins, (eg, Gi/o), and agonist stimulation of CB1 results in a reduction in cAMP accumulation, which was pertussis toxin–sensitive in a neuroblastoma cell line (Figure 2) (37, 38). Recently, Luttrell (39) reviewed “bias-based” signaling whereby multiple conformational states of a GPCR exist and activation of a GPCR can produce a multitude of signaling events. However, “biased” agonists might only have affinity for certain conformations and their subsequent coupling states (ie, with Gs, Gi, Gq/11, or β-arrestin), thus producing a distinct and limited array of activated second messengers. This functional selectivity has clinical value because one can potentially develop ligands that promote only certain desirable signaling events. Early evidence for biased signaling with CBRs was uncovered by Glass and Northup (40) using an analysis of both CB1 and CB2 receptors expressed in Sf9 cells into which either purified Gαi or Gαo proteins were reconstituted in situ. They found that different ligands (including Δ9-THC, AEA, and WIN55212-2) preferentially bound CBRs, depending on the precoupled G protein, thus supporting the existence of differing conformational states of CBRs that may be maintained by different G protein subunits (40).
Figure 2.
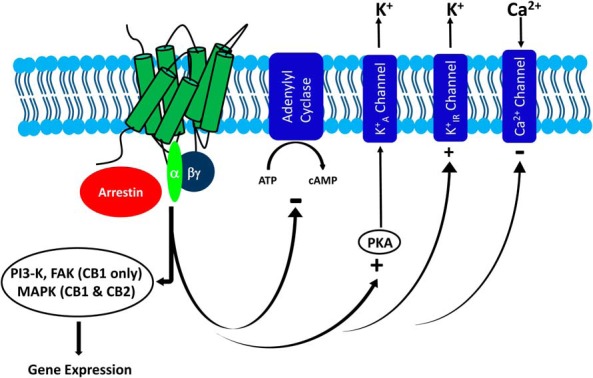
Cannabinoid receptor modulation has an impact on a host of cellular pathways. Activation of CB1 or CB2 receptors results in a release of G protein heterotrimer, which has a negative impact on cellular production of cAMP, activates K+A and K+IR channels, and inhibits Ca2+ channels. In addition, activated CB1 or CB2 receptors can recruit β-arrestin to the plasma membrane. When stimulated, CB1 has the ability to activate MAPK, phosphatidylinositol 3-kinase, and FAK, among other pathways, and although less is known about CB2, it is also capable of activating MAPK.
Consistent with complexity in signaling, McAllister and colleagues (41) reported that in Xenopus oocytes, cannabinoid agonists were capable of activating G protein–coupled inwardly rectifying potassium channels, which were believed to be mediated by the βγ subunits of the G protein heterotrimer. Moreover, it is becoming increasingly apparent in GPCR pharmacology that “all that signals, is not G protein–related,” and a role for β-arrestin–mediated signaling distinct from G protein–initiated cascades often converges upon ERK (42). Classically, β-arrestin has a role in associating with GPCRs that had been phosphorylated and assisting in subsequent receptor down-regulation and trafficking to proteolytic endosomes for recycling or degradation. Recently, Flores-Otero and colleagues (43) demonstrated β-arrestin–biased signaling in distinct cellular environments for CB1. Moreover, it was demonstrated in different cell types that 2-AG activates both G protein and arrestin pathways, yet with particular kinetic distinctions, whereas the synthetic ligands analyzed to date signal largely via G proteins (43, 44).
CB1 Allosteric Modulators
In addition to agonists, antagonists, and inverse agonists, allosteric modulators have been identified for CB1 receptors (42, 45–48). In contrast to the endogenous ligand-binding site, referred to as the orthosteric site, the allosteric site is structurally and topographically distinct (Figure 3). This presents an advantage in the design of agents targeting the allosteric site, because conserved topographical elements in the orthosteric site cross receptor subtypes (CB1 vs CB2) and even some different GPCRs (eg, cannabinoid vs adrenergic receptors). Moreover, allosteric ligands can augment (or inhibit) the temporal and spatial activity of the endogenous ligand, thus providing a mechanism for fine-tuning the receptor response with fewer side effects (49). The allosteric modulator binds the receptor, inducing a conformational change that may enhance (or inhibit) orthosteric ligand-binding affinity and/or attenuate orthosteric ligand efficacy, whereas some allosteric modulators may act independently of the orthosteric ligand (eg, ago-agonism). Regardless, the design of allosteric modulators could involve targeting the extracellular loops of the GPCR and for CB1 and CB2 could be accomplished with less lipophilic, water-soluble agents that are more amenable to being developed as clinically useful therapeutic agents and, potentially, could include selective antibodies.
Figure 3.

Allosteric ligands for CB1 are capable of modulating orthosteric ligand (agonist) activity. A, Schematic representation of the CB1 receptor TMD bundle illustrating the topographic distinction between allosteric and orthosteric binding sites. B, Structures of representative allosteric modulators for CB1 including ORG27569, PSNCBAM-1, lipoxin A4, RTI371, and LDK1256 and LDK1258 (also referred to as compounds 12d and 12f from Ref. 46).
Several allosteric modulators of the CB1 receptor have been identified, including ORG27569, PSNCBAM-1, RTI371, and the endogenous ligand lipoxin A4 (47, 50–52) (Figure 3). ORG27569 increases specific binding of the CB1 agonist CP55940 and decreases the binding of the inverse agonist SR141716A to CB1-containing cell membranes consistent with its activation of the receptor (47, 53). Furthermore, it induces robust cellular internalization of the CB1 receptor (53). However, ORG27569 antagonizes agonist-induced G protein coupling to the receptor yet it activates ERK signaling mediated instead by β-arrestins, further indicating that CB1 is in an activated form (42, 53, 54). Structure-activity relationship analysis has revealed that the indole-2-carboxamide scaffold is a viable template for developing CB1 allosteric modulators such as LDK1256 and LDK1258 (Figure 3) (42, 45, 46, 55). These data indicate that CB1 allosteric modulators offer the potential to develop drugs able to provide therapeutic effects including via ligand-biased signaling pathways. At present, there are no reports of allosteric modulators for CB2, but this remains an active area of research.
