

Abstract
Aims
H2S is known to confer cardioprotection; however, the pathways mediating its effects in vivo remain incompletely understood. The purpose of the present study is to evaluate the contribution of cGMP-regulated pathways in the infarct-limiting effect of H2S in vivo.
Methods and results
Anaesthetized rabbits were subjected to myocardial ischaemia (I)/reperfusion (R), and infarct size was determined in control or H2S-exposed groups. The H2S donor sodium hydrosulfide (NaHS, an agent that generates H2S) increased cardiac cGMP and reduced the infarct size. The cGMP-dependent protein kinase (PKG)-I inhibitor DT2 abrogated the protective effect of NaHS, whereas the control peptide TAT or l-nitroarginine methyl ester (l-NAME) did not alter the effect of NaHS. Moreover, the KATP channel inhibitor, glibenclamide, partially reversed the effects of NaHS, whereas inhibition of mitochondrial KATP did not modify the NaHS response. NaHS enhanced phosphorylation of phospholamban (PLN), in a PKG-dependent manner. To further investigate the role of PLN in H2S-mediated cardioprotection, wild-type and PLN KO mice underwent I/R. NaHS did not exert cardioprotection in PLN KO mice. Unlike what was observed in rabbits, genetic or pharmacological inhibition of eNOS abolished the infarct-limiting effect of NaHS in mice.
Conclusions
Our findings demonstrate (i) that administration of NaHS induces cardioprotection via a cGMP/PKG/PLN pathway and (ii) contribution of nitric oxide to the H2S response is species-specific.
Keywords: H2S, Ischaemia, Postconditioning, cGMP, Phospholamban
1. Introduction
Reperfusion of the ischaemic myocardium protects the tissue from necrotic death, but it is associated with a paradoxical injury, compromising the overall benefit of reinstating blood supply to the heart.1 Mechanical manoeuvres [ischaemic preconditioning (IPC) and postconditioning (PostC)] have been shown to be successful in limiting ischaemia/reperfusion (I/R) injury in preclinical models, but have failed to translate into therapeutically useful cardioprotective strategies that impact everyday clinical practice.2,3 A better understanding of the signal-transduction pathways implicated in cardiomyocyte survival could reveal potential targets, amenable to pharmacological modulation.4
Intensive investigation over the last two decades has led to the discovery of several signal-transduction pathways involved in ischaemic conditioning. Most investigators accept that these mediators converge on the mitochondria.5,6 Intracellular Ca2+ overload plays a prominent role in I–R injury by (i) activating calcium-dependent proteolytic enzymes leading to cytoskeletal and sarcolemmal fragility, (ii) inducing excessive contraction (hypercontracture), and (iii) promoting mitochondrial permeability transition pore (mPTP) opening that halts ATP production and leads to mitochondrial breakdown.7,8 Several pathways have been implicated in limiting Ca2+ oscillations and Ca2+ overload including the sodium–calcium exchanger (NCX), the sodium–hydrogen exchanger (NHE), and the sarcoplasmic reticulum (SR) Ca2+ regulators SERCA and ryanodine receptors.9,10
Hydrogen sulfide is an endogenously produced gaseous signalling molecule, with important roles in the regulation of cardiovascular function.11 Endogenously produced or exogenously supplied H2S has been reported to be cytoprotective during reperfusion injury in vitro or in isolated hearts.11–21 However, in most of the studies where H2S was exogenously administered to trigger conditioning, H2S donor compounds were not given in a manner that would be consistent with administration of a pharmacological agent in patients, as the donors were given hours to days prior to induction of ischaemia. In the limited number of in vivo studies, administration of H2S-producing agents during sustained ischaemia has been shown to be beneficial in infarct size limitation and in post-infarction myocardial function.22–25 This cytoprotection was associated with up-regulation of antioxidant pathways,25 inhibition of myocardial inflammation,22 preservation of mitochondrial structure and function22 after ischaemia, and inhibition of apoptosis.22,24 While the cardioprotective effects of H2S have been reported to be nitric oxide (NO)-dependent, this was claimed to occur in a cGMP-independent manner.23 In the studies where H2S was administered at a clinically relevant time (at the end of ischaemia and during reperfusion),22–25 the intracellular signalling pathways determining H2S-induced cardioprotection were not studied in detail. The positive preclinical findings with H2S donors have served as the impetus for a clinical trial designed to evaluate the cardioprotective potential of H2S in humans (NCT01989208). Given the increasing interest on the role of H2S, here we aimed to investigate the signalling components responsible for the beneficial effects of H2S in vivo focusing on the contribution of NO and cGMP-regulated pathways.
2. Methods
2.1. Animals
All animal procedures were in compliance with the European Community guidelines for the use of experimental animals; experimental protocols were approved by the Ethical Committee of the Prefecture of Athens. Animals received a standard laboratory diet. The animal cohort compromised of 92 New Zealand white male rabbits weighing 2.7–3 kg, 44 C57BL/6 male mice 13–15 weeks old, 12 eNOS KO male mice 13–15 weeks old, 29 129SvJ male and female mice 12–15 weeks old, and 27 PLN KO male and female mice 12–15 weeks old.
2.2. Surgical procedures
2.2.1. Rabbit in vivo model of I/R injury
Sodium thiopentone at a dose of 30 mg kg−1 was injected into a peripheral vein for anaesthesia. The depth of anaesthesia was monitored at a regular basis by eye reflex and haemodynamics. Then, the animals were intubated and connected to a respirator for small animals (MD Industries, Mobile, AL, USA) for mechanical ventilation at a rate adjusted to maintain normal blood gases. The chest was surgically opened with a left thoracotomy and the beating heart was exposed. The pericardium was opened and a 3-0 silk suture was passed around a prominent coronary artery. Ischaemia was induced by pulling the thread through a small piece of soft tubing, which was firmly positioned against the coronary arterial wall with the aid of a small clamp. Ischaemia resulted in ST elevation on the ECG (Power lab 4.0, Adinstruments, UK). At the end of ischaemic period the snare was opened, the artery refilled and the myocardium reperfused. At the end of reperfusion period, rabbits were sacrificed with a high-dose intravenous injection of the anaesthetic sodium thiopentone (150 mg kg−1); hearts were rapidly excised, mounted on an apparatus, and perfused with normal saline for 2 min for blood removal. Then, the coronary ligature was retightened at the same site and 10 mL of green fluorescent microspheres (Duke Scientific Corp., Palo Alto, CA, USA) were infused for the separation of the normally perfused area from the area at risk. Hearts were kept in the refrigerator for 24 h and cut into 3 mm thick sections. The slices were stained with triphenyltetrazolium chloride (TTC) at 37°C and immersed in formaldehyde. With a wavelength of 366 nm UV light, we identified the risk zone and separated it from the infarcted zone of the heart. The infarcted, the risk, and the normal areas were traced onto an acetate sheet, which had been placed over the top glass plate. The tracings were subsequently scanned with the Adobe Photoshop 6.0 and measured with the Scion Image program. The areas of myocardial tissue at risk and infarcted were automatically transformed into volumes. Infarct and risk area volumes were expressed in cm3 and the percentage of infarct-to-risk area ratio (%I/R) was calculated.26
