

Editor’s Highlight: A new initiative to better link scientific proceedings of the Society of Toxicology Annual Meeting with Toxicological Sciences involves the invitation of a review article from especially interesting and innovative symposia. Fariss and colleagues present an integration of new concepts from a symposium held at the 2012 Annual Meeting related to transient receptor potential (TRP) channels as sensors of xenobiotics and secondary intermediates and ultimately determinants of particulate matter toxicity. Compelling evidence sheds light on TRP channels mediating airway injury, hyperreactivity, and cardiovascular toxicity of inhaled pollutants — Matthew Campen.
Key Words: transient receptor potential channels, vehicle emissions, cigarette smoke, calcium, humic-like substances.
Abstract
The mechanism for biological effect following exposure to combustion-generated particles is incompletely defined. The identification of pathways regulating the acute toxicological effects of these particles provides specific targets for therapeutic manipulation in an attempt to impact disease following exposures. Transient receptor potential (TRP) cation channels were identified as “particle sensors” in that their activation was coupled with the initiation of protective responses limiting airway deposition and inflammatory responses, which promote degradation and clearance of the particles. TRPA1, V1, V4, and M8 have a capacity to mediate adverse effects after exposure to combustion-generated particulate matter (PM); relative contributions of each depend upon particle composition, dose, and deposition. Exposure of human bronchial epithelial cells to an organic extract of diesel exhaust particle was followed by TRPV4 mediating Ca++ influx, increased RAS expression, mitogen-activated protein kinase signaling, and matrix metalloproteinase-1 activation. These novel pathways of biological effect can be targeted by compounds that specifically inhibit critical signaling reactions. In addition to TRPs and calcium biochemistry, humic-like substances (HLS) and cell/tissue iron equilibrium were identified as potential mechanistic targets in lung injury after particle exposure. In respiratory epithelial cells, iron sequestration by HLS in wood smoke particle (WSP) was associated with oxidant generation, cell signaling, transcription factor activation, and release of inflammatory mediators. Similar to WSP, cytotoxic insoluble nanosized spherical particles composed of HLS were isolated from cigarette smoke condensate. Therapies that promote bioelimination of HLS and prevent the disruption of iron homeostasis could function to reduce the harmful effects of combustion-generated PM exposure.
Environmental air pollution is a global concern, and its effect on human health, particularly respiratory and cardiovascular disease, is firmly established. Despite epidemiological evidence linking ambient air pollutants to human morbidity and mortality, a significant void remains in the recognition of the cellular and molecular mechanisms through which these exposures produce specific adverse effects. This is especially true for particulate air pollution. Similar physiological responses and pathology in the lung following exposure to a variety of combustion-generated particles (e.g., diesel exhaust [DE], wood smoke, and cigarette smoke) suggest common mechanism(s) of toxicity. The identification of these mechanistic pathways can provide potential targets for therapeutic strategies aimed at reducing human morbidity and mortality following exposures.
We summarize a symposium session entitled “Emerging Mechanistic Targets in Lung Injury Induced by Combustion-Generated Particles,” which was presented on March 15, 2012 at the 51st Annual Meeting of the Society of Toxicology in San Francisco, California. Novel avenues of biological effects are provided for combustion-generated particles that focus on the participation of (1) transient receptor potential (TRP) cation channels and calcium transport and (2) humic-like substances (HLS) and iron homeostasis. These mechanistic pathways are proposed as targets for developing therapies to diminish human morbidity and mortality following exposures to combustion-generated particles.
COMBUSTION-GENERATED PARTICLES
Much of the anthropogenic air pollution across the globe is produced by emission of particles, gases, and vapors from biomass burning and fossil fuel combustion (Lighty et al., 2000). The actual physicochemical makeup of these mixtures is extremely complex and varies according to the type of fuel and combustion conditions. To illustrate some of these differences, Figure 1 shows how biomass such as tobacco and wood have approximately equal amounts of carbon and oxygen content amounting for more than 90% of the mass, with lower levels of hydrogen and nitrogen. Fossil fuels like coal or diesel (from oil), on the other hand, lose oxygen during their formation, resulting in a material composed of greater than 80% carbon with the hydrogen, nitrogen, and oxygen filling out the mass balance. It should be noted that while the content of carbon, hydrogen, nitrogen, and oxygen is a simple assessment of the primary makeup, these constituents are present in a vast array of simple and complex molecules in association with other elements such as sulfur, halides, and metals.
FIG. 1.
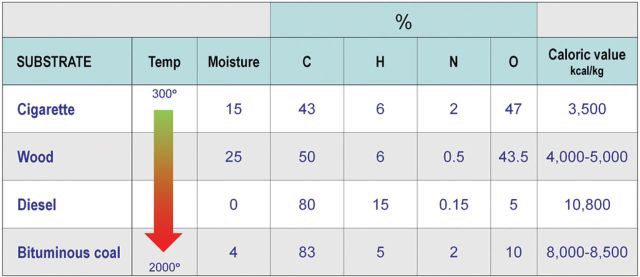
Chemical constituents, moisture content, calorific value, and approximate combustion temperature of cigarette tobacco, wood, diesel oil, and bituminous coal under normal operating conditions. (A color image is available in the online version.)
Combustion temperature is another major variable in how well the fuels burn or emit products of incomplete combustion (Lighty et al., 2000). Efficient combustion of biomass to carbon dioxide and water is largely hampered by low temperature, moisture content, and uneven oxygenation of the substrate. These factors are less problematic for the combustion of diesel and coal, where the temperature and mixing are engineered to be higher, and moisture content is closer to zero. Most of the mass of emissions from combustion processes is in the gas phase in the form of oxides of carbon, nitrogen, and sulfur. Organic gases and vapors usually make up the next largest constituent class and can include reactive compounds such as formaldehyde and acrolein. The particle phase makes up a smaller fraction of the mass and comprises ash, as well as both elemental and organic carbon moieties, the latter of which may be of low or intermediate volatility.
Inhalation exposure to combustion emissions has long been known to impact health depending upon the concentration and duration of exposure, as well as the chemistry of the aerosol. At the extreme end of the spectrum, smoke asphyxiation occurs through hypoxia and carbon monoxide poisoning. At lower levels, many of the health effects are reversible following cessation of exposure (e.g., an increased carboxyhemoglobin concentration), whereas other outcomes such as immune suppression in cigarette smokers may persist (Herr et al., 2009). This article discusses health impacts that occur at ambient, occupational or personal levels of exposure and the mechanisms that govern these effects.
The health hazards from both mainstream and side-stream cigarette smoke are well established, with smokers having a 2- to 4-fold increased incidence of coronary heart disease and stroke and a 12- to 13-fold increase in lung cancer and chronic obstructive pulmonary disease (COPD) (Centers for Disease Control and Prevention, 2012). These effects are thought to occur through the many toxic constituents of tobacco smoke that individually have been shown to have carcinogenic, fibrogenic, and cytotoxic properties. Not surprisingly, emissions from biomass burning contain many of these same components, and the incidences of COPD, lung cancer, and acute respiratory tract infections are all increased in association with use of solid household fuels in developing countries (Zhang and Smith, 2003). In addition to these chronic health effects, inhalation exposures can also have significant acute impacts. For example, the incidence of heart failure exacerbation and the number of hospital visits for asthma were increased in counties in North Carolina that experienced higher concentrations of air pollution from a wildfire in the eastern part of the state (Rappold et al., 2011).
Diesel and gasoline (GE) engines are other important sources of combustion emissions. The Mobile Source National Emissions Inventory estimates that each contributes millions of tons of volatile organic compounds, nitrogen oxides (NOx), and carbon monoxide and hundreds of thousands of tons of PM10, PM2.5 (particulate matter [PM] ≤ 10 µm, PM ≤ 2.5 µm, respectively), sulfur dioxide (SO2), and ammonia (U.S. Environmental Protection Agency, 2012). Diesel engines release more PM per mile travelled than GE engines, and many epidemiological, clinical, and animal studies have implicated diesel emissions in a broad array of health effects, from increased cancer and asthma incidence to elevated risk for cardiovascular disease. As a result of these concerns, diesel emissions are being dramatically reduced by the introduction of low-sulfur fuels and the phasing in of NOx catalysts and PM traps in contemporary engines.
Another large source of combustion air pollutants in the United States and across the world is coal-fired power plants that emit millions of tons of SO2 and NOx per year into the air. An estimated 36,000 total excess deaths in 2005 were attributed to PM2.5 and ozone formed from emissions from electrical generating units, with nonfatal health effects occurring in a far greater number of individuals (Fann et al., 2012). As with DE, emissions are predicted to decrease with tighter regulations such as the Mercury and Clean Air interstate rules although air pollution burdens will still be appreciable (U.S. Environmental Protection Agency, 2012).