CB2 Receptor Structure and Function
Since the identification of the CB2 receptor, there has been interest in developing therapeutic agents that would elicit the analgesic effects mediated by CB1 yet doing so outside of the central nervous system (CNS). The two receptor subtypes share 44% identity overall, whereas the TMDs are reported to have a higher degree of identity estimated to be 35% to 82% (56). Selectivity for CB2 has been achieved with AM630, an iodinated aminoalkylindole, along with the pyrazole SR144528, both behaving as CB2 inverse agonists, whereas highly selective and potent CB2 agonists AM1241, another aminoalkylindole, and the bicyclic nonclassical cannabinoid HU308 have also been reported (57–61) (Figure 1). The binding sites for the various ligands are not entirely identical to their corresponding CB1 sites whereby cannabinoids (Δ9-THC and CP55940) bind in TMDs 3-6-7 (33, 62), WIN55212-2 binds at TMDs 3-4-5-6 (32, 63–65), and the antagonist SR144528 minimally binds at TMDs 4-6 (56, 63). More recently, a report by Reggio and colleagues (66) has implicated TMDs 2-3-6-7 in the antagonist/inverse agonist interactions with novel CB2-selective carboxamides.
Coupled to Gi proteins, CB2 is also a pertussis toxin–sensitive GPCR, albeit with less promiscuity than CB1. Glass and Northup (40) in their in situ reconstituted system did not identify coupling at physiologically relevant ratios of G protein/CB2 for any G protein except Gαi (40). Further supporting this finding is a recent study by Mnpotra and colleagues (67) that used a tandem approach involving chemical cross-linking studies between G proteins and CB2 followed by proteolytic fingerprinting via mass spectrometry. They found no evidence of cross-linking except for sequences that corresponded to a CB2:Gαi complex, and this was supported by molecular dynamics simulations. CB2 activation has a negative impact on adenylyl cyclase activity (68) and has been shown to activate G protein–coupled inwardly rectifying potassium channels when expressed in Xenopus oocytes (41). Like CB1, CB2 also signals downstream to activate MAPK (ERK1/2) (69). As is the case with CB1 that displays some β-arrestin bias (42–44, 70), interestingly, biased signaling is also observed with CB2. Whereas CP55940 acts as an agonist to stimulate β-arrestin recruitment, induce MAPK activation, and inhibit voltage-gated calcium channels, WIN55212-2 promotes β-arrestin recruitment and activates MAPK with no detectable inhibition on voltage-gated calcium channels displaying profound functional selectivity (71).
Role of Cannabinoid Receptors in Human Physiology and Disease
It is now widely accepted that CB1 is dominantly expressed in the CNS where it influences the activity of other neuronal systems, making the endogenous cannabinoids neuromodulators. By using autoradiographic and genetic approaches, CB1 is found densely in the brain in regions of higher cognitive functions including the cerebral cortex. It is also in the hippocampus, basal ganglia, and cerebellum as well as in areas involved in reward, food, and drug intake, particularly the nucleus accumbens and ventral tegmental area (69, 72, 73). In fact, CB1 is among the most densely expressed GPCRs in the human brain. CB1 is typically found presynaptically where its activation can have a negative impact on the release of other key neurotransmitters, such as γ-aminobutyric acid and glutamate, by causing hyperpolarization of the neuron, thus stimulating K+ and inhibiting Ca2+ channels (ie, neuromodulatory activity). Outside of the CNS, expression of CB1 is found in the peripheral nervous system as well as testis, adrenal glands, pancreas, heart, and lung (69). Exploitation of the distribution patterns of the CBRs is fundamental to the design and development of clinically useful cannabinergic-based therapeutic agents (Figure 4).
Figure 4.

Cannabinoid receptors as therapeutic targets for human disease. Receptor activation (shown in green) and blockade (shown in red) have the ability to alter physiology both in rodent models of disease and in humans. A, CB1 receptor activation has been explored as a means to protect against excitotoxic insult in the brain and has been shown to occur during hyperglycemia, resulting in insulin release from the pancreas. Conversely, CB1 blockade has been investigated as a means to reduce addiction liability in rodent models and has been demonstrated to decrease food intake and improve surrogate markers of metabolic disease (liver and pancreas) while producing weight loss observed both in rodents and in human clinical trials. B, CB2 activation has been explored as a means to delay progression of ALS, selectively target HIV-infected microglial cells in the CNS, and inhibit HIV reverse transcriptase as well as restore the balance between osteoblasts and osteoclasts in a rodent model of postmenopausal osteoporosis while increasing trabecular bone mass.
CB1 Receptors in Neuroprotection and Addiction Liability
Several applications of cannabinergic agents, which include neuroprotection, medications for reducing addiction liability, and improving metabolism (including insulin release) and affecting obesity, among others, have been proposed for CB1 receptors. Neurophysiologically, in the face of excitotoxic injury such as what occurs in ischemic stroke, seizure, and even acute brain trauma, there is a remarkable increase in glutamate release. In hippocampal neurons, Zhao and colleagues (74) reported colocalization of CB1 with synaptic markers residing adjacent to glutamate GluR1-containing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR). Glutamate receptor activation is central to the excitotoxic insult that precipitates hippocampal damage associated with stroke, seizures, and traumatic brain injury. CB1 activation with WIN55212-2 was demonstrated to protect against this assault in vitro by preventing the increase in AMPAR surface expression through disruption of TNFα signaling. These effects were blocked by co-treatment of WIN55212-2 with the CB1 antagonist SR141716A, which would indicate that this is mediated through a CB1-dependent mechanism, an effect not observed with co-treatment of WIN55212-2 and the CB2-selective antagonist SR144528 (74). Trafficking of AMPAR to the neuronal membrane surfaces is thought to be an early event during toxic insult that precipitates cell death (75–77). Cannabinoid-selective blockade of this early event could be an important development in the acute treatment of CNS assaults to limit catastrophic damage. This suggests that CB1 activation protects neurons from excitotoxic stress in hippocampal neurons and renders the CB1 receptor a potential therapeutic target for stroke, seizures, or acute brain trauma accomplished through a CB1 receptor agonist.
With the use of marijuana recreationally and medicinally, possible addictive behaviors are of concern. Cannabinoids can induce a state of physical dependence, and administration of SR141716A in animals chronically treated with Δ9-THC precipitated a constellation of withdrawal symptoms (78, 79). In addition, a link in reward behavior between cannabinoid and opiate receptor systems has been identified experimentally in rodents (80). After a period of heroin abstinence, both CP55940 and WIN55212-2 independently caused a reinitiation of lever pressing, which was attenuated by SR141716A, arguing for a role of the CB1 receptor in facilitating the heroin-seeking behavior (81). Moreover, in CB1−/− mice, there was an apparent reduction in the reinforcing effects of morphine (82), cocaine (83), and alcohol (84, 85), and withdrawal symptoms were remarkably milder. Therapeutically, the ability to develop agents that could prevent relapse to addiction of abused substances could have an enormous impact on public health worldwide, and selective CB1 antagonism could provide a key to this dilemma.
