

Abstract
The insulin producing islet β-cells have increasingly gained attention for their role in the pathogeneses of virtually all forms of diabetes. Dysfunction, de-differentiation, and/or death of β-cells are pivotal features in the transition from normoglycemia to hyperglycemia in both animal models of metabolic disease and humans. In both type 1 and type 2 diabetes, inflammation appears to be a central cause of β-cell derangements, and molecular pathways that modulate inflammation or the inflammatory response are felt to be prime targets of future diabetes therapy. The lipoxygenases (LOs) represent a class of enzymes that oxygenate cellular polyunsaturated fatty acids to produce inflammatory lipid intermediates that directly and indirectly affect cellular function and survival. The enzyme 12-LO is expressed in all metabolically active tissues, including pancreatic islets, and has received increasing attention for its role in promoting cellular inflammation in the setting of diabetes. Genetic deletion models of 12-LO in mice reveal striking protection from metabolic disease and its complications and an emerging body of literature has implicated its role in human disease. This review focuses on the evidence supporting the proinflammatory role of 12-LO as it relates to islet β-cells, and the potential for 12-LO inhibition as a future avenue for the prevention and treatment of metabolic disease.
The crude prevalence of all forms of prediabetes and diabetes in the United States exceeds 40% (1). Worldwide, it is expected that up to 592 million people will develop diabetes by the year 2035 (2). These striking data reflect the fact that the major forms of diabetes, type 2 diabetes (T2D) and T1D, have been increasing in incidence in recent decades. The increase in T2D is closely linked to the high prevalence of obesity and prediabetes (3), whereas the reasons for the increase in T1D remain elusive (4–6). Diabetes is defined as the glycemic threshold (fasting plasma glucose ≥ 126 mg/dL or hemoglobin A1c ≥ 6.5%), at which microvascular complications, such as retinopathy and nephropathy, are observed (7). By contrast, cardiovascular complications, including stroke and myocardial infarction, increase even as blood sugars rise in the prediabetic phase (8). Therefore, the identification of drug targets that are common to all forms of diabetes is likely to have far-reaching implications for disorders of multiple organ systems. A key underlying feature of all forms of diabetes is the relative deficiency of insulin secretion from the islet β-cell. T2D arises primarily in the setting of long-standing insulin resistance, wherein the magnitude of insulin secretion by the β-cell fails to meet the peripheral tissue insulin demands (9). In T1D, insulin deficiency has traditionally (and perhaps too simplistically) been ascribed to autoimmune-mediated β-cell destruction; however, recent studies in both rodents and humans suggest that a “prodrome” may exist in T1D, in which insulin secretory capacity is diminished even before overt β-cell death (10–12). β-Cell loss, therefore, may represent a feature occurring very late in the pathogeneses of both T2D and T1D.
The β-cell is unique in its ability to synthesize and secrete physiologically relevant levels of insulin in response to ambient glucose concentrations. In recent years, a growing body of literature suggests that the pathways that initiate dysfunction of the β-cell in T2D and T1D may be similar, if not identical (13). Of particular relevance is inflammation, which leads to the development of oxidative stress, endoplasmic reticulum (ER) stress, and mitochondrial dysfunction in the β-cell (14, 15). In T2D, multiple cell types collaborate in the pathogenesis of β-cell inflammation, including adipocytes, macrophages and other immune cells (dendritic cells, T cells). In the setting of high-fat diets, macrophage polarization to a proinflammatory phenotype (“M1” type) within adipose tissue leads to the production of adipocytokines (eg, IL-6, TNFα), which signal systemically to β-cells (16, 17). This early innate immune response may give way to a later adaptive response, wherein the balance between proinflammatory, effector T cells in the fat and antiinflammatory, regulatory T cells determine the inflammatory features of adipose tissue (13, 18, 19). Moreover, M1 macrophage and auto-reactive T-cell trafficking into the islet itself may occur (20–23), leading to local cytokine release and cell-mediated immunity that directly trigger β-cell inflammation. This immune response leads to the loss of β-cell function through dysfunction, death, and/or dedifferentiation (24–26). More recently, the concept that β-cells might dedifferentiate and thereby diminish in function is particularly innovative. A decrease in the levels of β-cell-specific transcription factors (eg, MafA, pancreatic and duodenal homeobox [Pdx1], NK6 homeobox 1 [Nkx6.1]) was found in both mouse models of T2D and in islets isolated from human T2D donors (27), and expression of progenitor-like factors (eg, Neurogenin 3, NANOG, octamer-binding transcription factor 4) has been observed in mice (26).
The concept that β-cell dysfunction is a key early feature of T1D has seen increasing attention (12, 28–32). In the nonobese diabetic (NOD) mouse model of T1D, impaired glucose-stimulated insulin secretion, particularly first phase insulin secretion, precedes the loss of β-cell mass by several weeks (11, 12). Similar findings were also observed in humans with T1D, who exhibited defects in glucose-stimulated insulin release before the onset of diabetes (10, 33). Inflammation, possibly emanating from infection or primary autoimmunity, has been implicated as underlying β-cell dysfunction (30). More recently, it has been proposed that inflammation and its resultant oxidative and ER stresses act as triggers that initiate autoantigen and neoantigen exposure to drive autoimmunity (12, 34, 35). Taken together, these studies on β-cell function in T2D and T1D suggest that pathways that promote inflammation in β-cells represent potential targets to prevent or treat both diseases.
The lipoxygenase (LO) pathways
An important pathway recently implicated in diabetic inflammation involves a family of enzymes known as LOs. The LOs catalyze the oxygenation of cellular poly-unsaturated fatty acids (primarily arachidonic acid) and are classified according to both the specific carbon atom that is subjected to oxygenation (5, 12, and/or 15 positions) as well as the stereoselectivity of the reaction (the “S” type being the primary enantiomer) (see Figure 1, and reviewed in Ref. 36). The LOs are expressed in a variety of tissue types and often given common names based on the tissue types in which they were identified (Table 1). In humans, and the best studied LOs are “leukocyte” 5-LO, “platelet” 12-LO, “reticulocyte” 15-LO-1, and “epithelial 15-LO-2” (37). 5-LO, whose expression is primarily limited to bone marrow-derived cell types, has been studied in a variety of contexts with respect to inflammation, as the enzyme is required for the downstream production of proinflammatory leukotrienes (38). Accordingly, 5-LO knockout mice exhibit protection from atherogenesis and aortic aneurysms (39, 40), as well as diabetes-induced retinal capillary degeneration (41). Low-level production of 5-LO products has also been described in rodent pancreatic islets (42), and the mRNA encoding 5-LO is present in human islets (43). Notwithstanding evidence for reduced atherogenesis, whole-body 5-LO knockout mice exhibit reductions in glucose-stimulated insulin secretion, and islets isolated from these mice show reduced levels of mRNAs encoding for insulin and the key β-cell transcription factor Pdx1 (44). These data suggest a pleiotropic effect of 5-LO with respect to tissue-specific inflammation, with 5-LO conferring apparent protection in β-cell function. Nevertheless, the role of 5-LO in glycemic homeostasis has not been extensively investigated, and a cell-autonomous role for 5-LO in β-cells in vivo (using conditional knockout mice) is needed in follow-up studies.