Despite the large body of evidence showing associations between air pollution and excess cardiovascular mortality, source apportionment of these effects either to specific chemical components or to particular sources that have differing physicochemical properties has been difficult. Although there are numerous health studies of these various mixtures, the most thorough comparative toxicology program was developed at the National Environmental Respiratory Center at the Lovelace Respiratory Research Institute (LRRI) in Albuquerque, NM. Studies examined the relative toxicity of acute and subchronic inhalation of hardwood smoke (HWS) and DE, GE, and simulated downwind coal combustion atmospheres (SDCCA) in several animal models of human respiratory and cardiovascular disease. One of the first challenges encountered was how to compare these disparate emissions. Because PM atmospheres are compared (and regulated) by mass, the investigators first established target mass PM concentrations for the inhalation exposure studies. Setting a target concentration for PM, they discovered that 1000 µg/m3 SCCDA produced relatively low levels of gases, whereas the same concentration of HWS PM resulted in 20,000 µg/m3 of gases (excluding carbon dioxide, water, and methane) and approximately 90,000 µg/m3 of gases for DE (Jacob McDonald, LRRI, personal communication). At the same minimal dilution as DE (10:1), GE had even higher levels of accompanying gases but only 60 µg PM/m3. Despite these caveats, the group concluded that DE > GE > HWS >>>> SDCCA for most toxicological parameters measured. They also noted, however, that gas-phase pollutants in GE were responsible for promoting atherosclerosis in mice, whereas both gas-phase and PM components of DE contributed to this effect (Campen et al., 2010). They further reported that DE from engines operating under different load conditions could profoundly affect different pulmonary and cardiovascular outcomes (McDonald et al., 2011). It is clear from these types of studies that combustion emissions are highly complex and dynamic and that the chemistry and physicochemical properties and the concentration and duration of exposure influence the type and extent of health impacts. Multivariate statistical analyses of the relationship between chemical and physical components versus biological responses have been reported for cardiovascular responses, suggesting putative roles for gases such as SO2, NH3, CO, and NOx (Seilkop et al., 2012).
DIFFERENTIAL ACTIVATION OF TRP CATION CHANNELS ANKYRIN-1 (TRPA1), VANILLOID-1 (TRPV1), AND MELASTATIN-8 (TRPM8) BY COMBUSTION-DERIVED PM AND PM COMPONENTS
The elucidation of detailed mechanisms by which PM in ambient air causes adverse effects on human health has been hindered by the complexity and variability of these particles commonly used in in vitro and animal models. Here, a simplified approach and results identifying several members of the TRP family of cation channels as “PM sensors” (i.e., gene products that differentially detect and initiate specific responses to PM), which have been implicated by numerous studies to play important roles in PM effects in the respiratory system (Agopyan et al., 2003a , b; Ghelfi et al., 2008; Oortgiesen et al., 2000; Veronesi et al., 1999a , b; Wong et al., 2003), are summarized.
Cells were engineered to overexpress TRPA1, M8, V1, V2, V3, V4 (Deering-Rice et al., 2011), C4α, and M2 channels. These specific TRP channels were chosen because they detect and respond to a wide range of environmental variables, endogenous mediators, and xenobiotics including numerous prototypical irritants and pneumotoxicants (Venkatachalam and Montell, 2007; Voets et al., 2005; Vriens et al., 2009). These TRP channels play essential roles in airway reflex control and the regulation of inflammation (Agopyan et al., 2003a , b; Deering-Rice et al., 2012; Ghelfi et al., 2008; Oortgiesen et al., 2000; Reilly et al., 2003, 2005; Thomas et al., 2012; Veronesi et al., 1999a , b; Wong et al., 2003), and they potentially mediate PM toxicity (e.g., TRPV1) (Agopyan et al., 2003a , b; Deering-Rice et al., 2012; Ghelfi et al., 2008; Oortgiesen et al., 2000; Veronesi et al., 1999a , b; Willis et al., 2011; Wong et al., 2003). The PM used as agonists included ambient samples (NIST 1648 and fresh, size-fractionated PM collected in Salt Lake City, UT in December 2011), cigarette smoke and wood (pine and mesquite) smoke (WSP), coal fly ash (one collected from a local power plant and a second prepared in the laboratory by burning coal in a low oxygen atmosphere), Min-U-Sil 5 silica, a size-fractionated dust from Utah’s West Desert, and DE samples (DEP; NIST 2975 and PM collected from an on-road “black smoker” truck).
Screening of these particles for TRP channel agonist activity revealed that TRPA1, M8, V1, and V4 were differentially activated by ambient PM (A1 > M8 > V1 and V4), DEP (A1 > V1, V4, and M8) (Deering-Rice et al., 2011), cigarette and WSP (A1), authentic coal fly ash PM (V1 and M8 > A1) (Deering-Rice et al., 2012), and silica (V1 > M8).
TRPA1 was potently activated by chemicals associated with the DEP (aldehydes, quinones, and ketones) via modification of the electrophile/oxidant sensing site of TRPA1 (C621/C641/C665/K710) and multiple unsaturated dienes resembling 1,3-butadiene (Fig. 2A) (Deering-Rice et al., 2011). Two di-tert-butylphenol analogues, presumably arising on diesel PM from their use as a fuel/oil preservative, activated TRPA1 via the propofol/menthol binding site (S673/T674) (Fig. 2A), and the fully extracted residual “stripped” DEP mass activated TRPA1, V1, and M8 equivalently and to the same level as the authentic coal fly ash PM, whose potency at TRPA1 was unaffected by solvent extraction (Figs. 2A–C). These results, coupled with residual activity of the electrophile/oxidant site deletion mutant to the stripped DEP, supported a third minor mechanism of TRPA1 activation by PM, involving mechanical perturbation of extracellular receptor components. Finally, TRPA1 was activated by ambient air PM, cigarette smoke, and WSP via the electrophile/oxidant and menthol/propofol agonist recognition sites, presumably by similar and unique source-specific chemical components of these particles. Additional discussion of TRPV4 activation by organic extracts of DEP, and the potential role of this pathway in lung toxicology, is provided in the next section. Accordingly, future studies to differentiate the mechanistic basis for TRPA1 and TRPV4 by various chemical constituents of DEP and the elucidation of the relative importance in these seemingly redundant and complimentary pathways for pneumotoxicity are of great interest.
FIG. 2.



PM activation of TRPV1, M8, and A1. (A) Activation of TRPA1 via mechanical stimuli is proposed to be similar to that of TRPV1 and M8. TRPA1 is also activated by organic extracts of combustion-derived PM through the electrophilic and menthol/propofol binding sites. (B) TRPV1 activation by coal fly ash is proposed to occur through charge and mechanical interactions with the poor-loop region, as described in our most recent publication (Deering-Rice et al., 2012), resulting in cell-type specific responses (epithelial vs. neuronal). (C) Due to sequence similarity between TRPM8 and V1, mechanical activation by PM is proposed to occur in the poor-loop region. (A color image is available in the online version.)
TRPV1 was activated by coal fly ash also via a mechanism involving mechanical perturbation of extracellular components (Deering-Rice et al., 2012). Enrichment of TRPV1 at the cell surface exacerbated activation and neither repeated aqueous nor solvent extraction reduced coal fly ash potency. TRPV1 activation was equally and fully inhibited by the selective cell-permeable antagonist LJO-328 and by a combination of the nonselective cell-impermeable inhibitor ruthenium red and the chelator EGTA (Deering-Rice et al., 2012). Finally, site-directed mutagenesis of the capsaicin binding site on TRPV1 (Y511; capsaicin is the prototypical soluble agonist of TRPV1) did not inhibit activation by coal fly ash PM. However, neutralization of E649 on the pore-loop segment increased activity, and mutation of E600 (voltage/proton sensor), C578 (an ion channel helix component), and N604 (the glycosylation site adjacent to the pore-loop segment) inhibited activation (Deering-Rice et al., 2012). It was concluded that the pore-loop functions as a “mechanical switch,” where physical displacement by PM, particularly negatively charged PM, resulted in pore dilation and ion flux (Fig. 2B) (Deering-Rice et al., 2012). Results also indicate that TRPM8 activation by coal fly ash involves specific interactions at the extracellular surface (Fig. 2C). This mechanism also appears to be critical in determining TRPV1 and M8 responses to ambient PM, but not for more soluble PM such as the DEP, cigarette, or WSP that appear to have a greater capacity to release chemicals into the cells/cell culture treatment media.
It was proposed that the cellular expression of specific TRP channels would determine if, and how, a cell and/or the lungs are impacted by various PM. TRPA1 is primarily expressed by airway C-fiber neurons that also express TRPV1 and substance P, but recent studies indicate that TRPA1 may also be expressed in nonneuronal lung cells where they also participate in proinflammatory signaling (Mukhopadhyay et al., 2011; Nassini et al., 2012). Therefore, bronchoconstriction, cough, and inflammation/edema, which have been shown to be caused by PM such as diesel, cigarette, wood, and even ambient PM, should be, in part, TRPA1 mediated. Analysis of calcium flux elicited by the ethanol extract of DEP, which contains ~90% of the activity at TRPA1, demonstrated that TRPA1 was the primary mediator, based on near-complete inhibition by the TRPA1 selective antagonist HC-030031 and colocalization of neuronal responses to DEP extracts with those of the prototypical TRPA1 agonist allyl-isothiocyanate (AITC) (Deering-Rice et al., 2011). In agreement with this, DEP triggered action potentials in AITC-sensitive vagal C-fibers, but not in AITC-negative neurons, and instillation of DEP into lungs of rats rapidly decreased lung compliance, which was attenuated by cotreating with HC-030031. However, based on recent reports of TRPA1 in nonneuronal lung cells and its involvement in proinflammatory signaling, it is of great interest to better ascertain the relative contributions of neuronal TRPA1-mediated responses to those mediated by TRPA1, V1, M8, and V4 in lung epithelial cells.