CB1 Tone in Glucodynamics and Energy Balance
Metabolically, the presence of cannabinoid receptors on α- and β-cells of the endocrine pancreas (86–88), along with the identification of machinery for biosynthesis and catabolism of endogenous cannabinoids, fosters the question as to what role this system might play in glucodynamics (89). Agonist-stimulated activation of Gi proteins and molecular mechanisms underlying insulin release involve intracellular Ca2+ mobilization and converge on activation of Akt via phosphorylation. To explore this, an elegant series of experiments were undertaken in β-cells in culture and demonstrated that 1) CB1 agonists increased insulin release through a Ca2+-dependent mechanism, whereas blockade of CB1 with AM251 abrogated insulin release, 2) AEA and 2-AG biosynthesis was induced in settings of elevated glucose consistent with hyperglycemic conditions, and 3) inactivation of the catabolic enzymes responsible for AEA and 2-AG hydrolysis was also associated with increases in insulin secretion. These data provide strong support for the role of CB1 and endogenous cannabinoid tone in some aspect of insulin secretion, kinetically associated with first-phase insulin secretion from “predocked” insulin granules. Moreover, it was further illustrated that CB1 receptor activation was linked to cytoskeletal rearrangements via focal adhesion kinase (FAK) consistent with cellular dynamics associated with second-phase insulin release involving the trafficking of insulin granules to the plasma membrane of the β-cell. Importantly, it has been demonstrated in neurons that AEA stimulated cytoskeletal reorganization mediated by FAK via CB1 receptor activation (90). Malenczyk and colleagues (89) proposed a link between elevated endogenous cannabinoid levels and the adaptation of pancreatic β-cells in the development of insulin resistance through the associated impact of insulin hypersecretion. Development of a cannabinoid receptor antagonist might have utility in this situation as a means to reduce this hypersecretion of insulin.
Extending the observation of a functional endocannabinergic system and its potential role in insulin hypersecretion and insulin resistance and establishing a link to obesity is very attractive. A substantial body of evidence supports the role of the endogenous cannabinergic system in obesity, which is multifaceted. Metabolically, obesity can be thought of as a disease with peripheral manifestations driven, to a large degree, by CNS influence among highly redundant pathways designed to regulate energy balance. The hypothalamus is responsible for central control of hunger, and reward centers of the nucleus accumbens provide motivation to eat. Dysfunction of either of these centers is associated with obesity and the CB1 receptor is present in both centers of the brain. Using population genetic analyses, Russo and coworkers (91) investigated a link between polymorphisms in the CB1 gene CNR1 and obesity in a large cohort of Italian men from the Olivetti Prospective Heart Study. They found a high degree of correlation between anthropometric measures of obesity (body mass index and waist circumference) and the 3813 A/G polymorphism. This same finding was associated with the somewhat smaller cohort of white men from the Wandsworth Heart and Stroke Study (91). In addition, it had been well established that AEA, 2-AG, and Δ9-THC each stimulated hyperphagia in rodent models of obesity (92–94). This observation is supported by evidence from Martin and colleagues (95) that after administration of Δ9-THC to mice, there is a significant increase in food intake, an effect not observed when SR141716A was administered. In fact, SR141716A dose dependently decreased food intake as expected with an inverse agonist. Furthermore, Ravinet Trillou and colleagues (96) undertook a study in diet-induced obese mice and noted that chronic treatment with SR141716A dose dependently produced less weight gain (on a background of a high-fat diet) and improved insulin, and leptin levels and that this effect was not observed in CB1−/− mice. Added insight toward the role of CB1 in obesity comes from the observation that endocannabinoid levels tend to fluctuate relative to feeding status. With food deprivation, endocannabinoids tend to increase, whereas after feeding they decline, in contrast to leptin, making endocannabinoids orexigenic in nature (97).
With consistently observed effects of SR141716A on feeding behavior and weight in models of obesity, Sanofi-Aventis launched the Rimonabant in Obesity (RIO) clinical development program, enrolled more than 6000 obese or overweight subjects, and exposed them to SR141716A (clinically known as rimonabant) for up to 2 years. On average, a clinically and statistically meaningful weight loss of approximately 4 to 5 kg resulted (98). Among the associated surrogate markers of obesity, decreases in weight are concomitant with a decline in insulin levels in both rodents and in humans in the RIO-Diabetes trial. However, the authors deem this finding to be partially independent of weight loss (99, 100). Declining insulin levels from a hyperinsulinemic state is consistent with restoring insulin sensitivity, particularly in the face of weight loss, as was proposed by Malenczyk and colleagues (89) in their work with cultured pancreatic β-cells. Other peripheral actions of CB1 blockade that are associated with improvements in metabolism are related to direct and indirect effects on adiposity as well as on the liver. SR141716A stimulated the expression and secretion of adiponectin in obese Zucker (fa/fa) mice expressing CB1 as well as in cultured adipocytes. In these obese Zucker (fa/fa) mice, the increase in adiponectin levels was associated with weight loss and improvements in hyperglycemic status over the 10-week course of treatment (101). Recently, Boon and colleagues (102) demonstrated that SR141716A, through a CB1 receptor–dependent mechanism activated brown fat in diet-induced obese mice by increasing lipoprotein lipase, stimulating very low-density lipoprotein–triglyceride lipolysis, and increasing expression of Ucp1, which encodes the uncoupling protein that mediates thermogenesis (102). Animal studies have shown that inhibition of CB1 with SR141716A decreased fatty acid biosynthesis in the liver, an effect that was not seen in CB1−/− mice (103).