Figure 1.
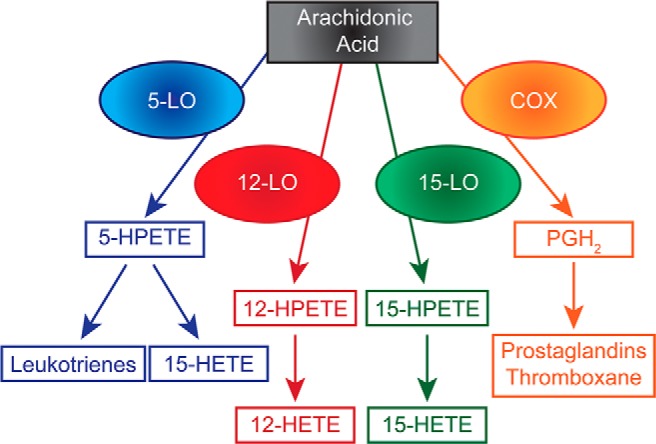
Arachidonic acid metabolism. Schematic diagram showing metabolism of arachidonic acid by LOs and COX. PGH2, prostaglandin H2.
Table 1.
Major LOs and Their General Expression Patterns
| Protein | Human gene (common name) | Mouse gene (common name) | Tissue expression (partial list) |
|---|---|---|---|
| 5-LO | ALOX5 | Alox5 | Leukocytes, macrophages, dendritic cells |
| Platelet-type 12-LO | ALOX12 | Alox12 | Platelets, skin, thrombocytes, islets (human) |
| (12(S)-LO) | |||
| Leukocyte-type 12-LO | ALOX15 | Alox15 | β-Cells, eosinophils, epithelial cells, macrophages, kidney, smooth muscle, islets (mouse) |
| (15-LO-1) | (12/15-LO) | ||
| Epidermal-type 12-LO | Not expressed | Alox12e | Skin |
| 12(R)-LO | ALOX12B | Alox12B | Skin |
| 15-LO | ALOX15B | Alox15B | Hair roots, prostate, lung, cornea |
| (15-LO-2) | (15-LO) | ||
| Epidermis-type LO-3 | ALOXE3 | Aloxe3 | Skin |
| (eLOX-3) | (eLOX-3) |
In contrast to the pleiotropic effects of 5-LO, 12-LO appears to have more uniform proinflammatory effects, and is broadly expressed in virtually all metabolically active cell types, including hepatocytes, adipose tissue, islets, and macrophages/monocytes. 12-LO converts arachidonic acid to 12-hydroperoxyeicosatetraenoic acid (12-HPETE), which is subsequently reduced to more stable 12-hydroxyeicosatetraenoic acid (12-HETE) by glutathione peroxidase. The mouse leukocyte 12-LO enzyme encoded by the gene Alox15 produces an approximately 6:1 ratio of 12-HETE:15-HETE from arachidonic acid, and is often referred to as 12/15-LO (37). Functionally, the mouse LO encoded by Alox15 is closest to the human platelet 12-LO encoded by ALOX12, which produces almost exclusively 12-HETE (45). The role of 12-LO and its major product 12-HETE has been studied extensively in the context of rodent models of diabetes and in normal and diabetic human tissues; in this review, the simplified term “12-LO” will refer to the enzyme encoded by the Alox15 and ALOX12 genes in mice and humans, respectively.
Role of 12-LO in Inflammation and Metabolic Disease
The role of 12-LO in inflammation has been studied best in the immune/inflammatory responses involving monocytes and macrophages and also, more recently, mouse and human pancreatic islets. Unlike 5-LO, whose downstream products include both the HETEs and leukotrienes, the effects of 12-LO are primarily attributed to the production of 12-HETE. Activation of 12-LO has been shown to accelerate inflammation via p38 mitogen-activated protein kinase (p38 MAPK) and nuclear factor κB pathways, cause oxidation of low density lipoprotein to promote foam cell formation, and promote oxidative stress (46–54). In macrophages, 12-LO activity increases production of proinflammatory cytokines such as TNFα, and IL-6, and also stimulates expression of inflammatory genes such as cyclooxygenase (Cox)2 (54, 55). Notably, 12-LO also stimulates production of IL-12, a pivotal cytokine that mediates microbial immunity, atherosclerosis, and the Type 1 helper T cell autoimmune response in T1D (56–58).
The expression and activity of 12-LO are up-regulated in a variety of metabolically active cell types (macrophages, adipose tissue, hepatocytes, and islet β-cells) in response to hyperglycemia (59–62), proinflammatory cytokines (46, 63), and saturated free fatty acids (64–67). 12-LO expression also increased in the whole brain of rats after treatment with a high sucrose diet (68). The cause-effect relationship of 12-LO in the context of vascular disease and metabolism in vivo initially arose from studies of whole-body Alox15−/− mice, which exhibit no overt phenotype, but show striking protection from disease upon challenges. In the setting of the atherosclerosis-prone Ldlr−/− and Apoe−/− mouse backgrounds, the absence of 12-LO confers protection against atherosclerosis and steatohepatitis (67, 69, 70). These effects likely result from the absence of 12-LO in macrophages/monocytes, because Apoe−/− mice receiving bone marrow from Alox15−/−;Apoe−/− mice were protected from the development of atherosclerosis (70) and macrophages from Alox15−/− mice have reduced ability form foam cells (47, 70)
A role for 12-LO in metabolic disease has been studied in the context of obesity/T2D and T1D. Studies of Nunemaker et al (71) examined the effects of Western high-fat diet (45% kcal from saturated fat) on Alox15−/− mice on the C57BL/6 background. Compared with control mice, the authors observed that Alox15−/− mice exhibited improved glucose tolerance, with reduced macrophage infiltration into fat, reduced proinflammatory cytokine levels (IL-6, TNFα), and improved β-cell function (as assessed by glucose-stimulated insulin secretion in vivo and in vitro). Using a similar feeding paradigm, Sears et al (72) observed that high fat-fed Alox15−/− mice displayed reduced levels of proinflammatory cytokines (IL-1β, IL-6, IL-12, and TNFα) accompanied by improved whole-body insulin sensitivity as assessed by hyperinsulinemic euglycemic clamp. Although these studies point to a role for 12-LO in proinflammatory macrophages, they must be reconciled with the known expression of 12-LO in white adipocytes, particularly after exposure to high-fat diets (64, 65, 67). 12-LO and its product 12-HETE are increased in visceral white adipose tissue of morbidly obese humans with T2D (73). High fat-fed adipose-specific Alox15−/− mice (Alox15lox/lox;Ap2-Cre) strikingly phenocopy whole-body Alox15−/− mice, exhibiting reduced macrophage infiltration into islets, improved insulin sensitivity, and protection from glucose intolerance (74). A caveat in these studies is the known expression of the Ap2 gene in macrophages as well as fat cells (75, 76), but these studies nevertheless raise the possibility that 12-LO in both cell types may contribute to insulin resistance and glucose intolerance seen during obesity/T2D.
