

Abstract
Background. Exophiala species are mostly responsible for skin infections. Invasive Exophiala dermatitidis disease is a rare and frequently fatal infection, with 42 cases reported. About half of these cases had no known risk factors. Similarly, invasive Exophiala spinifera disease is extremely rare, with only 3 cases reported, all in patients with no known immunodeficiency. Autosomal recessive CARD9 deficiency has recently been reported in otherwise healthy patients with severe fungal diseases caused by Candida species, dermatophytes, or Phialophora verrucosa.
Methods. We investigated an 8-year-old girl from a nonconsanguineous Angolan kindred, who was born in France and developed disseminated E. dermatitidis disease and a 26 year-old woman from an Iranian consaguineous kindred, who was living in Iran and developed disseminated E. spinifera disease. Both patients were otherwise healthy.
Results. We sequenced CARD9 and found both patients to be homozygous for loss-of-function mutations (R18W and E323del). The first patient had segmental uniparental disomy of chromosome 9, carrying 2 copies of the maternal CARD9 mutated allele.
Conclusions. These are the first 2 patients with inherited CARD9 deficiency and invasive Exophiala disease to be described. CARD9 deficiency should thus be considered in patients with unexplained invasive Exophiala species disease, even in the absence of other infections.
Keywords: Exophiala species, invasive fungal infection, central nervous system, osteomyelitis, autosomal recessive CARD9 deficiency, parental unidisomy
(See the editorial commentary by Lionakis and Holland on pages 1205–7.)
Exophiala dermatitidis, previously known as Wangiella dermatitidis, is an environmental black yeast [1, 2]. In Western countries, E. dermatitidis has been reported to colonize the respiratory tract of about 6% of patients with cystic fibrosis [3] and to be present in the stools of 0.5% of patients with diarrhea [4]. By contrast, invasive E. dermatitidis disease (ie, noncontiguous extracutaneous localization) is rare. Since 1959, only 42 cases have been reported worldwide, and for 23, no risk factors were identified (Table 1) [5–18]. Exophiala spinifera is rare, representing 2.7% of the Exophiala species among clinical isolates [1, 2]. Only 29 cases of E. spinifera diseases have been reported in the literature [19–24]). Only 3 patients, none of whom had any known risk factors, had invasive infection (ie, noncontiguous extra cutaneous localization) [13, 23, 24]. We describe 2 patients with isolated, unexplained invasive fungal disease caused by E. dermatitidis or E. spinifera. Because autosomal recessive (AR) CARD9 deficiency has recently been reported in 30 otherwise healthy patients from Iran, North Africa, Asia, and Canada with invasive fungal diseases [25–29], we sequenced CARD9 in the patients.
Table 1.
Demographic and Clinical Characteristics of Patients With Invasive Exophiala dermatitidis Infection Described in Previous Studies and the Patient Studied Here
| Characteristic | Age, y (Sex) | Location |
Outcome (Time From Diagnosis to Death, mo) | Reference | ||
|---|---|---|---|---|---|---|
| Brain | Liver | Other | ||||
| Patients without known risk factors | ||||||
| Country of origin | ||||||
| South Korea | 11 (F) | No | Yes | No | Death (14) | [9] |
| South Korea | 28 (M) | Yes | No | No | Death (0.5) | [10] |
| Turkey | 8 (M) | No | Yes | LN | Death (7) | [17] |
| Turkey | 24 (F) | No | Yes | LN | Cure | [11] |
| China | 3 (M) | Yes | No | No | Death (4) | [12] |
| China | 19 (F) | Yes | No | LN | Death (5) | [13] |
| China | 30 (F) | Yes | No | No | Death (0.1) | [13] |
| China | 3 (F) | Yes | No | No | Death (6) | [13] |
| China | 19 (F) | Yes | No | LN | Death (1) | [8] |
| China | 21 (F) | No | Yes | LN | Cure | [7] |
| Japan | 25 (F) | No | No | Palate, LN | Death (36) | [6] |
| Japan | 30 (F) | Yes | No | LN | Death (3) | [6] |
| Japan | 11 (M) | No | Yes | LN, dig | Death (ND) | [6] |
| Japan | 14 (F)a | No | Yes | No | Death (2) | [6] |
| Japan | 5 (F)a | Yes | No | No | Death (1) | [6] |
| Japan | NA (F) | Yes | No | No | Death (ND) | [6] |
| Japan | 26 (F) | Yes | No | No | Death (9) | [6] |
| Japan | 19 (M)a | Yes | Yes | LN | Death (2) | [6] |