Unfortunately, unexpected adverse events began to emerge during clinical development of CB1 antagonists, involving an increase in depressive disorders in subjects. Although the weight reductions observed in subjects exposed to rimonabant in RIO were clinically relevant, there was an association with neuropsychiatric effects, leading to withdrawal of rimonabant from the European marketplace (100). Peripheral restriction of a molecule by designing an agent that is not able to access the CNS has the ability to circumvent adverse impacts on the CNS and still maintain many of the beneficial effects of CB1 blockade on obesity. AM6545 is designed to be less lipophilic and is actively transported from the brain, thereby limiting central access considerably while preserving the overall selectivity for CB1 over CB2 much like SR141716A. Interestingly, with peripheral restriction of AM6545, there is an observed reduction in feeding behavior along with the expected array of improvements in insulin sensitivity, in glucodynamics, and in lipid parameters and adiposity that would be expected with improvements in metabolic syndrome (104). It is intriguing to hypothesize that the impact on feeding behavior might be attributed to an improvement in adiponectin levels, which would have a positive impact on energy balance, possibly through central actions of IL-6 because AM6545 does not appear to have access to the brain. Recently, AM6545 has been shown to activate brown fat similarly to SR141716A by increasing lipoprotein lipase, stimulating very low-density lipoprotein–triglyceride lipolysis, and increasing expression of Ucp1 (102). Alternatively, designing a molecule that does not cross the blood-brain barrier and targets an allosteric site might limit the untoward neuropsychiatric effects associated with cannabinoid receptor blockade while attenuating the effects of up-regulated signaling of endogenous cannabinergic agents such as in development of insulin resistance and adiposity associated with metabolic syndrome. A peripherally restricted allosteric modulator of CB1 would be a tremendous advance in combating this constellation of metabolic sequelae.
CB2 Receptors in Neuroinflammation: Peripheral Analgesia, Amyotrophic Lateral Sclerosis, and HIV Infection
As exemplified by the case of rimonabant, the development of clinically useful cannabinergic agents has been a challenge, due to, in part, the wide array of potentially unwanted central affects with the high level of expression of CB1 in the brain. In contrast, CB2, which is mostly localized to the periphery, largely on cells of immune origin, such as B lymphocytes, macrophages, mast cells, microglia, natural killer cells, peripheral mononuclear cells, and CD4/CD8 cells, as well as spleen, tonsils, and the thymus, provides additional promise for the realization of clinically useful cannabinergic agents (69). The most widely characterized application of CB2 is development of peripheral analgesics that might find use clinically in diabetic neuropathy and in acute and chronic inflammatory pain (105). A well-characterized agent for peripheral analgesia mediated through CB2 is AM1241, which has been studied in animal models of neuropathic, inflammatory, postsurgical, and chemical-induced pain (106). In each case, AM1241 was found to be effective and competitively antagonized by the CB2 selective antagonists AM630 or SR144528. Moreover, there is enantioselectivity with AM1241, which is racemic, whereby the (R)-isomer possesses greater affinity for rodent and human CB2 receptors, the (S)-isomer consistently acts as a CB2 agonist, and the racemic mixture can display mixed pharmacological effects in a species-dependent manner (107).
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that results in motor neuron death in the cortex, brain stem, and spinal cord (108, 109). As with many neurodegenerative diseases, the pathophysiology is not entirely clear beyond the likely involvement of a combination of neuroinflammation and oxidative stress (110, 111). High levels of microglia, the resident immune cells of the CNS, are present in affected brain regions of patients with ALS postmortem (112). A common mutation in humans who develop ALS occurs in Cu/Zn superoxide dismutase (SOD1) and a very useful animal model for the study of ALS has been developed by introducing mutations of SOD1 into transgenic mice. Recently, Moreno-Martet and colleagues (113) identified an elevated level of CB2 expression in spinal cords of SOD1 transgenic mice. Their data are consistent with those of Yiangou and colleagues (114) who examined human postmortem spinal cords via immunohistochemical analysis and found very intense CB2 staining of microglial cells in the dorsolateral white matter of degenerating corticospinal tracts not observed in healthy subjects. AM1241 was also demonstrated to delay progression of ALS in an SOD1 transgenic mouse model, presumably mediated through the CB2 receptor. Kim and colleagues (115) demonstrated in rodents that upon treatment with AM1241, motor neuron effects were preserved significantly longer in males, while a trend toward improvement was observed in females. Importantly, however, they noted that disease progression was delayed but AM1241 did not confer benefits on survival in this rodent model of ALS. Interestingly, Shoemaker and colleagues (116) also described a study in SOD1 mutant mice in which AM1241 was initiated at ALS symptom onset and found delayed motor neuron decline and a statistically significant increase in survival with treated mice living 56% longer than vehicle-treated control mice (116).
Since the identification of HIV infection as the causative mechanism underlying development of AIDS, retroviral therapies have evolved, essentially transitioning certain mortality to the current situation of managing a chronic viral infection. Still, there are consequences that are poorly understood in the pathophysiology of this infection. Among them is HIV-associated neurocognitive disorder, which is thought to stem from chronic inflammation in the brain despite a low level of HIV infection of macrophages and microglia residing centrally (117, 118). Ramirez and colleagues (119) identified an increased level of CB2 receptors on macrophages and microglia in postmortem subjects with HIV encephalitis, relative to that of noninfected age-matched control subjects, suggesting that HIV infection up-regulates CB2 expression. This is in agreement with other reports quantifying CB2 expression in HIV encephalitis (120). Using qualitative PCR, Ramirez and colleagues noted progressively more expression of CB2 in differentiated macrophages with the greatest expression in HIV-infected macrophages and the lowest in monocytes. This result was confirmed via fluorescence-activated cell sorting analysis using a CB2-selective antibody to correlate gene expression from the PCR analysis with that of protein at the cellular level. The observed expression of CB2 receptors on differentiated macrophages infected with HIV, in vitro, is also functional as treatment with a CB2-selective agonist produced a dose-dependent blockade of reverse transcriptase activity that was attenuated by the CB2-selective antagonist SR144528, an effect that is attributed to altering activity of the HIV long terminal repeat. The authors suggest that this impact on the HIV long terminal repeat may be attributed to a consequence of shared transcription factors between CB2 activation and the process of neuroinflammation (119). Collectively, these data indicate that the up-regulation of CB2 expression associated with HIV infection could allow for selective targeting of the CB2 receptor to minimize the neuroinflammatory effect of HIV infection and potentially limit further HIV replication in microglia due to attenuation of reverse transcriptase activity.