2.2.2. Murine in vivo model of I/R injury
The mice were anaesthetized by intraperitoneal injection (0.01 mL kg−1) with a combination of ketamine, xylazine, and atropine (final doses of ketamine, xylazine, and atropine were 100, 20, and 0.6 mg kg−1, respectively). Anaesthetic depth was evaluated by the loss of pedal reflex to toe-pinch stimulus and breathing rate. A tracheotomy was performed for artificial respiration at 120–150 breaths/min and positive end-expiratory pressure 2.0 (0.2 mL of tidal volume; Flexivent rodent ventilator, Scireq, Montreal, ON, Canada). A thoracotomy was then performed between the fourth and fifth ribs and the pericardium carefully retracted to visualize the left anterior descending (LAD), which was ligated using an 8-0 prolene monofilament polypropylene suture placed 1 mm below the tip of the left auricle. The heart was allowed to stabilize for 15 min prior to ligation to induce ischaemia. After the ischaemic period, the ligature was released allowing reperfusion of myocardium. Throughout experiments, body temperature was maintained at 37 ± 0.5°C by way of a heating pad and monitored via a thermocouple inserted rectally. After reperfusion, animals were sacrificed with an intraperitoneal injection of the ketamine (200 mg kg−1) and xylazine (50 mg kg−1) anaesthetic cocktail (0.02 mL g−1) hearts were rapidly excised from mice and directly cannulated and washed with 2.5 mL of saline heparin 1% for blood removal. 5 mL of 1% TTC phosphate buffer 37°C were infused into the coronary circulation; the tissue was incubated for 5 min in the same buffer. 2.5 mL of 1% Evans Blue (diluted in distilled water) was then infused into the heart. Hearts were kept at −20°C for 24 h and then sliced in 1 mm sections parallel to the atrio-ventricular groove, and then fixed in 4% formaldehyde overnight. Slices were then compressed between glass plates 1 mm apart and photographed with a Cannon Powershot A620 Digital Camera through a Zeiss 459300 microscope and measured with the Scion Image program. The areas of myocardial tissue at risk and infarcted were automatically transformed into volumes. Infarct and risk area volumes were expressed in cm3 and the percentage of infarct-to-risk area ratio (%I/R) was calculated.27
2.3. Chemicals
DT2: BIOLOG Life Science Institute (Bremen, Germany); Glibenclamibe: Tocris Bioscience (Bristol, UK); Phosphodiesterase (PDE) activity colorimetric assay kit and TAT: Abcam (Cambridge, UK); l-nitroarginine methyl ester (l-NAME): Cayman Chemicals (Lab Supplies P. Galanis & Co., Athens, Greece); the H2S donor sodium hydrosulfide (NaHS), 5-hydroxydecanoate and TTC: Sigma-Aldrich Co. (Life Science Chemilab S.A., Athens, Greece); Protein G Sepharose Fast flow beads: GE Healthcare Life Sciences (G. Kordopatis Ltd, Athens, Greece).
2.4. Experimental protocol
2.4.1. Experiments in rabbits
New Zealand white male rabbits weighing 2.7–3.2 kg were subjected to 30 min regional ischaemia of the myocardium, followed by 3 h of reperfusion, and were randomized into 10 groups as follows (experimental protocol presented in detail in Supplementary material online, Figure S1A):
Control group (n = 8): No additional intervention.
PostC group (n = 7): Application of eight cycles of 30 s I–R immediately after sustained ischaemia.
PostC + DT2 group (n = 6): Application of PostC and administration of the PKG inhibitor DT2 at a dose of 0.25 mg kg−1 iv bolus 10 min before sustained ischaemia, as previously described.28
NaHS group (n = 7): Animals were treated with NaHS at a dose of 100 μg kg−1 iv bolus on the 20th min of ischaemia followed by infusion of 1 mg kg−1 h−1 for the next 120 min, as previously described.26
NaHS + DT2 group (n = 6): Animals were treated with NaHS (as in the NaHS group) and the PKG inhibitor DT2 given at a dose of 0.25 mg kg−1 iv bolus 10 min before sustained ischaemia.28
NaHS + TAT group (n = 6): Animals were treated with NaHS (as in the NaHS group) and the control peptide of DT-2; TAT given at a dose of 0.143 mg kg−1 iv bolus 10 min before sustained ischaemia.
NaHS + PostC (n = 6): Animals were treated with NaHS at a dose of 100 μg kg−1 iv bolus on the 20th min of ischaemia followed by eight cycles of 30 s I–R immediately after sustained ischaemia.
NaHS + 5-HD group (n = 10): Animals were treated with NaHS (as in the NaHS group) and the mitoKATP channels inhibitor 5-hydroxydecanoic acid (5-HD) iv bolus 40 min before occlusion at a dose of 5 mg kg−1 as previously described.29
NaHS + Glibenclamide group (n = 9): Animals were treated with NaHS (as in the NaHS group) and the mitoKATP and sarcKATP channels inhibitor glibenclamide iv bolus at the 15th min of ischaemia at a dose of 0.3 mg kg−1 iv as previously described.30
NaHS + l-NAME (n = 6): Animals were treated with NaHS (as in the NaHS group) and the inhibitor of the synthase of NO (l-NAME) iv bolus at the 19th min of ischaemia at a dose of 10 mg kg−1 as previously described.26
In a second series of experiments, rabbits (five per group of Control, PostC, NaHS, and NaHS + DT2 groups) were subjected to the same interventions up to the 10th min of reperfusion, when tissue samples from the ischaemic area of myocardium were collected, snap-frozen in liquid nitrogen, and stored at −80°C for western blot analysis of vasodilator-stimulated phosphoprotein (VASP), Erk1/2, GSK3β, eNOS, and PLN.
2.4.2. Experiments in mice
Forty-four wild-type (WT) C57BL/6 male mice 13–15 weeks old and 12 eNOS KO male mice 13–15 weeks old were subjected to 30 min regional ischaemia of the myocardium followed by 2 h of reperfusion with the following interventions (experimental protocol presented in detail in Supplementary material online, Figure S1B).
WT group (n = 6): Vehicle (100 μL of water for injection) administered iv at the 19th min of ischaemia.
WT group + NaHS (n = 6): Administration of NaHS as an iv bolus dose of 100 μg kg−1 at the 20th min of ischaemia as previously described.14
WT group + DT2 + NaHS (n = 6): Co-administration of NaHS (as described above) plus DT2 given at a dose of 0.37 mg kg−1 iv bolus 10 min before sustained ischemia.28
WT group + l-NAME (n = 6): Administration of l-NAME as an iv bolus dose of 15 mg kg−1 at the 19th min of ischaemia.26
WT + NaHS + l-NAME group (n = 6): Co-administration of NaHS plus l-NAME as described above.
eNOS KO group (n = 6): Vehicle (100 μL of water for injection) administered iv at the 19th min of ischaemia.
eNOS KO + NaHS group (n = 6): Administration of NaHS as an iv bolus dose of 100 μg kg−1 at the 20th min of ischaemia.14
In another series of experiments, WT mice (four per group of Control, NaHS, and NaHS + DT2 groups) were subjected to the same interventions up to the 10th min of reperfusion, when tissue samples from the ischaemic area of myocardium were collected, snap-frozen in liquid nitrogen, and stored at −80°C for western blot analysis of pVASP.
In another series of experiments, 29 WT 129SvJ and 27 PLN knockout male and female mice were subjected to 30 min regional ischaemia of the myocardium followed by 2 h of reperfusion with the following interventions (experimental protocol presented in detail in Supplementary material online, Figure S1C):
WT group (n = 8): Vehicle (100 μL of water for injection) administered iv at the 20th min of ischaemia.