| Japan | NA (M)a | Yes | Yes | NA | Death (ND) | [6] |
| Japan | 10 (M) | Yes | No | Lung | Death (11) | [6] |
| Japan | 20 (M) | Yes | No | LN | Death (NA) | [6] |
| Japan | 17 (M) | Yes | Yes | LN | Death (48) | [6] |
| Pakistan | 70 (M) | Yes | No | No | Death (0.5) | [18] |
| Patients with predisposing factors other than PID | ||||||
| Predisposing factor | ||||||
| Contaminated steroid use | 77 (F) | Yes | No | No | Death | [5] |
| Contaminated steroid use | 61 (F) | Yes | No | No | Cure | [5] |
| Contaminated steroid use | 71 (F) | Yes | No | No | ND | [5] |
| Contaminated steroid use | 65 (F) | Yes | No | No | ND | [5] |
| Contaminated steroid use | 52 (F) | Yes | No | No | ND | [5] |
| Bronchiectasis | 81 (M) | No | No | Lung | Cure | [15] |
| Lung cancer, catheter use | 58 (F) | No | No | Blood | Cure | [5] |
| CAPD, catheter use | 55 (M) | No | No | Peritoneal | Cure | [5] |
| CAPD, catheter use | 39 (M) | No | No | Peritoneal | Cure | [5] |
| ALL | 62 (F) | No | No | LN | Cure | [5] |
| Cystic fibrosis | 29 (F) | No | No | Lung | Cure | [5] |
| Cystic fibrosis | 6 (F) | No | No | Lung | Cure | [5] |
| Cystic fibrosis | 54 (F) | No | No | Lung | ND | [5] |
| AIDS, catheter use | 3 (M) | No | No | Blood | Cure | [5] |
| Parenteral nutrition, catheter use | 53 (F) | No | No | Blood | Cure | [5] |
| ALL, catheter use | 5 (M) | No | No | Blood | Cure | [5] |
| Injection drug use | 63 (M) | No | No | Endocarditis | Death | [5] |
| Solid organ transplantation | 50 (F) | No | No | Endocarditis | ND | [5] |
| Patients with PID | ||||||
| CGD | 21 (F) | Yes | No | Lung | Cure | [14] |
| CARD9 deficiency | 6 (F) | Yes | Yes | No | Relapse | This report |
Abbreviations: AIDS, acquired immunodeficiency syndrome; ALL, acute lymphoblastic leukemia; CAPD, continuous ambulatory peritoneal dialysis; CGD, chronic granulomatous disease; dig, digestive tract; F, female; LN, lymph node; M, male; mo, months; NA, not available; ND, not determined; PID, primary immunodeficiency disease.
a Multiplex family.
CASE SUMMARY
The first patient studied was an 8-year-old girl (Figure 1A) born in France to nonconsanguineous parents originating from Angola. At the age of 5 years, she developed liver and brain E. dermatitidis infection (Figure 1B and 1C). E. dermatitidis grew in cultures and was identified on the basis of its phenotype, and this identification was confirmed by internal transcribed spacer (ITS) sequencing. Her case report is detailed in the Supplementary Materials. The patient was treated by irrigation of the biliary tract with amphotericin B, a high intravenous dose of liposomal amphotericin B, and oral voriconazole. A favorable outcome was achieved, but despite this antifungal maintenance treatment, the patient developed E. dermatitidis pachymeningitis (Figure 1D and Figure 2A–D). This patient had never had any other severe infectious disease. In addition, neither her parents nor her 2 older brothers, aged 13 and 16 years, had had any severe infectious disease.
Figure 1.
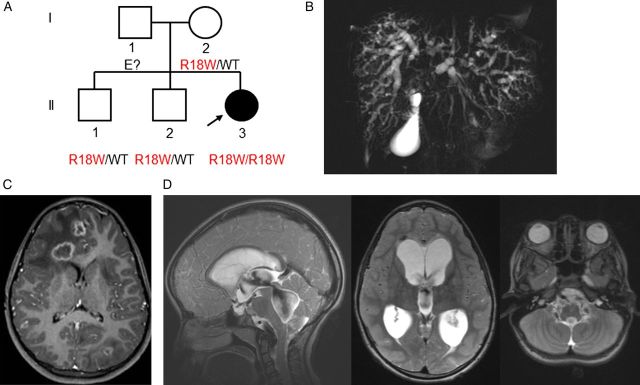
A, Pedigree of the patient with Exophiala dermatitidis infection and a CARD9 mutation. Each generation is represented by a Roman numeral, and each individual is represented by an Arabic numeral. The proband with E. dermatitidis infection is shown in black and indicated by an arrow. The CARD9 genotype is indicated below each individual. B–D, Radiological features of patient 1. B, Cholangio-magnetic resonance imaging showing a dilated and irregular biliary tree with diffuse biliary duct infiltration. C, Brain magnetic resonance imaging (MRI) showing 13 cerebral lesions (diameter, 10–16 mm). D, Brain MRI showing pachymeningitis and hydrocephalus. Abbreviation: WT, wild-type.
Figure 2.

A–D, Histological features of the meningeal biopsy specimen. A, Hematoxylin-eosin (H-E) staining showing fungi surrounded by an epithelioid and giant cell granuloma (original magnification ×200). B, Strong periodic acid-Schiff staining of the fungi within the granuloma (original magnification ×400). C, H-E staining revealing a huge infiltration of fungi without granuloma in another area of the biopsy sample (original magnification ×400). D, Grocott staining of this large area of fungi (original magnification ×400).
