

Abstract
We studied mechanisms of resistance to the novel taxane cabazitaxel in established cellular models of taxane resistance. We also developed cabazitaxel-resistant variants from MCF-7 breast cancer cells by stepwise selection in drug alone (MCF-7/CTAX) or drug plus the transport inhibitor PSC-833 (MCF-7/CTAX-P). Among multidrug resistant (MDR) variants, cabazitaxel was relatively less cross-resistant than paclitaxel and docetaxel (15 vs. 200-fold in MES-SA/Dx5 and 9 vs. 60-fold in MCF-7/TxT50, respectively). MCF-7/TxTP50 cells that were negative for MDR but had 9-fold resistance to paclitaxel were also 9-fold resistant to cabazitaxel. Selection with cabazitaxel alone (MCF-7/CTAX) yielded 33-fold resistance to cabazitaxel, 52-fold resistance to paclitaxel, activation of ABCB1, and 3-fold residual resistance to cabazitaxel with MDR inhibition. The MCF-7/CTAX-P variant did not express ABCB1, nor did it efflux rhodamine-123, BODIPY-labeled paclitaxel, and [3H]-docetaxel. These cells are hypersensitive to depolymerizing agents (vinca alkaloids and colchicine), have reduced baseline levels of stabilized microtubules, and impaired tubulin polymerization in response to taxanes (cabazitaxel or docetaxel) relative to MCF-7 parental cells. Class III β-tubulin (TUBB3) RNA and protein were elevated in both MCF-7/CTAX and MCF-7/CTAX-P. Decreased BRCA1 and altered epithelial-mesenchymal transition (EMT) markers are also associated with cabazitaxel resistance in these MCF-7 variants, and may serve as predictive biomarkers for its activity in the clinical setting. In summary, cabazitaxel resistance mechanisms include MDR (although at a lower level than paclitaxel and docetaxel), and alterations in microtubule dynamicity, as manifested by higher expression of TUBB3, decreased BRCA1, and by the induction of EMT.
Keywords: Drug resistance, taxanes, cabazitaxel, XRP6258, microtubules, EMT, BRCA1
Introduction
The taxanes paclitaxel (Taxol) and docetaxel (Taxotere) have substantial clinical activity in breast, ovarian, lung, and other cancers. These tubulin-active agents stabilize microtubules, blocking cells in the late G2/M phase of the cell cycle, resulting in cell death (1). Their clinical efficacy is limited by pre-existing or acquired drug resistance. We have previously derived multiple paclitaxel- and docetaxel-selected variants in human breast and ovarian cancer cell lines. In these studies, selection with taxanes alone induced multidrug resistance (MDR) related to expression of P-glycoprotein (P-gp) (2), and this resistance can be modulated in the presence of known transport inhibitors. Incorporation of the potent P-gp inhibitor PSC-833 during taxane selection has enabled us to establish non-MDR cellular models of taxane resistance. The taxane resistance observed in these variants is not associated with alterations in drug transport, but may include alterations in tubulin expression, apoptotic proteins, and cell cycle regulation (3–6).
Several semisynthetic taxane analogues have been developed with the goal of evading drug resistance, including cabazitaxel (Jevtana, XRP6258), which was selected for clinical development based on its in vivo activity in docetaxel-resistant MDR tumor models. Preclinical studies indicated that this taxane is as potent as docetaxel in cellular models, and more effective in variants selected for resistance to taxanes (7). In 2010 the FDA approved cabazitaxel in combination with prednisone/prednisolone for the treatment of patients with metastatic hormone-refractory prostate cancer previously treated with docetaxel (8–10).
This current study assessed cabazitaxel activity and resistance mechanisms in several taxane resistant variants, as well as two new cabazitaxel-selected variants of MCF-7 breast cancer cells, one selected with cabazitaxel alone and another co-selected with PSC-833.
Materials and Methods
Drugs and reagents
The anticancer drugs cisplatin, colchicine, daunorubicin, doxorubicin, paclitaxel, vinblastine, and vincristine were obtained from the drug repository of the National Cancer Institute (Bethesda, MD). Docetaxel and cabazitaxel (XRP6258, Jevtana, formerly RPR116258A, Supplementary Figure 1A) were gifts from Sanofi Oncology (Bridgewater, NJ). Novartis Pharmaceuticals (East Hanover, NJ) kindly provided the P-gp inhibitor PSC-833 (valspodar). All drugs were prepared in 100% ethanol as 1 mmol/L stock solutions and stored at −20 °C. All other chemicals were purchased from the Sigma-Aldrich Chemical Co. (St Louis, MO).
Cell culture and establishment of cabazitaxel-resistant MCF-7 variants
The MCF-7 human breast adenocarcinoma and OVCAR-3 human ovarian adenocarcinoma cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, purchased 6/1999). The human ovarian clear cell carcinoma cell line ES-2, human ovarian carcinoma MES-OV, and the human uterine sarcoma cell line MES-SA were established in our laboratory (MES-SA and ES-2 authenticated and submitted to the ATCC as CRL-1976 and 1978, respectively, and the MES-OV was submitted to the ATCC 7/2014). Cells were grown in McCoy 5A medium supplemented with 10% (v/v) fetal calf serum (NBCS), 100 U of penicillin/mL, and 100 μg of streptomycin/mL (Life Technologies, Carlsbad, CA) at 37 °C in a humidified atmosphere containing 5% CO2, and were routinely screened to rule out mycoplasma infection.
Parallel drug selections were initiated using 0.1 nmol/L cabazitaxel, a concentration that would inhibit growth in MCF-7 cells by 50% (IC50 value) with and without 2 μmol/L PSC-833. Selections continued by increasing the drug concentration in a stepwise manner up to a final concentration of 5 nmol/L cabazitaxel. Variants were grown drug-free for at least two passages prior to experiments.
The doxorubicin-selected human uterine sarcoma MDR variant MES-SA/Dx5 (authenticated and submitted to the ATCC as CRL-1977) was used as a positive control for transporter activity (11). In addition, two docetaxel-selected MCF-7 variants were used in this study. The MCF-7/TxT50 variant was selected with docetaxel alone, is positive for P-gp and demonstrates a typical MDR phenotype. The MCF-7/TxTP50 variant was co-selected with docetaxel and PSC-833, and its resistance is not due to transporters (4).
Growth inhibition assays
The in vitro activity of various anticancer drugs was tested using a modified sulforhodamine B (SRB) colorimetric assay following a 72 hr drug incubation representing approximately three cell divisions (4). Clonogenic assays were also used to assess cell survival and proliferation. In these assays, 5–10 × 103 cells were seeded in six-well tissue culture dishes and allowed to attach overnight. Cells were exposed to increasing concentrations of taxane (0.1 nmol/L to 1 μmol/L) for 24 hr, at which time the medium containing drug was aspirated and replaced with drug-free complete medium. Cells were incubated for an additional 14 days at 37 °C and 5% CO2, fixed in 10% (w/v) trichloroacetic acid overnight at 4 °C, stained with a 0.4% (w/v) SRB solution in 1% (v/v) acetic acid, and colonies greater than 50 cells per aggregate were scored. Drug effects were calculated as a percentage relative to untreated control survival, and response versus drug concentration was calculated using the Hill equation in KaleidaGraph software (Synergy Software, Reading, PA). Each drug concentration was tested in quadruplicate measurements per experiment, and data presented are the average of three independent experiments ± standard deviations.
