

Abstract
Objective. SLE is an autoimmune disease characterized by autoantibody generation, organ damage and an increased risk of cardiovascular disease. Generally considered an anti-inflammatory cytokine, IL-10 is increased in SLE and correlates with poor cardiovascular outcomes in the general population. The aim of this study was to explore the putative role of IL-10 in modulating endothelial function in SLE by examining the effects of this cytokine on endothelial progenitor cell/circulating angiogenic cell (EPC/CAC) differentiation.
Methods. Human and murine control and lupus EPCs/CACs were differentiated into mature endothelial cells (ECs) in the presence or absence of graded concentrations of recombinant IL-10 with or without recombinant IFN-α or a neutralizing antibody to IL-10. IL-10-deficient mice were examined to assess the role of this cytokine in type I IFN-mediated inhibition of EC differentiation and neo-angiogenesis using an in vivo Matrigel plug assay. Serum IL-10 concentrations were measured via ELISA.
Results. IL-10 hampers EC differentiation in a dose-dependent manner. In murine EPC cultures, IL-10 is required to observe the inhibitory effects of type I IFNs on EPC function and neo-angiogenesis. In human SLE EPC/CAC cultures, neutralization of IL-10 significantly improved the differentiation of EPCs, and IL-10 enhanced type I IFN-mediated EPC/CAC dysfunction. The presence of IL-10 in serum inversely correlated with EPC/CAC function in SLE but not in control cells.
Conclusion. IL-10 interferes with endothelial differentiation and may enhance the effects of type I IFN on vascular repair in SLE. IL-10 may be a relevant target for improving cardiovascular risk in SLE.
Keywords: IL-10, cardiovascular, lupus, endothelial progenitor, interferon α
Introduction
SLE is a multisystem disorder characterized by immune complex organ damage and a bimodal mortality secondary to an up to 50-fold increased risk of cardiovascular disease (CVD) [1]. In SLE, increased vascular damage and poor vascular repair may lead to endothelial dysfunction and promotion of plaque development. Previously we demonstrated that crucial cellular subsets for vascular repair are abnormal in SLE patients [2, 3] and in murine models of lupus [4–6]. These cells, bone marrow-derived endothelial progenitor cells (EPCs) and myeloid circulating angiogenic cells (CACs), are considered essential for vascular repair and maintenance of a healthy endothelium [3, 7]. Importantly, the health and function of EPCs and CACs correlate with cardiovascular (CV) events and outcomes [8, 9]. EPC/CAC numbers and function are decreased in human and murine SLE. In particular, the capacity of EPCs/CACs for differentiation into mature ECs is significantly impaired, and we proposed that this plays a role in the development of premature CVD in this disease [4]. In lupus mice there is impaired neo-angiogenesis and in human lupus tissue we have identified evidence of vascular rarefaction implicating abnormal vascular repair in the aetiology of premature vascular damage in this disease [10, 11].
The dysfunction of SLE EPC/CACs is likely mediated by several lupus-associated factors. Exposure to IFN-α or other type I IFNs, a cytokine family considered crucial in the pathogenesis of SLE, induces EPC/CAC dysfunction and impaired neo-angiogenesis in vitro and in vivo. Further, blockade of type I IFN signalling restores EPC/CAC differentiation of SLE patients and neo-angiogenesis in lupus-prone mice [4]. The mechanisms by which type I IFNs may influence EPC/CAC function include down-regulation of pro-angiogenic factors such as VEGF and IL-1β and up-regulation of IL-18 and its activator caspase-1 [6]. Furthermore, blockade of caspase-1 activation of IL-18 improves EPC/CAC differentiation in murine and human systems, suggesting that inflammasome activation is relevant in EPC/CAC dysfunction in SLE [6, 12].
IL-10 has received attention as a genetic risk factor for systemic lupus [13], is elevated in this disease and increases during periods of active nephritis [14]. Type I IFNs have been reported to induce IL-10 secretion [15], and the levels of IL-10 and IL-18 rise in parallel in SLE patients [14]. While IL-10 can play a protective role in atherosclerotic plaque formation in mice [16], levels of this cytokine predict death or recurrent CV events in patients with acute coronary syndrome [17]. However, a potential role for IL-10 in modulating CV risk in SLE has not previously been proposed.
We examined the role of IL-10 in influencing EPC/CAC function and the extent to which this is regulated by type I IFNs. We demonstrate that IL-10 serves as an intermediary of the deleterious effects of type I IFNs in murine systems but is not required in human cultures for IFN-α effects. Importantly, however, IL-10 enhances the effects of type I IFN on SLE EPC/CAC dysfunction and serum concentrations inversely correlate with EPC/CAC function in human SLE samples. Further, chronic inhibition of IL-10 improves lupus EPC/CAC differentiation, suggesting that this cytokine has negative consequences for vascular repair in this disease.
Methods
Mice
Ten-week-old wild-type Balb/c and IL-10−/− on a Balb/c background mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Mouse protocols were approved by the University of Michigan’s Committee on the Use and Care of Animals. Animals were housed in a specific pathogen-free facility at the University of Michigan.