Identification of a Homozygous CARD9 Missense Mutation
The coding exons of CARD9 were sequenced by the Sanger method. Patient 1 displayed a homozygous c.52C > T missense mutation in exon 2 of this gene, resulting in the replacement of the arginine in position 18 with a tryptophan residue (R18W), located in the N-terminal caspase-recruitment domain (CARD). The patient's healthy mother and her 2 brothers were heterozygous for the mutation (WT/R18W; Figure 1A). The patient's father could not be tested. An Affymetrix genome-wide human SNP Array 6.0 analysis combined to a CGH array analysis (Supplementary Materials) of the patient's DNA demonstrated that the patient had a segmental uniparental disomy (UPD) of chromosome 9, resulting in the carriage of 2 copies of the maternal CARD9 allele. The segregation of the mutation was consistent with AR CARD9 deficiency and complete clinical penetrance. The mutation reported here was not found in any of the various public databases or in any of the controls sequenced (Supplementary Materials). This mutation was predicted to be probably damaging (with the highest possible score of 1), by PolyPhen 2, and as damaging, by SIFT (with a score of 0). Collectively, these data strongly suggested that the patient was homozygous for a rare, deleterious CARD9 missense mutation.
The R18W Mutant CARD9 Protein Is Produced but Has Lost Its Function
We evaluated the consequences of the mutation at the CARD9 protein level, by Western blot analysis on whole-cell extracts of HEK-293 T cells transfected with a pcDNA3.1 V5 (C-terminally tagged) plasmid with no insert or carrying the WT or R18W allele of CARD9 (pcDNA3.1 V5 CARD9 WT and pcDNA3.1 V5 CARD9 R18W). Cells transfected with the CARD9 R18W allele had slightly higher CARD9 protein levels than those transfected with the WT allele, and the protein detected was of similar molecular weight (Figure 3A). Flow cytometry analysis of CARD9 protein in monocytes (Supplementary Figure A) and monocyte-derived macrophages (MDMs; Figure 3B) from the patient showed that the levels of this protein were comparable to those in monocytes and MDMs from a healthy control. We evaluated the functional consequence of this mutation by studying interleukin 6 (IL-6) production by whole-blood cells after 24 hours of stimulation with zymosan (an agonist of at least Dectin-1 and Toll-like receptor 2), heat-killed Saccharomyces cerevisiae, Candida albicans, E. dermatitidis, and Staphylococcus aureus, vesicular stomatitis virus (VSV), lipopolysaccharide (LPS; a TLR4 agonist), and PMA plus ionomycin. The cells from patient 1 produced much smaller amounts of IL-6 after stimulation with S. cerevisiae, E. dermatitidis, and, to a lesser extent, C. albicans, than cells from 6 healthy controls or the heterozygous mother tested in parallel (Figure 3C). The responses to zymosan, LPS, S. aureus, VSV, and PMA plus ionomycin were normal. We then assessed TNF-α production by monocyte-derived dendritic cells (MDDCs) obtained from the patient and 7 healthy controls and stimulated for 24 hours with curdlan (a dectin-1 agonist) and the same agonists used in the whole-blood assay (Figure 3D). The patient displayed a strong impairment of TNF-α production after stimulation with all fungal ligands used (curdlan, zymosan, heat-killed S. cerevisiae, C. albicans, and E. dermatitidis), whereas TNF-α production was similar to that in healthy controls after stimulation with S. aureus and LPS. In addition, 293 HEK cells transfected with the R18W CARD9 allele showed a strongly impaired NF-κB transcriptional activity, as assessed by NF-κB–luciferase reporter assays, compared with cells transfected with the WT CARD9 allele, either at the basal level or after stimulation with curdlan or E. dermatitidis (Figure 3E). Finally, we evaluated the proportion of ex vivo interleukin 17A (IL-17A)–producing T cells by flow cytometry, as low proportions of IL-17 T cells have been reported in some CARD9-deficient patients [25–27, 29]. No differences were observed between the patient and the 7 healthy controls tested in parallel (Supplementary Figure B). Furthermore, IL-17A production by whole-blood cells after 24 hours of stimulation with PMA and ionomycin, as measured by ELISA, was similar in the patient and the 2 healthy controls tested in parallel (Supplementary Figure B). We therefore conclude that the homozygous R18W mutation led to the production of normal amounts of a loss-of-function CARD9 protein, leading to the impairment of proinflammatory cytokine production by whole-blood cells and MDDCs after stimulation with various fungal ligands and impairment of NF-κB transcriptional activity in transfected HEK cells, whereas IL-17 T-cell production was normal.
Figure 3.