WT + PostC 3*10 s group (n = 7): Application of three cycles of 10 s ischaemia followed by 10 s of reperfusion immediately after prolonged ischaemia.31
WT + PostC 6*10 s group (n = 6): Application of six cycles of 10 s ischaemia followed by 10 s of reperfusion immediately after prolonged ischaemia.31
WT + NaHS group (n = 8): Administration of NaHS iv as a bolus dose of 100 μg kg−1 at the 20th min of ischaemia.14
PLN KO group (n = 6): Vehicle (100 μL of water for injection) administered iv at the 20th min of ischaemia.
PLN KO + PostC 3*10 s group (n = 9): Application of three cycles of 10 s ischaemia followed by 10 s of reperfusion immediately after prolonged ischaemia
PLN KO + PostC 6*10 s group (n = 6): Application of six cycles of 10 s ischaemia followed by 10 s of reperfusion immediately after prolonged ischaemia.31
PLN KO + NaHS group (n = 6): Administration of NaHS iv as a bolus dose of 100 μg kg−1 at the 20th min of ischaemia.14
2.5. Western blot analysis
Tissues from the ischaemic area of myocardium were pulverized and the sample powder formed was homogenized in lysis solution (1% Triton X-100, 20 mM Tris pH 7.4–7.6, 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 1 mM EGTA, 1 mM Glycerolphosphatase, 1% SDS, 100 mM PMSF, and 0.1% protease phosphatase inhibitor cocktail). After centrifugation at 11 000 × g for 15 min at 4°C, supernatants were collected. Supernatants were treated with protein G beads for rabbit IgG removal, in order to minimize the non-specific binding of the primary antibody. In brief, 40 μL of supernatants were incubated for 4 h with 400 μL of protein G beads. After centrifuging and aspiration of the supernatants, the samples were re-dissolved with lysis buffer and the protein concentration was measured. The protein concentration was determined based on the Bradford dye-binding procedure. The supernatant was mixed with Dave's buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenyl blue, and 0.125 M Tris–HCl). The samples were heated at 100°C for 10 min and stored at −80°C. An equal amount of protein was loaded into each well and then separated by SDS–PAGE 7.5–11% and transferred onto a polyvinylidene difluoride membrane. After blocking with 5% non-fat dry milk, membranes were incubated overnight at 4°C with the following primary antibodies: phospho-eNOS (ser1177), eNOS, phospho-GSK3β (ser9), GSK3β, phospho-p44/42 (Thr202/Tyr204), p44/42, phospho-VASP (Ser239), VASP, phospho-PLN (ser16, tyr17), and β-actin (Cell Signaling Technology, Beverly, MA, USA). Membranes were then incubated with secondary antibodies for 1–2 h at room temperature (goat anti-mouse and goat anti-rabbit HRP; Biorad, Interlab, Athens, Greece) and developed using the GE Healthcare ECL Western Blotting Detection Reagents (Thermo Scientific Technologies, Bioanalytica, Athens, Greece). Relative densitometry was determined using a computerized software package (NIH Image, National Institutes of Health, USA), and the values for phosphorylated eNOS, GSK3β, p44/42, VASP, and PLN (ser16/thr17) were normalized to the values for total eNOS, GSK3β, p44/42, VASP, and β-actin, respectively.26
2.6. cGMP determination
Powdered tissue samples from NaHS, NaHS + DT2, and control groups were homogenized with 0.1 N HCl 1 : 8 dilutions. cGMP content was measured in the lysates using a commercially available EIA kit (Assay Designs, Ann Arbor, MI, USA) following the manufacturer's instructions. Results were expressed as pmol cGMP/mg tissue.
2.7. Phosphodiesterase activity
Rabbit left ventricular tissue homogenized in buffer containing 10 mM Tris–HCl (pH 7.4) in the presence of protease inhibitors. The homogenates were centrifuged at 15 000 × g (10 min) and supernatants were desalted by gel filtration. PDE activity was measured following manufacturer's instruction. Briefly, 5 µg of protein was added to each well containing 3′,5′-cGMP as a substrate, with or without a H2S donor, and incubated at 37°C for 30 min. The reaction was terminated by adding green assay reagent and colour was allowed to develop for 30 min. Absorbance at 630 nm was read in a GENios micro-plate reader (Tecan). PDE activity was calculated using a 5′-GMP standard curve.
2.8. Statistical analysis
All results are presented as mean ± SEM. Comparisons of numeric variables among the groups were analysed using an one-way analysis of variance (ANOVA) model with Bonferroni correction and with Tukey post hoc analysis. A two- or three-way ANOVA with Bonferroni correction to adjust for multiple pair-wise comparisons was employed for Figures 1, 4, and 5A. Analyses were performed using a Stata 13.1 statistical software package (StataCorp, TX, USA). A calculated P-value of <0.05 was considered to be statistically significant.
Figure 1.

Mechanism of NaHS-induced reduction in infarct size in rabbits. Infarct-to-risk area ratio. *P < 0.05 vs. control group; #P < 0.05 vs. PostC group. PostC, postconditioning; DT2, PKG-I inhibitor; TAT, control peptide for DT2; 5-HD, mitoKATP channel inhibitor; Glibenclamide, mito/sarc KATP channel inhibitor; l-NAME, NOS inhibitor. Control (n = 8); PostC (n = 7); PostC + DT2 (n = 6); NaHS (n = 7); NaHS + DT2 (n = 6); NaHS + TAT (n = 6); NaHS + PostC (n = 6); NaHS + 5-HD (n = 10); NaHS + Glibenclamide (n = 9); NaHS + l-NAME (n = 6).
Figure 4.

NaHS-induced cardioprotection is PLN-dependent. Infarct-to-risk area ratio %I/R in WT and PLN KO mice after administration of NaHS or after application of different PostC algorithms (*P < 0.05 vs. WT). PLN, phospholamban; PostC, postconditioning; PostC 3*10 s, application of three cycles of 10 s ischaemia followed by 10 s of reperfusion immediately after prolonged ischaemia; PostC 6*10 s, application of six cycles of 10 s ischaemia followed by 10 s of reperfusion immediately after prolonged ischaemia. WT (n = 8); WT + PostC 3*10 s (n = 7); WT + PostC 6*10 s group (n = 6); WT + NaHS (n = 8); PLN KO (n = 6); PLN KO + PostC 3*10 s (n = 9); PLN KO + PostC 6*10 s (n = 6); PLN KO + NaHS (n = 6).
Figure 5.
