

Abstract
Helicobacter pylori strains that harbor the oncoprotein CagA increase gastric cancer risk, and this risk is augmented under iron-deficient conditions. We demonstrate here that iron depletion induces coccoid morphology in strains lacking cagA. To evaluate the stability of augmented H. pylori virulence phenotypes stimulated by low-iron conditions, H. pylori isolated from iron-depleted conditions in vivo were serially passaged in vitro. Long-term passage decreased the ability of hypervirulent strains to translocate CagA or induce interleukin 8, indicating that hypervirulent phenotypes stimulated by low-level iron conditions are reversible. Therefore, rectifying iron deficiency may attenuate disease among H. pylori–infected persons with no response to antibiotics.
Keywords: Helicobacter pylori, iron deficiency, CagA, gastric cancer
Helicobacter pylori is the strongest known risk factor for the development of gastric cancer, and H. pylori strains harboring the cag pathogenicity island further augment cancer risk [1]. The cag island encodes a bacterial type IV secretion system (T4SS), which translocates the bacterial protein CagA into host cells. Intracellular CagA aberrantly activates numerous signaling pathways that are mutated during gastric cancer, including β-catenin, resulting in cellular responses that lower the threshold for carcinogenesis, such as hyperproliferation and proinflammatory cytokine production [2, 3]. Further, transgenic mice overexpressing CagA have been shown to develop gastric adenocarcinoma [4], thus validating this effector as an oncoprotein.
Iron deficiency is also associated with an increased risk for neoplasms that arise within the gastrointestinal tract [5], and H. pylori is linked with iron deficiency [6]. CagA facilitates H. pylori colonization by mediating bacterial iron acquisition [7], suggesting that iron deficiency may influence the virulence of this pathogen. We recently demonstrated that iron deficiency augments and accelerates the development of gastric carcinogenesis within the context of H. pylori infection and that this is mediated by increased assembly and function of the cag secretion system [8]. Whether this phenotype is due to a stable genetic modification or is reversible remains unknown, which likely influences therapeutic strategies. Therefore, the aim of this study was to define the dynamics of H. pylori virulence phenotypes that are augmented by iron deficiency.
METHODS
H. pylori Strains
Wild-type carcinogenic H. pylori strain 7.13 and its cagA− isogenic mutant were minimally passaged and used to infect Mongolian gerbils maintained on iron-replete or iron-depleted diets, as previously described [8]. Gerbils were euthanized 12 weeks after challenge, and in vivo–adapted strains were harvested from gerbils maintained on iron-depleted diets [8]. The Vanderbilt University Institutional Animal Care and Use Committee approved all procedures.
Parental strain 7.13 or in vivo–adapted strains harvested from 5 iron-depleted gerbils were serially passaged every 24–48 hours for 1, 5, 10, 15, 20, 25, and 30 passages. At each passage, strains were grown in Brucella broth supplemented with 10% fetal bovine serum (FBS) for 16 hours at 37°C with 5% CO2 for coculture with gastric epithelial cells.
Gastric Epithelial Cells
Human AGS gastric epithelial cells were cocultured with H. pylori strains at a multiplicity of infection of 100:1 for 6 hours.
Immunofluorescence and Confocal and Scanning Electron Microscopy
Gastric tissue specimens from gerbils were processed for immunofluorescence as previously described [9]. Tissue samples were fixed in 2% paraformaldehyde and embedded in agar, and 100-µM sections were cut (Leica). Rabbit anti–H. pylori antibodies (Dako), Alexa Fluor 594 phalloidin (Invitrogen), and DAPI (Invitrogen) were used for visualization of H. pylori, actin, and nuclei, respectively. Samples were imaged with a Zeiss LSM 700 confocal microscope, and z-stacks were reconstructed using Volocity software (Improvision). For scanning electron microscopy, gastric tissue was fixed in paraformaldehyde phosphate buffer fixative and processed, as previously described [10].
Transmission Electron Microscopy
H. pylori were grown in Brucella broth supplemented with 10% FBS alone or with 100 µM FeCl3 (iron-replete), 100 µM dipyridyl (iron restricted), or 100 µM dipyridyl plus 100 µM FeCl3 (iron restricted with iron supplementation) for 16 hours at 37°C with 5% CO2. H. pylori were harvested in 0.05-M sodium cacodylate buffer, spotted onto Formvar-coated grids, and negatively stained with 1% ammonium molybdate. Samples were viewed with a Philips C-12 transmission electron microscope, as previous described [11].
Western Blot Analysis
H. pylori:AGS coculture protein lysates were harvested, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride (PVDF) membranes. Levels of total CagA (anti-CagA antibody; Austral Biologicals), phosphorylated CagA (anti-pY99 antibody; Santa Cruz) as a measure of translocated CagA, and β-actin (anti-β-actin antibody; Sigma) were determined. Protein intensities were quantified using ImageJ (National Institutes of Health) and standardized to levels of β-actin.