Gene expression profiling by quantitative PCR and NanoString Technology
Total RNA was extracted using the RNeasy Mini kit (Qiagen, Valencia, CA) and 1 μg was reverse transcribed (RT) into cDNA using the Super Script III First-Strand Synthesis kit with oligo(dT)20 primer (both from Life Technologies), and the resulting cDNA was stored at −20 °C. Expression of genes of interest was measured by qPCR (4) using a QuantStudio 12K Flex Real-Time PCR system (Life Technologies).
A highly sensitive, multiplexed measurement of gene expression was also employed. NanoString Technology (Seattle, WA) is based on a color-coded barcode (nCounter Reporter Probes) attached to a single target-specific probe corresponding to a gene of interest, which is hybridized directly to target molecules without the need for amplification (12). A custom-designed code set was used, and each reaction contained 250 ng of total RNA in a 5 μL aliquot. Probes were added in massive excess to target mRNA to ensure that each gene would be labeled, followed by a series of wash steps to remove unbound probes and non-target cellular transcripts. Color codes were counted and tabulated for each target molecule using the nCounter Digital Analyzer (NanoString Technology) at Stanford’s Functional Genomics Facility. Raw counts were normalized to internal levels of 7 reference genes, and a background count was estimated using the average count of 8 negative control probes in every reaction.
Western blotting
Total protein lysates were isolated from growing cells using 1x radioimmunoprecipitation assay buffer [RIPA, 1% (v/v) NP40, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS in 1x PBS buffer] with freshly added protease inhibitors (cocktail from Bio-Rad Laboratories, Hercules, CA). Total protein (10–25 μg) was separated by 4–20% (w/v) gradient polyacrylamide gels and transferred onto nitrocellulose membranes using the Trans-Blot Turbo transfer system (all Bio-Rad Laboratories). Membranes were blocked overnight at 4 °C in 1x TBS containing 5% (w/v) nonfat milk and 1% (w/v) bovine serum albumin, and then incubated with the following antibodies: anti-P-gp, BCRP, and MRP2 (Signet Laboratories, Dedham, MA), anti-MRP7 (Thermo Fisher Scientific, Rockford, IL), anti-class I, pan α- and β-tubulin (Sigma Aldrich), anti-class II and III β-tubulin (Covance, Berkeley, CA), class IV β-tubulin (Abcam, Cambridge, MA), anti-GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA), and specific antibodies for BRCA1, Bcl2, inhibitors of apoptosis, and p21 (Cell Signaling Technology, Danvers, MA). These primary antibodies were recognized by species-appropriate horseradish peroxidase-conjugated secondary antibodies, and detected using the Clarity Western ECL substrate (Bio-Rad).
Tubulin polymerization assays
Soluble and polymerized tubulin fractions were separated by centrifugation (20,000 × g) following a 5 minute incubation in hypotonic buffer with and without drug at 37 °C (13, 14). The soluble tubulin fractions were transferred to fresh microcentrifuge tubes and stored on ice, while fractions containing polymerized tubulin were sonicated for 10 seconds on ice prior to adding 4x Laemmli sample buffer (Bio-Rad). Equal volumes of soluble and polymerized fractions were resolved on gradient polyacrylamide gels and transferred to nitrocellulose as previously described. Immunoblotting with a pan α-tubulin antibody (clone DM1A, Sigma-Aldrich) isolated the tubulin fractions, and the percentage of tubulin polymer present in each fraction was calculated based on the total tubulin (soluble and polymerized) present in each experimental condition as determined by densitometry.
Functional assays for transporter activity
Cellular drug accumulation was determined using a published method (6). Briefly, 1 × 106 cells were seeded in six-well dishes and allowed to attach overnight. [3H]-docetaxel (10 nmol/L, American Radiolabeled Chemicals, St. Louis, MO) was allowed to accumulate for 1 hr at 37 °C with and without 2 μmol/L PSC-833, aspirated, and dishes were washed once with ice-cold PBS. Cells were lysed immediately using a 2% (w/v) SDS solution, and counts were determined upon the addition of EcoLite liquid scintillation cocktail (MP Biomedicals, Solon, OH), and normalized to protein content.
These data were confirmed by determining the accumulation of rhodamine-123 and BODIPY-paclitaxel (both Life Technologies) by flow cytometry. Cells were harvested, exposed to either rhodamine-123 or BODIPY-paclitaxel for 1 hr at 37 °C, drug was removed by centrifugation (200 × g) at 4 °C, and cells washed once in cold PBS. In order to correlate drug accumulation with P-gp content, cells were stained on ice using an anti-P-gp mouse monoclonal which detects an external epitope (clone UIC2, EMD Millipore, Billerica, Ma), and detected by a Texas-Red goat anti-mouse secondary antibody (Life Technologies) using an LSR II flow cytometer (BD Biosciences, San Jose, CA). The effects of 2 μmol/L PSC-833 and other known MDR modulators were accessed in separate experimental conditions.
Transient TUBB3 and BRCA1 silencing by small interfering RNA
Pools of four gene-specific siRNAs were designed using Dharmacon’s siDesign Center algorithm and synthesized (ON-TARGETplus reagents, GE Dharmacon, Lafayette, CO). Lipid-mediated siRNA delivery into cells was accomplished with DharmaFECT 1 transfection reagent (GE Dharmacon) according to the manufacturer’s protocol 24 hr after cells were seeded. Cells were allowed to incubate in siRNAs for 24 hr prior to the addition of cabazitaxel. Optimal concentrations of siRNAs were determined in order to avoid off-target effects, and the time course of gene silencing was evaluated relative to non-targeting controls (GE Dharmacon) from 24 to 96 hr after transfection by RT-qPCR and immunoblotting with specific antibodies.
Results
Cabazitaxel is more potent than paclitaxel and less cross-resistant in MDR variants
Cabazitaxel potency was measured in several taxane-sensitive cell lines using the SRB colorimetric cell proliferation assay. Cabazitaxel was 10-fold more potent than paclitaxel and comparable to docetaxel in MCF-7 breast cancer cells following a 72 hr drug incubation (Supplementary Figure 1B). These data were confirmed in several other cell lines including the human uterine sarcoma, MES-SA, human ovarian cancer cell lines ES-2, MES-OV, and OVCAR-3 cell lines (Supplementary Table S1).