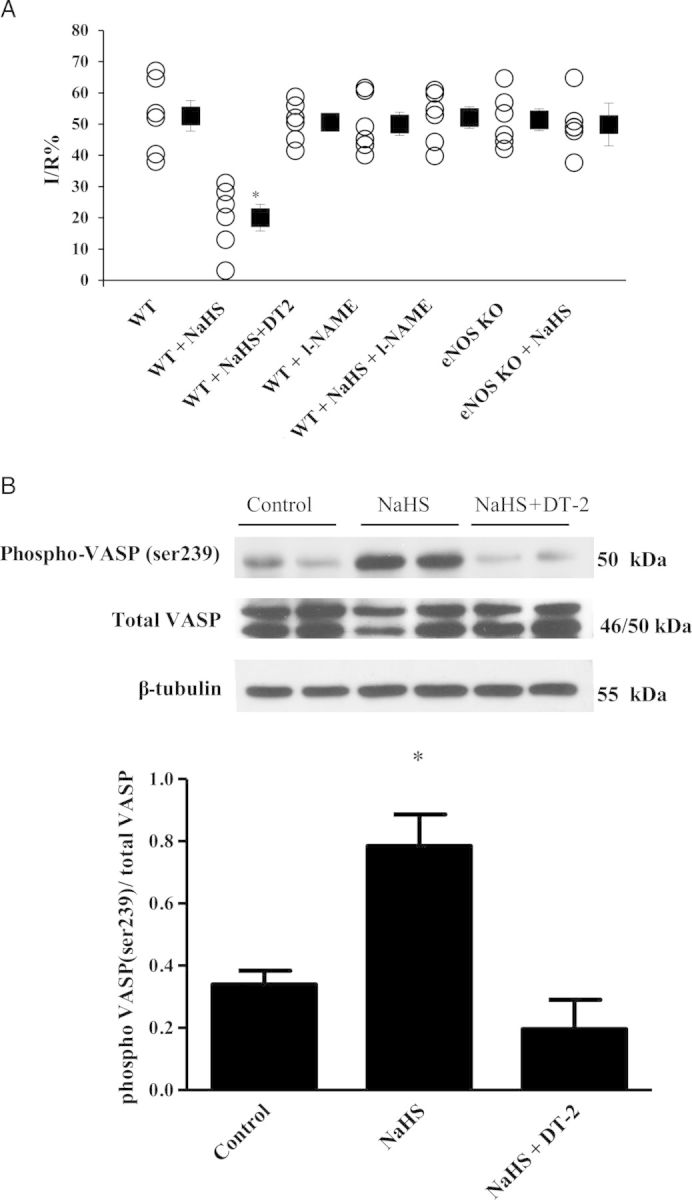
Inhibition of eNOS and PKG abolishes NaHS-induced cardioprotection in mice. (A) Infarct-to-risk area ratio %I/R in mice. *P < 0.05 vs. WT. WT (n = 6); WT + NaHS (n = 6); WT + DT2 + NaHS (n = 6); WT + l-NAME (n = 6); WT + NaHS + l-NAME (n = 6); eNOS KO (n = 6); eNOS KO + NaHS group (n = 6). (B) Representative blots along with the densitometric analysis of pVASP/VASP (n = 5; *P < 0.05 vs. control).
3. Results
3.1. Haemodynamic parameters
Characteristics and haemodynamic variables for all study rabbit groups are presented in Table 1. No significant differences were observed between the groups.
Table 1.
Characteristics and haemodynamic variables for the different study group
| Study group | HW (g) | Baseline |
20 min Ischaemia |
180 min Reperfusion |
|||
|---|---|---|---|---|---|---|---|
| HR (bpm) | MAP | HR | MAP (mmHg) | HR | MAP | ||
| Control | 7.3 ± 0.3 | 275 ± 10 | 85 ± 5 | 270 ± 12 | 81 ± 6 | 266 ± 11 | 77 ± 5 |
| PostC | 7.2 ± 0.2 | 280 ± 14 | 78 ± 6 | 273 ± 13 | 76 ± 5 | 269 ± 10 | 72 ± 6 |
| PostC + DT2 | 7.4 ± 0.4 | 278 ± 12 | 82 ± 4 | 271 ± 12 | 79 ± 4 | 268 ± 12 | 76 ± 4 |
| NaHS | 7.8 ± 0.3 | 282 ± 11 | 84 ± 3 | 274 ± 09 | 80 ± 3 | 270 ± 11 | 77 ± 7 |
| NaHS + DT2 | 7.3 ± 0.5 | 279 ± 10 | 88 ± 6 | 269 ± 11 | 82 ± 7 | 263 ± 09 | 79 ± 5 |
| NaHS + TAT | 7.4 ± 0.4 | 274 ± 09 | 81 ± 7 | 267 ± 10 | 77 ± 6 | 265 ± 13 | 74 ± 3 |
| NaHS + 5HD | 7.6 ± 0.3 | 283 ± 14 | 84 ± 4 | 272 ± 13 | 79 ± 4 | 269 ± 12 | 75 ± 4 |
| NaHS + Glibenclamide | 7.7 ± 0.2 | 271 ± 15 | 79 ± 6 | 267 ± 11 | 75 ± 5 | 264 ± 14 | 73 ± 5 |
| NaHS + l-NAME | 7.5 ± 0.4 | 279 ± 12 | 82 ± 5 | 272 ± 10 | 78 ± 4 | 268 ± 11 | 74 ± 3 |
HW, heart weight; HR, mean heart rate; MAP, mean arterial blood pressure.
3.2. Infarct size determination in rabbits following pharmacological manipulations
To evaluate the effects of NaHS on cardioprotection, we used a rabbit in vivo model of I–R injury. Rabbit heart is larger and exhibits slower heart rate than the mouse. In addition, the rabbit heart expresses the slow β-myosin heavy chain isoform, similar to the human heart, and its calcium cycling characteristics are similar to those of the human heart. Specifically, the rabbit and human heart share a major similarity in their reliance on SERCA and the NCX for the extrusion of calcium during diastole.32
In the rabbit cohort, no significant differences were detected in the areas at risk among the studied groups (see Supplementary material online, Figure S2). Application of PostC (as a positive control) limited infarct size compared with control (24.6 ± 1.2 vs. 47.5 ± 1.2%, P < 0.05), as expected (Figure 1). NaHS reduced the infarct size compared with the control group (11.9 ± 3.4 vs. 47.5 ± 1.2%, P < 0.05) and did so to a greater extent than PostC (P < 0.05).
DT2 is a membrane permeable peptide which has the ability to inhibit cGMP-dependent PKG-Iα activation.33 DT2 consists of peptide W45 (pseudosubstrate which cannot be phosphorylated by PKG-I) and a TAT peptide that is derived from the transactivator of transcription of HIV. TAT is a cell-penetrating peptide allowing W45, or any other peptide sequence fused to it, to gain intracellular access. Although DT2 is a selective PKG-Iα inhibitor, the TAT peptide has been reported to exert biological effects of its own in vivo.34 The addition of the PKG-Iα inhibitor DT2 abrogated the infarct size-limiting effect of NaHS (39.8 ± 3.5%, P = NS vs. control), whereas TAT did not modify the effect of NaHS (23.1 ± 2.7%, P = NS vs. NaHS group), suggesting that the effects of NaHS were PKG-Iα-mediated. In addition, administration of DT2 along with PostC application reversed the beneficial effects of PostC (42.6 ± 1.8 vs. 24.6 ± 1.2% in PostC, P < 0.05).
Administration of the mitoKATP inhibitor (5-HD) did not affect the NaHS response (14.8 ± 1.7%, P = NS). However, the mito/sarc KATP channel inhibitor, glibenclamide, partially reversed the cardioprotective effects of NaHS (30.7 ± 1.1, P < 0.05 vs. control and NaHS groups). Administration of pharmacological agents alone, (glibenclamide, 5-HD, and l-NAME), in rabbits under I/R, have been shown not to reduce infarct size (data not shown).30 In addition, application of PostC along with NaHS administration did not exhibit additional benefits to the ischaemic myocardium compared with NaHS alone (9.5 ± 1.2 vs. 11.9 ± 3.4%, P < 0.05).