CB2 Receptor Tone in Osteoporosis
A polymorphism in the CB2 encoding gene, CNR2, has been found to be linked to diminished bone mineral density and osteoporosis via a systematic genetic case-controlled association study in postmenopausal women (121). Zimmer and coworkers (121) concluded that there was a significant association of single polymorphisms encompassing the CNR2 gene on human chromosome 1p36. The balance of bone formation and resorption is attributed to the opposing actions of osteoblasts and osteoclasts, which are responsible for bone formation and resorption, respectively. A dysregulation of either cell type affects bone health and can lead to an increased risk of fracture with associated morbidity and mortality. Mechanistically, Ofek and coworkers (122) provided evidence to support a role of the CB2 receptor in bone health using rodent and cell culture models. Evaluating phenotypically normal CB2−/− mice, they identified lower trabecular bone volume density in both males and females, relative to those of age-matched control animals, as well as decreased trabecular bone mass with cortical expansion. They further found that in female CB2−/− mice, the osteoclast cell number per bone surface area was 40% higher, whereas the bone formation rate was increased, suggesting high bone turnover, akin to what is observed in postmenopausal osteoporosis. Expression of CB2 was confirmed in osteoclasts, osteoblasts, and osteocytes of wild-type animals, but not the corresponding CB2−/− mice. In addition, using a potent CB2-selective agonist, HU-308, an increase was noted in differentiation of stromal cells from a primary osteoblastogenic culture of bone marrow–derived stromal cells, something that was not observed in cells obtained from the CB2−/− mice. Moreover, this result was confirmed in MC3T3 E1 cells, an established osteoblastic cell line, in which HU-308 treatment stimulated proliferation, which was blocked by pertussis toxin treatment, a process that would be consistent with CB2 activation via the release of Gi-proteins. Next, the authors noted that HU-308 dose dependently reduced the formation of osteoclast-like cells from bone marrow–derived primary monocytic cultures undergoing osteoclastic differentiation. Finally, because HU-308 was found to have pro-osteoblastic and antiosteoclastic activity, its function was tested in a rodent model of postmenopausal osteoporosis by treating ovariectomized mice. As expected, HU-308 did attenuate the trabecular bone loss observed in nontreated ovariectomized mice, and the investigators concluded that this was associated with a decline in osteoclast number. Thus, this study illustrated that CB2 receptor activation plays a key role in the differentiation of osteoblasts and suppression of osteoclast expansion and indicates that CB2-targeted therapies could be clinically useful in restoring balance in bone metabolism lost after menopause (122).
Future Directions
There is considerable interest in GPCR drug discovery and in CB1 and CB2, in particular. Cannabinoid receptor endogenous ligands have been elucidated, and several synthetic agonists, neutral antagonists, inverse agonists, and allosteric modulators of the cannabinoid receptors have been developed, some of which are subtype specific, binding CB1 and CB2 with very different affinities. The downstream pathways that these orthosteric and allosteric ligands activate have been partly characterized and in several instances the pharmacological impact via in vivo analysis has been accomplished. Yet, for drug discovery via CB1 or CB2 targets to be successful and to avoid untoward effects, it is necessary to 1) gain more structural information of these receptors including the variety of conformations they can adopt, 2) develop orthosteric or allosteric ligands that are target and conformation specific, 3) elucidate more fully the role these receptors play in vivo so that it can be modulated, and 4) determine how to fine-tune desirable responses in a rheostat-like manner. These will involve correlating the profiles of ligand interaction with cell signaling and in vivo impact. Drug discovery will require a multidisciplinary, collaborative approach, which reveals overall patterns that link receptor activity with health and disease.
Acknowledgments
We gratefully acknowledge Kwang Hyun Ahn for critically reading this article.
We dedicate this Minireview to the memory of Donna J. Fournier, PhD, a mentor, colleague, and friend who was present when we began our studies of the cannabinoid receptors, shared our curiosities, and would be amazed with the progress made in understanding their role in human physiology and the potential for therapeutic intervention.
This study was supported in part by the National Institutes of Health Grant DA020763 (to D.A.K.).
Disclosure Summary: R.P.P. is an employee of Novo Nordisk Inc, and the opinions expressed in this article are his and not those of Novo Nordisk Inc. D.A.K. has nothing to disclose.
Footnotes
- AEA
- anandamide
- 2-AG
- 2-arachidonyl glycerol
- ALS
- amyotrophic lateral sclerosis
- AMPAR
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors
- CBR
- cannabinoid receptor
- CNS
- central nervous system
- FAK
- focal adhesion kinase
- GPCR
- G protein–coupled receptor
- RIO
- Rimonabant in Obesity
- SOD1
- Cu/Zn superoxide dismutase
- Δ9-THC
- (−)-Δ9-6a,10a-trans-tetrahydrocannabinol
- TMD
- α-helical transmembrane domain.
References
- 1. Adams R. Marihuana: Harvey Lecture, February 19, 1942. Bull NY Acad Med. 1942;18:705–730. [PMC free article] [PubMed] [Google Scholar]
- 2. Mechoulam R, Gaoni Y. A total synthesis of dl-Delta-1-tetrahydrocannabinol, the active constituent of hashish. J Am Chem Soc. 1965;87:3273–3275. [DOI] [PubMed] [Google Scholar]
- 3. Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 4. Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. [DOI] [PubMed] [Google Scholar]
- 5. Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. [DOI] [PubMed] [Google Scholar]
- 6. Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. [DOI] [PubMed] [Google Scholar]
- 7. Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci. 1996;16:3934–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Marzo V, De Petrocellis L, Sepe N, Buono A. Biosynthesis of anandamide and related acylethanolamides in mouse J774 macrophages and N18 neuroblastoma cells. Biochem J. 1996;316(Pt 3):977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ueda N, Kurahashi Y, Yamamoto K, Yamamoto S, Tokunaga T. Enzymes for anandamide biosynthesis and metabolism. J Lipid Mediat Cell Signal. 1996;14:57–61. [DOI] [PubMed] [Google Scholar]
- 10. Mechoulam R, Ben-Shabat S, Hanus L, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. [DOI] [PubMed] [Google Scholar]