Patient selection
This study was approved by the University of Michigan institutional review board. Subjects gave informed consent in accordance with the Declaration of Helsinki. Peripheral blood was obtained from patients enrolled in the University of Michigan outpatient rheumatology clinic who met the revised criteria for SLE according to the ACR. Healthy controls were recruited by advertisement. The SLEDAI was used to assess lupus disease activity. Patient and control demographics and clinical variables for EPC/CAC studies are reported in Table 1. Patients with anti-aPLs were excluded.
Table 1.
Demographic and clinical characteristics of healthy controls and SLE patients
| Variable | Controls (n = 21) | SLE (n = 37) | P- value |
|---|---|---|---|
| Females, % | 76.2 | 94.6 | 0.039 |
| Age, mean (s.e.m.), years | 32 (2.2) | 43 (2.1) | 0.002 |
| Disease activity by SLEDAI, mean (s.e.m.) | — | 4.6 (0.4) | |
| SLEDAI <2, % | — | 11.1 | |
| SLEDAI ≥2, % | — | 88.9 | |
| Medication, % | |||
| Antimalarials | — | 83.8 | |
| MTX | — | 5.4 | |
| AZA | — | 8.1 | |
| MMF | — | 29.7 | |
| CYC | — | 0 | |
| LEF | — | 0 | |
| Dapsone | — | 2.7 | |
| Prednisone | |||
| None | — | 27 | |
| <10 mg daily | — | 40.5 | |
| ≥10 mg daily | — | 32.4 | |
| Past or current LN, % | — | 32.4 | |
| Positive aPL, % | — | 0 |
Quantification of EPC/CAC differentiation
For human studies, differentiation of peripheral blood EPCs/CACs into mature ECs was assessed as previously described [4, 6]. The absolute numbers of EPCs in SLE patients have been shown to be both elevated and decreased depending on the study and the definition of markers for EPC populations [4, 18, 19]. Differentiation of ECs from peripheral blood mononuclear cell (PBMC) cultures is one of the primary ways to assess EPC/CAC function in vitro. All patients and controls were cultured individually and in triplicate for each treatment. Briefly, lupus or control PBMCs were isolated via density centrifugation on Ficoll and cultured in pro-angiogenic media (EGM Bulletkit, Lonza, Allendale, NJ, USA) supplemented with 20% heat-inactivated fetal bovine serum (FBS) on 48-well fibronectin-coated plates (BD Biosciences, Franklin Lakes, NJ, USA). Cells were then treated with LEAF-purified anti-human IL-10 antibody (BioLegend, San Diego, CA, USA) or mouse IgG2a κ isotype as a control (BD Biosciences Pharmingen, San Jose, CA, USA) at a concentration of 1 µg/ml for 30 min, at which time recombinant human IFN-α (Merck, Whitehouse Station, NJ, USA) was added to appropriate wells at a concentration of 1000 U/ml. Media were changed every 3 days and on day 14 the cells were incubated with 1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (diI) acetylated low-density lipoprotein (ac-LDL; Biomedical Technologies, Stoughton, MA, USA) and FITC–Ulex europaeus agglutinin 1 (UEA-1; Vector Labs, Burlingame, CA, USA). To assess expression of endothelial markers and cellular morphology, cells were analysed by fluorescent microscopy using an Olympus IX70 inverted microscope (Olympus, Center Valley, PA, USA) at the University of Michigan Center for Live Cell Imaging. Images of cells were acquired at room temperature at 100× total magnification (numeric aperture 0.3) in Ringer’s buffer. Three random fields of view were acquired for each triplicate well and images were analysed using the CellC program (http://www.cs.tut.fi/sgn/csb/cellc/) to quantify mature ECs, which were considered to be those that co-express UEA-1 and ac-LDL. Images were acquired with a CoolSNAP HQ2 14-bit charge-coupled device (CCD) camera (Photometrics, Tucson, AZ, USA) using the acquisition software Metamorph Premier version 6.3 (Molecular Devices, Downingtown, PA, USA). Final processing was done with Adobe Photoshop CS6 (Adobe, San Jose, CA, USA). Autologous serum was collected and stored at −80°C until use.
For murine EPC characterization, mononuclear fractions were isolated from bone marrow through gradient centrifugation on Histopaque 1083 (Sigma, St Louis, MO, USA) as previously described [6, 10]. Differentiation to ECs was induced by culture at a density of 1 × 106 cells/cm2 on fibronectin-coated plates (BD Biosciences) in pro-angiogenic EGM-2 media (Lonza) supplemented with 5% heat-inactivated FBS for 7 days. At the time of plating, 1000 U/ml of murine IFN-α (PBL Assay Science, Piscataway, NJ, USA), murine anti-IL-10 antibodies (1 µg/ml) (eBioscience, San Diego, CA, USA) or isotype controls were added when indicated. On the seventh day, live cells were incubated with Texas Red-conjugated ac-LDL and FITC-conjugated Bandeiraea (Griffonia) simplicifolia lectin 1 (BS-1; Vector Laboratories, Burlingame, CA, USA) for 3 h. Imaging was completed as noted for human studies. Mature ECs were identified as those cells that co-stained with BS-1 and ac-LDL and were quantified in triplicate in three random fields per well as described above.