3.3. Mechanisms of NaHS-induced pharmacological conditioning
To investigate whether NaHS administration results in increased cGMP levels in the heart, the levels of this cyclic nucleotide in the ischaemic part of rabbit myocardium were measured (Figure 2). In agreement with the results on infarct size suggesting involvement of PKG in the cardioprotective mechanism of H2S, cGMP levels in the myocardium were found to be higher in NaHS-treated animals compared with the control group (Figure 2). The increase in cGMP levels coincided with inhibition of phosphodiesterase activity in rabbit heart homogenates; addition of 100 μΜ of H2S donor reduced cGMP-inhibiting PDE activity to 65 ± 2.2% of control (P < 0.05). Activation of PKG after NaHS treatment and PostC was confirmed by determining the phosphorylation status of its surrogate marker VASP. Indeed, VASP phosphorylation on Ser239 was greater in NaHS-treated animals compared with control and NaHS + DT2 groups (Figure 3A). Phosphorylation of Erk1/2 was also found significantly higher in NaHS and PostC groups vs. control and NaHS + DT2 groups (Figure 3B), whereas no phosphorylation of GSK3β was observed in NaHS-treated animals; when PostC was used as a positive control, an increase in GSK3β phosphorylation was noted, in line with reports in the literature35 (Figure 3C). Since phospholamban (PLN) is a PKG substrate,36–38 we evaluated the ability of NaHS to alter its phosphorylation status. Levels of phospho-PLN were increased in the NaHS-treated group compared with control and NaHS + DT2 groups (Figure 3D). Similarly, PostC enhanced PLN phosphorylation.
Figure 3.

Continued.
Figure 2.

cGMP levels in heart homogenates. Following treatment, the ischaemic part of the heart was snap-frozen, pulverized, and cyclic nucleotides were extracted as detailed in the Methods section. Results are expressed as pmol/mg protein. n = 5/group (*P < 0.05 vs. control).
Figure 3.

Activation of intracellular signalling pathways associated with cardioprotection by NaHS in rabbits. Representative blots for each signalling protein are shown along with the densitometric analysis from the total number of animals per group (n = 5). (A) Ratio of pVASP/VASP; (B) ratio of p-p44/42/p44/42, (C) ratio of pGSK3β/GSK3β, (D) ratio of pPLN/β-actin, and (E) ratio of peNOS/eNOS. *P < 0.05 vs. control; #P < 0.05 vs. NaHS and PostC. PostC, postconditioning; DT2, PKG-I inhibitor. n = 5/group.
3.4. Genetic evidence for PLN involvement in NaHS-induced protection
To evaluate the functional relevance of our biochemical findings on PLN, we used mice with targeted disruption of the PLN locus (Figure 4). The infarct size in control WT mice (129SvJ background) was comparable to that of PLN KO animals (42.2 ± 2.6 vs. 41.3 ± 3.1%, P = NS). PostC application and NaHS administration were beneficial in WT animals compared with the control WT group (14.9 ± 2.2% (WT + PostC 3*10sec); 15.5 ± 1.1% (WT + NaHS) vs. 42.2 ± 2.6% (WT)). However, the cardioprotective effect of both NaHS and PostC was abolished in PLN KO animals (42.2 ± 1.5 and 45.2 ± 2.1%, respectively, P < 0.05 vs. NaHS and PostC in WT mice). To test whether PLN KO mice require a more robust PostC algorithm, we increased the PostC cycles. Applying six cycles of PostC did not lead to greater cardioprotection in WT mice, whereas in PLN KO animals we observed a trend towards a smaller infarct size that did not reach statistical significance. No significant differences were detected in the areas at risk among the studied groups (see Supplementary material online, Figure S4).
3.5. Evidence of species-specific eNOS activation following NaHS-induced protection
In our initial experiments in rabbits, administration of the NOS inhibitor (l-NAME) did not alter the infarct-limiting effects of NaHS (14.7 ± 2.2 vs. 11.9 ± 3.4%, respectively, P = NS vs. NaHS; Figure 1). In addition, no phosphorylation of eNOS was observed in NaHS-treated animals, in contrast to what has been reported in the literature.23,39–41 It should be noted that eNOS phosphorylation was evident in the positive control PostC group (Figure 3E). To reproduce the published results on the role of NO in H2S cardioprotection in mice, we co-administered NaHS and l-NAME in mice. Such treatment resulted in complete abrogation of the NaHS-infarct size-limiting effects (52.1 ± 3.5 vs. 20.1 ± 4.3%, respectively, P < 0.05), with animals that received NaHS + l-NAME presenting infarct size similar to WT and WT + l-NAME groups (52.7 ± 4.7 and 50.1 ± 3.7%, respectively, P = NS). The pharmacological findings were confirmed in the eNOS KO mice, where administration of NaHS had no protective results vs. vehicle-treated eNOS KO animals (49.9 ± 3.6 vs. 51.4 ± 3.5%, respectively, P = NS; Figure 5A). No significant differences were detected in the areas at risk among the studied groups (see Supplementary material online, Figure S5).
3.6. PKG-dependent cardioprotection of NaHS in murine myocardium
Since species differences were observed between rabbits and mice, we sought to confirm the importance of PKG-Iα in cardioprotection in mice. Administration of a single iv bolus dose of NaHS in mice (C57Bl6 background) resulted in a significant reduction on infarct size compared with the control group (20.1 ± 4.3 vs. 52.7 ± 4.9% respectively, P < 0.05). Inhibition of PKG-Iα before NaHS administration abrogated the infarct limiting effects of NaHS (50.4 ± 2.8 vs. 20.1 ± 4.3%, respectively, P < 0.05) (Figure 5A). No significant differences were detected in the areas at risk among the studied groups (see Supplementary material online, Figure S3). Additionally, the administration of NaHS resulted in increased phosphorylation of VASP (ser239) compared with WT control in the ischaemic area of the myocardium at the 10th min of reperfusion, whereas no phosphorylation was observed in the group treated with DT2 (Figure 5B).
4. Discussion
The current study confirms the cardioprotective effect of H2S in vivo and emphasizes the importance of a novel cGMP/PKG/PLN signalling cascade in the pharmacological PostC afforded by H2S. Several studies have suggested a beneficial role of cGMP/PKG-regulated pathways in PostC.9,36,42,43 PKG activation exerts anti-apoptotic effects through PKCε activation and inhibition of mPTP channel opening.36 In addition, increased cGMP levels and PKG activation lead to attenuated contractility during reperfusion through altered calcium handling.9,36,44 In two recent studies where the mechanism of PKG-mediated protection in PostC was studied, it was shown that the beneficial effects of PKG result from (i) delayed normalization of intracellular pH during reperfusion through altered NHE function43 and (ii) antioxidant effects that prevent eNOS uncoupling.42 Since cGMP has been shown to contribute to the biological effects of H2S,39,45 we evaluated the role of cGMP/PKG pathways in H2S-induced pharmacological conditioning. We observed that DT2 blocked the infarct-limiting effect of NaHS in both rabbits and mice. In line with this finding, we noted that cGMP levels and VASP phosphorylation were increased in the ischaemic myocardium.