In the airways, TRPV1 is expressed by substance P-positive C-fibers and epithelial cells of the conducting bronchi and alveoli. Coal fly ash activated capsaicin-sensitive neurons in culture, which was inhibited by the antagonist LJO-328 (Deering-Rice et al., 2012). Activation of TRPV1-expressing C-fibers by select forms of PM would also be predicted to elicit comparable neuronally mediated responses as those discussed for TRPA1, including cough and neurogenic inflammation, provided that the PM can physically interact with TRPV1 on nerve termini. Conversely, particles that activate TRPV1 also have the ability to initiate epithelial cell–mediated immune responses that are often described by PM toxicology studies (Agopyan et al., 2003a , b; Deering-Rice et al., 2012; Reilly et al., 2003, 2005; Veronesi et al., 1999a , b). Studies using airway epithelial cells demonstrated a role for TRPV1 in coal fly ash–induced calcium flux, as well as immunomodulatory cytokine/chemokine (IL-6 and IL-8) production; both calcium influx and cytokine/chemokine production were inhibited by LJO-328 and EGTA + ruthenium red (Deering-Rice et al., 2012). Furthermore, instillation of coal fly ash into mouse lungs increased IL-6, MIP2α/CXCL2, and KC/CXCL1 mRNA expression in the bronchioles (and much less in the trachea, bronchi, or alveoli), comparable to the prototypical soluble TRPV1 agonist nonivamide, which were attenuated in TRPV1˗/˗ mice (Deering-Rice et al., 2012).
The collective results outlined above suggest that TRPA1, V1, and M8 are important sensors of various environmental PM in the airways. Additionally, TRPV4 may be an important determinant of responses to select forms of PM, as detailed in the following section. The activation of various TRP channels, alone or possibly in combination, appears to be coupled with the initiation of protective neuronal reflex-type responses intended to limit deposition of materials in the airways and the initiation of protective inflammatory responses intended to promote degradation and clearance of foreign substances from the lung. Although these physiological responses are essential for human survival, they can also underlie many of the more severe toxicological manifestations that have been associated with human exposure to PM.
Finally, although it is likely that TRPA1, V1, M8, and presumably V4 will continue to emerge as vital molecular sensors and mediators of adverse effects for inhaled environmental PM, it must be emphasized that the relative contributions of each of these receptors (and likely other PM sensors yet to be characterized) to coordinate airway and systemic responses will depend upon several inherent variables that must be considered in experimental design and data interpretation. First, one must consider the PM source and the relative chemical and physical composition of the PM because variations will influence both the relative potency and specificity of PM at these, and presumably other, PM sensors. Second, individual variations in the expression and function of these PM sensors, such as gain- (e.g., TRPA1-E179K and others) or loss-of-function (e.g., TRPV1-I585V) polymorphic variants, should be evaluated (Cantero-Recasens et al., 2010; May et al., 2012; Smit et al., 2012). Third, the relative dose/deposition of the PM in the lung will determine which cells are most likely to be responsive, as a factor of relative expression levels of PM sensors, and which physiological responses are most likely to transpire.
ROLE OF TRPV4-MEDIATED CALCIUM INFLUX IN DE PARTICLE-INDUCED LUNG TOXICITY
DEP is a critical component of urban smog. It constitutes PM that is environmentally relevant and exerts a strong impact on human health. Inhalation of DEP has been implicated in worldwide increases in COPD and chronic asthma, both of them sharing a hyperreactive bronchial system. DEP has also been suggested to enhance the likelihood of malignant transformation of human airway epithelia given their stimulating effects upon cell proliferation (Bayram et al., 2006; Boland et al., 1999; Cao et al., 2007; Doornaert et al., 2003; Ghio et al., 2000; Hashimoto et al., 2000; Koike et al., 2004; McClellan, 1987; McClellan et al., 1985; Nikula et al., 1995; Parent et al., 2007; Sydbom et al., 2001). DEP exerts its effect also on bronchial epithelial cells, eliciting a number of signaling events that are very likely to contribute to respiratory toxicity, especially long term (Fahy et al., 2000; Ghio et al., 2000; McClellan et al., 1985; Parent et al., 2007; Sydbom et al., 2001). Upon exposure to DEP, human primary airway epithelia show reduced ciliary beat frequency, increased oxidative damage, an activation of the NF-κB pathway, and secretion of proinflammatory cytokines with some of the secreted mediators also sensitizing airway sensory neurons (Bayram et al., 1998, 2006; Bonvallot et al., 2001; Costa et al., 2010; Daniel and O’Byrne, 1991; Li et al., 2009, 2011; Takizawa et al., 1999, 2003; Tal et al., 2010; Totlandsdal et al., 2010). Specific signal transduction pathways underlying these DEP-evoked events are incompletely understood.
The underlying mechanism of biological effect by DEP has been addressed in two recent publications, where a novel and specific signaling mechanism is described in human respiratory epithelia (Fig. 3) (Li et al., 2009, 2011). This molecular mechanism involves proteinase-receptor 2, a G-protein coupled receptor, the G-protein Gi/o, membrane bound enzymes PI 3-kinase and phospholipase-C (β3 isoform), the TRP ion channel, TRPV4, and via TRPV4-mediated Ca++ influx, mitogen-activated protein (MAP)–kinase signaling via RAS, RAF, MEK, and ERK. The ERK is rapidly phosphorylated and translocates to the nucleus, where it activates the matrix metalloproteinase-1 gene (collagenase; MMP-1), and the product, MMP-1, functions as a propathogenic mediator in human airways (Fig. 4). In addition, the secretion of chemokines RANTES and IP-10 was also found to be increased with DEP exposure. Both of these are “airway tropic” with proinflammatory actions on specific cells in airways, and, importantly, with sensitizing effect on airway-innervating sensory neurons. Highlighting the relevance of MMP-1 in COPD, immunohistochemistry of human lung specimens showed strongly upregulated MMP-1 expression, especially in bronchial epithelial cells (Fig. 4) (Segura-Valdez et al., 2000). There is no orthologue in rodent genomes for MMP-1, which is found only in primates; Mmp-2 and Mmp-9 are believed to be functional MMP-1 orthologues in rodents (Chung, 2001; Joos et al., 2002; Schütz et al., 2002; Segura-Valdez et al., 2000). This will be relevant for future work in mice that seeks to confirm the findings obtained with human cells. It is clear that increased MMP secretion by respiratory epithelia exposed to PM is significantly linked to airway diseases, at least in part, because of a neurotropic effect on MMP-1 that likely will lead to sensitization of airway innervating sensory neurons, augmenting the function of RANTES and IP-10. Also, evidence that MMP-1 secretion, or that of other MMPs, leads to direct activation of PAR-2 similar to the effects of secreted MMP-1 on PAR-1 activation in breast cancer was not found. Rather, the effects were on MAP-kinase signaling, downstream of Ca++ influx via TRPV4, yet these effects were critical because pan-β-arrestin knockout (using siRNA) completely eliminated MMP-1 activation by DEPs.
FIG. 3.

Signaling cascades, as outlined by our two recent papers (Li et al., 2009, 2011). (A–C) Membrane-proximal signaling evoked by DEP-organics (DEP-OE) leading to Ca++ influx via TRPV4 and (D) activation of RAS (by Ca++) and subsequent MAP-kinase signaling that leads to MMP-1 activation. (A color image is available in the online version.)
FIG. 4.
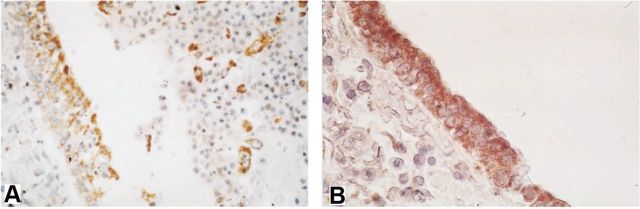
Expression of MMP-1 in bronchial epithelial cells of human COPD lungs (Segura-Valdez et al., 2000). (A) Differentiated bronchial epithelium (control; also note MMP-1+ macrophages in the bronchial lumen); (B) in striking contrast, a strongly MMP-1-expressing bronchial epithelium from a COPD case. (A color image is available in the online version.)
These findings have relevance on human disease beyond the characterization of a novel and pathophysiologically relevant signaling pathway. They highlight the known susceptibility of different human individuals to air pollution by demonstrating that the TRPV4P19S human polymorphism augments TRPV4-mediated Ca++ influx and MMP-1 activation strikingly. Importantly, TRPV4P19S has been shown to significantly predispose individual subjects to develop COPD (Zhu et al., 2009). TRPV4 was found expressed on respiratory cilia in human primary airway epithelia (Li et al., 2009, 2011) and in the same location on mouse tracheal epithelia (Lorenzo et al., 2008). Furthermore, with regard to human health, this investigation encompasses yet another human genetic polymorphism linked to respiratory health, namely the promoter polymorphism, MMP-1(-1607G/GG), thus highlighting the concept of disease susceptibility as a function of genetic (epigenetic) “makeup” in combination with environmental insults.