Studies of 12-LO in the context of T1D are more limited but nevertheless compelling in terms of its effect on disease outcome. In studies of McDuffie et al (121), Alox15−/− mice were backcrossed onto the NOD background. NOD mice are the only mouse strain to exhibit spontaneous development of diabetes as a result of β-cell autoimmunity (77). NOD mice exhibit immune cell infiltration into islets (insulitis) as early as 4 weeks of age (consisting mostly of macrophages at this age), with subsequent diabetes development after the age of 12 weeks (12, 78). Female NOD mice generally exhibit higher rates of spontaneous diabetes compared with males, for reasons that remain unclear. NOD-Alox15−/− female mice showed nearly complete protection from T1D, whereas approximately 60% of control females developed diabetes; similarly, NOD-Alox15−/− males were completely protected, compared with approximately 20% of control males that developed diabetes. A notable finding was the virtual absence of macrophage insulitis in NOD-Alox15−/− mice, a finding suggesting a possible role for 12-LO in macrophages during diabetes pathogenesis in this model. In a follow up study, Green-Mitchell et al (79) demonstrated that 12-LO is expressed in macrophages, but not T and B cells, of NOD mice. Splenocytes from Alox15−/− mice were unable to adoptively transfer T1D to recipient mice, whereas those from control mice adoptively transferred diabetes within 4 weeks. These findings suggested a primary role for macrophage 12-LO in T1D disease pathogenesis.
12-LO in the Pancreatic Islet
A major limitation to the global deletion models of 12-LO is the inability to definitively attribute its effects in specific tissues or cell types. Because characterization of 12-LO focused primarily on cells derived from the bone marrow, particularly cells of the macrophage/monocyte origin, much of the literature is arguably biased towards attributing effects of 12-LO in these cell types. However, an increasing body of literature suggests that 12-LO may also play an intrinsic role in islet inflammation and dysfunction. The leukocyte isoform of 12-LO has been identified in the both the rodent islets (46, 80–82) and human islets (43, 46, 49). Similar to macrophages and adipose tissue, 12-LO expression and/or activity are up-regulated in mouse islets in vitro under conditions of hyperglycemia (61) and cytokine exposure (46, 63, 81, 83), and in vivo after high-fat diet feeding (63). In isolated human islets, 12-LO protein and activity levels are up-regulated by incubation with proinflammatory cytokines (49). Notably, no expression of 12-LO in nonendocrine pancreatic cell types have been observed in these studies. Within the islet, 12-LO expression has been reported in β-cells (46, 63) and in α-cells (84). With respect to the latter, overexpression of 12-LO in an α-cell line enhanced glucagon secretion, suggesting that the promotion of glucagon secretion by 12-LO might contribute to hyperglycemia in the setting of diabetes. More recently, 12-LO expression has been documented in pancreatic polypeptide (PP) cells of human diabetic pancreas (85), an observation that may have implications for 12-LO in postnatal islet cell dedifferentiation (see below).
12-LO in the Islet β-Cell
12-LO and its products appear to affect islet β-cell function, survival, and possibly differentiation. The major product of 12-LO, 12-HETE, reduces glucose-stimulated insulin secretion in human islets at low concentrations (1nM) and induce islet death at higher concentrations (100nM), whereas 15-HETE and the inactive form 12(R)-HETE have no effect (49). Similar findings have been observed in mouse islets (46). These findings in vitro suggest that the up-regulation of 12-LO seen in response to cytokines exposure or hyperglycemia may correlate closely to the dysfunction of β-cells observed in T2D and T1D. Recently, studies of Grzesik et al (85) using pancreas from donors with T2D and T1D revealed that 12-LO immunoreactivity is increased in islets of these individuals, but curiously with expression colocalizing with PP-staining cells. In light of recent provocative studies suggesting that dedifferentiation of β-cells to PP-expressing cells may underlie diabetes pathogenesis (86), the findings of Grzesik et al (85) suggest a potential role for 12-LO and its products in the dedifferentiation of β-cells in disease.
The earliest studies implicating a causative role for 12-LO in islet dysfunction in vivo involved low-dose streptozotocin (STZ) treatment of whole-body 12-LO knockout mice (87). STZ is a β-cell toxin that is taken up via glucose transporter 2 glucose transporters (88). When given in multiple low doses, STZ results in the influx of proinflammatory leukocytes into islets, initiating a cascade of events resulting in the local release of proinflammatory cytokines, β-cell dysfunction, and eventual β-cell death (89–91). Bleich et al (87) demonstrated that whole-body 12-LO knockout mice are protected from hyperglycemia and β-cell loss after multiple low-dose STZ, suggesting an inherent resistance of β-cells to stress and death in the absence of 12-LO. Nevertheless, the loss of 12-LO in macrophages and other leukocytes might also have contributed to the observed phenotype. More definitive evidence supporting a causative role for 12-LO in islet dysfunction arose from recent studies of pancreas-specific Alox15−/− knockout mice (Alox15lox/lox;Pdx1-Cre). In this model, Tersey et al (63) demonstrated that loss of 12-LO in the pancreas (including islets) resulted in protection from both low-dose STZ-induced hyperglycemia and high-fat diet-induced glucose intolerance. Unlike whole-body knockout mice, however, the high-fat diet-fed pancreas-specific knockouts exhibited no protection from insulin resistance or macrophage infiltration into fat, emphasizing a previously unappreciated role for islet 12-LO in the deterioration of metabolic homeostasis. These findings suggest that phenotypes observed in whole-body 12-LO knockouts likely reflect a complexity of 12-LO action in multiple metabolically active tissue types. In the context of T1D, 12-LO enzyme levels are known to increase in islets of NOD mice in the prediabetic phase (79), suggesting a possible contribution of 12-LO to islet autoimmunity and dysfunction, however, a definitive role for islet 12-LO in T1D must await studies of tissue-specific knockouts on the NOD background.