KATP channel opening is known to mediate many of the biological activities of H2S.46 Given the key role of these channels in pharmacological conditioning mediated by cGMP/PKG, we evaluated their contribution in H2S-induced cardioprotection using two different inhibitors, namely 5-HD and glibenclamide. These agents target distinct populations of KATP channels, as 5-HD inhibits mitoKATP, while glibenclamide blocks KATP channels irrespectively of their subcellular localization inhibiting both sarcKATP and mitoKATP. Ex vivo studies on the role of mitoKATP vs. sarcKATP have been inconclusive, with some investigators claiming that mitoKATP, sarcKATP, or both types of channels mediate the protective effects of H2S12,17,18,21 depending on the preparation and conditions used. We found that 5-HD did not limit the beneficial effects of NaHS, indicating that mitoKATP channels do not mediate H2S-induced cardioprotection in vivo in rabbits. Interestingly, administration of glibenclamide in vivo reduced the infarct-limiting effects of NaHS, in line with the ex vivo observations.12,17,18,21 Our data, taken together, indicate that sarcKATP channel activation participates in the cardioprotective signalling of H2S. Sarcolemmal KATP channels were recently shown to mediate cGMP-dependent natriuretic peptide-induced cardioprotection.47 Sarcolemmal KATP channels are involved in intracellular Ca2+ handling and their opening results in resistance to apoptosis during I/R injury.48
We next focused on the possible role of Ca2+-handling proteins in H2S pharmacological conditioning. We decided to systematically investigate the contribution of PLN, as this protein is a PKG substrate.36–38 Moreover, a number of publications have shown altered PLN phosphorylation in the context of I/R.44,49–51 In a recent study, Inserte et al. showed that phosphorylation of PLN (ser16/thr17) peaked in the control group 3 min after the onset of reperfusion and declined thereafter. On the other hand, following PostC, increased PLN phosphorylation was evident in the 5th min of reperfusion. It was, thus, proposed that delayed PLN phosphorylation limits reperfusion-triggered Ca2+ oscillations leading to protection.44 PLN once phosphorylated by PKG reduces free intracellular Ca2+ concentration by dissociating from SERCA; SERCA is then able to pump Ca2+ ions back into the SR.52 Since no pharmacological inhibitors of PLN exist, the only way to determine the functional relevance of PLN in PostC or pharmacological conditioning would be through the use of PLN KO. Ablation of the PLN gene in mice is associated with greatly enhanced cardiac Ca2+ cycling and performance. The Ca2+ affinity (KCa) of SERCA2a and the overall sensitivity of this transport system for Ca2+ is increased. Ca2+ uptake rates are higher in PLN KO hearts than WT, especially at low [Ca2+], whereas there is no effect on Vmax.53 Intriguingly, this hyperdynamic cardiac function is maintained throughout the lifetime of the mouse without observable pathological consequences. The body weight, the heart weight, and the heart/body weight, along with heart rate, mean aortic pressure, cardiac output (venous return), stroke volume, and cardiac power (left ventricle) is unaltered compared with WT mice. Significant changes are, however, observed in the left intraventricular pressure (systolic diastolic and end-diastolic), as well as the cardiac contraction and relaxation properties (decreased time to peak pressure, increased absolute values of +dP/dt, and decreased half-relaxation time).53 In addition to the direct effect of the PLN ablation on SERCA2a, various other compensatory changes in protein expression and/or phosphorylation take place, as demonstrated by global proteome studies, affecting mostly structural and energy-related proteins.54 All of the above should be kept in mind when interpreting data obtained with the PLN KO animals.
To study the role of PLN in H2S pharmacological conditioning, we measured PLN phosphorylation in NaHS-treated animals. We observed that PLN was phosphorylated on Ser16/Thr17 after NaHS treatment in rabbits in a PKG-I-dependent manner. Since we only examined a single time-point, we cannot comment on the kinetics of phosphorylation during reperfusion or PostC. To determine the contribution of PLN in H2S-induced cardioprotection, we used PLN KO mice. We found that NaHS failed to restrict infarct size in PLN KO mice after I/R, suggesting that a PKG/PLN pathway mediates the H2S response. Moreover, our experiments with PLN KO mice indicate that inhibition of PLN abolished the effects of a commonly used PostC algorithm. Since it has been described that during IPC, it is possible to overcome the lost protection of eNOS KO by increasing the number of conditioning cycles,55 we tested a second algorithm of PostC in PLN KO mice. However, increasing the number of cycles from 3 to 6 in the PostC, although it showed a trend to limit infarct size, this did not reach statistical significance. Taken together, our results indicate that in addition to its established role in heart failure,52 PLN plays an important role in I/R injury. Although the mechanism of action of PLN in the context of cardioprotection was not studied in detail in the course of our studies, relieving the inhibition on SERCA-mediated Ca2+ uptake into the SR following PLN phosphorylation would be expected to lower free cytosolic Ca2+ levels and, thus, limit mitochondrial Ca2+ entry56,57 and preventing mPTP opening and hypercontracture. Indeed, H2S has been reported to accelerate SR-Ca2+ uptake rate in single ventricular myocytes.58
Erk1/2 has been shown to exert a protective role in conditioning. In line with this observation, we found that in rabbits treated with NaHS, Erk1/2 phosphorylation was increased. Erk1/2 activation following exposure to H2S has been noted before in endothelial cells.59 Since DT2 treatment reduced Erk1/2 phosphorylation in the rabbit heart under NaHS treatment, Erk1/2 must lie downstream of PKG in the H2S signalling pathway. Activated Erk1/2 can activate downstream protective pathways to limit infarct size. One of the downstream targets of Erk1/2 is GSK-3β, which once phosphorylated inhibits mPTP opening.60 In our study, no evidence for GSK3β phosphorlyation was observed on in H2S-treated animals, suggesting that Ekr1/2 acts independently of GSK-3β. In contrast, the beneficial effect of NaHS in limiting I/R injury was GSK-3β-mediated in pigs.24 Divergent results have been noted in experiments in pigs vs. rodents have been noted before.61,62 The targets of Erk1/2 in the context of H2S cardioprotection remain to be elucidated. Since PostC utilizes additional pathways to those triggered by NaHS (such as GSK3β and mKATP), we tested whether application of PostC and administration of NaHS have additive effects. In these experiments, we observed that the combination of treatments did not confer any benefits over NaHS administration alone. This finding indicates that although PostC and H2S utilize partially overlapping signalling pathways, they likely target a common end effector.
Several lines of evidence suggest that H2S requires eNOS to exert its protective effects in the cardiovascular system. For example, pharmacological inhibition or genetic ablation of eNOS has been shown to reverse the beneficial effects of H2S in vasorelaxation,39 angiogenesis,39 wound healing,39 and resuscitation.63 In the heart, H2S donor administration in the context of I/R23,41 or heart failure40,64 results in increased eNOS phosphorylation and enhanced NO bioavailability. In the most recent study published on H2S in I/R,23 H2S donors administered at the end of ischaemia resulted in no significant change in infarct size in eNOS KO and eNOS S1179A mice. However, in our initial studies, we found no biochemical or functional evidence for eNOS involvement in the H2S cardioprotection in rabbits. Given that H2S is being tested as a cardioprotective agent in human (NCT01989208), we studied whether there are species differences in the mode of action of H2S. We, thus, repeated the I/R injury experiments in mice. In agreement to what others observed in mice23,39–41,64 we found that eNOS inhibition abolished the effects of NaHS administration. Our data collectively demonstrate that the protective pathway of H2S in I/R is dependent on NO in mice, but not in rabbits.
In summary, we have shown that the cardioprotective effects of H2S are mediated through a cGMP/PKG pathway, independently of mitoKATP channel opening and GSK-3β inhibition. The pathways responsible for increased cGMP levels include eNOS activation and/or PDE inhibition (Figure 6). Depending on (i) the expression levels of eNOS vs. PDE and (ii) the presence and activity of other signalling components (for example, phosphatases that dephosphorylate eNOS on Ser1177 to restrict its activity), the contribution of these two pathways to cardioprotective cGMP signalling will vary. Thus, H2S might employ eNOS activation, PDE inhibition, or both depending on the species investigated, the experimental conditions used, and whether interventions are performed in healthy individuals or in the context of disease. Since different pools of cGMP exist in cells, it is possible that cGMP generated through eNOS activation and cGMP derived from PDE inhibition might exhibit subcellular compartmentalization, recruiting different or overlapping downstream signalling pathways to confer cardioprotection. Furthermore, our study highlights the participation of a novel mediator, namely PLN, in the cardioprotective signalling cascade. The present study expands our knowledge on the molecular mechanisms of action of H2S in pharmacological conditioning and strengthens the rational on which clinical testing and use of H2S-releasing compounds can be based.