WOOD SMOKE PARTICLES AND SEQUESTRATION OF MITOCHONDRIAL IRON
The burning of wood impacts domestic, occupational, and environmental exposures with elevations in PM to levels exceeding milligrams per cubic meter (Ezzati and Kammen, 2001; Kocbach Bølling et al., 2009; Larson and Koenig, 1994; Naeher et al., 2007; Riojas-Rodríguez et al., 2001). Respiratory symptoms, lower respiratory tract infections, exacerbations of chronic disease with hospitalizations, and systemic inflammation can all be associated with exposures to wood smoke (Bruce et al., 2000; Dennis et al., 1996; Koenig et al., 1993; Ozbay et al., 2001; Pérez-Padilla et al., 1996; Sharma et al., 1998; Tan et al., 2000; van Eeden et al., 2001). This clinical presentation following wood smoke exposure is similar to that after exposure to many diverse particles including those of cigarette smoke, environmental tobacco smoke, gas stoves, and mining/processing of coal and mineral oxides. These particle-related exposures can cause cough and phlegm production (Aditama, 2000; Jarvis et al., 1996; Jinot and Bayard, 1996; Larson and Koenig, 1994; Riojas-Rodríguez et al., 2001; Sydbom et al., 2001), and wheeze (Chilmonczyk et al., 1993; Donaldson et al., 2000; Gamble et al., 1987; Larson and Koenig, 1994); an acute, reversible decrement in pulmonary function and elevation in bronchial hyperreactivity (Chen et al., 2001; Koenig et al., 1993; Larson and Koenig, 1994; Morgan et al., 1997; Pope et al., 1991; Sheppard et al., 1986; Sherman et al., 1989); an induction of an acute neutrophilic inflammation, an emphysema, and a parenchymal fibrosis (Ghio et al., 2012b; Ozbay et al., 2001; Pinkerton et al., 2000); inflammatory changes in the peripheral blood (e.g., elevations in white blood cell counts and increases in C-reactive protein, fibrinogen, and blood viscosity) (Pekkanen et al., 2000; Seaton et al., 1999); and cardiovascular, cerebrovascular, and peripheral vascular disease (Glantz and Parmley, 1995; Havas et al., 1989; Melius, 1995). This shared presentation after exposure to dissimilar particles suggests a common mechanism for their biological effect. That mechanism is widely considered to be a generation of an oxidative stress that provokes increasing kinase activity, transcription factor activation, and mediator release with inflammation expressed histopathologically (Ghio et al., 2012a). Therefore, a common pathway may exist for all PM by which an oxidative stress and subsequent biological effects are generated.
One postulate that can explain a common pathway for oxidative stress and the clinical presentation following PM exposure is an altered iron homeostasis. Life evolved with an absolute dependency on iron availability, but those same chemical properties that allow this metal to function as a catalyst in reactions involving molecular oxygen can make it a threat to life via the generation of oxygen-based free radicals. Iron will react with oxygen-containing functional groups at particle surfaces (Ghio et al., 2004). In the lower respiratory tract, retained particles accordingly accumulate metal from available sources in a cell and tissue via complexation (Koerten et al., 1986). During such mobilization of iron from host sources, oxidants may be produced. Cells and tissues attempt to transfer iron away from the particle where it is catalytically active and capable of presenting an injurious stress. The resultant disruption in iron homeostasis, and the attempt to re-establish normal metal equilibrium in the host, can be observed following the introduction of any particle into the lung as a ferruginous body (Ghio et al., 2004).
With wood smoke exposure, there is formation of ferruginous bodies supporting an involvement of iron in the biological response to this specific PM (Fig. 5). Therefore, we evaluated the postulate that WSP sequesters host cell iron resulting in disruption of normal homeostasis and that this loss of essential metal results in both an oxidative stress and biological effect. With exposure to WSP, respiratory epithelial cells were observed to import iron. RNA for divalent metal transporter 1 (a major iron importer) increased after WSP exposure. With 24-h exposure to WSP, cell ferritin concentrations were also elevated. Using 57Fe labeling, sequestration of mitochondrial iron by WSP was confirmed. Oxidant generation, the activation of a transcription factor controlling the expression of genes involved in the antioxidant and inflammatory response, and release of proinflammatory mediators following cell exposure to WSP were all shown to be dependent on the sequestration of cell iron by the particle.
FIG. 5.

Ferruginous body following exposure to wood smoke. This is human lung tissue collected at autopsy, stained with hematoxylin and eosin, and viewed at approximately 100× magnification. (A color image is available in the online version.)
As a result of a capacity of WSP to bind cell iron, host sources of this metal are sequestered (Fig. 6). Accordingly, the cell responds to the particle, its sequestration of host iron, and the relative iron deficiency by upregulating import of iron. The attempts by the cell to reacquire iron necessitate ferrireduction including superoxide generated by the mitochondria. A new iron homeostasis is determined by the interaction between the cell and the particle. Total cell iron is increased, ferritin is increased, and organelle concentrations of metal return to normal. Subsequently, the cell has sufficient iron to meet its requirements for function despite the particle sequestering portions of this pool. If sufficient iron concentrations cannot be obtained by the cell, the response proceeds to include inflammation. Should this fail to re-establish an iron homeostasis adequate to meet all needs in the cell, apoptosis will be initiated.
FIG. 6.

Schematic of interaction between particle and airway epithelial cells in altering iron homeostasis. Endocytosed particle will complex available iron from intracellular sites (A). The cell recognizes a functional deficiency in the metal and responds by generating more ferrireductant (e.g., superoxide) and increasing importers (e.g., divalent metal transporter; rectangular box spanning basolateral membrane [left side of cell]) (B). Following import of iron, a new homeostasis is achieved in the respiratory epithelial cells with higher intracellular iron concentrations allowing the cell to function while the particle sequesters some portion of the metal (C). Ferritin (rectangular box in the cell) is increased as a result of elevated cellular concentrations of iron. (A color image is available in the online version.)
A component of WSP responsible for complexation and sequestration of cell iron is likely to be humic acid (Ghio et al., 1994). This is a naturally occurring heterogeneous organic substance, which is ubiquitous and occurs in all terrestrial and aqueous environments. As a result of a variety of acidic functional groups, humic acid complexes metal cations to facilitate their mobilization, transport, and deposition in soils and waters. A variety of cations are bound by humic acid, but of those metals with kinetics favoring complexation, iron is that trace metal in the highest available concentrations. In addition to soils and waters, other sources of humic acid can include coal, coal fly ash, tobacco smoke particulate, oil fly ash, DEPs, ambient air particles, and WSP (Ghio et al., 1996). An oxidative stress is initiated by WSP exposure following sequestration of host cell iron via humic acid. This pilfering of host iron by the humic acid ultimately results in a pulmonary and systemic inflammation.
CYTOTOXIC INSOLUBLE NANOSIZED PARTICLES IN REFERENCE CIGARETTE SMOKE CONDENSATE
Humans have smoked tobacco for centuries. Consequently, there exists a wealth of epidemiological data indicating that cigarette smoking can cause human disease. Repeated human exposure to smoke generated during the combustion of a variety of different materials appear to result in similar physiological responses and pathologies in the lung. For example, though tobacco smoking is clearly an important risk factor for developing COPD, approximately 25–45% of patients with COPD, worldwide, have never smoked tobacco (Salvi and Barnes, 2009). Many of these nontobacco smokers with COPD have been chronically exposed to biomass smoke. Such observations suggest a shared mechanism for the development of combustion material–induced lung disease. Surely, a better understanding of the critical cellular events and smoke constituents that are responsible for combustion material–induced lung disease may lead to novel strategies to reduce the potential harmful effects of smoke exposure.
It is difficult to overstate the chemical complexity of cigarette smoke. The basic principles of cigarette smoke formation and the properties that influence smoke complexity have been well summarized (Baker, 1999; Thielen et al., 2008). Smoke generated from a lit cigarette is a dense aerosol composed of microscopic liquid droplets, known as the particulate, dispersed in a vapor of air and other gases (Fig. 7). The particulate component of cigarette smoke is often referred to as wet total particulate matter (TPM) or cigarette smoke condensate (CSC). There are approximately 10 billion liquid droplet particles per cubic centimeter in mainstream cigarette smoke with particle sizes ranging from 0.1 to 1.0 µm in diameter (0.35 µm, average). Mainstream cigarette smoke is composed of more than 5000 chemicals distributed between gas/vapor phase (GVP) and TPM (Haussmann, 2012).
FIG. 7.
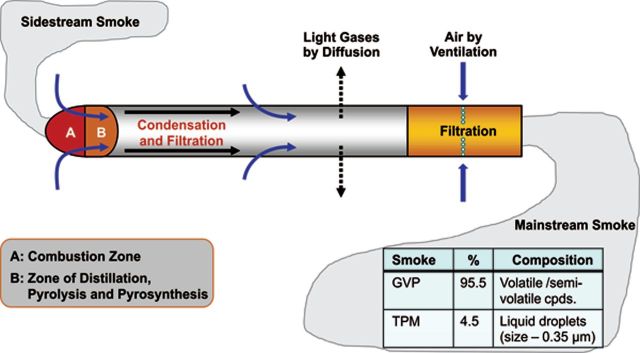
Burning cigarette processes. (Adapted from Thielen et al., 2008. Reproduced with permission of Urban & Fischer Verlang via Copyright Clearance Center.) (A color image is available in the online version.)