We evaluated cabazitaxel activity in P-gp expressing cell variants, including the doxorubicin-selected variant, MES-SA/Dx5, and the docetaxel-selected MCF-7/TxT50. The expression of ABCB1 in these variants and other resistant models is shown in Figure 1A. The MES-SA/Dx5 cell line is approximately 200-fold resistant to the taxanes, paclitaxel and docetaxel, but 15-fold resistant to cabazitaxel (Table 1). Likewise, the MCF-7/TxT50 is 60-fold resistant to paclitaxel and docetaxel, and 8.6-fold resistant to cabazitaxel. Co-incubation with the P-gp inhibitor PSC-833 (2 μmol/L) completely restored sensitivity to parental levels for all taxanes tested.
Figure 1.

The expression of ABCB1 was determined by RT-PCR following 35 cycles of amplification, with ribosomal RNA-specific amplimers used as an internal control (panel A). Parental MES-SA cells and doxorubicin-selected MES-SA/Dx5 variants were included as negative and positive controls for ABCB1 expression, respectively. In addition the docetaxel-selected variant MCF-7/TxT50 was included as another positive control for ABCB1 transcripts, and the docetaxel and PSC-833 co-selected MCF-7/TxTP50 variant was included as a negative control. UIC2 staining recognized by a Texas Red-conjugated secondary antibody was used to screen for P-gp status in MCF-7/CTAX (panel B, gray shaded area) and MCF-7/CTAX-P (dashed gray line) compared to MCF-7 parental cells (solid black line) by flow cytometry. [3H]-docetaxel levels were determined following a 1 hr accumulation at 37 °C with and without 2 μmol/L PSC-833 and normalized to protein content (panel C). The average of three determinations is presented ± standard deviation. BODIPY-paclitaxel binding was accessed following a 1 hr efflux at 37 °C after drug accumulation in MCF-7/CTAX-P (gray shaded area) relative to the MCF-7 control (solid black line) by flow cytometry (panel D). Untreated MCF-7 (dashed gray line) is included for reference.
Table 1.
Cabazitaxel activity in two ABCB1 variants, the doxorubicin-selected MES-SA/Dx5 and docetaxel-selected MCF-7/TxT50, and in the non-ABCB1 MCF-7/TxTP50 variant which was selected for resistance to docetaxel in the presence of the P-gp inhibitor PSC-833. SRB assays were run following 72 hr drug incubations with and without PSC-833 (2 μmol/L). Data are expressed as the mean of three independent determinations ± standard deviations.
| Relative Resistance1 (Modulation by PSC-833)2 | |||
|---|---|---|---|
| ABCB1 and P-gp | (+) | (+) | (−) |
| MES-SA/Dx5 | MCF-7/TxT50 | MCF-7/TxTP50 | |
| Docetaxel | 190 ± 20 (2.2 ± 0.76) | 60 ± 3.0 (1.1 ± 0.23) | 8.3 ± 0.76 (9.3 ± 0.51) |
| Paclitaxel | 210 ± 11 (1.5 ± 0.20) | 55 ± 2.0 (1.3 ± 0.32) | 8.5 ± 0.25 (9.1 ± 0.76) |
| Cabazitaxel | 15 ± 1.7 (0.80 ± 0.30) | 8.6 ± 1.6 (0.93 ± 0.32) | 9.2 ± 0.36 (8.5 ± 0.64) |
| Vinblastine | 150 ± 21 (1.5 ± 0.25) | 75 ± 4.0 (2.3 ± 0.20) | 0.76 ± 0.12 (0.90 ± 0.10) |
| Vincristine | 150 ± 26 (1.3 ± 0.20) | 70 ± 4.5 (1.5 ± 0.49) | 0.73 ± 0.21 (0.80 ± 0.10) |
| Colchicine | 170 ± 29 (1.2 ± 0.23) | 73 ± 7.6 (1.2 ± 0.10) | 0.87 ± 0.21 (0.93 ± 0.15) |
| Daunorubicin | 35 ± 2.5 (1.5 ± 0.20) | 15 ± 2.0 (1.9 ± 0.40) | 1.2 ± 0.10 (1.1 ± 0.32) |
| Doxorubicin | 32 ± 2.0 (1.3 ± 0.20) | 15 ± 2.1 (1.5 ± 0.25) | 1.1 ± 0.12 (1.3 ± 0.20) |
| Cisplatin | 2.0 ± 0.5 (1.5 ± 0.30) | 2.3 ± 0.3 (1.7 ± 0.15) | 1.2 ± 0.25 (1.1 ± 0.23) |
The relative resistance was calculated by dividing the IC50 value of the variant by the IC50 of the wild-type cell line.
Relative resistance following co-incubation with the P-gp inhibitor PSC-833 at 2 μmol/L.
A non-MDR docetaxel-selected variant, MCF-7/TxTP50, was developed by co-selection with docetaxel and PSC-833. These cells have elevated TUBB3 levels and altered tubulin dynamicity as a mechanism of resistance, and do not have mutations in beta-1 tubulin (Class I, M40) and alpha-tubulin (K-α1). The MCF-7/TxTP50 cell line is cross-resistant to cabazitaxel (9.2-fold), comparable to the other taxanes tested, and this resistance to cabazitaxel was not affected by the MDR modulator PSC-833 (Table 1).
Development of cabazitaxel-resistant MCF-7 variants
Two cabazitaxel-resistant variants were established by long-term selection with clinically relevant concentrations of cabazitaxel in the MCF-7 breast cancer cell line. The MCF-7/CTAX was derived by cabazitaxel exposure up to 5 nmol/L, and the predominant mechanism in this variant is activation of the ABCB1 gene. We confirmed the presence of ABCB1 transcripts by RT-PCR using several sets of ABCB1-specific amplimers (Figure 1A). MCF-7/CTAX cells were positive for P-gp using the monoclonal antibody UIC2 by flow cytometry (Figure 1B). Since taxanes are substrates for other ATP binding cassette MDR transporters, we also screened for ABCC2/MRP2, ABCC10/MRP7, and ABCG2/BCRP and found no activation of these transporters relative to parental content following cabazitaxel selection (Supplementary Figure S2). Of interest, ABCG2 (BCRP, MXR) was activated following docetaxel selection in both of the MCF-7 variants, MCF-7/TxT50 and MCF-7/TXTP50 (Supplementary Figure S2).
In a parallel selection, we co-selected MCF-7 cells with cabazitaxel and PSC-833 and established the MCF-7/CTAX-P variant, which is approximately 9-fold resistant to cabazitaxel and docetaxel, and 4-fold resistant to paclitaxel by SRB assays (Table 2). This variant is negative for P-gp expression (Supplementary Figure S2).
Table 2.