In vivo Matrigel plug assay
In order to determine the effects of IL-10 on type I IFN-mediated repression of angiogenesis, a Matrigel plug assay was used as previously described [20]. Briefly, 0.5 ml of cold growth factor reduced Matrigel (BD Biosciences, San Jose, CA, USA) embedded with 20 nM basic fibroblast growth factor (R&D Systems, Minneapolis, MN, USA) with or without 1000 U/ml murine IFN-α (PBL) was injected s.c. into wild-type or IL-10−/− mice. Seven days after injection, Matrigel plugs were isolated, weighed and homogenized and haemoglobin (HgB) was quantified via incubation in a 1:1 ratio with 3,3′,5,5′-tetramethylbenzidine (Pierce) compared with a standard curve of known HgB concentrations. Results were calculated as milligrams of HgB per gram of Matrigel plug.
IL-10 quantification
Human serum IL-10 levels were measured using a commercially available ELISA kit (eBioscience) according to the manufacturer’s instructions.
Statistical analysis
Student’s t-test analysis was completed to determine whether differences between experimental conditions for EPC differentiation were statistically different. Linear regression analysis was used to compare and determine the correlation between endothelial cell (EC) numbers and IL-10 concentration in patient and control sera. These data were then validated via quasi-Poisson regression. Statistical comparison of concentrations of IL-10 in control and SLE serum was made using the Mann–Whitney U-test. All analysis was completed using GraphPad Prism version 6.02 (GraphPad Software, La Jolla, CA, USA) except for quasi-Poisson regression, which was completed via R stats package version 3.1.0 (R Project for Statistical Computing, Vienna, Austria).
Results
IL-10 hampers EPC differentiation and mediates type I IFN-induced EPC dysfunction
EPC/CACs are considered crucial for repair of injured endothelium [7] and their numbers and function correlate significantly with CV risk in various disease states [8, 9]. In order to understand the effects of IL-10 on EPC function, control human EPCs/CACs was cultured in the presence of recombinant human IFN-α or graded concentrations of recombinant IL-10. As shown in Fig. 1a and b, IL-10 represses EPC/CAC differentiation in a dose-dependent manner, reaching 50% suppression of EC formation at 30 ng/ml, similar to the suppression seen with IFN-α treatment. These results suggest that IL-10 may be detrimental to EPC function, similar to what has been reported for type I IFNs [4, 10].
Fig. 1.
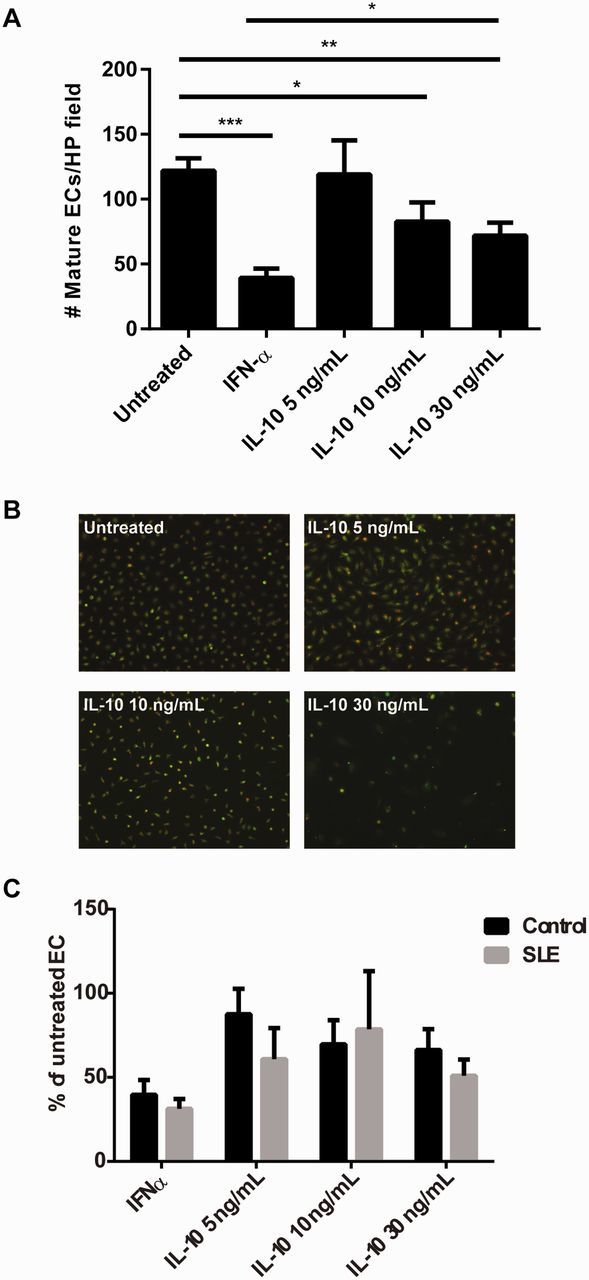
IL-10 inhibits human EPC differentiation in a dose-dependent manner
Human control EPCs were incubated with 1000 U/ml recombinant IFN-α or graded concentrations of recombinant IL-10 and cultured under pro-angiogenic conditions. After 2 weeks in culture, mature ECs were quantified as those that co-stained with ac-LDL and FITC-UEA lectin using live fluorescent microscopy imaging. (A) Results represent the mean (s.e.m.) of double-positive ECs per high-power field (n = 18). (B) Representative microscopic images displaying double-stained mature ECs. (C) SLE (n = 18) and control (n = 18) EPC/CACs were cultured in the presence of graded concentrations of recombinant IL-10 and results are reported as in Fig. 1A. *P < 0.05, **P < 0.01, ***P < 0.001. ac-LDL: acetylated low-density lipoprotein; CAC: circulating angiogenic cell; EC: endothelial cell; EPC: endothelial progenitor cell; UEA: Ulex europaeus agglutinin 1.