Molecular Mechanisms of 12-LO Contributing to β-Cell Dysfunction
The mechanisms by which 12-LO activity causes β-cell dysfunction in the setting of diabetogenic stress (proinflammatory cytokines, hyperglycemia, saturated free fatty acids) remain incompletely defined, but recent evidence points to involvement of reactive oxygen species (ROS) generated by its major products 12-HPETE and 12-HETE (see Figure 2). Islet β-cells are particularly sensitive to oxidative stress, because levels of antioxidant enzymes are low in these cells relative to other metabolically active tissues (92). In addition to the previously discussed activation of stress kinases c-Jun N-terminal kinase and p38 MAPK by 12-HETE (46, 49), 12-HETE also activates NADPH oxidase-1 (NOX-1) in mouse and human islets (93). Inhibition of 12-LO activity using specific inhibitors attenuates NOX-1 expression, reduces ROS and restores glucose-stimulated insulin secretion in response to proinflammatory cytokines (93). Studies of Tersey et al (63) also link 12-LO/12-HETE to the inactivation (ie, cytoplasmic sequestration) of the nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor, which is a major transcriptional activator of antioxidant genes. Pancreas-specific 12-LO knockout mice that were fed a high-fat diet exhibited greater nuclear levels of Nrf2 in β-cells, with concomitant increases in antioxidant enzymes superoxide dismutase and glutathione peroxidase (63).
Figure 2.

12-LO pathway in the β-cell. The figure depicts the pathways activated by 12-LO in the islet β-cell in response to elevated glucose levels, saturated free fatty acids, or proinflammatory cytokines. Activation of 12-LO leads to the production of proinflammatory lipid intermediates (12-HPETE and 12-HETE), which subsequent trigger inflammatory pathways mediated by c-Jun N-terminal kinase (JNK), p38-MAPK, and NOX. 12-HETE also prevents the translocation of Nrf2. Collectively, these pathways lead to increased ROS, oxidative stress, ER stress, and ultimately β-cell dysfunction and death. FFAR, free fatty acid receptor; PLA, phospholipase A; PGE2, prostaglandin E2; MCP1, monocyte chemoattractant protein 1.
Excessive ROS can induce perturbations in ER homeostasis, leading to protein misfolding, β-cell dysfunction, and eventual β-cell death (when the ER stress cascade is initiated) (reviewed in Ref. 94). In this respect, excessive 12-HETE (via ROS generation) leads to the development of β-cell ER stress, as evidenced by increased expression of Chop and spliced Xbp1, and increased production of unprocessed proinsulin (63). These and other effects of 12-HETE may be mediated via interaction with a G protein-coupled receptor (95). Recently, the orphan G protein-coupled receptor 31 (GPR31) was shown to interact with 12-HETE at low nanomolar concentrations (96). Activation of GPR31 receptor by 12-HETE was associated with stress kinase activation (96). However, a direct role in vivo for GPR31 in the proinflammatory effects of 12-LO, particularly in the β-cell, has yet to be elucidated. Other putative HETE receptors (although not specific for 12-HETE) include the peroxisome proliferator-activated receptors (97) and an eicosatetraenoic receptor (98).
Apart from the production of ROS, other mechanisms of 12-LO activity have also been proposed to contribute to β-cell dysfunction. Arachidonic acid levels are exceptionally high in pancreatic islets (∼30% of total islet glycerolipid fatty acid mass) (99) and is a potentiator of insulin secretion (43, 100, 101). In this respect, increased β-cell activation of 12-LO in the setting of diabetogenic stressors may cause metabolic shunting of arachidonic acid, providing less stimulus for insulin secretion. Additionally, 12-LO has been shown to activate Cox2 in β-cells, converting arachidonic acid to prostaglandin E2 (55), which is a potent inhibitor of insulin secretion (102–104). 12-HETE has been shown to induce macrophage chemoattractant protein 1 in β-cells (93), promoting the influx of proinflammatory macrophages into islets as part of a noncell autonomous role of 12-LO in inducing β-cell dysfunction. Finally, in hepatocytes, it was recently demonstrated that the absence or inhibition of 12-LO leads to an increase in the appearance of autophagy (105), a finding that suggests that 12-LO may suppress a potentially protective clearing mechanism that is otherwise required during periods of stress.
Discovery and Application of Small Molecule 12-LO Inhibitors
For their role in a variety of inflammatory disorders and malignancies, the LOs have been prime targets for the development of chemical inhibitors. To date, the only FDA-approved inhibitor is targeted against 5-LO (Zileuton) for use in asthma (106). Baicalein was used in early studies as a 12-LO inhibitor, but was later shown to be nonspecific and to inhibit both 12- and 15-LO (107). Early efforts to discover novel potent and selective 12-LO inhibitors through traditional medicinal chemistry (108–115), computational chemistry (116) and natural product isolation (117) were largely unsuccessful. The compounds discovered in these attempts were promiscuous and/or reductive in nature and not drug-like, chemically tractable, or selective. However, high throughput screening attempts followed by medicinal chemistry optimization resulted in an 8-hydroxyquinoline based compound, N-((5-bromo-8-hydroxyquinolin-7-yl)(thiophen-2-yl)methyl)acetamide (ML127) (Figure 3), which exhibits micromolar potency and over 50-fold selectivity over LO isozymes and COX (118). However, a subsequent molecule, N-(benzo[d]thiazol-2-yl)-4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide (Figure 3), exhibited slightly improved potency (submicromolar potency) and comparable selectivity with ML127 but is less likely to chelate metals and has improved drug-like qualities (119). Taylor-Fishwick et al (120) demonstrated that 8-hydroxyquinoline-based 12-LO inhibitors blocked 12-HETE production from cytokine-stimulated human islets, led to improved insulin release, and enhanced islet survival. Additionally, the authors demonstrated that one of the compounds (ML127) could reduce plasma 12-HETE levels when administered orally to mice. As such, 8-hydroxyquinoline compounds represent strong leads as clinically tractable 12-LO inhibitors.
Figure 3.

Small molecule inhibitors of 12-LO. Shown are the structures of the 8-hydroxyquinoline-based inhibitors of 12-LO, ML127 and N-(benzo[d]thiazol-2-yl)-4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide (ML355).