Figure 6.

Proposed molecular pathways of H2S-induced cardioprotection. Administration of H2S activates endothelial NOS resulting in NO release that in turn stimulates guanyl cyclase. H2S can also inhibit PDE. Both pathways (eNOS activation and PDE inhibition) operate in the mouse, whereas only PDE inhibition occurs in rabbits.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work has been co-financed by the European Union (European Social Fund—ESF) and Greek National Funds through the Operational Program ‘Education and Lifelong Learning’ of the National Strategic Reference Framework (NSRF)—Research Funding Program: Aristeia 2011 (1436) to A.P. and by the COST Action BM1005 (ENOG: European network on gasotransmitters). C.C. is supported by a postdoctoral fellowship of the American Heart Association. C.S. is supported by a grant from the National Institutes of Health. This work has been supported in part by a scholarship of the Experimental Research Center of ELPEN Pharmaceuticals.
Acknowledgments
Conflict of interest: none declared.
References
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 2.Heusch G. Cardioprotection: chances and challenges of its translation to the clinic. Lancet 2013;381:166–175. [DOI] [PubMed] [Google Scholar]
- 3.Sluijter JP, Condorelli G, Davidson SM, Engel FB, Ferdinandy P, Hausenloy DJ, Lecour S, Madonna R, Ovize M, Ruiz-Meana M, Schulz R, Van Laake LW. Novel therapeutic strategies for cardioprotection. Pharmacol Ther 2014;144:60–70. [DOI] [PubMed] [Google Scholar]
- 4.Hausenloy DJ, Erik Botker H, Condorelli G, Ferdinandy P, Garcia-Dorado D, Heusch G, Lecour S, van Laake LW, Madonna R, Ruiz-Meana M, Schulz R, Sluijter JP, Yellon DM, Ovize M. Translating cardioprotection for patient benefit: position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res 2013;98:7–27. [DOI] [PubMed] [Google Scholar]
- 5.Di Lisa F, Canton M, Carpi A, Kaludercic N, Menabo R, Menazza S, Semenzato M. Mitochondrial injury and protection in ischemic pre- and postconditioning. Antioxid Redox Signal 2011;14:881–891. [DOI] [PubMed] [Google Scholar]
- 6.Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 2009;104:189–202. [DOI] [PubMed] [Google Scholar]
- 7.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 2013;123:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shintani-Ishida K, Inui M, Yoshida K. Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca(2)(+) overload. J Mol Cell Cardiol 2012;53:233–239. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Dorado D, Agullo L, Sartorio CL, Ruiz-Meana M. Myocardial protection against reperfusion injury: the cGMP pathway. Thromb Haemost 2009;101:635–642. [PubMed] [Google Scholar]
- 10.Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M, Inserte J, Agullo L, Cabestrero A. The end-effectors of preconditioning protection against myocardial cell death secondary to ischemia-reperfusion. Cardiovasc Res 2006;70:274–285. [DOI] [PubMed] [Google Scholar]
- 11.Polhemus DJ, Lefer DJ. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res 2014;114:730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY, Zhou S, Moore PK. Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J Pharmacol Exp Ther 2006;316:670–678. [DOI] [PubMed] [Google Scholar]
- 13.Bliksoen M, Kaljusto ML, Vaage J, Stenslokken KO. Effects of hydrogen sulphide on ischaemia-reperfusion injury and ischaemic preconditioning in the isolated, perfused rat heart. Eur J Cardiothorac Surg 2008;34:344–349. [DOI] [PubMed] [Google Scholar]
- 14.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res 2009;105:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu LF, Li Y, Neo KL, Yong QC, Lee SW, Tan BK, Bian JS. Hydrogen sulfide regulates Na+/H+ exchanger activity via stimulation of phosphoinositide 3-kinase/Akt and protein kinase G pathways. J Pharmacol Exp Ther 2011;339:726–735. [DOI] [PubMed] [Google Scholar]
- 16.Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin ES, Moore PK, Bian JS. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch 2008;455:607–616. [DOI] [PubMed] [Google Scholar]
- 17.Ji Y, Pang QF, Xu G, Wang L, Wang JK, Zeng YM. Exogenous hydrogen sulfide postconditioning protects isolated rat hearts against ischemia-reperfusion injury. Eur J Pharmacol 2008;587:1–7. [DOI] [PubMed] [Google Scholar]
- 18.Johansen D, Ytrehus K, Baxter GF. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury—evidence for a role of K ATP channels. Basic Res Cardiol 2006;101:53–60. [DOI] [PubMed] [Google Scholar]
- 19.Yao LL, Huang XW, Wang YG, Cao YX, Zhang CC, Zhu YC. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3beta-dependent opening of mPTP. Am J Physiol Heart Circ Physiol 2010;298:H1310–H1319. [DOI] [PubMed] [Google Scholar]
- 20.Yong QC, Lee SW, Foo CS, Neo KL, Chen X, Bian JS. Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. Am J Physiol Heart Circ Physiol 2008;295:H1330–H1340. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Huang H, Liu P, Tang C, Wang J. Hydrogen sulfide contributes to cardioprotection during ischemia-reperfusion injury by opening K ATP channels. Can J Physiol Pharmacol 2007;85:1248–1253. [DOI] [PubMed] [Google Scholar]
- 22.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA 2007;104:15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci USA 2014;111:3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osipov RM, Robich MP, Feng J, Liu Y, Clements RT, Glazer HP, Sodha NR, Szabo C, Bianchi C, Sellke FW. Effect of hydrogen sulfide in a porcine model of myocardial ischemia-reperfusion: comparison of different administration regimens and characterization of the cellular mechanisms of protection. J Cardiovasc Pharmacol 2009;54:287–297. [DOI] [PubMed] [Google Scholar]
- 25.Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, Szabo C, Stahl GL, Sellke FW. Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J Thorac Cardiovasc Surg 2009;138:977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andreadou I, Farmakis D, Prokovas E, Sigala F, Zoga A, Spyridaki K, Papalois A, Papapetropoulos A, Anastasiou-Nana M, Kremastinos DT, Iliodromitis EK. Short-term statin administration in hypercholesterolaemic rabbits resistant to postconditioning: effects on infarct size, endothelial nitric oxide synthase, and nitro-oxidative stress. Cardiovasc Res 2012;94:501–509. [DOI] [PubMed] [Google Scholar]
- 27.Bell RM, Kunuthur SP, Hendry C, Bruce-Hickman D, Davidson S, Yellon DM. Matrix metalloproteinase inhibition protects CyPD knockout mice independently of RISK/mPTP signalling: a parallel pathway to protection. Basic Res Cardiol 2013;108:331. [DOI] [PubMed] [Google Scholar]
- 28.Koika V, Zhou Z, Vasileiadis I, Roussos C, Finetti F, Monti M, Morbidelli L, Papapetropoulos A. PKG-I inhibition attenuates vascular endothelial growth factor-stimulated angiogenesis. Vascul Pharmacol 2010;53:215–222. [DOI] [PubMed] [Google Scholar]
- 29.Andreadou I, Iliodromitis EK, Tsovolas K, Aggeli IK, Zoga A, Gaitanaki C, Paraskevaidis IA, Markantonis SL, Beis I, Kremastinos DT. Acute administration of vitamin E triggers preconditioning via K(ATP) channels and cyclic-GMP without inhibiting lipid peroxidation. Free Radic Biol Med 2006;41:1092–1099. [DOI] [PubMed] [Google Scholar]