Resistance phenotype in the ABCB1-positive MCF-7/CTAX, and in the non-MDR MCF-7/CTAX-P variant which was co-selected with cabazitaxel and PSC-833. Assays were run after 72 hr drug incubations with and without PSC-833 (2 μmol/L). Data are expressed as the mean of three independent determinations ± standard deviation.
| Relative Resistance1 (Modulation by PSC-833)2 | ||
|---|---|---|
| ABCB1 and P-gp | (+) | (−) |
| MCF-7/CTAX | MCF-7/CTAX-P | |
| Cabazitaxel | 33 ± 3.8 (2.6 ± 0.11) | 8.6 ± 0.53 (9.3 ± 0.42) |
| Docetaxel | 58 ± 4.5 (2.8 ± 0.25) | 9.0 ± 1.0 (8.5 ± 0.5) |
| Paclitaxel | 52 ± 2.5 (3.2 ± 0.15) | 4.1 ± 0.81 (4.8 ± 0.76) |
| Vinblastine | 35 ± 5.0 (0.87 ± 0.15) | 0.82 ± 0.046 (0.87 ± 0.21) |
| Vincristine | 37 ± 3.6 (0.90 ± 0.17) | 0.80 ± 0.10 (0.90 ± 0.10) |
| Colchicine | 4.3 ± 0.40 (0.77 ± 0.23) | 0.78 ± 0.076 (0.58 ± 0.19) |
| Daunorubicin | 3.2 ± 0.29 (1.3 ± 0.20) | 1.1 ± 0.15 (1.3 ± 0.21) |
| Doxorubicin | 3.0 ± 0.50 (1.1 ± 0.20) | 1.1 ± 0.12 (1.1 ± 0.15) |
| Cisplatin | 1.3 ± 0.20 (1.3 ± 0.20) | 1.2 ± 0.29 (1.0 ± 0.10) |
The relative resistance was calculated by dividing the IC50 value of the variant by the IC50 of the wild-type cell line.
Relative resistance following co-incubation with the P-gp inhibitor PSC-833 at 2 μmol/L.
The functional status of the transporter was accessed by accumulation assays using known P-gp substrates. The P-gp positive MCF-7/CTAX cells had lower levels of [3H]-docetaxel following a one hour accumulation at 37 °C (Figure 1C), and these levels could be restored to parental MCF-7 levels in the presence of 2 μmol/L PSC-833 and other MDR modulators. These findings were confirmed with Rhodamine-123 accumulation by flow cytometry in MCF-7/CTAX cells (Supplementary Figure S3, panel A), compared with P-gp negative MCF-7/CTAX-P cells (Supplementary Figure S3, panel B). MCF-7/CTAX also demonstrates reduced BODIPY-paclitaxel accumulation under identical experiment conditions (Supplementary Figure S3, panel C), and is cross-resistant to a number of P-gp substrates including other taxanes, vinca alkaloids, colchicine, and the anthracyclines, doxorubicin and daunorubicin (Table 2). MCF-7/CTAX cells were 33-fold resistant to cabazitaxel, 52-fold resistant to paclitaxel, and 58-fold resistant to docetaxel. This resistance to taxanes was modulated in the presence of PSC-833, but 3-fold residual resistance remained to all taxanes tested, indicating that MDR was not the sole mechanism of cabazitaxel in these cells.
Taxane resistance in the MCF-7/CTAX-P variant does not appear to be transporter-mediated since it accumulates levels of [3H]-docetaxel similar to parental cells (Figure 1C), as well as accumulation of rhodamine-123 and BODIPY-paclitaxel as measured by flow cytometry (Supplementary Figure S3, panels B and D, respectively). PSC-833 did not modulate taxane resistance or the accumulation of drugs in MCF-7/CTAX-P cells. These cells are hypersensitive to the vinca alkaloids and colchicine indicating an alteration in microtubule dynamic instability. No change in sensitivity to anthracyclines or platinum agents was detected by SRB assays.
Although we did not detect any differences in taxane accumulation in MCF-7/CTAX-P relative to parental controls, we did observe a reduction in bound fluorescent-labeled paclitaxel using flow cytometry. After accumulation in BODIPY-paclitaxel for 1 hour at 37 °C, cells were allowed to efflux in drug-free complete medium for an additional hour and washed several times to insure that no unbound drug remained. MCF-7/CTAX-P cells had a 34% decrease in residual bound BODIPY-paclitaxel as measured by average fluorescence intensity compared to bound drug measured in parental MCF-7 cells (Figure 1D). Incorporating PSC-833 in the drug treatment did not impact the degree of drug binding in this cell line.
Both cabazitaxel-selected MCF-7 variants have elevated TUBB3
An analysis of the expression of all members of the β-tubulin gene family was conducted by RT-qPCR using isotype-specific amplimers. Both cabazitaxel-resistant MCF-7 variants have elevated TUBB3 content relative to parental controls, and this finding was confirmed at the protein level by immunoblotting with the TUJ-1 class III β-tubulin-specific monoclonal antibody (Figure 2A). The degree of overexpressed TUBB3 protein content detected was comparable in both cabazitaxel-resistant cell models regardless of ABCB1 status. No mutations were detected in beta-1 tubulin (Class I, M40) and alpha-tubulin (K-α1) by sequencing PCR-amplified products.
Figure 2.

Expression of the class III (TUBB3) β-tubulin isotype, total α- and β-tubulin levels, and BRCA1 was evaluated by immunoblotting total cell lysates from MCF-7, the ABCB1-positive MCF-7/CTAX and the co-selected MCF-7/CTAX-P cell lines (panel A). Protein loading was confirmed by screening for β-actin expression. Tubulin polymer was separated from soluble tubulin by centrifugation (20,000 × g) following a 5 minute incubation in hypotonic buffer with and without drug at 37 °C, and fractions were resolved on 4–20% gradient polyacrylamide gels and transferred to nitrocellulose. Immunoblotting with a pan α-tubulin antibody (clone DM1A, Sigma-Aldrich) isolated the tubulin fractions in MCF-7 parental cells and the MCF-7/CTAX-P variant (panel B).
Under the experimental conditions of our tubulin polymerization assay, the majority of the tubulin was present in the soluble form in our cell lines. However, we detected reduced tubulin polymer in MCF-7/CTAX-P cells compared to MCF-7 cells at baseline, and the ratio of polymerized to soluble tubulin was markedly decreased (12% versus 37%, respectively, Figure 2B and Supplementary Figure S4). In order to determine the effects of taxane treatment on tubulin polymerization, we exposed cells to either cabazitaxel or docetaxel from 1.0 nmol/L to 10 μmol/L. We observed a dose-dependent increase in levels of polymerized tubulin in response to taxane treatment in parental cells, but there was a significant difference in polymerized tubulin after cabazitaxel or docetaxel treatment at concentrations less than 1 μmol/L in MCF-7/CTAX-P versus the parental cell line (Figure 2B). This impaired tubulin polymerization in response to drug treatment correlated with resistance to the taxanes in cytotoxicity assays, and may be due to the reduced binding of taxanes to microtubules observed in MCF-7/CTAX-P.