Considerable resources over the past half century have been directed at understanding the critical role of chemicals in cigarette smoke on lung injury. Despite these research efforts, the individual smoke constituent(s) responsible for pulmonary disease have yet to be identified. However, it is interesting to note the persistence of oxidative stress and inflammation in the lung, even after prolonged smoking cessation (Brusselle et al., 2011). One possible explanation for these findings is the accumulation of biopersistent material in the lung that may play an important role in cigarette smoke–induced lung disease. Examples of substances reported to accumulate in smokers’ lungs include iron (Ghio et al., 2008; Weinberg, 2009), HLS (Ghio et al., 1994), and elemental carbon (Saxena et al., 2011).
Another possible biopersistent substance to consider may be the presence of insoluble nanotube-like material in CSC. Hajaligol and coworkers (Baliga et al., 2012) observed the formation of nanotube-like structures during tobacco combustion (see Fig. 8). We hypothesized that these structures are insoluble nanotubes that survive combustion and are captured in the liquid droplet particles that make up CSC. To test this hypothesis, we isolated insoluble material from CSC followed by physical and chemical characterization (manuscript in preparation). CSC was produced from 180 research cigarettes (2R4F) smoked mechanically using the FTC method. The CSC underwent multiple extractions with methanol and heptanes, followed by centrifugation at 104,000 × g after each extraction to capture potential nanosized insoluble material. Results from these studies demonstrated that approximately 0.25% of CSC (5.6mg/2.24g CSC) appears to be insoluble material. In fact, the ratio of insoluble material to CSC produced under varying smoking parameters (number of cigarettes, ventilation, and puffing intensity) was remarkably constant at 0.25%.
FIG. 8.

Scanning electron photomicrograph of cigarette coal morphology at the base of the combustion zone. Shreds from the puff burn coal base with a smoking regime of 70cm3 puff volume, 2 s puff duration, and puffed once every minute were extinguished with nitrogen gas, 1 s into the fourth puff. Coal base morphology shows the growth of tube-like projections.
The physical properties of the isolated, insoluble material were determined by transmission electron microscopy. Though nanotube structures were not found, numerous spherical particles with a median diameter of 34nm were observed (Fig. 9). These particles were composed of predominately amorphous carbon; their chemical properties were determined by elemental analysis, fourier transform infrared spectroscopy (FTIR), and electron paramagnetic resonance spectroscopy (EPR). The findings from the elemental analysis and FTIR studies indicated a striking similarity to HLS that was previously reported by Ghio and coworkers to be present in CSC (Ghio et al., 1994). In addition, very stable, carbon-centered radicals were observed in these isolated particles of HLS, as determined by EPR analysis. A similar EPR signal (g value of about 2.0028, consistent with a semiquinone) was previously observed in CSC generated from cigarette smoke (Pryor et al., 1983; Wooten et al., 2006) and from humic acid (Jezierski et al., 2000). The cytotoxicity of these insoluble particles was examined in human BEAS-2B cells in culture. Cell death was observed at levels as low as 2.5 µg/ml and was found as early as 3h following exposure. After 24-h exposure, 100% cell death was observed. In contrast, exposure to 50 µg/ml carbon black particles (16nm) induced approximately 25% cell death after 24-h exposure.
FIG. 9.

Transmission electron photomicrograph of insoluble material isolated from cigarette smoke condensate shows numerous spherical particles with a median diameter of 34nm.
In conclusion, insoluble material has been isolated and quantified from CSC and appears to be composed of HLS and nanosized spherical particles. As previous studies have clearly demonstrated potent toxic characteristics for HLS (Ghio et al., 1996), humic acids (Paciolla et al., 1999; van Eijl et al., 2011), and nanoparticles (Maynard et al., 2012; Renwick et al., 2001), the existence of HLS in the form of nanoparticles in cigarette smoke appears to represent a potential novel mechanistic target for lung injury.
FUTURE DIRECTIONS
The identification of TRP ion channels (i.e., TRPA1, M8, V1, and V4) as cellular sensors for and mediators of discrete, pathologically relevant molecular processes in lung cells represents an advancement in the understanding of how particulate matter might cause adverse effects in the respiratory tract. Much research remains to define a role for TRP channels in the cell and tissue response to combustion-generated particles. Biomarkers reflecting the selective activation of different TRP-mediated events such that the precise contributions of different pathways can be appropriately linked to the composite response in respiratory tissue after exposure to combustion-derived PM should be identified. Cellular expression of TRP channels and variants, including gain- or loss-of-function polymorphisms and splice variants, must be accounted for in assessing cellular responses to a particle. Dynamic changes in the expression of TRP channels and individual variation in activity must be considered in order to establish plausible links between the environment exposure and specific genes and clinical phenotypes.
Modifying the expression and activity of TRP channels in cell and animal models will allow insight into potential therapeutic benefit. Nonspecific agents with a capacity to alter TRP channel activity include mustard oil, capsaicin, and chloroquine. Alternatively, channels can be activated by signaling molecules released by other receptors (e.g., muscarinic, histamine, and Mas-related G-protein coupled receptors). The advent of specific therapeutics that target TRP channels will be instrumental in linking TRP channel function with disease by facilitating animal-based and clinical trials. Combining comprehensive chemical and biochemical, genetic, in vitro cellular, animal, and epidemiological data with clinical characterization of phenotypes attributable to exposure to complex mixtures resulting from incomplete combustion of various materials will confirm or refute the hypothesis that TRP channels, individually or as a group, are important early mediators of processes that culminate in the well-established adverse effects of these materials on human health.
The existence of HLS in the form of nanoparticles in cigarette smoke clearly represents a potentially important mechanistic target for lung injury. However, as this report is the first to describe the existence of HLS nanoparticles (34nm), confirmation by other investigators is required. For example, are these nanosized particles an artifact of the isolation procedure or are they formed in cigarette smoke and deposited in the lung during smoking? Once their presence in cigarette smoke is confirmed, it would be interesting to determine the similarities of these particles to those generated during combustion of other materials (e.g., wood and biomass). Other important questions to answer include whether these HLS nanoparticles play a critical role in smoke-induced lung disease?
To address these questions, determinants of HLS in combustion-derived particles must be defined. Likewise the relationship between HLS and its complexation of host iron with indices of biological effect must be determined. It is possible that sequestration of host iron by this organic compound results in oxidant generation, cell signaling, transcription factor activation, and inflammatory mediator release. If true, the cascade of reactions which culminate in inflammation after WSP, cigarette smoking, and other combustion-generated particles can be interrupted with the provision of excess iron through either oral or inhaled routes. In addition, utilization of HLS in in vitro and in vivo exposures (cell, animal, and human) may function as a model for combustion-derived particles and allow assays for therapeutic interventions.
ACKNOWLEDGMENTS
Funding of Dr Marc Fariss’ effort was provided by Philip Morris USA and Altria Client Services. The authors also acknowledge the editorial assistance of Eileen Y. Ivasauskas of Accuwrit Inc.