- 30.Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol 2004;44:1103–1110. [DOI] [PubMed] [Google Scholar]
- 31.Skyschally A, van Caster P, Iliodromitis EK, Schulz R, Kremastinos DT, Heusch G. Ischemic postconditioning: experimental models and protocol algorithms. Basic Res Cardiol 2009;104:469–483. [DOI] [PubMed] [Google Scholar]
- 32.Pattison JS, Waggoner JR, James J, Martin L, Gulick J, Osinska H, Klevitsky R, Kranias EG, Robbins J. Phospholamban overexpression in transgenic rabbits. Transgenic Res 2008;17:157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dostmann WR, Taylor MS, Nickl CK, Brayden JE, Frank R, Tegge WJ. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Ialpha inhibit NO-induced cerebral dilation. Proc Natl Acad Sci USA 2000;97:14772–14777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon JS, Jung YT, Hong SK, Kim SH, Shin MC, Lee DG, Shin WS, Min WS, Paik SY. Characteristics of HIV-Tat protein transduction domain. J Microbiol 2004;42:328–335. [PubMed] [Google Scholar]
- 35.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation 2008;117:2761–2768. [DOI] [PubMed] [Google Scholar]
- 36.Burley DS, Ferdinandy P, Baxter GF. Cyclic GMP and protein kinase-G in myocardial ischaemia-reperfusion: opportunities and obstacles for survival signaling. Br J Pharmacol 2007;152:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karczewski P, Kuschel M, Baltas LG, Bartel S, Krause EG. Site-specific phosphorylation of a phospholamban peptide by cyclic nucleotide- and Ca2+/calmodulin-dependent protein kinases of cardiac sarcoplasmic reticulum. Basic Res Cardiol 1997;92(Suppl 1):37–43. [DOI] [PubMed] [Google Scholar]
- 38.Sabine B, Willenbrock R, Haase H, Karczewski P, Wallukat G, Dietz R, Krause EG. Cyclic GMP-mediated phospholamban phosphorylation in intact cardiomyocytes. Biochem Biophys Res Commun 1995;214:75–80. [DOI] [PubMed] [Google Scholar]
- 39.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, Asimakopoulou A, Gero D, Sharina I, Martin E, Szabo C. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA 2012;109:9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polhemus DJ, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, Lefer DJ, Calvert JW. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail 2013;6:1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Predmore BL, Kondo K, Bhushan S, Zlatopolsky MA, King AL, Aragon JP, Grinsfelder DB, Condit ME, Lefer DJ. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am J Physiol Heart Circ Physiol 2012;302:H2410–H2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inserte J, Hernando V, Vilardosa U, Abad E, Poncelas-Nozal M, Garcia-Dorado D. Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J Am Heart Assoc 2013;2:e005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inserte J, Ruiz-Meana M, Rodriguez-Sinovas A, Barba I, Garcia-Dorado D. Contribution of delayed intracellular pH recovery to ischemic postconditioning protection. Antioxid Redox Signal 2011;14:923–939. [DOI] [PubMed] [Google Scholar]
- 44.Inserte J, Hernando V, Ruiz-Meana M, Poncelas-Nozal M, Fernandez C, Agullo L, Sartorio C, Vilardosa U, Garcia-Dorado D. Delayed phospholamban phosphorylation in post-conditioned heart favours Ca2+ normalization and contributes to protection. Cardiovasc Res 2014;103:542–553. [DOI] [PubMed] [Google Scholar]
- 45.Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P, Cantalupo A, Dhayade S, Karalis KP, Wang R, Feil R, Cirino G. cGMP-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation. PLoS ONE 2012;7:e53319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 2012;92:791–896. [DOI] [PubMed] [Google Scholar]
- 47.Burley DS, Cox CD, Zhang J, Wann KT, Baxter GF. Natriuretic peptides modulate ATP-sensitive K(+) channels in rat ventricular cardiomyocytes. Basic Res Cardiol 2014;109:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown DA, Chicco AJ, Jew KN, Johnson MS, Lynch JM, Watson PA, Moore RL. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. J Physiol 2005;569:913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abdallah Y, Gkatzoflia A, Pieper H, Zoga E, Walther S, Kasseckert S, Schafer M, Schluter KD, Piper HM, Schafer C. Mechanism of cGMP-mediated protection in a cellular model of myocardial reperfusion injury. Cardiovasc Res 2005;66:123–131. [DOI] [PubMed] [Google Scholar]
- 50.Gorbe A, Giricz Z, Szunyog A, Csont T, Burley DS, Baxter GF, Ferdinandy P. Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res Cardiol 2010;105:643–650. [DOI] [PubMed] [Google Scholar]
- 51.Vittone L, Mundina-Weilenmann C, Said M, Ferrero P, Mattiazzi A. Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts. Dissociation of phospholamban phosphorylation pathways. J Mol Cell Cardiol 2002;34:39–50. [DOI] [PubMed] [Google Scholar]
- 52.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 2012;110:1646–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res 1994;75:401–409. [DOI] [PubMed] [Google Scholar]
- 54.Chu G, Kerr JP, Mitton B, Egnaczyk GF, Vazquez JA, Shen M, Kilby GW, Stevenson TI, Maggio JE, Vockley J, Rapundalo ST, Kranias EG. Proteomic analysis of hyperdynamic mouse hearts with enhanced sarcoplasmic reticulum calcium cycling. FASEB J 2004;18:1725–1727. [DOI] [PubMed] [Google Scholar]
- 55.Bell RM, Yellon DM. The contribution of endothelial nitric oxide synthase to early ischaemic preconditioning: the lowering of the preconditioning threshold. An investigation in eNOS knockout mice. Cardiovasc Res 2001;52:274–280. [DOI] [PubMed] [Google Scholar]
- 56.Davis J, Westfall MV, Townsend D, Blankinship M, Herron TJ, Guerrero-Serna G, Wang W, Devaney E, Metzger JM. Designing heart performance by gene transfer. Physiol Rev 2008;88:1567–1651. [DOI] [PubMed] [Google Scholar]
- 57.Murgia M, Giorgi C, Pinton P, Rizzuto R. Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J Mol Cell Cardiol 2009;46:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan TT, Neo KL, Hu LF, Yong QC, Bian JS. H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am J Physiol Cell Physiol 2008;294:C169–C177. [DOI] [PubMed] [Google Scholar]
- 59.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabo C. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA 2009;106:21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cohen MV, Downey JM. Ischemic postconditioning: from receptor to end-effector. Antioxid Redox Signal 2011;14:821–831. [DOI] [PubMed] [Google Scholar]
- 61.Heusch G, Skyschally A, Schulz R. The in-situ pig heart with regional ischemia/reperfusion—ready for translation. J Mol Cell Cardiol 2011;50:951–963. [DOI] [PubMed] [Google Scholar]
- 62.Skyschally A, van Caster P, Boengler K, Gres P, Musiolik J, Schilawa D, Schulz R, Heusch G. Ischemic postconditioning in pigs: no causal role for RISK activation. Circ Res 2009;104:15–18. [DOI] [PubMed] [Google Scholar]
- 63.Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, Lefer DJ, Bloch KD, Ichinose F. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation 2009;120:888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Sr, Gojon G, Jr, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013;127:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]