Conclusion and Future Directions
The LOs and their lipid products have been studied extensively for their roles in a variety of diseases from allergic/immunologic disorders to metabolism to cancer. 12-LO has been shown to be almost uniformly proinflammatory in all metabolically active tissues studied. To the extent that whole-body Alox15−/− mice exhibit no phenotype when unstressed, 12-LO represents an attractive, yet still somewhat underdeveloped target in metabolic disease. The near-parallel expression patterns and functions of 12-LO in mouse and human tissues provides some level of confidence that successes with next-generation 12-LO inhibitors in mouse models will portend potential utility in human disease. Nevertheless, several crucial questions still remain unanswered with respect to the role of 12-LO in different tissue types, and precisely how 12-LO products (such as 12-HETE) exert their downstream effects and via which receptor types. Moreover, it is presently unknown if inhibition or elimination of 12-LO after the establishment of T2D or T1D will allow for reversal of disease. These and other crucial questions can be fairly readily addressed in mouse models, because both conditional knockout mice and specific inhibitors are now available.
Acknowledgments
We thank Dr B. Maier for his helpful discussions and critiques.
This work was supported by an American Diabetes Junior Faculty Award (S.A.T.); an American Physiological Society Porter Fellowship (E.B.); National Institutes of Health Grants R01 DK083583 and R01 DK060581 (to R.G.M.), R01 AG047986 (to T.R.H.), and R01 HL112605 (to J.L.N.); a grant from the Juvenile Diabetes Research Foundation (J.L.N. and T.R.H.); and grants from the Ball Brothers Foundation and the George and Francis Ball Foundation (R.G.M.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Cox
- cyclooxygenase
- ER
- endoplasmic reticulum
- 12-HETE
- 12-hydroxyeicosatetraenoic acid
- 12-HPETE
- 12-hydroperoxyeicosatetraenoic acid
- GPR31
- G protein-coupled receptor 31
- LO
- lipoxygenase
- ML127
- N-((5-bromo-8-hydroxyquinolin-7-yl)(thiophen-2-yl)methyl)acetamide
- NOD
- nonobese diabetic
- NOX-1
- NADPH oxidase-1
- Nrf2
- nuclear factor erythroid 2-related factor 2
- ROS
- reactive oxygen species
- Pdx1
- pancreatic and duodenal homeobox
- P38 MAPK
- p38 mitogen-activated protein kinase
- PP
- pancreatic polypeptide
- STZ
- streptozotocin
- T2D
- type 2 diabetes.
References
- 1. Cowie CC, Rust KF, Ford ES, et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care. 2009;32:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. International Diabetes Federation. IDF Diabetes Atlas. 6th ed Brussels, Belgium: International Diabetes Federation; 2013. [Google Scholar]
- 3. Narayan KM, Boyle JP, Thompson TJ, Sorensen SW, Williamson DF. Lifetime risk for diabetes mellitus in the United States. JAMA. 2003;290:1884–1890. [DOI] [PubMed] [Google Scholar]
- 4. Dabelea D, Mayer-Davis EJ, Saydah S, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014;311(17):1778–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forlenza GP, Rewers M. The epidemic of type 1 diabetes: what is it telling us? Curr Opin Endocrinol Diabetes Obes. 2011;18:248–251. [DOI] [PubMed] [Google Scholar]
- 6. Lawrence JM, Imperatore G, Dabelea D, et al. Trends in incidence of type 1 diabetes among non-Hispanic white youth in the U.S., 2002–2009. Diabetes. 2014;63(11):3938–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanas R, John G, International HBA1c Consensus Committee. 2010 consensus statement on the worldwide standardization of the hemoglobin A1C measurement. Diabetes Care. 2010;33:1903–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lawes CM, Parag V, Bennett DA, et al. Blood glucose and risk of cardiovascular disease in the Asia Pacific region. Diabetes Care. 2004;27:2836–2842. [DOI] [PubMed] [Google Scholar]
- 9. Kahn SE, Prigeon RL, McCulloch DK, et al. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects: evidence for a hyperbolic function. Diabetes. 1993;42(11):1663–1672. [DOI] [PubMed] [Google Scholar]
- 10. Ferrannini E, Mari A, Nofrate V, Sosenko JM, Skyler JS, DPT-1 Study Group. Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes 2010;59:679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ize-Ludlow D, Lightfoot YL, Parker M, et al. Progressive erosion of β-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes. 2011;60:2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tersey SA, Nishiki Y, Templin AT, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Velloso LA, Eizirik DL, Cnop M. Type 2 diabetes mellitus-an autoimmune disease? Nat Rev Endocrinol. 2013;9:750–755. [DOI] [PubMed] [Google Scholar]
- 14. Evans-Molina C, Hatanaka M, Mirmira RG. Lost in translation: endoplasmic reticulum stress and the decline of β-cell health in diabetes mellitus. Diabetes Obes Metab. 2013;15(suppl 3):159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imai Y, Dobrian AD, Morris MA, Nadler JL. Islet inflammation: a unifying target for diabetes treatment? Trends Endocrinol Metab. 2013;24:351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nishimura S, Manabe I, Nagai R. Adipose tissue inflammation in obesity and metabolic syndrome. Discov Med. 2009;8:55–60. [PubMed] [Google Scholar]
- 17. Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. [DOI] [PubMed] [Google Scholar]
- 18. Morris DL, Cho KW, Delproposto JL, et al. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes. 2013;62(8):2762–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr Opin Clin Nutr Metab Care. 2011;14:341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brooks-Worrell BM, Boyko EJ, Palmer JP. Impact of islet autoimmunity on the progressive β-cell functional decline in type 2 diabetes. Diabetes Care. 2014;37(12):3286–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brooks-Worrell BM, Iyer D, Coraza I, et al. Islet-specific T-cell responses and proinflammatory monocytes define subtypes of autoantibody-negative ketosis-prone diabetes. Diabetes Care. 2013;36(12):4098–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eguchi K, Manabe I, Oishi-Tanaka Y, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012;15:518–533. [DOI] [PubMed] [Google Scholar]
- 23. Ehses JA, Perren A, Eppler E, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–2370. [DOI] [PubMed] [Google Scholar]