- 11. Sugiura T, Kondo S, Sukagawa A, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. [DOI] [PubMed] [Google Scholar]
- 12. Gonsiorek W, Lunn C, Fan X, Narula S, Lundell D, Hipkin RW. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol Pharmacol. 2000;57:1045–1050. [PubMed] [Google Scholar]
- 13. Sugiura T. Physiological roles of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Biofactors. 2009;35:88–97. [DOI] [PubMed] [Google Scholar]
- 14. Bisogno T. Endogenous cannabinoids: structure and metabolism. J Neuroendocrinol. 2008;20(Suppl 1):1–9. [DOI] [PubMed] [Google Scholar]
- 15. Desarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem. 1995;270:6030–6035. [DOI] [PubMed] [Google Scholar]
- 16. Bisogno T, Sepe N, Melck D, Maurelli S, De Petrocellis L, Di Marzo V. Biosynthesis, release and degradation of the novel endogenous cannabimimetic metabolite 2-arachidonoylglycerol in mouse neuroblastoma cells. Biochem J. 1997;322(Pt 2):671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. [DOI] [PubMed] [Google Scholar]
- 18. Hurst DP, Schmeisser M, Reggio PH. Endogenous lipid activated G protein-coupled receptors: emerging structural features from crystallography and molecular dynamics simulations. Chem Phys Lipids. 2013;169:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahmoud MM, Olszewska T, Liu H, et al. (4-(Bis(4-fluorophenyl)methyl)piperazin-1-yl)(cyclohexyl)methanone hydrochloride (LDK1229): a new cannabinoid CB1 receptor inverse agonist from the class of benzhydryl piperazine analogs. Mol Pharmacol. 2015;87:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shim JY, Ahn KH, Kendall DA. Molecular basis of cannabinoid CB1 receptor coupling to the G protein heterotrimer Gαiβγ: identification of key CB1 contacts with the C-terminal helix α5 of Gαi. J Biol Chem. 2013;288:32449–32465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scott CE, Abrol R, Ahn KH, Kendall DA, Goddard WA., 3rd Molecular basis for dramatic changes in cannabinoid CB1 G protein-coupled receptor activation upon single and double point mutations. Protein Sci. 2013;22:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang K, Zhang J, Gao ZG, et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014;509:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tan Q, Zhu Y, Li J, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47(Suppl 1):345–358. [DOI] [PubMed] [Google Scholar]
- 26. Mella-Raipán J, Hernández-Pino S, Morales-Verdejo C, Pessoa-Mahana D. 3D-QSAR/CoMFA-based structure-affinity/selectivity relationships of aminoalkylindoles in the cannabinoid CB1 and CB2 receptors. Molecules. 2014;19:2842–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wiley JL, Marusich JA, Huffman JW. Moving around the molecule: relationship between chemical structure and in vivo activity of synthetic cannabinoids. Life Sci. 2014;97:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wierucka-Rybak M, Wolak M, Bojanowska E. The effects of leptin in combination with a cannabinoid receptor 1 antagonist, AM 251, or cannabidiol on food intake and body weight in rats fed a high-fat or a free-choice high sugar diet. J Physiol Pharmacol. 2014;65:487–496. [PubMed] [Google Scholar]
- 29. Wiley JL, Selley DE, Wang P, Kottani R, Gadthula S, Mahadeven A. 3-Substituted pyrazole analogs of the cannabinoid type 1 (CB1) receptor antagonist rimonabant: cannabinoid agonist-like effects in mice via non-CB1, non-CB2 mechanism. J Pharmacol Exp Ther. 2012;340:433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lan R, Liu Q, Fan P, et al. Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem. 1999;42:769–776. [DOI] [PubMed] [Google Scholar]
- 31. Picone RP, Khanolkar AD, Xu W, et al. (−)-7′-Isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol Pharmacol. 2005;68:1623–1635. [DOI] [PubMed] [Google Scholar]
- 32. Chin CN, Murphy JW, Huffman JW, Kendall DA. The third transmembrane helix of the cannabinoid receptor plays a role in the selectivity of aminoalkylindoles for CB2, peripheral cannabinoid receptor. J Pharmacol Exp Ther. 1999;291:837–844. [PubMed] [Google Scholar]
- 33. Tao Q, McAllister SD, Andreassi J, et al. Role of a conserved lysine residue in the peripheral cannabinoid receptor (CB2): evidence for subtype specificity. Mol Pharmacol. 1999;55:605–613. [PubMed] [Google Scholar]
- 34. Chin CN, Lucas-Lenard J, Abadji V, Kendall DA. Ligand binding and modulation of cyclic AMP levels depend on the chemical nature of residue 192 of the human cannabinoid receptor 1. J Neurochem. 1998;70:366–373. [DOI] [PubMed] [Google Scholar]
- 35. Song ZH, Bonner TI. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol Pharmacol. 1996;49:891–896. [PubMed] [Google Scholar]
- 36. Shire D, Calandra B, Delpech M, et al. Structural features of the central cannabinoid CB1 receptor involved in the binding of the specific CB1 antagonist SR 141716A. J Biol Chem. 1996;271:6941–6946. [DOI] [PubMed] [Google Scholar]
- 37. Howlett AC. Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol Pharmacol. 1985;27:429–436. [PubMed] [Google Scholar]
- 38. Howlett AC, Qualy JM, Khachatrian LL. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol. 1986;29:307–313. [PubMed] [Google Scholar]
- 39. Luttrell LM. Minireview: More than just a hammer: ligand “bias” and pharmaceutical discovery. Mol Endocrinol. 2014;28:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1999;56:1362–1369. [DOI] [PubMed] [Google Scholar]
- 41. McAllister SD, Griffin G, Satin LS, Abood ME. Cannabinoid receptors can activate and inhibit G protein-coupled inwardly rectifying potassium channels in a xenopus oocyte expression system. J Pharmacol Exp Ther. 1999;291:618–626. [PubMed] [Google Scholar]
- 42. Ahn KH, Mahmoud MM, Samala S, Lu D, Kendall DA. Profiling two indole-2-carboxamides for allosteric modulation of the CB1 receptor. J Neurochem. 2013;124:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Flores-Otero J, Ahn KH, Delgado-Peraza F, Mackie K, Kendall DA, Yudowski GA. Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat Commun. 2014;5:4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laprairie RB, Bagher AM, Kelly ME, Dupré DJ, Denovan-Wright EM. Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J Biol Chem. 2014;289:24845–24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mahmoud MM, Ali HI, Ahn KH, et al. Structure-activity relationship study of indole-2-carboxamides identifies a potent allosteric modulator for the cannabinoid receptor 1 (CB1). J Med Chem. 2013;56:7965–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khurana L, Ali HI, Olszewska T, et al. Optimization of chemical functionalities of indole-2-carboxamides to improve allosteric parameters for the cannabinoid receptor 1 (CB1). J Med Chem. 2014;57:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Price MR, Baillie GL, Thomas A, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. [DOI] [PubMed] [Google Scholar]