TUBB3 gene silencing sensitizes MCF-7/CTAX-P cells to cabazitaxel
In order to test the role of TUBB3 expression on cabazitaxel activity, we transfected MCF-7/CTAX-P cells with a pool of four siRNAs specific for the class III β-tubulin. We achieved greater than 90% silencing, reversing the TUBB3 content back to parental levels (Figure 3A). Clonogenic assays demonstrated that cells were approximately 2-fold more sensitive to cabazitaxel than the control transfected with a pool of non-targeting siRNAs (Figure 3B). In a parallel experiment, we silenced TUBB3 in parental MCF-7 cells using the same pool of siRNAs under the same transfection conditions. TUBB3 was undetectable by immunoblotting following transfection, and these cells were 1.3-fold more sensitive to both cabazitaxel and docetaxel compared to the non-targeting control (data not shown). Data from these experiments indicate that TUBB3 is at least partially responsible for the resistance to cabazitaxel in MCF-7/CTAX-P cells.
Figure 3.

Transient silencing of the class III β-tubulin (TUBB3) and BRCA1 genes was accomplished by transfecting gene-specific ON-TARGETplus SMARTpools of four siRNAs (GE Dharmacon). Briefly, MCF-7/CTAX-P cells were transfected with 25 nM TUBB3-specific siRNAs using Dharmafect 1, and silencing was monitored by immunoblotting 48 hr post-transfection relative to non-targeting SMARTpool control siRNAs under identical experimental conditions (panel A). Cabazitaxel activity was determined following TUBB3-silencing by clonogenic assays with colonies greater than 50 cells scored 14 days after drug exposure (panel B). MCF-7 wild-type cells were transfected with BRCA1-specific siRNAs (25 nM) using the same transfection conditions, and BRCA1 content was evaluated by immunoblotting 48 hr post-transfection (panel C). SRB assays were used to screen for cabazitaxel activity following a 72 hr drug incubation in BRCA1-silenced MCF-7 cells (panel D). Cabazitaxel was added 24 hr post-transfection with siRNAs, and each drug concentration was tested in quadruplicate measurements. All data are expressed as the average percent survival values relative to an untreated control ± standard deviation, with significance determined between the non-targeting control and gene-silenced cells per cabazitaxel concentration tested (unpaired t-test, *p < 0.05, **p < 0.01, ***p < 0.001).
Reduced BRCA1 expression in response to cabazitaxel drug selection, and resistance to cabazitaxel by BRCA1 silencing in MCF-7 parental cells
We have previously reported down-regulation of BRCA1 resulting from either paclitaxel or docetaxel selection in several models of non-ABCB1 taxane resistance. Reduced BRCA1 content was observed early in taxane selections, usually during the first round of drug treatment, and these alterations were associated with decreased G2-M arrest and apoptosis induced by taxane treatment (15). Selection with cabazitaxel in MCF-7 cells also resulted in BRCA1 down-regulation in both MCF-7 variants, but we observed lower levels of BRCA1 in the non-ABCB1 MCF-7/CTAX-P (70% reduced compared to MCF-7) than in MCF-7/CTAX (35% reduced, Figure 2A). In order to evaluate the role of BRCA1 down-regulation in cabazitaxel resistance, we silenced the gene in the parental MCF-7 cell line and measured effects on taxane cytotoxicity. Treatment with a BRCA1-specific pool of four siRNAs achieved greater than 90% silencing compared to parental and a non-targeting control by immunoblotting (Figure 3C), and BRCA1-silenced MCF-7 cells were approximately 4-fold resistant to cabazitaxel (Figure 3D) relative to the non-targeting control. These observations were confirmed following treatment with docetaxel after BRCA1 silencing (data not shown). Thus, BRCA1 expression is directly implicated in cellular responses to taxanes in MCF-7 cells.
Altered epithelial-mesenchymal and apoptosis regulating genes
Gene expression profiling using the NanoString Technology platform identified alterations in the expression of epithelial-mesenchymal transition markers in both cabazitaxel-selected variants, with elevated levels of the mesenchymal marker, Vimentin (VIM), and decreased expression of the epithelial cell-cell adhesion glycoprotein E-cadherin (CDH1) compared to parental cells (Figure 4). Therefore, cabazitaxel selection induces epithelial-mesenchymal transition, implicating the mesenchymal phenotype in resistance to cabazitaxel.
Figure 4.
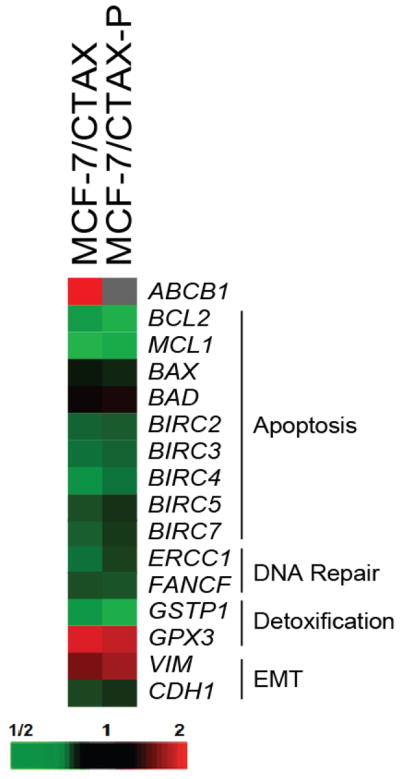
Gene expression profiling using a color-coded barcode technology was performed. Briefly, each reaction contained 250 ng of total RNA isolated from each cell line. A custom-designed probe set specific for genes of interest was added in excess to ensure that each gene was labeled. Following a series of wash steps to remove unbound probes and non-target cellular transcripts, bar codes were counted using NanoString’s Digital Analyzer. Counts were normalized to 7 reference genes, and a background count was estimated using 8 separate negative controls probes in every reaction. Data is presented as a heatmap of gene expression comparing the ABCB1-positive MCF-7/CTAX and the non-MDR MCF-7/CTAX-P with parental MCF-7 cells. The map is organized by function including genes involved in the regulation of apoptosis, DNA repair, detoxification, and epithelial-mesenchymal transition (EMT).
Although altered expression of apoptotic regulators has been associated with taxane resistance in other published models (16, 17), gene expression profiling using the NanoString platform indicated that there were no significant changes in the apoptotic promoters BAX and BAD, and we observed reduced expression of the anti-apoptotic regulators BCL2, MCL1, and several inhibitors of apoptosis (IAPs) including cIAP-1/BIRC2, cIAP-2/BIRC3, XIAP/BIRC4, Survivin/BIRC5, and Livin/BIRC7 in the cabazitaxel-selected variants compared to MCF-7 parental cells (Figure 4). These findings were confirmed by immunoblotting with specific antibodies, and we did not detect a difference in Bcl-XL protein levels in either cabazitaxel variant compared to MCF-7 levels (Supplementary Figure S5). Furthermore, several DNA repair genes were down-regulated in both cabazitaxel variants such as the DNA excision repair ERCC1 and Fanconi anemia group F (FANCF) genes, and we observed alterations in genes associated with detoxification including reduced glutathione S-transferase P1 (GSTP1) and elevated glutathione peroxidase 3 (GPX3) genes (Figure 4).