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR)
RNA was extracted (Qiagen) from H. pylori:AGS cocultures, and quantitative RT-PCR was performed according to the manufacturer's instructions. IL-8 messenger RNA (mRNA) expression was normalized to levels of GAPDH mRNA expression (TaqMan; Applied Biosystems).
Interleukin 8 (IL-8) Enzyme-Linked Immunosorbent Assay (ELISA)
H. pylori:AGS coculture supernatants were collected, and levels of IL-8 were determined by the Quantikine IL-8 ELISA (R&D Systems) and analyzed with Gen5 software (Synergy4; BioTek).
Statistical Analysis
Mean values with standard errors are shown from experiments performed on at least 3 independent occasions. Mann–Whitney U t tests and 1-way analysis of variance were used for comparisons. A P value of ≤ .05 was considered statistically significant.
RESULTS
We previously demonstrated that (1) iron deficiency augments gastric carcinogenesis within the context of H. pylori infection in Mongolian gerbils [8], and (2) loss of cagA significantly decreases H. pylori colonization under iron-deficient conditions but not normal iron conditions [7], suggesting that CagA provides a survival advantage in vivo that is revealed under conditions of iron deficiency. To define mechanisms that may account for these findings, we assessed the morphology of the cagA− isogenic mutant in gastric tissue under iron-replete and iron-depleted conditions, using confocal microscopy (Figure 1A) and scanning electron microscopy (Figure 1B). Under iron-replete conditions, the cagA− isogenic mutant exhibited an elongated and spiral morphology; however, iron depletion led to the development of coccoid phenotypes. This phenotype was not observed with wild-type H. pylori, which maintained its spiral morphology under both iron-replete and iron-depleted conditions (data not shown). To determine whether the transition to coccoid morphology under conditions of iron depletion could be replicated in vitro, we next assessed morphologic phenotypes of wild-type strain 7.13 and its cagA− mutant under control, iron-replete, iron-restricted, or iron-restricted conditions with iron supplementation, by transmission electron microscopy (Figure 1C). Loss of cagA led to coccoid morphology but only under conditions of iron depletion, confirming our ex vivo results (Figure 1A and 1B). Of note, this phenotype was prevented by the addition of exogenous iron (Figure 1C).
Figure 1.
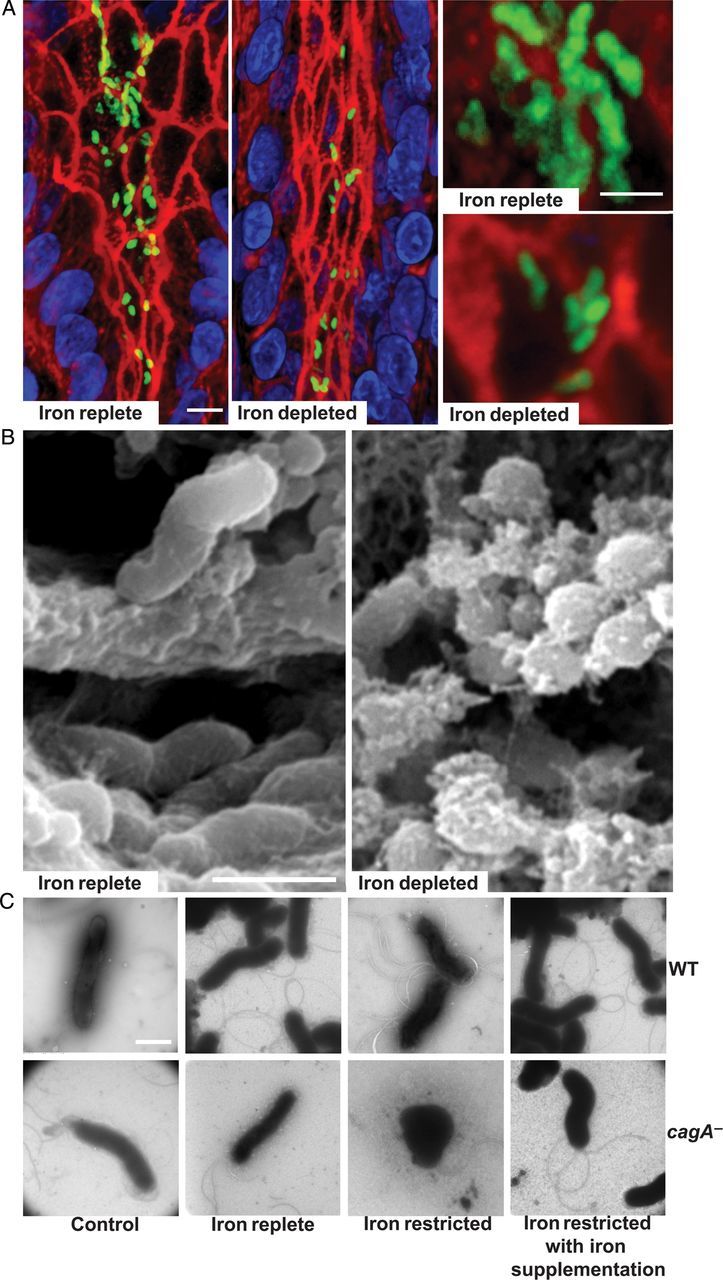
Loss of cagA alters Helicobacter pylori morphology in an iron-dependent manner. Immunofluorescence and confocal microscopy (A) and scanning electron microscopy (B) were performed on gastric tissue specimens recovered from gerbils infected with the cagA− isogenic mutant under iron-replete or iron-depleted conditions. A, H. pylori (green), actin (red), and nuclei (blue). Bars = 10 µm. C, Transmission electron microscopy was performed on wild-type H. pylori strain 7.13 (WT) and the cagA− isogenic mutant (cagA−) under control, iron-replete, iron-restricted, or iron-restricted conditions with iron supplementation. Bars = 1 µm.