Because IL-10 may have pathogenic effects in lupus, we assessed whether the anti-angiogenic responses to IL-10 differed between control and SLE cells. EPC/CACs from healthy controls and SLE subjects were cultured in the presence or absence of graded concentrations of recombinant IL-10. No significant differences in the inhibitory effects of IL-10 on EPC/CAC differentiation were noted between control and SLE samples (Fig. 1c).
In murine systems, type I IFNs induce synthesis of IL-10 [15]. Indeed, IL-10 is considered an intermediary in the pathway of type I IFN-induced repression of pro-angiogenic IL-1β. In order to assess whether type I IFNs require IL-10 as a downstream mediator for repression of EPC differentiation, murine EPC cultures were isolated and incubated with IFN-α in the presence or absence of neutralizing antibodies to IL-10. As shown in Fig. 2a, the blockade of IL-10 reversed the effects of IFN-α on murine EPC cultures. Further, EPC cultures from IL-10 knock-out mice displayed no differentiation abnormalities in the presence of IFN-α (Fig. 2b and c). These results indicate that IL-10 is required for the effects of IFN-α on murine EPC cultures. In order to evaluate whether the inhibitory effects of IFN-α on human EPC/CAC differentiation also required IL-10, neutralizing antibodies to IL-10 were added to healthy control EPC cultures incubated with or without recombinant IFN-α. As shown in Fig. 2d, neutralization of IL-10 in human control EPC/CAC cultures exposed to IFN-α did not improve EC differentiation. Further, real-time PCR analysis of human control EPC/CAC cultures demonstrated that IFN-α did not up-regulate IL-10 (data not shown), suggesting that the effects of type I IFNs on human and murine systems may diverge in downstream mediators required to alter EPC differentiation.
Fig. 2.

IL-10 is required for the inhibitory effects of type I IFN on murine EPC differentiation and in vivo neo-angiogenesis
(A) Wild-type (WT) murine EPCs were incubated with 1000 U/ml IFN-α in the presence or absence of a neutralizing IL-10 antibody or isotype control. Following 7 days of culture, mature ECs were quantified as in Fig. 1 using BS-1 instead of UEA-lectin. Results represent the mean (s.e.m.) of mature ECs quantified in triplicate from three different mice. (B) WT (n = 4) or IL-10−/− (n = 6) EPCs were incubated with IFN-α and mature ECs were quantified as in Fig. 2A. (C) Representative images (total magnification 100 × ) of murine EC cultures from Fig. 2B (green: BS-lectin; red: ac-LDL). (D) Human EPC/CAC cultures (n = 6) were treated as in Fig. 2A utilizing a neutralizing antibody specific for human IL-10. On day 14 of culture, mature ECs were quantified as in Fig. 1. **P ≤ 0.01, NS: non-significant. (E and F) Matrigel embedded with 20 nM fibroblast growth factor with or without 1000 U/ml IFN-α was injected subcutaneously into WT (n = 13) or IL-10−/− (n = 8) mice. Seven days after injection, Matrigel plugs were harvested and their HgB content was quantified as an estimate of neo-angiogenesis within the plug. Results in Fig. 2A represent HgB quantification. Results in Fig. 2B show the percentage inhibition of angiogenesis in plugs containing IFN-α as compared with plugs not exposed to IFN-α. *P < 0.05. BS-1: Bandeiraea (Griffonia) simplicifolia lectin 1; CAC: circulating angiogenic cell; EPC: endothelial progenitor cell; HgB: haemoglobin; UEA: Ulex europaeus agglutinin 1.
IL-10 is required for neo-angiogenesis and may mediate effects of type I IFN in vivo
In addition to negative effects on EPC differentiation, type I IFNs inhibit in vivo neo-angiogenesis [11]. To determine whether IL-10 impacts IFN-α-mediated repression of angiogenesis, an in vivo Matrigel plug neo-angiogenesis assay was performed in wild-type and IL-10−/− mice. IL-10−/− mice had a reduction in total weight-normalized HgB concentration within their Matrigel plugs when compared with wild-type (Fig. 2e), suggesting a basal deficiency in angiogenesis in mice lacking IL-10. When IFN-α was included in the plug matrix, wild-type but not IL-10−/− mice had significant repression of angiogenesis (Fig. 2f). These results suggest that IL-10 is important for neo-angiogenesis under basal conditions and is required for further inhibitory effects of type I IFNs on neo-angiogenesis in murine systems.