- 48. Gamage TF, Ignatowska-Jankowska BM, Wiley JL, et al. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behav Pharmacol. 2014;25:182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burford NT, Traynor JR, Alt A. Positive allosteric modulators of the μ-opioid receptor: a novel approach for future pain medications. Br J Pharmacol. 2015;172:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horswill JG, Bali U, Shaaban S, et al. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pamplona FA, Ferreira J, Menezes de Lima O, Jr, et al. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc Natl Acad Sci USA. 2012;109:21134–21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem. 2012;287:12070–12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baillie GL, Horswill JG, Anavi-Goffer S, et al. CB1 receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2013;83:322–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Piscitelli F, Ligresti A, La Regina G, et al. Indole-2-carboxamides as allosteric modulators of the cannabinoid CB1 receptor. J Med Chem. 2012;55:5627–5631. [DOI] [PubMed] [Google Scholar]
- 56. Shire D, Calandra B, Bouaboula M, et al. Cannabinoid receptor interactions with the antagonists SR 141716A and SR 144528. Life Sci. 1999;65:627–635. [DOI] [PubMed] [Google Scholar]
- 57. Bolognini D, Cascio MG, Parolaro D, Pertwee RG. AM630 behaves as a protean ligand at the human cannabinoid CB2 receptor. Br J Pharmacol. 2012;165:2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kotsikorou E, Navas F, 3rd, Roche MJ, et al. The importance of hydrogen bonding and aromatic stacking to the affinity and efficacy of cannabinoid receptor CB2 antagonist, 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528). J Med Chem. 2013;56:6593–6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ibrahim MM, Deng H, Zvonok A, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003;100:10529–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ma L, Jia J, Liu X, Bai F, Wang Q, Xiong L. Cannabinoid CB2 receptor agonist AM1241 attenuates microglial activation by modulating the activated state in N9 microglia exposed to LPS plus IFNγ. Biochem Biophys Res Commun. 2015;485:92–97. [DOI] [PubMed] [Google Scholar]
- 61. Hanus L, Breuer A, Tchilibon S, et al. HU-308: a specific agonist for CB2, a peripheral cannabinoid receptor. Proc Natl Acad Sci USA. 1999;96:14228–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pei Y, Mercier RW, Anday JK, et al. Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem Biol. 2008;15:1207–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nebane NM, Hurst DP, Carrasquer CA, Qiao Z, Reggio PH, Song ZH. Residues accessible in the binding-site crevice of transmembrane helix 6 of the CB2 cannabinoid receptor. Biochemistry. 2008;47:13811–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Song ZH, Slowey CA, Hurst DP, Reggio PH. The difference between the CB1 and CB2 cannabinoid receptors at position 5.46 is crucial for the selectivity of WIN55212-2 for CB2. Mol Pharmacol. 1999;56:834–840. [PubMed] [Google Scholar]
- 65. Reggio PH, Basu-Dutt S, Barnett-Norris J, et al. The bioactive conformation of aminoalkylindoles at the cannabinoid CB1 and CB2 receptors: insights gained from (E)- and (Z)-naphthylidene indenes. J Med Chem. 1998;41:5177–5187. [DOI] [PubMed] [Google Scholar]
- 66. Lucchesi V, Hurst DP, Shore DM, et al. CB2-selective cannabinoid receptor ligands: synthesis, pharmacological evaluation, and molecular modeling investigation of 1,8-naphthyridin-2(1H)-one-3-carboxamides. J Med Chem. 2014;57:8777–8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mnpotra JS, Qiao Z, Cai J, et al. Structural basis of G protein-coupled receptor-Gi protein interaction: formation of the cannabinoid CB2 receptor-Gi protein complex. J Biol Chem. 2014;289:20259–20272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Felder CC, Joyce KE, Briley EM, et al. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- 69. Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. [DOI] [PubMed] [Google Scholar]
- 70. Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J Biol Chem. 2013;288:9790–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Atwood BK, Wager-Miller J, Haskins C, Straiker A, Mackie K. Functional selectivity in CB2 cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB2 ligands. Mol Pharmacol. 2012;81:250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Onaivi ES. Cannabinoid receptors in brain: pharmacogenetics, neuropharmacology, neurotoxicology, and potential therapeutic applications. Int Rev Neurobiol. 2009;88:335–369. [DOI] [PubMed] [Google Scholar]
- 74. Zhao P, Leonoudakis D, Abood ME, Beattie EC. Cannabinoid receptor activation reduces TNFα-induced surface localization of AMPAR-type glutamate receptors and excitotoxicity. Neuropharmacology. 2010;58:551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16:281–287. [DOI] [PubMed] [Google Scholar]
- 77. Oguro K, Oguro N, Kojima T, et al. Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci. 1999;19:9218–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Aceto MD, Scates SM, Lowe JA, Martin BR. Cannabinoid precipitated withdrawal by the selective cannabinoid receptor antagonist, SR 141716A. Eur J Pharmacol. 1995;282:R1–R2. [DOI] [PubMed] [Google Scholar]
- 79. Tsou K, Patrick SL, Walker JM. Physical withdrawal in rats tolerant to Δ9-tetrahydrocannabinol precipitated by a cannabinoid receptor antagonist. Eur J Pharmacol. 1995;280:R13–R15. [DOI] [PubMed] [Google Scholar]
- 80. Fattore L, Cossu G, Spano MS, et al. Cannabinoids and reward: interactions with the opioid system. Crit Rev Neurobiol. 2004;16:147–158. [DOI] [PubMed] [Google Scholar]
- 81. Fattore L, Spano MS, Cossu G, Deiana S, Fratta W. Cannabinoid mechanism in reinstatement of heroin-seeking after a long period of abstinence in rats. Eur J Neurosci. 2003;17:1723–1726. [DOI] [PubMed] [Google Scholar]
- 82. Ledent C, Valverde O, Cossu G, et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. [DOI] [PubMed] [Google Scholar]
- 83. De Vries TJ, Shaham Y, Homberg JR, et al. A cannabinoid mechanism in relapse to cocaine seeking. Nat Med. 2001;7:1151–1154. [DOI] [PubMed] [Google Scholar]
- 84. Serra S, Carai MA, Brunetti G, et al. The cannabinoid receptor antagonist SR 141716 prevents acquisition of drinking behavior in alcohol-preferring rats. Eur J Pharmacol. 2001;430:369–371. [DOI] [PubMed] [Google Scholar]
- 85. Gessa GL, Serra S, Vacca G, Carai MA, Colombo G. Suppressing effect of the cannabinoid CB1 receptor antagonist, SR147778, on alcohol intake and motivational properties of alcohol in alcohol-preferring sP rats. Alcohol Alcohol. 2005;40:46–53. [DOI] [PubMed] [Google Scholar]