- 24. Weir GC, Bonner-Weir S. Five stages of evolving β-cell dysfunction during progression to diabetes. Diabetes. 2004;53(suppl 3):S16–S21. [DOI] [PubMed] [Google Scholar]
- 25. White MG, Marshall HL, Rigby R, et al. Expression of mesenchymal and α-cell phenotypic markers in islet β-cells in recently diagnosed diabetes. Diabetes Care. 2013;36(11):3818–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo S, Dai C, Guo M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest. 2013;123(8):3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Atkinson MA, Bluestone JA, Eisenbarth GS, et al. How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited. Diabetes. 2011;60:1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. [DOI] [PubMed] [Google Scholar]
- 30. Maganti A, Evans-Molina C, Mirmira RG. From immunobiology to β-cell biology: the changing perspective on type 1 diabetes. Islets. 2014;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Sullivan-Murphy B, Urano F. ER stress as a trigger for β-cell dysfunction and autoimmunity in type 1 diabetes. Diabetes. 2012;61:780–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soleimanpour SA, Stoffers DA. The pancreatic β cell and type 1 diabetes: innocent bystander or active participant? Trends Endocrinol. Metab. 2013;24(7):324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keskinen P, Korhonen S, Kupila A, et al. First-phase insulin response in young healthy children at genetic and immunological risk for type I diabetes. Diabetologia. 2002;45:1639–1648. [DOI] [PubMed] [Google Scholar]
- 34. Engin F, Yermalovich A, Nguyen T, et al. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci Transl Med. 2013;5:211ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marhfour I, Lopez XM, Lefkaditis D, et al. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia. 2012;55:2417–2420. [DOI] [PubMed] [Google Scholar]
- 36. Powell WS, Rokach J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim Biophys Acta. 2015;1851(4):340–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111(10):5866–5898. [DOI] [PubMed] [Google Scholar]
- 38. Rådmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim Biophys Acta. 2015;1851(4):331–339. [DOI] [PubMed] [Google Scholar]
- 39. Mehrabian M, Allayee H, Wong J, et al. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91(2):120–126. [DOI] [PubMed] [Google Scholar]
- 40. Zhao L, Moos MP, Gräbner R, et al. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat Med. 2004;10(9):966–973. [DOI] [PubMed] [Google Scholar]
- 41. Gubitosi-Klug RA, Talahalli R, Du Y, Nadler JL, Kern TS. 5-Lipoxygenase, but not 12/15-lipoxygenase, contributes to degeneration of retinal capillaries in a mouse model of diabetic retinopathy. Diabetes. 2008;57(5):1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morgan RO, Laychock SG. Biosynthesis of peptidyl leukotrienes and other lipoxygenase products by rat pancreatic islets. Comparison with macrophages and neutrophils. Prostaglandins. 1988;35(4):609–623. [DOI] [PubMed] [Google Scholar]
- 43. Persaud SJ, Muller D, Belin VD, et al. The role of arachidonic acid and its metabolites in insulin secretion from human islets of langerhans. Diabetes. 2007;56:197–203. [DOI] [PubMed] [Google Scholar]
- 44. Mehrabian M, Schulthess FT, Nebohacova M, et al. Identification of ALOX5 as a gene regulating adiposity and pancreatic function. Diabetologia. 2008;51(6):978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen XS, Brash AR, Funk CD. Purification and characterization of recombinant histidine-tagged human platelet 12-lipoxygenase expressed in a baculovirus/insect cell system. Eur J Biochem. 1993;214(3):845–852. [DOI] [PubMed] [Google Scholar]
- 46. Chen M, Yang ZD, Smith KM, Carter JD, Nadler JL. Activation of 12-lipoxygenase in proinflammatory cytokine-mediated β cell toxicity. Diabetologia. 2005;48:486–495. [DOI] [PubMed] [Google Scholar]
- 47. Funk CD, Cyrus T. 12/15-lipoxygenase, oxidative modification of LDL and atherogenesis. Trends Cardiovasc Med. 2001;11:116–124. [DOI] [PubMed] [Google Scholar]
- 48. Kühn H, O'Donnell VB. Inflammation and immune regulation by 12/15-lipoxygenases. Prog Lipid Res. 2006;45:334–356. [DOI] [PubMed] [Google Scholar]
- 49. Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 12-Lipoxygenase products reduce insulin secretion and β-cell viability in human islets. J Clin Endocrinol Metab. 2010;95:887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Natarajan R, Nadler JL. Lipid inflammatory mediators in diabetic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1542–1548. [DOI] [PubMed] [Google Scholar]
- 51. Nieves D, Moreno JJ. Enantioselective effect of 12(S)-hydroxyeicosatetraenoic acid on 3T6 fibroblast growth through ERK 1/2 and p38 MAPK pathways and cyclin D1 activation. Biochem Pharmacol. 2008;76(5):654–661. [DOI] [PubMed] [Google Scholar]
- 52. Parthasarathy S, Wieland E, Steinberg D. A role for endothelial cell lipoxygenase in the oxidative modification of low density lipoprotein. Proc Natl Acad Sci USA. 1989;86(3):1046–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tang DG, Honn KV. 12-Lipoxygenase, 12(S)-HETE, and cancer metastasis. Ann NY Acad Sci. 1994;744:199–215. [DOI] [PubMed] [Google Scholar]
- 54. Wen Y, Gu J, Chakrabarti SK, et al. The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-α in macrophages. Endocrinology. 2007;148:1313–1322. [DOI] [PubMed] [Google Scholar]
- 55. Han X, Chen S, Sun Y, Nadler JL, Bleich D. Induction of cyclooxygenase-2 gene in pancreatic β-cells by 12-lipoxygenase pathway product 12-hydroxyeicosatetraenoic acid. Mol Endocrinol. 2002;16:2145–2154. [DOI] [PubMed] [Google Scholar]
- 56. Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat Immunol. 2002;3(1):76–82. [DOI] [PubMed] [Google Scholar]
- 57. Middleton MK, Rubinstein T, Puré E. Cellular and molecular mechanisms of the selective regulation of IL-12 production by 12/15-lipoxygenase. J Immunol. 2006;176(1):265–274. [DOI] [PubMed] [Google Scholar]
- 58. Zhao L, Cuff CA, Moss E, et al. Selective interleukin-12 synthesis defect in 12/15-lipoxygenase-deficient macrophages associated with reduced atherosclerosis in a mouse model of familial hypercholesterolemia. J Biol Chem. 2002;277(38):35350–35356. [DOI] [PubMed] [Google Scholar]
- 59. Laybutt DR, Sharma A, Sgroi DC, Gaudet J, Bonner-Weir S, Weir GC. Genetic regulation of metabolic pathways in β-cells disrupted by hyperglycemia. J Biol Chem. 2002;277:10912–10921. [DOI] [PubMed] [Google Scholar]