Impact of the CARD9 R18W mutation on protein level and function. A, Immunoblot analysis of CARD9 in whole-cell extracts of HEK-293T cells cotransfected with pcDNA3.1 V5 (C-terminally tagged), either empty or carrying the wild-type (WT) or mutant (R18W) CARD9 allele, together with a CFP plasmid, as a transfection control. Antibodies against CARD9, V5, CFP, and GAPDH (as a loading control) were used. B, Flow cytometry analysis of CARD9 in monocyte-derived macrophages from a control (left panel) or the patient (right panel). C, Interleukin 6 (IL-6) production by whole-blood cells, as measured by enzyme-linked immunosorbent assay (ELISA), after 24 hours of stimulation with zymosan (5 µg/mL), heat-killed Saccharomyces cerevisiae (106 particles/mL), heat-killed Candida albicans (106 particles/mL), heat-killed Exophiala dermatitidis (106 particles/mL), heat-killed Staphylococcus aureus (5 × 108 particles/mL), vesicular stomatitis virus (VSV), lipopolysaccharide (LPS; 100 ng/mL), and PMA plus ionomycin for 6 controls, the CARD9-heterozygous (R18W/WT) mother, and the CARD9-homozygous (R18W/R18W) patient. Results are expressed as mean ± standard error of the mean (SEM) of 3 independent experiments. D, Tumor necrosis factor α (TNF-α) production by monocyte-derived dendritic cells, measured by ELISA, after 24 hours of stimulation with curdlan (25 µg/mL), zymosan (25 µg/mL), heat-killed S. cerevisiae (106 particles/mL), heat-killed C. albicans (106 particles/mL), heat-killed E. dermatitidis (107 particles/mL and 106 particles/mL), heat-killed S. aureus (5 × 108 particles/mL), and LPS (100 ng/mL) for 7 controls and the CARD9-homozygous (R18W/R18W) patient. Results are expressed as mean ± SEM of 4 independent experiments. E, The CARD9 R18W allele impairs downstream NF-κB activation. NF-κB–luciferase assay in 293 HEK cells transfected with NF-κB–luciferase and pRL-SV40 vectors alone (A); with DECTIN1, SYK, and BCL10 constructs (B); with DECTIN-1, SYK, BCL10, and CARD9 WT constructs (C); and with DECTIN-1, SYK, BCL10, and CARD9 R18W constructs (D). Cells were stimulated or not stimulated with 25 µg/mL curdlan or 107 particles/mL E. dermatitidis. Results are representative of 2 independent experiments performed and are expressed as mean ± SEM of the ratio between Renilla luciferase and firefly control luciferase activities adjusted to 1. *P ≤ .05, **P ≤ .01, and ***P ≤ .001 by the Student t test. Abbreviation: RLU, relative light units.
Autosomal Recessive CARD9 Deficiency in Another Patient With Invasive E. spinifera Infection
We recently studied a 26-year-old woman (Figure 4A) from an Iranian consanguineous kindred living in Iran who presented recurrent fungal infections from the age of 18 years onward, with E. spinifera subcutaneous (Figure 4B), bone (Figure 4C), and lung (Figure 4D) infection. E. spinifera grew in cultures and was identified on the basis of its phenotype, and this identification was confirmed by ITS sequencing. The case report is detailed in the Supplementary Materials. The patient's parents and siblings were all healthy and had never had a severe infectious fungal disease. The sequencing of CARD9 in the patient's DNA showed a homozygous in-frame deletion, c.GAG967-969del or p.E323del, located in the coiled-coil domain of CARD9. This deletion was not found in any of the various public databases or in any of the controls sequenced (Supplementary Materials). Collectively, these data strongly suggest that this patient is homozygous for a rare, deleterious CARD9 deletion mutation. Parental DNA testing was possible only for the father, who was found to be heterozygous for the mutation.
Figure 4.

A, Pedigree of the patient with Exophiala spinifera infection and a CARD9 mutation. Each generation is represented by a Roman numeral, and each individual is represented by an Arabic numeral. The proband with E. spinifera infection is shown in black and indicated by an arrow. The CARD9 genotype is indicated below the individuals sequenced. B–D, Clinical and radiological features of the patient. B, Subcutaneous skin infection. C, Hyperfixation revealing osteomyelitis on scintigraphy. D, Computed tomography of the chest. Abbreviation: WT, wild type.
DISCUSSION
We identified a homozygous R18W CARD9 mutation in a child with cholangitis and central nervous system (CNS) infection caused by E. dermatitidis. We showed that this was a loss-of-function mutation and that the child had functional CARD9 deficiency. The mutation was homozygous because of segmental UPD of the maternal chromosome 9q, which harbored the mutant R18W CARD9 allele. The segmental UPD accounts for the autosomal recessive form of primary immunodeficiency disease (PID) in this patient, with the CARD9 germline mutation being present in a heterozygous state in the mother. UPD is defined as the inheritance of a pair of duplicated chromosomes from a single parent, and it is associated with 2 main types of developmental risk: the occurrence of imprinting disorders and the inheritance of a recessive trait [30]. Some autosomal recessive PIDs due to UPD have already been reported, such as interferon γR1 deficiency (Mendelian susceptibility to mycobacterial disease), perforin deficiency (familial hemophagocytic lymphohistiocytosis type 2), lysosomal trafficking regulator deficiency (Chediak-Higashi syndrome), or T-lymphocyte–specific protein tyrosine kinase deficiency [31–34]. Patients with these conditions often display other clinical signs unrelated to the mutated PID-causing gene, but due to the extent of the UPD [31]. UPD of chromosome 9 region 9q34 has already been reported for the homozygous sea urchin retroposon family 1 mutation responsible for Leigh syndrome, a mitochondrial subacute necrotizing encephalomyelopathy [35]. Other homozygous variants of the segment of chromosome 9 that the patient inherited from her mother may have clinical impacts that are not immediately evident.