REFERENCES
- Aditama T. Y. 2000. Impact of haze from forest fire to respiratory health: Indonesian experience. Respirology 5, 169–174. [DOI] [PubMed] [Google Scholar]
- Agopyan N., Bhatti T., Yu S., Simon S. A. (2003). Vanilloid receptor activation by 2- and 10-µm particles induces responses leading to apoptosis in human airway epithelial cells. Toxicol. Appl. Pharmacol. 192, 21–35. [DOI] [PubMed] [Google Scholar]
- Agopyan N., Li L., Yu S., Simon S. A. (2003). Negatively charged 2- and 10-µm particles activate vanilloid receptors, increase cAMP, and induce cytokine release. Toxicol. Appl. Pharmacol. 186, 63–76. [DOI] [PubMed] [Google Scholar]
- Baker R. R. 1999. Smoke chemistry. In Tobacco: Production, Chemistry and Technology (Davis L. D., Nielson M. T, Eds.), pp. 308–439. Blackwell, London. [Google Scholar]
- Baliga V. L., Thurston M. E., Miser D. E., Sharma R. K., Chan W. G., Hajalingol M. R. 2012. Physical characterization of the cigarette coal: Part II. Puff burn. J. Anal. Appl. Pyrolysis 72, 83–96. [Google Scholar]
- Bayram H., Devalia J. L., Sapsford R. J., Ohtoshi T., Miyabara Y., Sagai M., Davies R. J. 1998. The effect of diesel exhaust particles on cell function and release of inflammatory mediators from human bronchial epithelial cells in vitro. Am. J. Respir. Cell Mol. Biol. 18, 441–448. [DOI] [PubMed] [Google Scholar]
- Bayram H., Ito K., Issa R., Ito M., Sukkar M., Chung K. F. 2006. Regulation of human lung epithelial cell numbers by diesel exhaust particles. Eur. Respir. J. 27, 705–713. [DOI] [PubMed] [Google Scholar]
- Boland S., Baeza-Squiban A., Fournier T., Houcine O., Gendron M. C., Chevrier M., Jouvenot G., Coste A., Aubier M., Marano F. 1999. Diesel exhaust particles are taken up by human airway epithelial cells in vitro and alter cytokine production. Am. J. Physiol. 276, L604–L613. [DOI] [PubMed] [Google Scholar]
- Bonvallot V., Baeza-Squiban A., Baulig A., Brulant S., Boland S., Muzeau F., Barouki R., Marano F. 2001. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am. J. Respir. Cell Mol. Biol. 25, 515–521. [DOI] [PubMed] [Google Scholar]
- Bruce N., Perez-Padilla R., Albalak R. 2000. Indoor air pollution in developing countries: A major environmental and public health challenge. Bull. World Health Organ. 78, 1078–1092. [PMC free article] [PubMed] [Google Scholar]
- Brusselle G. G., Joos G. F., Bracke K. R. 2011. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 378, 1015–1026. [DOI] [PubMed] [Google Scholar]
- Campen M. J., Lund A. K., Doyle-Eisele M. L., McDonald J. D., Knuckles T. L., Rohr A. C., Knipping E. M., Mauderly J. L. 2010. A comparison of vascular effects from complex and individual air pollutants indicates a role for monoxide gases and volatile hydrocarbons. Environ. Health Perspect. 118, 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero-Recasens G., Gonzalez J. R., Fandos C., Duran-Tauleria E., Smit L. A., Kauffmann F., Anto J. M., Valverde M. A. 2010. Loss of function of transient receptor potential vanilloid 1 (TRPV1) genetic variant is associated with lower risk of active childhood asthma. J. Biol. Chem. 285, 27532–27535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D., Tal T. L., Graves L. M., Gilmour I., Linak W., Reed W., Bromberg P. A., Samet J. M. 2007. Diesel exhaust particulate-induced activation of Stat3 requires activities of EGFR and Src in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L422–L429. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. 2012. CDC Tobacco Fact Sheet Available at: http://www.cdc.gov/tobacco/data_statistics/fact_sheets/health_effects/effects_cig_smoking Accessed December 20, 2012
- Chen R., Tunstall-Pedoe H., Tavendale R. 2001. Environmental tobacco smoke and lung function in employees who never smoked: The Scottish MONICA study. Occup. Environ. Med. 58, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilmonczyk B. A., Salmun L. M., Megathlin K. N., Neveux L. M., Palomaki G. E., Knight G. J., Pulkkinen A. J., Haddow J. E. 1993. Association between exposure to environmental tobacco smoke and exacerbations of asthma in children. N. Engl. J. Med. 328, 1665–1669. [DOI] [PubMed] [Google Scholar]
- Chung K. F. 2001. Cytokines in chronic obstructive pulmonary disease. Eur. Respir. J. Suppl. 34, 50s–59s. [PubMed] [Google Scholar]
- Costa S. K., Teles A. M., Kumagai Y., Brain S. D., Teixeira S. A., Varriano A. A., Barreto M. A., de Lima W. T., Antunes E., Muscará M. N, et al. 2010. Involvement of sensory nerves and TRPV1 receptors in the rat airway inflammatory response to two environment pollutants: Diesel exhaust particles (DEP) and 1,2-naphthoquinone (1,2-NQ). Arch. Toxicol. 84, 109–117. [DOI] [PubMed] [Google Scholar]
- Daniel E. E., O’Byrne P. 1991. Effect of inflammatory mediators on airway nerves and muscle. Am. Rev. Respir. Dis. 143(3 Pt 2), S3–S5. [DOI] [PubMed] [Google Scholar]
- Deering-Rice C. E., Johansen M. E., Roberts J. K., Thomas K. C., Romero E. G., Lee J., Yost G. S., Veranth J. M., Reilly C. A. 2012. Transient receptor potential vanilloid-1 (TRPV1) is a mediator of lung toxicity for coal fly ash particulate material. Mol. Pharmacol. 81, 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deering-Rice C. E., Romero E. G., Shapiro D., Hughen R. W., Light A. R., Yost G. S., Veranth J. M., Reilly C. A. 2011. Electrophilic components of diesel exhaust particles (DEP) activate transient receptor potential ankyrin-1 (TRPA1): A probable mechanism of acute pulmonary toxicity for DEP. Chem. Res. Toxicol. 24, 950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis R. J., Maldonado D., Norman S., Baena E., Martinez G. 1996. Woodsmoke exposure and risk for obstructive airways disease among women. Chest 109, 115–119. [DOI] [PubMed] [Google Scholar]
- Donaldson K., Gilmour M. I., MacNee W. 2000. Asthma and PM10. Respir. Res. 1, 12–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doornaert B., Leblond V., Galiacy S., Gras G., Planus E., Laurent V., Isabey D., Lafuma C. 2003. Negative impact of DEP exposure on human airway epithelial cell adhesion, stiffness, and repair. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L119–L132. [DOI] [PubMed] [Google Scholar]
- Ezzati M., Kammen D. 2001. Indoor air pollution from biomass combustion and acute respiratory infections in Kenya: An exposure-response study. Lancet 358, 619–624. [DOI] [PubMed] [Google Scholar]
- Fahy O., Hammad H., Sénéchal S., Pestel J., Tonnel A. B., Wallaert B., Tsicopoulos A. 2000. Synergistic effect of diesel organic extracts and allergen Der p 1 on the release of chemokines by peripheral blood mononuclear cells from allergic subjects: Involvement of the map kinase pathway. Am. J. Respir. Cell Mol. Biol. 23, 247–254. [DOI] [PubMed] [Google Scholar]
- Fann N., Lamson A. D., Anenberg S. C., Wesson K., Risley D., Hubbell B. J. 2012. Estimating the national public health burden associated with exposure to ambient PM2.5 and ozone. Risk Anal. 32, 81–95. [DOI] [PubMed] [Google Scholar]
- Gamble J., Jones W., Minshall S. 1987. Epidemiological-environmental study of diesel bus garage workers: Chronic effects of diesel exhaust on the respiratory system. Environ. Res. 44, 6–17. [DOI] [PubMed] [Google Scholar]
- Ghelfi E., Rhoden C. R., Wellenius G. A., Lawrence J., Gonzalez-Flecha B. 2008. Cardiac oxidative stress and electrophysiological changes in rats exposed to concentrated ambient particles are mediated by TRP-dependent pulmonary reflexes. Toxicol. Sci. 102, 328–336. [DOI] [PubMed] [Google Scholar]
- Ghio A. J., Carraway M. S., Madden M. C. (2012). Composition of air pollution particles and oxidative stress in cells, tissues, and living systems. J. Toxicol. Environ. Health. B. Crit. Rev. 15, 1–21. [DOI] [PubMed] [Google Scholar]
- Ghio A. J., Churg A., Roggli V. L. 2004. Ferruginous bodies: Implications in the mechanism of fiber and particle toxicity. Toxicol. Pathol. 32, 643–649. [DOI] [PubMed] [Google Scholar]
- Ghio A. J., Hilborn E. D., Stonehuerner J. G., Dailey L. A., Carter J. D., Richards J. H., Crissman K. M., Foronjy R. F., Uyeminami D. L., Pinkerton K. E. 2008. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am. J. Respir. Crit. Care Med. 178, 1130–1138. [DOI] [PubMed] [Google Scholar]
- Ghio A. J., Richards J. H., Carter J. D., Madden M. C. 2000. Accumulation of iron in the rat lung after tracheal instillation of diesel particles. Toxicol. Pathol. 28, 619–627. [DOI] [PubMed] [Google Scholar]
- Ghio A. J., Soukup J. M., Case M., Dailey L. A., Richards J., Berntsen J., Devlin R. B., Stone S., Rappold A. (2012). Exposure to wood smoke particles produces inflammation in healthy volunteers. Occup. Environ. Med. 69, 170–175. [DOI] [PubMed] [Google Scholar]
- Ghio A. J., Stonehuerner J., Pritchard R. J., Piantadosi C. A., Quigley D. R., Dreher K. L., Costa D. L. 1996. Humic-like substances in air pollution particulates correlate with concentrations of transition metals and oxidant generation. Inhal. Toxicol. 8, 479–494. [Google Scholar]