H. pylori strains grown under iron-limiting conditions develop an increased capacity to assemble the cag type IV secretion system, which is accompanied by increased expression and translocation of CagA, and increased production of IL-8 by infected gastric epithelial cells [8]. Our data (Figure 1C) demonstrated that altered morphology of the cagA− mutant under conditions of iron deficiency was reversible. Therefore, we next sought to determine whether augmented cag-dependent virulence phenotypes were stable or reversible.
The parental strain 7.13 or in vivo–adapted strains harvested from gerbils maintained on iron-depleted diets were serially passaged in vitro and then cocultured with AGS gastric epithelial cells at each passage number. Compared with minimally passaged H. pylori isolates, long-term in vitro passage resulted in significantly decreased levels of CagA expression and translocation (Figure 2A and 2B). This occurred rapidly, after only 10 passages in vitro, indicating that heightened CagA expression and translocation is reversible in H. pylori strains that have been exposed to iron-deficient conditions.
Figure 2.

CagA translocation and interleukin 8 (IL-8) induction by in vivo–adapted iron-depleted strains is attenuated following long-term passage. Parental Helicobacter pylori strain 7.13 or in vivo–adapted 7.13 strains harvested from iron-depleted gerbils (N = 5) were serially passaged (1, 5, 10, 15, 20, 25, and 30 passages). At each passage, strains were cocultured with AGS for 6 hours. A, Levels of total CagA, phosphorylated CagA (as a measure of translocated CagA), and β-actin were determined. B, Protein intensities quantified using ImageJ (National Institutes of Health) are represented as fold-changes over values for the parental strain 7.13. C, IL-8 messenger RNA expression levels are represented as fold-changes over values for uninfected control cells. D, Levels of secreted IL-8 protein are represented as fold-changes over values for uninfected control cells. Mann–Whitney U t tests and 1-way analysis of variance were used to determine statistical significance. **P < .01 and ***P < .001.
Consistent with the pattern observed for CagA expression and translocation, low-passage in vivo–adapted strains isolated from gerbils maintained on iron-depleted diets induced significantly higher levels of IL-8 mRNA and protein expression in gastric epithelial cells (Figure 2C and 2D), when compared to the parental strain 7.13. However, following in vitro passage of these strains (>5 passages), increased levels of IL-8 mRNA and protein expression decreased to levels observed with the parental strain 7.13 (Figure 2C and 2D). Collectively, these data demonstrate that the hypervirulent phenotypes stimulated by iron depletion are reversible.
DISCUSSION
In the setting of iron deficiency, H. pylori accelerates the development of inflammation, dysplasia, and adenocarcinoma in an animal model of gastric cancer that closely resembles human disease [8]. This phenotype is mediated by enhanced deployment of the cag T4SS, increased CagA translocation into host cells, and increased proinflammatory immune responses. In addition to inducing pathogenic responses, CagA also facilitates H. pylori colonization through iron acquisition, indicating that CagA provides a survival advantage in vivo under iron-limiting conditions [7]. Our current results demonstrate that loss of cagA under conditions of iron depletion led to a conversion from a spiral morphology to a coccoid morphology, a phenotype that is prevented by iron supplementation. H. pylori typically exists in a helical form but can convert to a coccoid morphology during environmental stress. The functional status exerted by coccoid H. pylori, however, remains controversial. Data have shown that this morphological manifestation leads to nonviable H. pylori [12], while other results have demonstrated that coccoid forms are viable and maintain cell structure and virulence expression profiles [13, 14]. Coccoid forms of H. pylori have also been implicated in relapses of infection after antimicrobial treatment [15]. Our data have provided fresh insights into these questions as we have demonstrated that CagA, an oncoprotein, is required for normal growth and spiral morphology under iron-restricted conditions. Further, the ability of CagA to regulate bacterial fitness in the absence of host cell contact is a novel finding, thus providing new insights into how pathogenic factors may regulate microbial homeostasis.