When IL-10 signalling was inhibited in the presence of IFN-α in human control EPC cultures, no improvement in EPC differentiation was detected (Fig. 2d). This suggests that in healthy individuals, IL-10 could have protective effects on the vasculature in the presence of type I IFNs. Indeed, when control EPC cultures were exposed to both IFN-α and IL-10 together, a protective effect on EPC/CAC differentiation was noted in control cultures, resulting in differentiation near the levels of untreated wells. In contrast, the opposite effect was detected in cultures from SLE patients, where the combination of IFN-α and IL-10 was significantly more detrimental to EPC/CAC differentiation than either cytokine alone (Fig. 3b). These results suggest that while IL-10 may be protective in the presence of IFN-α in controls, IL-10 and IFN-α are both detrimental to lupus EPC function and likely utilize distinct signalling pathways.
Fig. 3.

IFN-α + IL-10 is detrimental to SLE but not control EPC differentiation
EPC/CACs from SLE (n = 18) and control (n = 18) subjects were incubated with 1000 U/ml IFN-α, 30 ng/ml IL-10 or both and mature EC differentiation was assessed as in Fig. 1. Results are presented as the mean (s.e.m.) percentage inhibition of EPC differentiation compared with autologous untreated cells. *P < 0.05, **P < 0.01. CAC: circulating angiogenic cell; EC: endothelial cell; EPC: endothelial progenitor cell.
SLE is characterized by an elevated type I IFN signature [21], thus patient endothelium is chronically exposed to type I IFNs, resulting in dysfunction of repair pathways [4, 11]. Because IL-10 is detrimental to SLE EPC cultures, we hypothesized that IL-10 blockade may improve EPC differentiation in SLE patients. As shown in Fig. 4a and b, neutralization of IL-10 resulted in a modest, but significant, improvement in EPC differentiation in SLE but not control cultures. Quantification of IL-10 in supernatants of EPC cultures did not show detectable levels (data not shown), suggesting that the limited improvement observed with IL-10 neutralization may be secondary to low levels of IL-10 production in culture. Additionally, the effects of type I IFNs or IL-18 remaining in the culture are not overcome by the addition of IL-10 blockade, thus potentially explaining the modest response [4, 6].
Fig. 4.
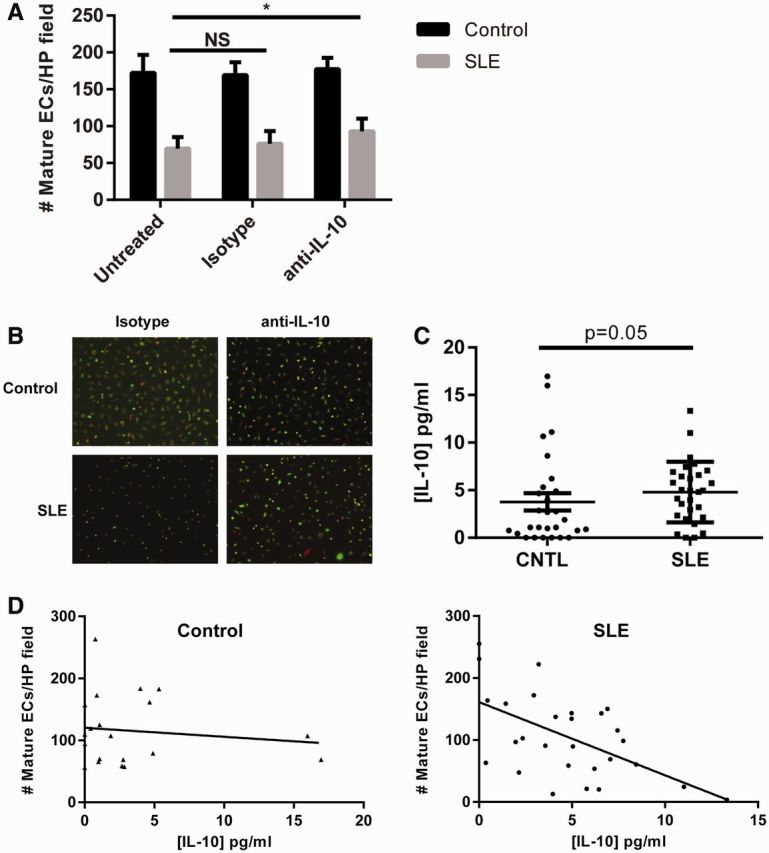
Endogenous IL-10 impairs SLE EPC function and negatively correlates with EPC differentiation in SLE but not control samples
(A) Control (n = 5) or SLE (n = 11) EPC/CACs were cultured in the presence of a neutralizing antibody to IL-10 or an isotype control. On day 14, mature EC differentiation was assessed as in Fig. 1A. (B) Representative photomicrographs of day 14 mature EC cultures from Fig. 4A (red: ac-LDL; green: UEA-lectin). (C) Serum concentrations of IL-10 of control (n = 28) and SLE (n = 29) serum were determined by ELISA. Comparisons between serum levels were made via Mann–Whitney U-test. (D) IL-10 as determined in Fig. 2C was correlated with mature EC numbers derived from untreated control and SLE EPCs/CACs from each subject and linear regression was performed. For control samples, P = 0.5597 and Pearson’s r = − 0.1249. For SLE samples, P = 0.0016 and Pearson’s r = −0.5674. Statistical significance was validated via quasi-Poisson regression: for control, P = 0.598; for SLE, P = 0.0015. ac-LDL: acetylated low-density lipoprotein; CAC: circulating angiogenic cell; CNTL: control; EC: endothelial cell; EPC: endothelial progenitor cell; UEA: Ulex europaeus agglutinin 1.