- 86. Juan-Picó P, Fuentes E, Bermúdez-Silva FJ, et al. Cannabinoid receptors regulate Ca2+ signals and insulin secretion in pancreatic β-cell. Cell Calcium. 2006;39:155–162. [DOI] [PubMed] [Google Scholar]
- 87. Bermudez-Silva FJ, Sanchez-Vera I, Suárez J, et al. Role of cannabinoid CB2 receptors in glucose homeostasis in rats. Eur J Pharmacol. 2007;565:207–211. [DOI] [PubMed] [Google Scholar]
- 88. Starowicz KM, Cristino L, Matias I, et al. Endocannabinoid dysregulation in the pancreas and adipose tissue of mice fed with a high-fat diet. Obesity (Silver Spring). 2008;16:553–565. [DOI] [PubMed] [Google Scholar]
- 89. Malenczyk K, Jazurek M, Keimpema E, et al. CB1 cannabinoid receptors couple to focal adhesion kinase to control insulin release. J Biol Chem. 2013;288:32685–32699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Derkinderen P, Toutant M, Burgaya F, et al. Regulation of a neuronal form of focal adhesion kinase by anandamide. Science. 1996;273:1719–1722. [DOI] [PubMed] [Google Scholar]
- 91. Russo P, Strazzullo P, Cappuccio FP, et al. Genetic variations at the endocannabinoid type 1 receptor gene (CNR1) are associated with obesity phenotypes in men. J Clin Endocrinol Metab. 2007;92:2382–2386. [DOI] [PubMed] [Google Scholar]
- 92. Kirkham TC, Williams CM, Fezza F, Di Marzo V. Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br J Pharmacol. 2002;136:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Williams CM, Kirkham TC. Anandamide induces overeating: mediation by central cannabinoid (CB1) receptors. Psychopharmacology (Berl). 1999;143:315–317. [DOI] [PubMed] [Google Scholar]
- 94. Williams CM, Kirkham TC. Reversal of Δ9-THC hyperphagia by SR141716 and naloxone but not dexfenfluramine. Pharmacol Biochem Behav. 2002;71:333–340. [DOI] [PubMed] [Google Scholar]
- 95. Wiley JL, Burston JJ, Leggett DC, et al. CB1 cannabinoid receptor-mediated modulation of food intake in mice. Br J Pharmacol. 2005;145:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ravinet Trillou C, Arnone M, Delgorge C, et al. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am J Physiol Regul Integr Comp Physiol. 2003;284:R345–R353. [DOI] [PubMed] [Google Scholar]
- 97. Di Marzo V, Goparaju SK, Wang L, et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–825. [DOI] [PubMed] [Google Scholar]
- 98. Scheen AJ. CB1 receptor blockade and its impact on cardiometabolic risk factors: overview of the RIO programme with rimonabant. J Neuroendocrinol. 2008;20(Suppl 1):139–146. [DOI] [PubMed] [Google Scholar]
- 99. Scheen AJ, Finer N, Hollander P, Jensen MD, Van Gaal LF. Efficacy and tolerability of rimonabant in overweight or obese patients with type 2 diabetes: a randomised controlled study. Lancet. 2006;368:1660–1672. [DOI] [PubMed] [Google Scholar]
- 100. Di Marzo V. CB1 receptor antagonism: biological basis for metabolic effects. Drug Discov Today. 2008;13:1026–1041. [DOI] [PubMed] [Google Scholar]
- 101. Bensaid M, Gary-Bobo M, Esclangon A, et al. The cannabinoid CB1 receptor antagonist SR141716 increases Acrp30 mRNA expression in adipose tissue of obese fa/fa rats and in cultured adipocyte cells. Mol Pharmacol. 2003;63:908–914. [DOI] [PubMed] [Google Scholar]
- 102. Boon MR, Kooijman S, van Dam AD, et al. Peripheral cannabinoid 1 receptor blockade activates brown adipose tissue and diminishes dyslipidemia and obesity. FASEB J. 2014;28:5361–5375. [DOI] [PubMed] [Google Scholar]
- 103. Osei-Hyiaman D, DePetrillo M, Pacher P, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tam J, Vemuri VK, Liu J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dhopeshwarkar A, Mackie K. CB2 Cannabinoid receptors as a therapeutic target-what does the future hold? Mol Pharmacol. 2014;86:430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Anand P, Whiteside G, Fowler CJ, Hohmann AG. Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Res Rev. 2009;60:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yao BB, Mukherjee S, Fan Y, et al. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB2 receptor? Br J Pharmacol. 2006;149:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Brown RH., Jr Amyotrophic lateral sclerosis. Insights from genetics. Arch Neurol. 1997;54:1246–1250. [DOI] [PubMed] [Google Scholar]
- 109. Nicholson SJ, Witherden AS, Hafezparast M, Martin JE, Fisher EM. Mice, the motor system, and human motor neuron pathology. Mamm Genome. 2000;11:1041–1052. [DOI] [PubMed] [Google Scholar]
- 110. Ludolph AC, Meyer T, Riepe MW. The role of excitotoxicity in ALS—what is the evidence? J Neurol. 2000;247(Suppl 1):I7–I16. [DOI] [PubMed] [Google Scholar]
- 111. Robberecht W. Genetics of amyotrophic lateral sclerosis. J Neurol. 2000;247(Suppl 6):VI/2–VI/6. [DOI] [PubMed] [Google Scholar]
- 112. Turner MR, Cagnin A, Turkheimer FE, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–609. [DOI] [PubMed] [Google Scholar]
- 113. Moreno-Martet M, Espejo-Porras F, Fernández-Ruiz J, de Lago E. Changes in endocannabinoid receptors and enzymes in the spinal cord of SOD1(G93A) transgenic mice and evaluation of a Sativex(®)-like combination of phytocannabinoids: interest for future therapies in amyotrophic lateral sclerosis. CNS Neurosci Ther. 2014;20:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yiangou Y, Facer P, Durrenberger P, et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kim K, Moore DH, Makriyannis A, Abood ME. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol. 2006;542:100–105. [DOI] [PubMed] [Google Scholar]
- 116. Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem. 2007;101:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kraft-Terry SD, Buch SJ, Fox HS, Gendelman HE. A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron. 2009;64:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol. 2011;179:1623–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ramirez SH, Reichenbach NL, Fan S, et al. Attenuation of HIV-1 replication in macrophages by cannabinoid receptor 2 agonists. J Leukoc Biol. 2013;93:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Cosenza-Nashat MA, Bauman A, Zhao ML, Morgello S, Suh HS, Lee SC. Cannabinoid receptor expression in HIV encephalitis and HIV-associated neuropathologic comorbidities. Neuropathol Appl Neurobiol. 2011;37:464–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Karsak M, Cohen-Solal M, Freudenberg J, et al. Cannabinoid receptor type 2 gene is associated with human osteoporosis. Hum Mol Genet. 2005;14:3389–3396. [DOI] [PubMed] [Google Scholar]
- 122. Ofek O, Karsak M, Leclerc N, et al. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA. 2006;103:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]