Among the 42 cases reported with invasive E. dermatitidis disease, a risk factor was identified for 19, while the other 23 cases remained unexplained (Table 1) [5–13, 16–18]. These 23 patients belonged to 21 families, all from Asia. Twelve patients came from 2 multiplex families from Japan, 3 patients were from South Korea, 5 were from China, 1 was from Pakistan, and 2 were from Turkey [10, 12, 17, 18, 36–40]. Consanguinity was not reported in these families. The clinical outcome was poor, particularly in patients with no known risk factors. In these patients, the disease typically affected the brain and/or liver (Table 1) [5–13, 16, 17], with onset at a median age of 19 years [range, 3–70 years]. For the patients who are known to have an immunodeficiency disease or who are receiving immunosuppressive treatment, brain involvement was only reported in the patient with chronic granulomatous disease [14] and in 5 patients with meningitis secondary to the intrathecal injection of contaminated steroids [41]. E. dermatitidis has been shown to be neurotropic in a mouse model [42]. However, the absence of brain involvement in most of the patients with immunodeficiency disease and E. dermatitidis infection suggests that predisposing genetic factors are also required for E. dermatitidis infection of the CNS in humans. None of the patients with a known immunodeficiency disease had liver involvement. Mortality in the group of patients without known risk factors was much higher than that in patients with known risk factors (91% vs 11%).
E. spinifera infections are also very rare. To date, 29 cases of E. spinifera infections have been reported in the literature, 3 corresponding to chromoblastomycosis and 26 to phaeohyphomycosis [19–22]. The diseases were cutaneous for all patients, with frequent subcutaneous and bone extension. Among the 26 patients with phaeohyphomycosis, only 3 without known immunodeficiency disease had invasive infections with lymph node involvement or positive blood culture results [13, 23, 24]. E. spinifera infections reported in children without any known risk factors usually started as subcutaneous abscesses and frequently disseminated, with a poor outcome [19–24]. In contrast, the 23 E. spinifera infections reported in adults were usually localized and occurred in patients with known risk factors, such as systemic lupus erythematous, corticosteroid treatments, solid organ transplant receipt, or cancer [19–22]. Patients with E. spinifera–related phaeohyphomycosis were from China (n = 5), Brazil (n = 2), India (n = 8), El Salvador (n = 1), Pakistan (n = 1), the United States (n = 5), Argentina (n = 1), Japan (n = 1), and France (n = 2). The 3 patients with invasive E. spinifera disease were a 9-year-old boy from China who developed fungemia associated with subcutaneous and bone infections, leading to his death [13], and a 12-year-old girl and 41-year-old woman from India and Argentina, respectively, who had lymph node and subcutaneous infections [23, 24].
The similarity of clinical presentation between the 2 patients reported here and the patients with idiopathic E. dermatitidis or E. spinifera disease reported in the literature suggests that some of these patients may have had CARD9 deficiency. Furthermore, the identification of multiplex families is also consistent with this hypothesis [6]. The identification of CARD9 deficiency in an Angolan patient homozygous for the CARD9 R18W allele and with isolated invasive E. dermatitidis disease and in an Iranian patient homozygous for the CARD9 E323del mutation and with recurrent invasive E. spinifera disease broadens the spectrum of invasive fungal diseases known to be associated with CARD9 deficiency. The previously reported patients with other CARD9 mutations displayed other invasive fungal infections. Three patients from a consanguineous Iranian kindred homozygous for the Q295X CARD9 allele [26], 1 Korean girl compound heterozygous for the CARD9 G72S and R373S alleles [27], and 1 French-Canadian male patient homozygous for the CARD9 Y91H allele [28] had Candida species infections of the CNS (all had meningitis/meningoencephalitis and 2, the Korean girl and an Iranian girl, also had brain abscesses). In addition, 6 members of the Iranian kindred were reported to have chronic mucocutaneous candidiasis, and 3 were found to have superficial dermatophytosis [26]. Two related Moroccan patients homozygous for the CARD9 R101C allele and 15 North African patients (11 from 5 Algerian kindreds and 4 patients from 2 Tunisian kindreds) homozygous for the CARD9 Q289X allele developed deep dermatophytosis [25]. Finally, 4 unrelated Chinese patients, 1 with compound heterozygous mutations (c.191-192insTGCT encoding L64fsX59 and c.472C > T encoding Q158X) and 3 with the same homozygous frameshift mutation (c.819-820insG encoding D274fsX60) were reported to have subcutaneous phaeohyphomycosis caused by Phialophora verrucosa [29].