To determine whether EPC/CAC exposure to IL-10 in vivo prior to isolation and culture could influence EC differentiation, serum levels of IL-10 were measured from healthy controls and SLE patients and concurrent EPC differentiation was assessed. As shown in Fig. 4c and d, while the average concentration of IL-10 did not differ significantly between control and SLE serum samples, the ability of EPC/CACs to differentiate to mature ECs inversely correlated with IL-10 concentrations in SLE patients but not in healthy controls. These results suggest that circulating IL-10 in SLE patients may promote EPC/CAC dysfunction, whereas elevated IL-10 levels do not impact differentiation of control EPC/CACs.
Discussion
The relevance of IL-10 in CVD has remained controversial, with evidence existing for both a preventive and deleterious role in vascular health. Our observations indicate that IL-10 can hamper endothelial differentiation and neo-angiogenesis, especially in the context of IFN-mediated disease such as SLE. In vitro, IL-10 negatively impacts EPC differentiation starting at 10 ng/ml. Thus if the effective local concentration of IL-10 in vivo reaches this level, IL-10 may be detrimental to vascular repair. However, in control and SLE serum, IL-10 is found at picogram per millilitre levels, suggesting that the impact of IL-10 on the vasculature may depend on co-existing factors, such as chronic activation of type I IFNs.
The function of EPC/CACs in SLE is impaired secondary to the effects of type I IFNs. Type I IFNs promote apoptosis of EPCs [4] and also suppress their differentiation by inhibiting transcription of IL-1β while promoting inflammasome activation of IL-18 [6, 10]. In murine macrophages, type I IFNs require IL-10 as a downstream intermediary, which signals to repress IL-1β but not IL-18 [22] transcription [15]. Similarly, in murine EPC cultures we found that type I IFN repression of EPC differentiation required the presence of functional IL-10, as neutralization of this cytokine and the use of IL-10−/− mice prevented inhibitory effects of IFN-α on EPC growth. IL-10 was required for efficient neo-angiogenesis, confirming a previously documented requirement for IL-10 in EPC-mediated myocardial infarct repair [23]. In contrast, in human cultures we did not observe a similar requirement for IL-10 on IFN-α-mediated repression of EPC/CAC differentiation. Rather, IL-10 and IFN-α appear to operate in parallel pathways where both cytokines in isolation are able to inhibit EPC/CAC differentiation to mature ECs and may have synergistic or additive effects in SLE. This difference may reflect that the primary induction of IL-10 in response to type I IFNs has been reported primarily in myeloid cells. Murine EPC cultures are derived directly from the bone marrow, where myeloid progenitors are prevalent, potentially allowing for signalling reflective of a macrophage environment. Alternatively, the effects of type I IFN on human EPCs/CACs do not reflect up-regulation of IL-10 in response to type I IFN signalling or dependence on IL-10 for type I IFN function.
The ability of type I IFN to inhibit EPC/CAC differentiation was abrogated in control cells when IL-10 was added concurrently. However, in SLE patients the combination of IFN-α and IL-10 resulted in poor EC numbers in culture. This suggests that the chronic environment to which SLE EPCs are exposed alters their responses such that the combination of IL-10 and IFN-α remains detrimental. This was not secondary to altered sensitivity to IL-10 in SLE cultures, as the dose response was similar in both control and SLE populations. One mechanism to explain this discrepancy may rely on the data that IL-10 is able to inhibit certain type I IFN responses but is unable to block those generated by Toll-like receptor 7 stimulation [24], such as is observed in SLE patients with circulating RNA-containing immune complexes. Alternatively, pre-exposure to IFN-α is able to enhance IL-10 responses [25]. Another consideration is that IL-18, which is increased in SLE EPCs secondary to type I IFN effects [6] and rises in parallel with IL-10 expression [14, 26], is able to up-regulate suppressor of cytokine signalling 3 expression [27], which can suppress the anti-inflammatory signals mediated by IL-10 [28]. Our control and SLE populations differed in average age by about 11 years, which could potentially contribute to differences in EPC response. However, in our experience, significant discrepancies in EPC growth are not seen in persons of between 30 and 50 years old and regression analysis of age vs EC number does not show a significant correlation for control or SLE populations (supplementary Fig. S1, available at Rheumatology Online), thus we would not anticipate that age alone would explain the differences in response to IL-10.
Contrary to previous reports [29], we did not detect a difference in serum IL-10 concentration between SLE patients and controls. The use of immunosuppressants has been reported to lower IL-10 levels [30]. A substantial number of our patients were on DMARD immunosuppressants (43.2%) and 73% were taking prednisone at the time of sampling, thus potentially explaining this discrepancy. Importantly, despite similar IL-10 concentrations between control and SLE samples, we noted that serum IL-10 levels inversely correlated with the differentiation capacity of EPCs from SLE and not control samples. One explanation for this may be the chronic type I IFN exposure in SLE [21]. Because IL-10 synergizes with type I IFN to impair EPC/CAC differentiation, the presence of circulating IL-10 in SLE patients may result in increased EPC/CAC dysfunction, whereas in healthy controls without chronic type I IFN exposure, IL-10 is not detrimental in the concentrations found in serum. This is further supported by our observation that neutralization of IL-10 in SLE but not control cultures improved EPC/CAC differentiation. Our data agree with previous reports that IL-10 can inhibit cellular proliferation in SLE patients [31]. Taken together, these data suggest that IL-10 plays a relevant and likely pathological role in endothelial dysfunction in SLE and should be investigated further as a potential therapeutic target for CV prevention in this disease.