In addition to defining the dynamics of bacterial morphology under changing iron conditions, we have now also demonstrated that H. pylori hypervirulent phenotypes mediated by iron deficiency are reversible. One potential explanation is that in vivo, there is positive selection for enhanced T4SS activity, whereas in vitro growth results in negative selection for such activity. However, we have previously demonstrated that when hypervirulent strains isolated from iron-depleted gerbils are reintroduced into iron-replete gerbils, they no longer induce increased frequency of disease [10], indicating that this is not simply due to negative selection in vitro.
Global and indiscriminate test-and-treat strategies for H. pylori have not been fully embraced, primarily because of the relatively low incidence of cancer among infected individuals and studies reporting that carriage of H. pylori is inversely related to the development of esophageal and atopic diseases [1]. These data underscore the importance of carefully identifying colonized persons who are highly predisposed to pathologic outcomes. Our earlier studies provided an important initial step in this direction by indicating that iron deficiency increases the virulence of H. pylori, thereby identifying a high-risk group of persons who may warrant therapy directed against H. pylori. Our new findings have extended these observations and indicate that hypervirulent H. pylori phenotypes stimulated by iron deficiency are reversible. Because many studies have reported that the efficacy of first-line anti–H. pylori therapy is around 80%–85%, a clinical strategy that focuses on treatment of iron deficiency among H. pylori–colonized individuals who do not respond therapy may ultimately attenuate disease. Collectively, these studies should enable clinicians to more appropriately focus diagnostic testing and eradication therapies on high-risk populations to effectively prevent the development of gastric cancer.
Notes
Financial support. This work was supported by the National Institutes of Health (grant F32CA153539 to J. M. N.; grants R01AI068009 and R01AI039657 to T. L. C.; and grants to R01CA077955, R01DK058587, P01CA116087, and P30DK058404 R. M. P.), the Department of Veterans Affairs (grant IK2BX001701 to J. A. G. and grant 2I01BX000627 to T. L. C.), the R. Robert and Sally Funderburg Research Award in Gastric Cancer (to M. R. A.), and the Morgridge Faculty Scholar Award (to M. R. A.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 2010; 10:403–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000; 287:1497–500. [DOI] [PubMed] [Google Scholar]
- 3.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003; 300:1430–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A 2008; 105:1003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pra D, Rech Franke SI, Pegas Henriques JA, Fenech M. A possible link between iron deficiency and gastrointestinal carcinogenesis. Nutr Cancer 2009; 61:415–26. [DOI] [PubMed] [Google Scholar]
- 6.Muhsen K, Cohen D. Helicobacter pylori infection and iron stores: a systematic review and meta-analysis. Helicobacter 2008; 13:323–40. [DOI] [PubMed] [Google Scholar]
- 7.Tan S, Noto JM, Romero-Gallo J, Peek RM, Jr, Amieva MR. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog 2011; 7:e1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noto JM, Gaddy JA, Lee JY, et al. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest 2013; 123:479–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pentecost M, Otto G, Theriot JA, Amieva MR. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog 2006; 2:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan S, Tompkins LS, Amieva MR. Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog 2009; 5:e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radin JN, Gaddy JA, Gonzalez-Rivera C, Loh JT, Algood HM, Cover TL. Flagellar localization of a Helicobacter pylori autotransporter protein. MBio 2013; 4:e00613–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kusters JG, Gerrits MM, Van Strijp JA, Vandenbroucke-Grauls CM. Coccoid forms of Helicobacter pylori are the morphologic manifestation of cell death. Infect Immun 1997; 65:3672–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azevedo NF, Almeida C, Cerqueira L, Dias S, Keevil CW, Vieira MJ. Coccoid form of Helicobacter pylori as a morphological manifestation of cell adaptation to the environment. Appl Environ Microbiol 2007; 73:3423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monstein HJ, Jonasson J. Differential virulence-gene mRNA expression in coccoid forms of Helicobacter pylori. Biochem Biophys Res Commun 2001; 285:530–6. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein NS. Chronic inactive gastritis and coccoid Helicobacter pylori in patients treated for gastroesophageal reflux disease or with H. pylori eradication therapy. Am J Clin Pathol 2002; 118:719–26. [DOI] [PubMed] [Google Scholar]