Discussion
Cabazitaxel was selected for clinical development due to its activity in tumor models that were demonstrated to be poorly sensitive or insensitive to docetaxel treatment, including lung, pancreatic, colon, gastric, and mammary cancers, and a melanoma model (B16/TXT) with acquired in vivo resistance to docetaxel (7). Preclinical data indicated that the drug may be more active than paclitaxel and docetaxel in models that express the ABCB1/P-gp transporter, and our study confirms that this is the case. We tested cabazitaxel activity in several MDR variants established in our laboratory after selection with either doxorubicin or docetaxel, and these cell models demonstrate cross-resistance to a variety of P-gp substrates that can be modulated in the presence of known inhibitors. In the MES-SA/Dx5 human sarcoma cell model, cabazitaxel was 13-fold more active than the first generation taxanes, paclitaxel and docetaxel, and 7-fold more active in the docetaxel-selected MCF-7/TxT50 variant. The cross-resistance to cabazitaxel in these P-gp positive cell models could be completely modulated to parental levels in the presence of 2 μmol/L PSC-833.
A major mechanism of resistance in response to selection with taxanes in vitro is the activation of MDR transporters including ABCB1/P-gp (1, 2, 5, 18–20). Although cabazitaxel is more active in P-gp expressing tumor cells than paclitaxel and docetaxel, long-term selection with cabazitaxel alone in the human breast cancer MCF-7 cell line resulted in ABCB1 activation, but not ABCC2, ABCC10, or ABCG2. Moreover, even though taxane accumulation was restored to parental levels by including MDR modulators, 3-fold residual taxane resistance remained in MCF-7/CTAX. It is likely that this low level of residual resistance to cabazitaxel resulted from elevated TUBB3 and reduced BRCA1 content, similar to the co-selected ABCB1-negative MCF-7/CTAX-P variant.
Alterations in microtubule composition and dynamics have been reported in taxane variants (21–24). Previous data from our laboratory indicated that an increased expression of the class IVa (TUBB4) β-tubulin isotype was associated with non-ABCB1 taxane resistance in a human leukemia variant co-selected with paclitaxel and PSC-833 (6). No other changes in β-tubulin isotype content including mutations in the putative taxane binding region were observed in this cell line that was exclusively resistant to taxanes, and total α- and β-tubulin levels remained unchanged. In addition, the majority of our taxane-resistant non-ABCB1 breast and ovarian cancer models have elevated class III (TUBB3) content following selection with either docetaxel or paclitaxel in the presence of PSC-833, and we demonstrated that the MCF-7/TxTP50 variant is also cross-resistant to cabazitaxel.
Others have reported that elevated levels of class III β-tubulin conferred resistance to taxanes (25–28). Hari et al. generated clones of the Chinese hamster ovary cell line that had tetracycline-controlled expression of the class III β-tubulin isotype (25). Stable clones with high levels of class III had significantly lower proliferation rates compared to wild-type cells and to cells grown in the presence of tetracycline. These high levels of class III β-tubulin were found to be cytotoxic, diminishing microtubule assembly in transfected cells, and were found to confer modest resistance (1.5- to 2-fold) to paclitaxel over controls. As with our results in the MCF-7 breast cancer cell line, several others have observed slight sensitization to taxanes following transient TUBB3 silencing using specific siRNAs or antisense oligonucleotides (29–31). Although several published reports have questioned the importance of β-tubulin isotype expression as a significant mechanism of resistance to paclitaxel (32–34), TUBB3 content appears to predict paclitaxel response in breast, non-small cell lung and other cancers, with elevated TUBB3 levels correlated with poor prognosis and clinical outcome in patients treated with taxanes (30, 35–39). Our study demonstrated that transient TUBB3 silencing resulted in modest sensitization to cabazitaxel in the MCF-7/CTAX-P cell line.
Furthermore, expression of cell cycle regulators has been implicated in models of taxane resistance. In experiments testing the functional significance of these alterations, transfection of active, wild-type BRCA1 in BRCA1-negative HCC1937 human breast cancer cells sensitized cells to taxanes (40), whereas both transient and stable silencing of BRCA1 resulted in taxane resistance in a number of cell models including breast, ovarian, and lung cancer cell lines (40–45). A recent study found that BRCA1 regulates microtubule dynamics, with reduced taxane binding impairing drug-induced microtubule stabilization, resulting in resistance to taxanes in A549 lung cancer cells transfected with a BRCA1-specific shRNA relative to parental and empty vector controls (45). Kurebayashi et al. found that loss of BRCA1 expression may predict a shorter progression-free survival time in breast cancer patients treated with taxanes (46). Reduced BRCA1 expression was also associated with cabazitaxel resistance in our MCF-7 variants, and transient silencing of BRCA1 by siRNA transfection in MCF-7 parental cells resulted in 4-fold resistance to cabazitaxel. The altered taxane binding observed in the MCF-7/CTAX-P variant may result from its reduced tubulin polymer content compared to parental MCF-7 cells (14, 47).
The acquisition of mesenchymal properties has been associated with chemoresistance, including upregulation of TUBB3 and resistance to taxanes (48–50). EMT is characterized by the loss of the epithelial and the gain of the mesenchymal markers, and we observed these alterations in both MCF-7 variants following cabazitaxel selection, indicating that resistance to cabazitaxel is associated with a more aggressive and invasive phenotype. The functional significance of these changes in marker expression is being studied in the laboratory. The blockade of EMT may modulate taxane resistance in these cell models of cabazitaxel resistance.
We found substantial cross-resistance between cabazitaxel and the first generation taxanes, paclitaxel and docetaxel, despite the higher potency of cabazitaxel and its lower degree of resistance in P-gp positive resistant variants. Thus, in lieu of data from appropriate clinical trials, our data should not be interpreted to support the use of cabazitaxel in docetaxel-refractory breast cancers.
This study has identified several potential biomarkers for predicting cabazitaxel effectiveness in the clinical setting, including reduced BRCA1 and elevated class III β-tubulin isotype expression. In addition, a mesenchymal phenotype is associated with resistance to cabazitaxel in these cell lines. Although this new taxane has more activity than paclitaxel and docetaxel in the MDR models tested, substantial cross-resistance remained, and MDR status may also be a key marker for its efficacy.
Supplementary Material
Acknowledgments
Grant Support: G. E. Duran and B. I. Sikic received grant support from Sanofi, and G. E. Duran, Y. C. Wang, E. B. Francisco, F. J. Martinez, J. Coller, and B. I. Sikic received grant support from the U. S. National Institutes of Health (NIH) through the National Cancer Institute (R01 CA184968).
Footnotes
Disclosure of Potential Conflicts of Interest: The Stanford authors received grant support from Sanofi, while Drs. Brassard and Vrignaud are employees of Sanofi Oncology. They have no other potential financial conflicts of interest.