Conclusions
In summary, our data suggest that IL-10 negatively impacts EPC/CAC differentiation, but the context of the exposure determines the severity of these effects. In murine systems, IL-10 acts as a downstream mediator of the anti-angiogenic effects of type I IFNs. In human systems, IL-10 is not required for type I IFN-mediated effects (illustrated in Fig. 5). Importantly, in healthy controls, IL-10 may confer protective effects on the vasculature upon acute IFN-α exposure, such as during viral infections. In contrast, in SLE, IL-10 may contribute to EPC/CAC dysfunction in the presence of IFN-α, thus affecting vascular repair. In summary, our data suggest that IL-10 should be further explored as a potential target for improvement of vascular health in SLE.
Fig. 5.

Summation of the effects of IL-10 in murine and human EPC cultures
In mice, IL-10 acts as an intermediary for the inhibitory effects of IFN-α on vasculogenesis and EPC differentiation. In contrast, in human EPC cultures, IL-10 is not required for the effects of IFN-α on EPC function. In control cultures, IL-10 is protective in the presence of IFN-α, but in SLE patients, IL-10 and IFN-α promote EPC/CAC dysfunction and the combination of the cytokines remains detrimental. CAC: circulating angiogenic cell; EPC: endothelial progenitor cell.
Rheumatology key messages.
IL-10 can hamper endothelial progenitor cell function in control and SLE populations.
Combined IL-10 and IFN-α is detrimental to EPC function in SLE but not in control EPCs.
Serum IL-10 correlates with EPC function in SLE but not in control samples.
Supplementary Material
Acknowledgements
We would like to thank Tamra J. Reed for her excellent technical help. E.M.M. was supported by the US Department of Veterans Affairs. Support to C.K.S. was via a National Institutes of Health Research Training in Experimental Immunology grant (T32AI007413). J.M.K was partially supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number K09AR063668. This article was prepared while M.J.K. was employed at the University of Michigan. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the US government. A.M.C. carried out the design of assays, EPC growth assays, ELISAs and data analysis and participated in the drafting of the manuscript. V.I.H. carried out murine EPC assays and participated in design of the study and manuscript revision. E.M.M. participated in patient data collection and analysis and manuscript revision. C.K.S. participated in the design and conduct of murine EPC assays and manuscript revision. M.J.K. participated in design of the assays, data analysis and drafting of the manuscript. J.M.K. participated in design of the assays, human and murine EPC experiments and data analysis and drafted the manuscript. All authors read and approved the final manuscript.
Funding: This work was supported by the Alliance for Lupus Research (to M.J.K.) and the ACR Rheumatology Research Foundation Physician Scientist Development Award (to J.M.K.).
Disclosure statement: The authors have declared no conflicts of interest.
Supplementary data
Supplementary data are available at Rheumatology Online.
References
- 1.Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol. 1997;145:408–15. doi: 10.1093/oxfordjournals.aje.a009122. [DOI] [PubMed] [Google Scholar]
- 2.Briasoulis A, Tousoulis D, Antoniades C, Papageorgiou N, Stefanadis C. The role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque development. Cardiovasc Ther. 2011;29:125–39. doi: 10.1111/j.1755-5922.2009.00131.x. [DOI] [PubMed] [Google Scholar]
- 3.Vasa M, Fichtlscherer S, Aicher A, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 4.Denny MF, Thacker S, Mehta H, et al. Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood. 2007;110:2907–15. doi: 10.1182/blood-2007-05-089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thacker SG, Duquaine D, Park J, Kaplan MJ. Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelial dysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus. 2010;19:288–99. doi: 10.1177/0961203309353773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahlenberg JM, Thacker SG, Berthier CC, et al. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol. 2011;187:6143–56. doi: 10.4049/jimmunol.1101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briasoulis A, Tousoulis D, Antoniades C, Papageorgiou N, Stefanadis C. The role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque development. Cardiovasc Ther. 2011;29:125–39. doi: 10.1111/j.1755-5922.2009.00131.x. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt-Lucke C, Rössig L, Fichtlscherer S, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111:2981–7. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- 9.George J, Herz I, Goldstein E, et al. Number and adhesive properties of circulating endothelial progenitor cells in patients with in-stent restenosis. Arterioscler Thromb Vasc Biol. 2003;23:e57–60. doi: 10.1161/01.ATV.0000107029.65274.db. [DOI] [PubMed] [Google Scholar]