The identification and characterization of 32 patients from 17 families in 8 countries and with 11 different morbid alleles indicate that CARD9 deficiency is associated with invasive fungal infection due to at least 4 genera (Candida species, Exophiala species, P. verrucosa, Trichophyton species), with a tropism for the CNS [25–29] (this report). CARD9 deficiency may thus be associated with a specific predisposition to fungal infections of the CNS. As an initial hypothesis, we suggest that CARD9-deficient patients may display an impairment of fungal elimination by monocytes, macrophages, and/or microglial cells at the blood-brain barrier [43]. On the other hand, CARD9-deficient cells have been shown to require fungal opsonization by serum for the efficient killing of fungi [27]. The limited access of plasma proteins to the CNS results in fungi not being opsonized, potentially accounting for the impairment of fungal killing specifically in the CNS of CARD9-deficient patients [27]. No other severe infections have been reported in these patients, with a particular absence of reported infections due to Aspergillus species, bacteria, and parasites. It is also intriguing that the CARD9-deficient patients identified to date, including in this report, have each displayed a single type of invasive fungal disease [25–29].
The diverse and expanding clinical presentations of CARD9 deficiency suggest that this protein may control multiple molecular pathways in multiple cell types involved in immunity to various fungi. Interestingly, like patients homozygous for the R101C or Q289X allele [25], the patient homozygous for the R18W CARD9 allele displayed impaired production of both IL-6 by whole-blood cells and TNF-α by MDDCs in response to stimulation with fungal ligands (heat-killed C. albicans and S. cerevisiae, curdlan, and zymosan). In addition, whole-blood cells and MDDCs also displayed impaired responses to E. dermatitidis, potentially accounting for the susceptibility of the patient. Similarly, macrophages and dendritic cells from patients with subcutaneous phaeohyphomycosis and the L64fsX59/Q158X or the D274fsX60 CARD9 alleles displayed impaired IL-6, TNF-α, interleukin 1β (IL-1β), and interleukin 23 production in response to P. verrucosa stimulation [29], and peripheral blood mononuclear cells from the patient with Candida dubliniensis meningoencephalitis and the G72S/R373P CARD9 alleles showed impaired IL-6 and IL-1β production in response to C. albicans. All these CARD9 alleles are deleterious for at least some leukocyte and dendritic cell responses. In addition, there seems to be a correlation between the in vitro defect and the clinical presentation. However, we hypothesize that CARD9 mutations confer a predisposition to infections caused by dermatophytes, Candida species, P. verrucosa, or Exophiala species, with the phenotype depending on the nature and amount of the infecting fungus.
The R18W mutation affects the CARD domain of the protein, which is known to create heterodimers with the CARD domain of BcL10. It may therefore impair this interaction, decreasing downstream NF-κB activation and target gene transcription [44]. In particular, patients with CARD9 deficiency have been reported to have low proportions of IL-17 T cells, probably resulting from impaired IL-17–inducing cytokine production [25–27, 29]. However, patient 1 (patient 2 could not be tested) had a normal proportion of these cells, consistent with her lack of mucocutaneous candidiasis [45–47]. Similar observations have recently been reported for another patient homozygous for the Y91H CARD9 allele with recurrent C. albicans meningoencephalitis [28]. These findings suggest that poor IL-17 T-cell development is not an intrinsic feature, or at least not a general feature, of human CARD9 deficiency. Finally, there seems to be no correlation between the type of mutation and the type of fungal disease. Indeed, we recently identified a patient with CNS candidiasis bearing the homozygous Q289X CARD9 mutation (Lanternier et al, unpublished data), an allele previously identified in patients with deep dermatophytosis [25]. Our findings indicate that CARD9 deficiency should be considered as a possible diagnosis in children and young adults with disseminated, isolated, unexplained Exophiala disease and in cases of fungal infection of the CNS. More generally, this study suggests that idiopathic invasive fungal infections should lead to a search for underlying inborn errors of immunity [48, 49].
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the patients and their families, for participating in this study; Dr Daniele Pariente (Pediatric Radiology, Bicêtre Hospital), for performing biliary drainage and liver biopsy; Sylvain Poirée and Daniele Pariente, for radiological interpretation; Prof Catherine Guettier, for the pathological study of the liver; Dea Garcia-Hermoso (Institut Pasteur, National Reference Center for Invasive Mycoses and Antifungals, Molecular Mycology Unit), for identification and susceptibility testing of the E. dermatitidis isolate; members of both branches of the Laboratory of Human Genetics of Infectious Diseases, for helpful discussions; Malik Bensifi; and Yuval Itan.