- Ghio A. J., Stonehuerner J., Quigley D. R. 1994. Humic-like substances in cigarette smoke condensate and lung tissue of smokers. Am. J. Physiol. 266(4 Pt 1), L382–L388. [DOI] [PubMed] [Google Scholar]
- Glantz S. A., Parmley W. W. 1995. Passive smoking and heart disease. Mechanisms and risk. JAMA 273, 1047–1053. [PubMed] [Google Scholar]
- Hashimoto S., Gon Y., Takeshita I., Matsumoto K., Jibiki I., Takizawa H., Kudoh S., Horie T. 2000. Diesel exhaust particles activate p38 MAP kinase to produce interleukin 8 and RANTES by human bronchial epithelial cells and N-acetylcysteine attenuates p38 MAP kinase activation. Am. J. Respir. Crit. Care Med. 161, 280–285. [DOI] [PubMed] [Google Scholar]
- Haussmann H. J. 2012. Use of hazard indices for a theoretical evaluation of cigarette smoke composition. Chem. Res. Toxicol. 25, 794–810. [DOI] [PubMed] [Google Scholar]
- Havas S., Wozenski S., Deprez R., Miller L., Charman R., Hamrell M., Green L., Benn S. 1989. Report of the New England task force on reducing heart disease and stroke risk. Public Health Rep. 104, 134–142. [PMC free article] [PubMed] [Google Scholar]
- Herr C., Beisswenger C., Hess C., Kandler K., Suttorp N., Welte T., Schroeder J. M., Vogelmeier C. R Bals for the CAPNETZ Study Group. 2009. Suppression of pulmonary innate host defence in smokers. Thorax 64, 144–149. [DOI] [PubMed] [Google Scholar]
- Jarvis D., Chinn S., Luczynska C., Burney P. 1996. Association of respiratory symptoms and lung function in young adults with use of domestic gas appliances. Lancet 347, 426–431. [DOI] [PubMed] [Google Scholar]
- Jezierski A., Czechowski F., Jerzykiewicz M., Chen Y., Drozd J. 2000. Electron paramagnetic resonance (EPR) studies on stable and transient radicals in humic acids from compost, soil, peat and brown coal. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 56A, 379–385. [DOI] [PubMed] [Google Scholar]
- Jinot J., Bayard S. 1996. Respiratory health effects of exposure to environmental tobacco smoke. Rev. Environ. Health 11, 89–100. [DOI] [PubMed] [Google Scholar]
- Joos L., He J. Q., Shepherdson M. B., Connett J. E., Anthonisen N. R., Paré P. D., Sandford A. J. 2002. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum. Mol. Genet. 11, 569–576. [DOI] [PubMed] [Google Scholar]
- Kocbach Bølling A., Pagels J., Yttri K. E., Barregard L., Sallsten G., Schwarze P. E., Boman C. 2009. Health effects of residential wood smoke particles: The importance of combustion conditions and physicochemical particle properties. Part. Fibre Toxicol. 6, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig J. Q., Larson T. V., Hanley Q. S., Rebolledo V., Dumler K., Checkoway H., Wang S. Z., Lin D., Pierson W. E. 1993. Pulmonary function changes in children associated with fine particulate matter. Environ. Res. 63, 26–38. [DOI] [PubMed] [Google Scholar]
- Koerten H. K., Brederoo P., Ginsel L. A., Daems W. T. 1986. The endocytosis of asbestos by mouse peritoneal macrophages and its long-term effect on iron accumulation and labyrinth formation. Eur. J. Cell Biol. 40, 25–36. [PubMed] [Google Scholar]
- Koike E., Hirano S., Furuyama A., Kobayashi T. 2004. cDNA microarray analysis of rat alveolar epithelial cells following exposure to organic extract of diesel exhaust particles. Toxicol. Appl. Pharmacol. 201, 178–185. [DOI] [PubMed] [Google Scholar]
- Larson T. V., Koenig J. Q. 1994. Wood smoke: Emissions and noncancer respiratory effects. Annu. Rev. Public Health 15, 133–156. [DOI] [PubMed] [Google Scholar]
- Li J., Ghio A. J., Cho S. H., Brinckerhoff C. E., Simon S. A., Liedtke W. 2009. Diesel exhaust particles activate the matrix-metalloproteinase-1 gene in human bronchial epithelia in a beta-arrestin-dependent manner via activation of RAS. Environ. Health Perspect. 117, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Kanju P., Patterson M., Chew W. L., Cho S. H., Gilmour I., Oliver T., Yasuda R., Ghio A., Simon S. A, et al. 2011. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ. Health Perspect. 119, 784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lighty J. S., Veranth J. M., Sarofim A. F. 2000. Combustion aerosols: Factors governing their size and composition and implications to human health. J. Air Waste Manag. Assoc. 50, 1565–618; discussion 1619. [DOI] [PubMed] [Google Scholar]
- Lorenzo I. M., Liedtke W., Sanderson M. J., Valverde M. A. 2008. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 105, 12611–12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May D., Baastrup J., Nientit M. R., Binder A., Schünke M., Baron R., Cascorbi I. 2012. Differential expression and functionality of TRPA1 protein genetic variants in conditions of thermal stimulation. J. Biol. Chem. 287, 27087–27094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard R. L., Donaldson K., Tetley T. D. 2012. Type 1 pulmonary epithelial cells: a new compartment involved in the slow phase of particle clearance from alveoli. Nanotoxicology, 10.3109/17435390.2012.658446. [DOI] [PubMed]
- McClellan R. O. 1987. Health effects of exposure to diesel exhaust particles. Annu. Rev. Pharmacol. Toxicol. 27, 279–300. [DOI] [PubMed] [Google Scholar]
- McClellan R. O., Mauderly J. L., Jones R. K., Cuddihy R. G. 1985. Health effects of diesel exhaust. A contemporary air pollution issue. Postgrad. Med. 78, 199–201, 204. [DOI] [PubMed] [Google Scholar]
- McDonald J. D., Campen M. J., Harrod K. S., Seagrave J., Seilkop S. K., Mauderly J. L. 2011. Engine-operating load influences diesel exhaust composition and cardiopulmonary and immune responses. Environ. Health Perspect. 119, 1136–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melius J. M. 1995. Cardiovascular disease among firefighters. Occup. Med. 10, 821–827. [PubMed] [Google Scholar]
- Morgan W. K., Reger R. B., Tucker D. M. 1997. Health effects of diesel emissions. Ann. Occup. Hyg. 41, 643–658. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay I., Gomes P., Aranake S., Shetty M., Karnik P., Damle M., Kuruganti S., Thorat S., Khairatkar-Joshi N. 2011. Expression of functional TRPA1 receptor on human lung fibroblast and epithelial cells. J. Recept. Signal Transduct. Res. 31, 350–358. [DOI] [PubMed] [Google Scholar]
- Naeher L. P., Brauer M., Lipsett M., Zelikoff J. T., Simpson C. D., Koenig J. Q., Smith K. R. 2007. Woodsmoke health effects: A review. Inhal. Toxicol. 19, 67–106. [DOI] [PubMed] [Google Scholar]
- Nassini R., Pedretti P., Moretto N., Fusi C., Carnini C., Facchinetti F., Viscomi A. R., Pisano A. R., Stokesberry S., Brunmark C, et al. 2012. Transient receptor potential ankyrin 1 channel localized to non-neuronal airway cells promotes non-neurogenic inflammation. PLoS ONE 7, e42454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikula K. J., Snipes M. B., Barr E. B., Griffith W. C., Henderson R. F., Mauderly J. L. 1995. Comparative pulmonary toxicities and carcinogenicities of chronically inhaled diesel exhaust and carbon black in F344 rats. Fundam. Appl. Toxicol. 25, 80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oortgiesen M., Veronesi B., Eichenbaum G., Kiser P. F., Simon S. A. 2000. Residual oil fly ash and charged polymers activate epithelial cells and nociceptive sensory neurons. Am. J. Physiol. Lung Cell Mol. Physiol. 278, L683–L695. [DOI] [PubMed] [Google Scholar]
- Ozbay B., Uzun K., Arslan H., Zehir I. 2001. Functional and radiological impairment in women highly exposed to indoor biomass fuels. Respirology 6, 255–258. [DOI] [PubMed] [Google Scholar]
- Paciolla M. D., Davies G., Jansen S. A. 1999. Generation of hydroxyl radicals from metal-loaded humic acids. Environ. Sci. Technol. 33, 1814–1818. [Google Scholar]
- Parent M. E., Rousseau M. C., Boffetta P., Cohen A., Siemiatycki J. 2007. Exposure to diesel and gasoline engine emissions and the risk of lung cancer. Am. J. Epidemiol. 165, 53–62. [DOI] [PubMed] [Google Scholar]
- Pekkanen J., Brunner E. J., Anderson H. R., Tiittanen P., Atkinson R. W. 2000. Daily concentrations of air pollution and plasma fibrinogen in London. Occup. Environ. Med. 57, 818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Padilla R., Regalado J., Vedal S., Paré P., Chapela R., Sansores R., Selman M. 1996. Exposure to biomass smoke and chronic airway disease in Mexican women. A case-control study. Am. J. Respir. Crit. Care Med. 154(3 Pt 1), 701–706. [DOI] [PubMed] [Google Scholar]
- Pinkerton K. E., Green F. H., Saiki C., Vallyathan V., Plopper C. G., Gopal V., Hung D., Bahne E. B., Lin S. S., Ménache M. G, et al. 2000. Distribution of particulate matter and tissue remodeling in the human lung. Environ. Health Perspect. 108, 1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C. A., 3rd, Dockery D. W., Spengler J. D., Raizenne M. E. 1991. Respiratory health and PM10 pollution. A daily time series analysis. Am. Rev. Respir. Dis. 144(3 Pt 1), 668–674. [DOI] [PubMed] [Google Scholar]