- 60. Natarajan R, Gerrity RG, Gu JL, Lanting L, Thomas L, Nadler JL. Role of 12-lipoxygenase and oxidant stress in hyperglycaemia-induced acceleration of atherosclerosis in a diabetic pig model. Diabetologia. 2002;45:125–133. [DOI] [PubMed] [Google Scholar]
- 61. Natarajan R, Gu JL, Rossi J, et al. Elevated glucose and angiotensin II increase 12-lipoxygenase activity and expression in porcine aortic smooth muscle cells. Proc Natl Acad Sci USA. 1993;90:4947–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tokuyama Y, Sturis J, DePaoli AM, et al. Evolution of β-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes. 1995;44:1447–1457. [DOI] [PubMed] [Google Scholar]
- 63. Tersey SA, Maier B, Nishiki Y, Maganti AV, Nadler JL, Mirmira RG. 12-Lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol Cell Biol. 2014;34(19):3735–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chakrabarti SK, Wen Y, Dobrian AD, et al. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am J Physiol Endocrinol Metab. 2011;300:E175–E187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chakrabarti SK, Cole BK, Wen Y, Keller SR, Nadler JL. 12/15-lipoxygenase products induce inflammation and impair insulin signaling in 3T3-L1 adipocytes. Obesity. 2009;17:1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ferré N, Martínez-Clemente M, López-Parra M, et al. Increased susceptibility to exacerbated liver injury in hypercholesterolemic ApoE-deficient mice: potential involvement of oxysterols. Am J Physiol Gastrointest Liver Physiol. 2009;296(3):G553–G562. [DOI] [PubMed] [Google Scholar]
- 67. Martínez-Clemente M, Ferré N, Titos E, et al. Disruption of the 12/15-lipoxygenase gene (Alox15) protects hyperlipidemic mice from nonalcoholic fatty liver disease. Hepatology. 2010;52:1980–1991. [DOI] [PubMed] [Google Scholar]
- 68. Taha AY, Gao F, Ramadan E, Cheon Y, Rapoport SI, Kim H-W. Upregulated expression of brain enzymatic markers of arachidonic and docosahexaenoic acid metabolism in a rat model of the metabolic syndrome. BMC Neurosci. 2012;13:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. George J, Afek A, Shaish A, et al. 12/15-Lipoxygenase gene disruption attenuates atherogenesis in LDL receptor-deficient mice. Circulation. 2001;104:1646–1650. [DOI] [PubMed] [Google Scholar]
- 70. Huo Y, Zhao L, Hyman MC, et al. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–2031. [DOI] [PubMed] [Google Scholar]
- 71. Nunemaker CS, Chen M, Pei H, et al. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab. 2008;295:E1065–E1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sears DD, Miles PD, Chapman J, et al. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS One. 2009;4:e7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lieb DC, Brotman JJ, Hatcher MA, et al. Adipose tissue 12/15 lipoxygenase pathway in human obesity and diabetes. J Clin Endcrinol Metab. 2014;99(9):E1713–E1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cole BK, Morris MA, Grzesik WJ, Leone KA, Nadler JL. Adipose tissue-specific deletion of 12/15-lipoxygenase protects mice from the consequences of a high-fat diet. Mediat. Inflamm. 2012;2012:851798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fu Y, Luo N, Lopes-Virella MF. Oxidized LDL induces the expression of ALBP/aP2 mRNA and protein in human THP-1 macrophages. J Lipid Res. 2000;41:2017–2023. [PubMed] [Google Scholar]
- 76. Makowski L, Boord JB, Maeda K, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. [DOI] [PubMed] [Google Scholar]
- 78. Reddy S, Liu W, Elliott RB. Distribution of pancreatic macrophages preceding and during early insulitis in young NOD mice. Pancreas. 1993;8:602–608. [DOI] [PubMed] [Google Scholar]
- 79. Green-Mitchell SM, Tersey SA, Cole BK, et al. Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15(null) mice, leading to protection against type 1 diabetes development. PLoS One. 2013;8:e56763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bleich D, Chen S, Gu JL, Nadler JL. The role of 12-lipoxygenase in pancreatic -cells (review). Int J Mol Med. 1998;1:265–272. [PubMed] [Google Scholar]
- 81. Bleich D, Chen S, Gu JL, et al. Interleukin-1 β regulates the expression of a leukocyte type of 12-lipoxygenase in rat islets and RIN m5F cells. Endocrinology. 1995;136:5736–5744. [DOI] [PubMed] [Google Scholar]
- 82. Shannon VR, Ramanadham S, Turk J, Holtzman MJ. Selective expression of an arachidonate 12-lipoxygenase by pancreatic islet β-cells. Am J Physiol. 1992;263:E828–E836. [DOI] [PubMed] [Google Scholar]
- 83. Ma Z, Ramanadham S, Corbett JA, et al. Interleukin-1 enhances pancreatic islet arachidonic acid 12-lipoxygenase product generation by increasing substrate availability through a nitric oxide-dependent mechanism. J Biol Chem. 1996;271:1029–1042. [DOI] [PubMed] [Google Scholar]
- 84. Kawajiri H, Zhuang D, Qiao N, et al. Expression of arachidonate 12-lipoxygenase in rat tissues: a possible role in glucagon secretion. J Histochem Cytochem. 2000;48(10):1411–1419. [DOI] [PubMed] [Google Scholar]
- 85. Grzesik WJ, Nadler JL, Machida Y, Nadler JL, Imai Y, Morris MA. Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J Clin Endocrinol Metab. 2015;100(3):E387–E395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. El-Gohary Y, Tulachan S, Wiersch J, et al. A Smad signaling network regulates islet cell proliferation. Diabetes. 2014;63(1):224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bleich D, Chen S, Zipser B, Sun D, Funk CD, Nadler JL. Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J Clin Invest. 1999;103:1431–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang Z, Gleichmann H. GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice. Diabetes. 1998;47:50–56. [DOI] [PubMed] [Google Scholar]
- 89. Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of Langerhans constitutively present β cell-derived peptides bound to their class II MHC molecules. Proc Natl Acad Sci USA. 2008;105:6121–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Luki ML, Stosi-Grujici S, Shahin A. Effector mechanisms in low-dose streptozotocin-induced diabetes. Dev Immunol. 1998;6:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Maier B, Ogihara T, Trace AP, et al. The unique hypusine modification of eIF5A promotes islet β cell inflammation and dysfunction in mice. J Clin Invest. 2010;120:2156–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. [DOI] [PubMed] [Google Scholar]