Financial support. This work was supported by the ANR (grant GENCMCD 11-BSV3-005-01 to A. P.); the Jeffrey Modell Foundation (grant to A. P.); the French government's Investissement d'Avenir program (ANR-10-LABX-62-IBEID); the Clinical and Translational Science Award Program, National Center for Advancing Translational Sciences, National Institutes of Health (grant UL1TR000043); Rockefeller University, including the St. Giles Foundation; INSERM; and Paris Descartes University.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Sudhadham M, Prakitsin S, Sivichai S, et al. The neurotropic black yeast Exophiala dermatitidis has a possible origin in the tropical rain forest. Stud Mycol 2008; 61:145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeng JS, Sutton DA, Fothergill AW, Rinaldi MG, Harrak MJ, de Hoog GS. Spectrum of clinically relevant Exophiala species in the United States. J Clin Microbiol 2007; 45:3713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horre R, Schaal KP, Siekmeier R, Sterzik B, de Hoog GS, Schnitzler N. Isolation of fungi, especially Exophiala dermatitidis, in patients suffering from cystic fibrosis. A prospective study. Respiration 2004; 71:360–6. [DOI] [PubMed] [Google Scholar]
- 4.de Hoog GS, Matos T, Sudhadham M, Luijsterburg KF, Haase G. Intestinal prevalence of the neurotropic black yeast Exophiala (Wangiella) dermatitidis in healthy and impaired individuals. Mycoses 2005; 48:142–5. [DOI] [PubMed] [Google Scholar]
- 5.Patel AK, Patel KK, Darji P, Singh R, Shivaprakash MR, Chakrabarti A. Exophiala dermatitidis endocarditis on native aortic valve in a postrenal transplant patient and review of literature on E. dermatitidis infections. Mycoses 2013; 56:365–72. [DOI] [PubMed] [Google Scholar]
- 6.Hiruma M, Kawada A, Ohata H, et al. Systemic phaeohyphomycosis caused by Exophiala dermatitidis. Mycoses 1993; 36:1–7. [DOI] [PubMed] [Google Scholar]
- 7.Jiang Y, Zhang F. Systemic phaeohyphomycosis caused by Wangiella dermatitidis: a case report. J Clin Dermatol 1994; 20:137–9. [Google Scholar]
- 8.Bai Y, Zhang Y. Chromomycosis: one case report. Chin J Dermatol 1964; 10:172–4. [Google Scholar]
- 9.Hong KH, Kim JW, Jang SJ, Yu E, Kim EC. Liver cirrhosis caused by Exophiala dermatitidis. J Med Microbiol 2009; 58:674–7. [DOI] [PubMed] [Google Scholar]
- 10.Chang CL, Kim DS, Park DJ, Kim HJ, Lee CH, Shin JH. Acute cerebral phaeohyphomycosis due to Wangiella dermatitidis accompanied by cerebrospinal fluid eosinophilia. J Clin Microbiol 2000; 38:1965–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oztas E, Odemis B, Kekilli M, et al. Systemic phaeohyphomycosis resembling primary sclerosing cholangitis caused by Exophiala dermatitidis. J Med Microbiol 2009; 58:1243–6. [DOI] [PubMed] [Google Scholar]
- 12.Chang X, Li R, Yu J, Bao X, Qin J. Phaeohyphomycosis of the central nervous system caused by Exophiala dermatitidis in a 3-year-old immunocompetent host. J Child Neurol 2009; 24:342–5. [DOI] [PubMed] [Google Scholar]
- 13.Li DM, Li RY, de Hoog GS, Sudhadham M, Wang DL. Fatal Exophiala infections in China, with a report of seven cases. Mycoses 2010; 54:e136–42. [DOI] [PubMed] [Google Scholar]
- 14.Kenney RT, Kwon-Chung KJ, Waytes AT, et al. Successful treatment of systemic Exophiala dermatitidis infection in a patient with chronic granulomatous disease. Clin Infect Dis 1992; 14:235–42. [DOI] [PubMed] [Google Scholar]
- 15.Ozawa Y, Suda T, Kaida Y, et al. A case of bronchial infection of Wangiella dermatitidis. Nihon Kokyuki Gakkai Zasshi 2007; 45:907–11. [PubMed] [Google Scholar]
- 16.Urabe H, Yasumoto K, Nakashima K. A case of chromoblastomycosis: probable involvement of central nervous system. Jap J Dermatol Urol 1967; 29:1012–21. [Google Scholar]
- 17.Alabaz D, Kibar F, Arikan S, et al. Systemic phaeohyphomycosis due to Exophiala (Wangiella) in an immunocompetent child. Med Mycol 2009; 47:653–7. [DOI] [PubMed] [Google Scholar]
- 18.Ajanee N, Alam M, Holmberg K, Khan J. Brain abscess caused by Wangiella dermatitidis: case report. Clin Infect Dis 1996; 23:197–8. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, She X, Lv G, et al. Cutaneous and mucosal phaeohyphomycosis caused by Exophiala spinifera in a pregnant patient: case report and literature review. Mycopathologia 2013; 175:331–8. [DOI] [PubMed] [Google Scholar]
- 20.Harris JE, Sutton DA, Rubin A, Wickes B, De Hoog GS, Kovarik C. Exophiala spinifera as a cause of cutaneous phaeohyphomycosis: case study and review of the literature. Med Mycol 2009; 47:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daboit TC, Duquia RP, Magagnin CM, et al. A case of Exophiala spinifera infection in Southern Brazil: Molecular identification and antifungal susceptibility. Med Mycol Case Rep 2012; 1:72–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baubion E, Desbois N, Durox H, et al. Ulcerated nodular lesions on extremities of patient undergoing treatment for glioblastoma on French Caribbean island of Martinique. Med Trop (Mars) 2008; 68:537–40. [PubMed] [Google Scholar]