References
- 1.Horwitz S, Cohen D, Rao S, Ringel I, Shen H, Yang C. Taxol: mechanisms of action and resistance. Journal of the National Cancer Institute. 1993;15:55–61. [PubMed] [Google Scholar]
- 2.Wang YC, Juric D, Francisco B, Yu RX, Duran GE, Chen GK, et al. Regional activation of chromosomal arm 7q with and without gene amplification in taxane-selected human ovarian cancer cell lines. Genes Chromosomes Cancer. 2006 Apr;45(4):365–74. doi: 10.1002/gcc.20300. [DOI] [PubMed] [Google Scholar]
- 3.Schaner ME, Ross D, Ciaravino G, Sorlie T, Troyanskaya O, Diehn M, et al. Gene expression patterns in ovarian carcinomas. Molecular Biology of the Cell. 2003;14(11):4376–86. doi: 10.1091/mbc.E03-05-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spicakova T, O’Brien MM, Duran GE, Sweet-Cordero A, Sikic BI. Expression and silencing of the microtubule-associated protein Tau in breast cancer cells. Mol Cancer Ther. 2010;9(11):2970–81. doi: 10.1158/1535-7163.MCT-10-0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dumontet C, Duran GE, Steger KA, Beketic-Oreskovic L, Sikic BI. Resistance mechanisms in human sarcoma mutants derived by single-step exposure to paclitaxel (Taxol) Cancer Research. 1996;56:1091–7. [PubMed] [Google Scholar]
- 6.Jaffrezou J-P, Dumontet C, Derry W, Duran G, Chen G, Tsuchiya E, et al. Novel mechanism of resistance to paclitaxel (Taxol) in human K562 leukemia cells by combined selection with PSC 833. Oncology Research. 1995;7:517–27. [PubMed] [Google Scholar]
- 7.Vrignaud P, Sémiond D, Lejeune P, Bouchard H, Calvet L, Combeau C, et al. Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clinical Cancer Research. 2013;19(11):2973–83. doi: 10.1158/1078-0432.CCR-12-3146. [DOI] [PubMed] [Google Scholar]
- 8.Summary of product characteristics, EMA. 2011. Sanofi. Jevtana (cabazitaxel) injection. [Google Scholar]
- 9.Prescribing Information, FDA. 2010. Sanofi. Jevtana (cabazitaxel) injection. [Google Scholar]
- 10.Calcagno F, Nguyen T, Dobi E, Villanueva C, Curtit E, Kim S, et al. Safety and efficacy of cabazitaxel in the docetaxel-treated patients with hormone-refractory prostate cancer. Clin Med Insights Oncol. 2013;7:1–12. doi: 10.4137/CMO.S7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harker WG, Sikic BI. Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line, MES-SA. Cancer Research. 1985;45:4091–96. [PubMed] [Google Scholar]
- 12.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 13.Giannakakou P, Sackett DL, Kang Y-K, Zhan Z, Buters JT, Fojo T, et al. Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. Journal of Biological Chemistry. 1997;272:17118–25. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 14.Minotti AM, Barlow SB, Cabral F. Resistance to antimitotic drugs in chinese hamster ovary cells correlates with changes in the level of polymerized tubulin. Journal of Biological Chemistry. 1991;266:3987–94. [PubMed] [Google Scholar]
- 15.Duran GE, Wang YC, Francisco EB, Yu RX, Sikic BI. Identification of genes associated with non-MDR1 taxane resistance in human ovarian carcinoma cell lines by microarray analysis [abstract]. Proceedings of the 94th Annual Meeting of the American Association for Cancer Research; 2003, July 11–14; Washington, DC. 2003. p. Abstract No 5760. [Google Scholar]
- 16.Huang Y, Ibrado AM, Reed JC, Bullock G, Ray S, Tang C, et al. Co-expression of several molecular mechanisms of multidrug resistance and their significance for paclitaxel cytotoxicity in human AML HL-60 cells. Leukemia. 1997;11:253–7. doi: 10.1038/sj.leu.2400557. [DOI] [PubMed] [Google Scholar]
- 17.Zapata JM, Krajewska M, Krajewski S, Huang R-P, Takayama S, Wang H-G, et al. Expression of multiple apoptosis-regulatory genes in human breast cancer cell lines and primary tumors. Breast Cancer Research and Treatment. 1998;47:129–40. doi: 10.1023/a:1005940832123. [DOI] [PubMed] [Google Scholar]
- 18.Boyerinas B, Park SM, Murmann AE, Gwin K, Montag AG, Zillardt MR, et al. Let-7 modulates acquired resistance of ovarian cancer to Taxanes via IMP-1-mediated stabilization of MDR1. Int J Cancer. 2012 Apr 15;130(8):1787–1797. doi: 10.1002/ijc.26190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malofeeva EV, Domanitskaya N, Gudima M, Hopper-Borge EA. Modulation of the ATPase and transport activities of broad-acting multidrug resistance factor ABCC10 (MRP7) Cancer Res. 2012 Dec 15;72(24):6457–67. doi: 10.1158/0008-5472.CAN-12-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huisman MT, Chhatta AA, van Tellingen O, Beijnen JH, Schinkel AH. MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. int J Cancer. 2005 Sep 20;116(5):824–9. doi: 10.1002/ijc.21013. [DOI] [PubMed] [Google Scholar]
- 21.Kavallaris M, Kuo D, Kurkhart C, Regl D, Norris M, Haber M, et al. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. Journal of Clinical Investigation. 1997;100(5):1282–93. doi: 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol resistance related to microtubules. Oncogene. 2003 Oct 20;22(47):7280–95. doi: 10.1038/sj.onc.1206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goncalves A, Braguer D, Kamath K, Martello L, Briand C, Horwitz S, et al. Resistance to Taxol in lung cancer cells associated with increased microtubule dynamics. Proc Natl Acad Sci U S A. 2001 Sep 25;98(20):11737–42. doi: 10.1073/pnas.191388598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horwitz SB, Cohen D, Rao S, Ringel I, Shen HJ, Yang CP. Taxol: mechanisms of action and resistance. J Natl Cancer Inst Monogr. 1993;(15):55–61. [PubMed] [Google Scholar]
- 25.Hari M, Yang H, Zeng C, Canizales M, Cabral F. Expression of class III beta-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil Cytoskeleton. 2003;56:45–56. doi: 10.1002/cm.10132. [DOI] [PubMed] [Google Scholar]
- 26.Ferlini C, Raspaglio G, Mozzetti S, Cicchillitti L, Filippetti F, Gallo D, et al. The seco-taxane IDN5390 is able to target class III beta-tubulin and to overcome paclitaxel resistance. Cancer Research. 2005 Mar 15;65(6):2397–405. doi: 10.1158/0008-5472.CAN-04-3065. [DOI] [PubMed] [Google Scholar]