- 93. Weaver JR, Holman TR, Imai Y, et al. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet β cell dysfunction. Mol Cell Endocrinol. 2012;358(1):88–95. [DOI] [PubMed] [Google Scholar]
- 94. Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. [DOI] [PubMed] [Google Scholar]
- 95. Liu B, Khan WA, Hannun YA, et al. 12(S)-hydroxyeicosatetraenoic acid and 13(S)-hydroxyoctadecadienoic acid regulation of protein kinase C-α in melanoma cells: role of receptor-mediated hydrolysis of inositol phospholipids. Proc Natl Acad Sci USA. 1995;92(20):9323–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Guo Y, Zhang W, Giroux C, et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem. 2011;286:33832–33840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shappell SB, Gupta RA, Manning S, et al. 15S-hydroxyeicosatetraenoic acid activates peroxisome proliferator-activated receptor γ and inhibits proliferation in PC3 prostate carcinoma cells. Cancer Res. 2001;61(2):497–503. [PubMed] [Google Scholar]
- 98. Hosoi T, Koguchi Y, Sugikawa E, et al. Identification of a novel human eicosanoid receptor coupled to Gi/o. J Biol Chem. 2002;277(35):31459–31465. [DOI] [PubMed] [Google Scholar]
- 99. Ramanadham S, Bohrer A, Mueller M, Jett P, Gross RW, Turk J. Mass spectrometric identification and quantitation of arachidonate-containing phospholipids in pancreatic islets: prominence of plasmenylethanolamine molecular species. Biochemistry. 1993;32(20):5339–5351. [DOI] [PubMed] [Google Scholar]
- 100. Jones PM, Persaud SJ. Arachidonic acid as a second messenger in glucose-induced insulin secretion from pancreatic β-cells. J Endocrinol. 1993;137(1):7–14. [DOI] [PubMed] [Google Scholar]
- 101. Turk J, Gross RW, Ramanadham S. Amplification of insulin secretion by lipid messengers. Diabetes. 1993;42(3):367–374. [DOI] [PubMed] [Google Scholar]
- 102. Kimple ME, Keller MP, Rabaglia MR, et al. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes. 2013;62(6):1904–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Parazzoli S, Harmon JS, Vallerie SN, Zhang T, Zhou H, Robertson RP. Cyclooxygenase-2, not microsomal prostaglandin E synthase-1, is the mechanism for interleukin-1β-induced prostaglandin E2 production and inhibition of insulin secretion in pancreatic islets. J Biol Chem. 2012;287(38):32246–32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tran PO, Gleason CE, Poitout V, Robertson RP. Prostaglandin E2 mediates inhibition of insulin secretion by interleukin-1β. J Biol Chem. 1999;274(44):31245–31248. [DOI] [PubMed] [Google Scholar]
- 105. Jang I, Park S, Cho JW, Yigitkanli K, van Leyen K, Roth J. Genetic ablation and short-duration inhibition of lipoxygenase results in increased macroautophagy. Exp Cell Res. 2014;321(2):276–287. [DOI] [PubMed] [Google Scholar]
- 106. Berger W, De Chandt MT, Cairns CB. Zileuton: clinical implications of 5-lipoxygenase inhibition in severe airway disease. Int J Clin Pract. 2007;61(4):663–676. [DOI] [PubMed] [Google Scholar]
- 107. Deschamps JD, Kenyon VA, Holman TR. Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg Med Chem. 2006;14:4295–4301. [DOI] [PubMed] [Google Scholar]
- 108. Amagata T, Whitman S, Johnson TA, et al. Exploring sponge-derived terpenoids for their potency and selectivity against 12-human, 15-human, and 15-soybean lipoxygenases. J Nat Prod. 2003;66:230–235. [DOI] [PubMed] [Google Scholar]
- 109. Cichewicz RH, Kenyon VA, Whitman S, et al. Redox inactivation of human 15-lipoxygenase by marine-derived meroditerpenes and synthetic chromanes: archetypes for a unique class of selective and recyclable inhibitors. J Am Chem Soc. 2004;126(45):14910–14920. [DOI] [PubMed] [Google Scholar]
- 110. Malterud KE, Rydland KM. Inhibitors of 15-lipoxygenase from orange peel. J Agric Food Chem. 2000;48(11):5576–5580. [DOI] [PubMed] [Google Scholar]
- 111. Moreau RA, Agnew J, Hicks KB, Powell MJ. Modulation of lipoxygenase activity by bacterial hopanoids. J Nat Prod. 1997;60(4):397–398. [DOI] [PubMed] [Google Scholar]
- 112. Sailer ER, Schweizer S, Boden SE, Ammon HP, Safayhi H. Characterization of an acetyl-11-keto-β-boswellic acid and arachidonate-binding regulatory site of 5-lipoxygenase using photoaffinity labeling. Eur J Biochem. 1998;256(2):364–368. [DOI] [PubMed] [Google Scholar]
- 113. Segraves EN, Shah RR, Segraves NL, et al. Probing the activity differences of simple and complex brominated aryl compounds against 15-soybean, 15-human, and 12-human lipoxygenase. J Med Chem. 2004;47(16):4060–4065. [DOI] [PubMed] [Google Scholar]
- 114. Vasquez-Martinez Y, Ohri RV, Kenyon V, Holman TR, Sepúlveda-Boza S. Structure-activity relationship studies of flavonoids as potent inhibitors of human platelet 12-hLO, reticulocyte 15-hLO-1, and prostate epithelial 15-hLO-2. Bioorg Med Chem. 2007;15(23):7408–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Whitman S, Gezginci M, Timmermann BN, Holman TR. Structure-activity relationship studies of nordihydroguaiaretic acid inhibitors toward soybean, 12-human, and 15-human lipoxygenase. J Med Chem. 2002;45(12):2659–2661. [DOI] [PubMed] [Google Scholar]
- 116. Kenyon V, Chorny I, Carvajal WJ, Holman TR, Jacobson MP. Novel human lipoxygenase inhibitors discovered using virtual screening with homology models. J Med Chem. 2006;49(4):1356–1363. [DOI] [PubMed] [Google Scholar]
- 117. Deschamps JD, Gautschi JT, Whitman S, et al. Discovery of platelet-type 12-human lipoxygenase selective inhibitors by high-throughput screening of structurally diverse libraries. Bioorg Med Chem. 2007;15(22):6900–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kenyon V, Rai G, Jadhav A, et al. Discovery of potent and selective inhibitors of human platelet-type 12- lipoxygenase. J Med Chem. 2011;54(15):5485–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Luci DK, Jameson JB, 2nd, Yasgar A, et al. Synthesis and structure-activity relationship studies of 4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide derivatives as potent and selective inhibitors of 12-lipoxygenase. J Med Chem. 2014;57(2):495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Taylor-Fishwick DA, Weaver J, Glenn L, et al. Selective inhibition of 12-lipoxygenase protects islets and β cells from inflammatory cytokine-mediated β cell dysfunction. Diabetologia. 2015;58(3):549–557. [DOI] [PubMed] [Google Scholar]
- 121. McDuffie M, Maybee NA, Keller SR, Stevens BK, Germey JC, Morris MA, et al. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes. 2008;57(1):199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]