- 23.Negroni R, Helou SH, Petri N, Robles AM, Arechavala A, Bianchi MH. Case study: posaconazole treatment of disseminated phaeohyphomycosis due to Exophiala spinifera. Clin Infect Dis 2004; 38:e15–20. [DOI] [PubMed] [Google Scholar]
- 24.Rajendran C, Khaitan BK, Mittal R, Ramam M, Bhardwaj M, Datta KK. Phaeohyphomycosis caused by Exophiala spinifera in India. Med Mycol 2003; 41:437–41. [DOI] [PubMed] [Google Scholar]
- 25.Lanternier F, Pathan S, Vincent Q, et al. Human deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med 2013; 369:1704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glocker EO, Hennigs A, Nabavi M, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 2009; 361:1727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drewniak A, Gazendam RP, Tool AT, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 2013; 121:2385–92. [DOI] [PubMed] [Google Scholar]
- 28.Gavino C, Cotter A, Lichtenstein D, et al. CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy [manuscript published online ahead of print 9 May 2014]. Clin Infect Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Wang W, Lin Z, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J Allergy Clin Immunol 2014; 133:905–8 e3. [DOI] [PubMed] [Google Scholar]
- 30.Kotzot D. Complex and segmental uniparental disomy (UPD): review and lessons from rare chromosomal complements. J Med Genet 2001; 38:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prando C, Boisson-Dupuis S, Grant AV, et al. Paternal uniparental isodisomy of chromosome 6 causing a complex syndrome including complete IFN-gamma receptor 1 deficiency. Am J Med Genet A 2010; 152A:622–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Jasmi F, Abdelhaleem M, Stockley T, Lee KS, Clarke JT. Novel mutation of the perforin gene and maternal uniparental disomy 10 in a patient with familial hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2008; 30:621–4. [DOI] [PubMed] [Google Scholar]
- 33.Dufourcq-Lagelouse R, Lambert N, Duval M, et al. Chediak-Higashi syndrome associated with maternal uniparental isodisomy of chromosome 1. Eur J Hum Genet 1999; 7:633–7. [DOI] [PubMed] [Google Scholar]
- 34.Hauck F, Randriamampita C, Martin E, et al. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol 2012; 130:1144–52 e11. [DOI] [PubMed] [Google Scholar]
- 35.van Riesen AK, Antonicka H, Ohlenbusch A, Shoubridge EA, Wilichowski EK. Maternal segmental disomy in Leigh syndrome with cytochrome c oxidase deficiency caused by homozygous SURF1 mutation. Neuropediatrics 2006; 37:88–94. [DOI] [PubMed] [Google Scholar]
- 36.Brandt ME, Warnock DW. Epidemiology, clinical manifestations, and therapy of infections caused by dematiaceous fungi. J Chemother 2003; 15(suppl 2):36–47. [DOI] [PubMed] [Google Scholar]
- 37.Silveira F, Nucci M. Emergence of black moulds in fungal disease: epidemiology and therapy. Curr Opin Infect Dis 2001; 14:679–84. [DOI] [PubMed] [Google Scholar]
- 38.Revankar SG, Sutton DA, Rinaldi MG. Primary central nervous system phaeohyphomycosis: a review of 101 cases. Clin Infect Dis 2004; 38:206–16. [DOI] [PubMed] [Google Scholar]
- 39.Kantarcioglu AS, de Hoog GS. Infections of the central nervous system by melanized fungi: a review of cases presented between 1999 and 2004. Mycoses 2004; 47:4–13. [DOI] [PubMed] [Google Scholar]
- 40.Li DM, de Hoog GS. Cerebral phaeohyphomycosis--a cure at what lengths? Lancet Infect Dis 2009; 9:376–83. [DOI] [PubMed] [Google Scholar]
- 41.Exophiala infection from contaminated injectable steroids prepared by a compounding pharmacy--United States, July-November 2002. MMWR Morb Mortal Wkly Rep 2002; 51:1109–12. [PubMed] [Google Scholar]
- 42.Calvo E, Rodriguez MM, Marine M, Mayayo E, Pastor FJ, Guarro J. Comparative virulence of three species of Exophiala in mice. Med Mycol 2010; 48:853–7. [DOI] [PubMed] [Google Scholar]
- 43.Vinh DC. Insights into human antifungal immunity from primary immunodeficiencies. Lancet Infect Dis 2011; 11:780–92. [DOI] [PubMed] [Google Scholar]
- 44.Hara H, Ishihara C, Takeuchi A, et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol 2007; 8:619–29. [DOI] [PubMed] [Google Scholar]
- 45.Puel A, Picard C, Cypowyj S, Lilic D, Abel L, Casanova JL. Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr Opin Immunol 2010; 22:467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011; 332:65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu L, Okada S, Kong XF, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011; 208:1635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann N Y Acad Sci 2010; 1214:18–33. [DOI] [PubMed] [Google Scholar]
- 49.Casanova JL, Abel L. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genomics Hum Genet 2013; 14:215–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.