- Pryor W. A., Prier D. G., Church D. F. 1983. Electron-spin resonance study of mainstream and sidestream cigarette smoke: Nature of the free radicals in gas-phase smoke and in cigarette tar. Environ. Health Perspect. 47, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappold A. G., Stone S. L., Cascio W. E., Neas L. M., Kilaru V. J., Carraway M. S., Szykman J. J., Ising A., Cleve W. E., Meredith J. T, et al. 2011. Peat bog wildfire smoke exposure in rural North Carolina is associated with cardiopulmonary emergency department visits assessed through syndromic surveillance. Environ. Health Perspect. 119, 1415–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly C. A., Johansen M. E., Lanza D. L., Lee J., Lim J. O., Yost G. S. 2005. Calcium-dependent and independent mechanisms of capsaicin receptor (TRPV1)-mediated cytokine production and cell death in human bronchial epithelial cells. J. Biochem. Mol. Toxicol. 19, 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly C. A., Taylor J. L., Lanza D. L., Carr B. A., Crouch D. J., Yost G. S. 2003. Capsaicinoids cause inflammation and epithelial cell death through activation of vanilloid receptors. Toxicol. Sci. 73, 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renwick L. C., Donaldson K., Clouter A. 2001. Impairment of alveolar macrophage phagocytosis by ultrafine particles. Toxicol. Appl. Pharmacol. 172, 119–127. [DOI] [PubMed] [Google Scholar]
- Riojas-Rodríguez H., Romano-Riquer P., Santos-Burgoa C., Smith K. R. 2001. Household firewood use and the health of children and women of Indian communities in Chiapas, Mexico. Int. J. Occup. Environ. Health 7, 44–53. [DOI] [PubMed] [Google Scholar]
- Salvi S. S., Barnes P. J. 2009. Chronic obstructive pulmonary disease in non-smokers. Lancet 374, 733–743. [DOI] [PubMed] [Google Scholar]
- Saxena R. K., McClure M. E., Hays M. D., Green F. H., McPhee L. J., Vallyathan V., Gilmour M. I. 2011. Quantitative assessment of elemental carbon in the lungs of never smokers, cigarette smokers, and coal miners. J. Toxicol. Environ. Health Part A 74, 706–715. [DOI] [PubMed] [Google Scholar]
- Schütz A., Schneidenbach D., Aust G., Tannapfel A., Steinert M., Wittekind C. 2002. Differential expression and activity status of MMP-1, MMP-2 and MMP-9 in tumor and stromal cells of squamous cell carcinomas of the lung. Tumour Biol. 23, 179–184. [DOI] [PubMed] [Google Scholar]
- Seaton A., Soutar A., Crawford V., Elton R., McNerlan S., Cherrie J., Watt M., Agius R., Stout R. 1999. Particulate air pollution and the blood. Thorax 54, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura-Valdez L., Pardo A., Gaxiola M., Uhal B. D., Becerril C., Selman M. 2000. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 117, 684–694. [DOI] [PubMed] [Google Scholar]
- Seilkop S. K., Campen M. J., Lund A. K., McDonald J. D., Mauderly J. L. 2012. Identification of chemical components of combustion emissions that affect pro-artherosclerotic vascular responses in mice. Inhal. Toxicol. 24, 270–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Sethi G. R., Rohtagi A., Chaudhary A., Shankar R., Bapna J. S., Joshi V., Sapir D. G. 1998. Indoor air quality and acute lower respiratory infection in Indian urban slums. Environ. Health Perspect. 106, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard D., Distefano S., Morse L., Becker C. 1986. Acute effects of routine firefighting on lung function. Am. J. Ind. Med. 9, 333–340. [DOI] [PubMed] [Google Scholar]
- Sherman C. B., Barnhart S., Miller M. F., Segal M. R., Aitken M., Schoene R., Daniell W., Rosenstock L. 1989. Firefighting acutely increases airway responsiveness. Am. Rev. Respir. Dis. 140, 185–190. [DOI] [PubMed] [Google Scholar]
- Smit L. A., Kogevinas M., Antó J. M., Bouzigon E., González J. R., Le Moual N., Kromhout H., Carsin A. E., Pin I., Jarvis D, et al. 2012. Transient receptor potential genes, smoking, occupational exposures and cough in adults. Respir. Res. 13, 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydbom A., Blomberg A., Parnia S., Stenfors N., Sandström T., Dahlén S. E. 2001. Health effects of diesel exhaust emissions. Eur. Respir. J. 17, 733–746. [DOI] [PubMed] [Google Scholar]
- Takizawa H., Abe S., Okazaki H., Kohyama T., Sugawara I., Saito Y., Ohtoshi T., Kawasaki S., Desaki M., Nakahara K, et al. 2003. Diesel exhaust particles upregulate eotaxin gene expression in human bronchial epithelial cells via nuclear factor-kappa B-dependent pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L1055–L1062. [DOI] [PubMed] [Google Scholar]
- Takizawa H., Ohtoshi T., Kawasaki S., Kohyama T., Desaki M., Kasama T., Kobayashi K., Nakahara K., Yamamoto K., Matsushima K, et al. 1999. Diesel exhaust particles induce NF-kappa B activation in human bronchial epithelial cells in vitro: Importance in cytokine transcription. J. Immunol. 162, 4705–4711. [PubMed] [Google Scholar]
- Tal T. L., Simmons S. O., Silbajoris R., Dailey L., Cho S. H., Ramabhadran R., Linak W., Reed W., Bromberg P. A., Samet J. M. 2010. Differential transcriptional regulation of IL-8 expression by human airway epithelial cells exposed to diesel exhaust particles. Toxicol. Appl. Pharmacol. 243, 46–54. [DOI] [PubMed] [Google Scholar]
- Tan W. C., Qiu D., Liam B. L., Ng T. P., Lee S. H., van Eeden S. F., D’Yachkova Y., Hogg J. C. 2000. The human bone marrow response to acute air pollution caused by forest fires. Am. J. Respir. Crit. Care Med. 161(4 Pt 1)1213–1217. [DOI] [PubMed] [Google Scholar]
- Thielen A., Klus H., Müller L. 2008. Tobacco smoke: Unraveling a controversial subject. Exp. Toxicol. Pathol. 60, 141–156. [DOI] [PubMed] [Google Scholar]
- Thomas K. C., Roberts J. K., Deering-Rice C. E., Romero E. G., Dull R. O., Lee J., Yost G. S., Reilly C. A. 2012. Contributions of TRPV1, endovanilloids, and endoplasmic reticulum stress in lung cell death in vitro and lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 302, L111–L119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totlandsdal A. I., Cassee F. R., Schwarze P., Refsnes M., Låg M. 2010. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Part. Fibre Toxicol. 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency. 2012. EPA National Emissions Inventory Available at: http://www.epa.gov/ttnchie1/net/2005inventory.html Accessed December 20, 2012 [PubMed]
- van Eeden S. F., Tan W. C., Suwa T., Mukae H., Terashima T., Fujii T., Qui D., Vincent R., Hogg J. C. 2001. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)). Am. J. Respir. Crit. Care Med. 164, 826–830. [DOI] [PubMed] [Google Scholar]
- van Eijl S., Mortaz E., Ferreira A. F., Kuper F., Nijkamp F. P., Folkerts G., Bloksma N. 2011. Humic acid enhances cigarette smoke-induced lung emphysema in mice and IL-8 release of human monocytes. Pulm. Pharmacol. Ther. 24, 682–689. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K., Montell C. 2007. TRP channels. Annu. Rev. Biochem. 76, 387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veronesi B., Carter J. D., Devlin R. B., Simon S. A., Oortgiesen M. (1999). Neuropeptides and capsaicin stimulate the release of inflammatory cytokines in a human bronchial epithelial cell line. Neuropeptides 33, 447–456. [DOI] [PubMed] [Google Scholar]
- Veronesi B., Oortgiesen M., Carter J. D., Devlin R. B. (1999). Particulate matter initiates inflammatory cytokine release by activation of capsaicin and acid receptors in a human bronchial epithelial cell line. Toxicol. Appl. Pharmacol. 154, 106–115. [DOI] [PubMed] [Google Scholar]
- Voets T., Talavera K., Owsianik G., Nilius B. 2005. Sensing with TRP channels. Nat. Chem. Biol. 1, 85–92. [DOI] [PubMed] [Google Scholar]
- Vriens J., Appendino G., Nilius B. 2009. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 75, 1262–1279. [DOI] [PubMed] [Google Scholar]
- Weinberg E. D. 2009. Tobacco smoke iron: An initiator/promoter of multiple diseases. Biometals 22, 207–210. [DOI] [PubMed] [Google Scholar]
- Willis D. N., Liu B., Ha M. A., Jordt S. E., Morris J. B. 2011. Menthol attenuates respiratory irritation responses to multiple cigarette smoke irritants. FASEB J. 25, 4434–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. S., Sun N. N., Keith I., Kweon C. B., Foster D. E., Schauer J. J., Witten M. L. 2003. Tachykinin substance P signaling involved in diesel exhaust-induced bronchopulmonary neurogenic inflammation in rats. Arch. Toxicol. 77, 638–650. [DOI] [PubMed] [Google Scholar]
- Wooten J. B., Chouchane S., McGrath T. E. 2006. Tobacco smoke constituents affecting oxidative stress. In Cigarette Smoke and Oxidative Stress (Halliwell B. B., Poulsen H. E, Eds.), pp. 5–46. Springer, Germany. [Google Scholar]
- Zhang J., Smith K. R. 2003. Indoor air pollution: A global health concern. Br. Med. Bull. 68, 209–225. [DOI] [PubMed] [Google Scholar]
- Zhu G., Gulsvik A., Bakke P., Ghatta S., Anderson W., Lomas D. A., Silverman E. K., Pillai S. G. ICGN Investigators. 2009. Association of TRPV4 gene polymorphisms with chronic obstructive pulmonary disease. Hum. Mol. Genet. 18, 2053–2062. [DOI] [PubMed] [Google Scholar]