- 10.Thacker S, Berthier CC, Mattinzoli D, et al. The detrimental effects of IFN-α on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J Immunol. 2010;185:4457–69. doi: 10.4049/jimmunol.1001782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thacker SG, Zhao W, Smith CK, et al. Type I interferons modulate vascular function, repair, thrombosis and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum. 2012;64:2975–85. doi: 10.1002/art.34504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahlenberg JM, Yalavarthi S, Zhao W, et al. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthritis Rheumatol. 2014;66:152–62. doi: 10.1002/art.38225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu P, Song J, Su H, et al. IL-10 gene polymorphisms and susceptibility to systemic lupus erythematosus: a meta-analysis. PLoS One. 2013;8:e69547. doi: 10.1371/journal.pone.0069547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koenig KF, Groeschi I, Pesickova SS, et al. Serum cytokine profile in patients with active lupus nephritis. Cytokine. 2012;60:410–6. doi: 10.1016/j.cyto.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Mallat Z, Besnard S, Duriez M, et al. Protective role of interleukin-10 in atherosclerosis. Circulation Res. 1999;85:e17–24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 17.Cavusoglu E, Marmur JD, Hojjati MR, et al. Plasma interleukin-10 levels and adverse outcomes in acute coronary syndrome. Am J Med. 2011;124:724–30. doi: 10.1016/j.amjmed.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 18.Rodríguez-Carrio J, Prado C, de Paz B, et al. Circulating endothelial cells and their progenitors in systemic lupus erythematosus and early rheumatoid arthritis patients. Rheumatology. 2012;51:1775–84. doi: 10.1093/rheumatology/kes152. [DOI] [PubMed] [Google Scholar]
- 19.Castejon R, Jimenez-Ortiz C, Valero-Gonzalez S, et al. Decreased circulating endothelial progenitor cells as an early risk factor of subclinical atherosclerosis in systemic lupus erythematosus. Rheumatology. 2014;53:631–8. doi: 10.1093/rheumatology/ket367. [DOI] [PubMed] [Google Scholar]
- 20.Rabquer BJ, Amin MA, Teegala N, et al. Junctional adhesion molecule-C Is a soluble mediator of angiogenesis. J Immunol. 2010;85:1777–85. doi: 10.4049/jimmunol.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crow MK, Kirou KA. Interferon-alpha in systemic lupus erythematosus. Curr Opin Rheumatol. 2004;16:541–7. doi: 10.1097/01.bor.0000135453.70424.1b. [DOI] [PubMed] [Google Scholar]
- 22.Zediak VP, Hunter CA. IL-10 fails to inhibit the production of IL-18 in response to inflammatory stimuli. Cytokine. 2003;21:84–90. doi: 10.1016/s1043-4666(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 23.Krishnamurthy P, Thai M, Verma S, et al. Interleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in ischemic myocardium. Circ Res. 2011;109:1280–9. doi: 10.1161/CIRCRESAHA.111.248369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waibler Z, Anzaghe M, Konur A, et al. Excessive CpG 1668 stimulation triggers IL-10 production by cDC that inhibits IFN-alpha responses by pDC. Eur J Immunol. 2008;38:3127–37. doi: 10.1002/eji.200838184. [DOI] [PubMed] [Google Scholar]
- 25.Liu B-S, Janssen HLA, Boonstra A. Type I and III interferons enhance IL-10R expression on human monocytes and macrophages, resulting in IL-10-mediated suppression of TLR-induced IL-12. Eur J Immunol. 2012;42:2431–40. doi: 10.1002/eji.201142360. [DOI] [PubMed] [Google Scholar]
- 26.Pestka JJ, Vines LL, Bates MA, He K, Langohr I. Comparative effects of n-3, n-6 and n-9 unsaturated fatty acid-rich diet consumption on lupus nephritis, autoantibody production and CD4+ T cell-related gene responses in the autoimmune NZBWF1 mouse. PLoS One. 2014;9:e100255. doi: 10.1371/journal.pone.0100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsui F, Rhee A, Hile KL, Zhang H, Meldrum KK. IL-18 induces profibrotic renal tubular cell injury via STAT3 activation. Am J Physiol Renal Physiol. 2013;305:F1014–21. doi: 10.1152/ajprenal.00620.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehlting C, Lai WS, Schaper F, et al. Regulation of suppressor of cytokine signaling 3 (SOCS3) mRNA stability by TNF-alpha involves activation of the MKK6/p38MAPK/MK2 cascade. J Immunol. 2007;178:2813–26. doi: 10.4049/jimmunol.178.5.2813. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Zhu T, Cai G, et al. Elevated circulating CD4+ICOS+Foxp3+ T cells contribute to overproduction of IL-10 and are correlated with disease severity in patients with systemic lupus erythematosus. Lupus. 2011;20:620–7. doi: 10.1177/0961203310392431. [DOI] [PubMed] [Google Scholar]
- 30.Yu SL, Wong CK, Wong PT, et al. Down-regulated NOD2 by immunosuppressants in peripheral blood cells in patients with SLE reduces the muramyl dipeptide-induced IL-10 production. PLoS One. 2011;6:e23855. doi: 10.1371/journal.pone.0023855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lauwerys BR, Garot N, Renauld JC, Houssiau FA. Interleukin-10 blockade corrects impaired in vitro cellular immune responses of systemic lupus erythematosus patients. Arthritis Rheum. 2000;43:1976–81. doi: 10.1002/1529-0131(200009)43:9<1976::AID-ANR8>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.