- 27.Ganguly A, Yang H, Cabral F. Class III beta-tubulin counteracts the ability of paclitaxel to inhibit cell migration. Oncotarget. 2011 May;2(5):368–77. doi: 10.18632/oncotarget.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kavallaris M, Kuo DY-S, Burkhart CA, Regl DL, Norris MD, Haber M, et al. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta tubulin isotypes. Journal of Clinical Investigation. 1997;100:1282–93. doi: 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kavallaris M, Burkhart CA, Horwitz SB. Antisense oligonucleotides to class III beta-tubulin sensitize drug-resistant cells to Taxol. British Journal of Cancer. 1999 Jun;80(7):1020–5. doi: 10.1038/sj.bjc.6690507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gan PP, Pasquier E, Kavallaris M. Class III beta-tubulin mediates sensitivity to chemotherapeutic drugs in non small cell lung cancer. Cancer Research. 2007 Oct 1;67(19):9356–63. doi: 10.1158/0008-5472.CAN-07-0509. [DOI] [PubMed] [Google Scholar]
- 31.Don S, Verrills NM, Liaw TY, Liu ML, Norris MD, Haber M, et al. Neuronal-associated microtubule proteins class III beta-tubulin and MAP2c in neuroblastoma: role in resistance to microtubule-targeted drugs. Molecular Cancer Therapeutics. 2004 Sep;3(9):1137–46. [PubMed] [Google Scholar]
- 32.Blade K, Menick D, Cabral F. Overexpression of class I, II or IVb β-tubulin isotypes in CHO cells is insufficient to confer resistance to paclitaxel. Journal of Cell Science. 1999;112:2213–21. doi: 10.1242/jcs.112.13.2213. [DOI] [PubMed] [Google Scholar]
- 33.Ranganathan S, McCauley R, Dexter D, Hudes G. Modulation of endogenous β-tubulin isotype expression as a result of human βIII cDNA transfection into prostate carcinoma cells. British Journal of Cancer. 2001;85(5):735–40. doi: 10.1054/bjoc.2001.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicoletti M, Valoti G, Giannakakou P, Zhan Z, Kim J-H, Lucchini V, et al. Expression of beta-tubulin isotypes in human ovarian carcinoma xenografts and in a sub-panel of human cancer cell lines from the NCI-anticancer drug screen: correlation with sensitivity to microtubule active agents. Clinical Cancer Research. 2001;7:2912–22. [PubMed] [Google Scholar]
- 35.Hasegawa S, Miyoshi Y, Egawa C, Ishitobi M, Taguchi T, Tamaki Y, et al. Prediction of response to docetaxel by quantitative analysis of class I and III beta-tubulin isotype mRNA expression in human breast cancers. Clin Cancer Res. 2003 Aug 1;9(8):2992–7. [PubMed] [Google Scholar]
- 36.Mozzetti S, Ferlini C, Concolino P, Filippetti F, Raspaglio G, Prislei S, et al. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin Cancer Res. 2005 Jan 1;11(1):298–305. [PubMed] [Google Scholar]
- 37.Dumontet C, Isaac S, Souquet PJ, Bejui-Thivolet F, Pacheco Y, Peloux N, et al. Expression of class III beta tubulin in non-small cell lung cancer is correlated with resistance to taxane chemotherapy. Bull Cancer. 2005 Feb;92(2):E25–30. [PubMed] [Google Scholar]
- 38.Ferrandina G, Zannoni GF, Martinelli E, Paglia A, Gallotta V, Mozzetti S, et al. Class III beta-tubulin overexpression is a marker of poor clinical outcome in advanced ovarian cancer patients. Clin Cancer Res. 2006 May 1;12(9):2774–9. doi: 10.1158/1078-0432.CCR-05-2715. [DOI] [PubMed] [Google Scholar]
- 39.Hetland TE, Hellesylt E, Florenes VA, Trope C, Davidson B, Kaern J. Class III beta-tubulin expression in advanced-stage serous ovarian carcinoma effusions is associated with poor survival and primary chemoresistance. Human Pathology. 2011 Jul;42(7):1019–26. doi: 10.1016/j.humpath.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 40.Quinn JE, Kennedy RD, Mullan PB, Gilmore PM, Carty M, Johnston PG, et al. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Research. 2003;63:6221–8. [PubMed] [Google Scholar]
- 41.Chabalier C, Lamare C, Racca C, Privat M, Valette A, Larminat F. BRCA1 downregulation leads to premature inactivation of spndle checkpoint and confers paclitaxel resistance. Cell Cycle. 2006;5(9):1001–7. doi: 10.4161/cc.5.9.2726. [DOI] [PubMed] [Google Scholar]
- 42.Lafarge S, Sylvain V, Ferrara M, Bignon YJ. Inhibition of BRCA1 leads to increased chemoresistance to microtubule-interfering agents, an effect that involves the JNK pathway. Oncogene. 2001;20(45):6597–606. doi: 10.1038/sj.onc.1204812. [DOI] [PubMed] [Google Scholar]
- 43.Mullan PB, Quinn PE, Gilmore PM, McWilliams S, Andrews H, Gervin C, et al. BRCA1 and GADD45 mediated G2/M cell cycle arrest in response to antimicrotubule agents. Oncogene. 2001;20:6123–31. doi: 10.1038/sj.onc.1204712. [DOI] [PubMed] [Google Scholar]
- 44.Quinn JE, James CR, Stewart GE, Mulligan JM, White P, Chang GKF, et al. BRCA1 mRNA expression levels predict for overall survival in ovarian cancer after chemotherapy. Clinical Cancer Research. 2007;13(24):7413–20. doi: 10.1158/1078-0432.CCR-07-1083. [DOI] [PubMed] [Google Scholar]
- 45.Sung M, Giannakakou P. BRCA1 regulates microtubule dynamics and taxane-induced apoptotic cell signaling. Oncogene. 2013:1–11. doi: 10.1038/onc.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurebayashi J, Yamamoto Y, Kurosumi M, Okubo S, Nomura T, Tanaka K, et al. Loss of BRCA1 expression may predict shorter time-to-progression in metastatic breast cancer patients treated with taxanes. Anticancer Research. 2006;26:695–702. [PubMed] [Google Scholar]
- 47.Parness J, Horwitz SB. Taxol binds to polymerized tubulin in vitro. J Cell Biology. 1981 Nov 1;91(2):479–87. doi: 10.1083/jcb.91.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. int J Cancer. 2007 Aug;31(2):277–83. [PubMed] [Google Scholar]
- 49.Işeri OD, Kars MD, Arpaci F, Atalay C, Pak I, Gündüz U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed Pharmacother. 2011 Feb;65(1):40–5. doi: 10.1016/j.biopha.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Yang Q, Huang J, Wu Q, Cai Y, Zhu L, Lu X, et al. Acquisition of epithelial–mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. British J Cancer. 2014 Mar 18;110:1958–67. doi: 10.1038/bjc.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.