

Abstract
Photolyase, a class of flavoproteins, uses blue light to repair two types of ultraviolet-induced DNA damage, cyclobutane pyrimidine dimer (CPD) and pyrimidine-pyrimidone (6–4) photoproduct (6–4PP). In this perspective, we review the recent progress on the repair dynamics and mechanisms of both types of DNA restoration by photolyases. We first report the spectroscopic characterization of flavin in various redox states and the active-site solvation dynamics in photolyases. We then systematically summarize the detailed repair dynamics of damaged DNA by photolyases and a biomimetic system through resolving all elementary steps on the ultrafast timescales, including multiple intermolecular electron- and proton-transfer reactions and bond-breaking and -making processes. We determined the unique electron tunneling pathways, identified the key functional residues and revealed the molecular origin of high repair efficiency, and thus elucidate the molecular mechanisms and repair photocycles at the most fundamental level. We finally conclude that the active sites of photolyases, unlike aqueous solution for the biomimetic system, provide a unique electrostatic environment and local flexibility and thus a dedicated synergy for all elementary dynamics to maximize the repair efficiency. This repair photomachine is the first enzyme that the entire functional evolution is completely mapped out in real time.
1. Introduction
The ultraviolet (UV) irradiation in sunlight can cause damages of DNA by inducing formation of a cyclobutane pyrimidine dimer (CPD, ~80%) and a less-frequently pyrimidine-pyrimidone (6–4) photoproduct (6–4PP, ~20%) (Fig. 1).1 Both photoproducts lead to mutagenesis and eventually to skin cancer.2–4 Photolyase, a class of flavoproteins, restores damaged DNA through absorption of blue light.1,5–7 Two different kinds of photolyases, usually classified as CPD photolyase and (6–4) photolyase based on their functions, share similar primary sequences and folding structures but a photolyase repairs one photoproduct and cannot repair another. Both photolyases contain a noncovalently bound, fully reduced flavin adenine dinucleotide (FADH−) molecule as the active cofactor. In the recent years, the crystal structures of photolyases,8–13 especially of the enzyme-substrate (CPD or 6–4PP) complexes, were solved (Fig. 2),9,10 and the FADH− cofactor adopts an unusual U-shape folding configuration in the active sites of both enzymes.
Fig. 1.

Chemical structures of undamaged thymine bases and damaged DNA photolesions of cyclobutane pyrimidine dimer (CPD) and 6–4 photoproduct (6–4PP).
Fig. 2.

Crystal structures of CPD photolyase and 6–4 photolyase. (A) X-ray complex structure of A. nidulans photolyase with DNA containing a repaired photoproduct of thymine dimer. E. coli photolyase has a similar structure. The thymine dimer is flipped out of DNA and inserted into the active site. (B) A close-up view shows the relative positions of the catalytic cofactor FADH− and the repaired substrate with the electron tunneling pathways in repair. (C) X-ray structure of D. melanogaster (6–4) photolyase (blue) bound to DNA (yellow) containing a (6–4) photoproduct. A. thaliana photolyase has a similar structure and a conserved histidine residue in the active site (His 364 in A. thaliana and His 365 in D. melanogaster).The 6–4PP is flipped out of DNA and inserted into the active site. (D) The close-up view shows the relative positions of the catalytic cofactor FADH−, the conserved His 364 (His 365) residue and the 6–4PP substrate.
The mechanism of CPD photorepair has been proposed and examined in the past thirty years1,14–17 until we resolved a cyclic electron-transfer (ET) reaction photocycle in 2005 with femtosecond (fs) spectroscopy.18 On the other side, several hypotheses of 6–4PP repair were proposed19–26 but the detailed mechanism remained elusive until very recently we reported an ET-induced proton transfer photocycle in 2010.27 In this review, we first give our recent characterization of the steady-state spectra of flavin at various redox states,28 important to understanding the flavin properties in photolyase and other flavoprotiens.29,30 We then report the active-site solvation dynamics in photolyases,31 a critical factor in understanding of the repair reactions and high efficiency. With single-residue spatial resolution, we subsequently present the complete mapping of the entire dynamic functional evolution from the reactants, to various intermediates and to the final products in real time and thus elucidate the complete repair photocycles for both CPD and 6–4PP photolyases.32–35 We also present the dynamics of a similar CPD biomimetic system but with low repair efficiency36 and reveal the molecular mechanism of the high repair quantum yield by CPD photolyases.35,37
2. Absorption and emission spectra of flavins in various redox states
The flavin molecule is one of the most important cofactors in enzymatic functions.38–47 Flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) are the most commonly occurring flavins in flavoproteins. Flavin can have three different redox states: oxidized form, one-electron reduced radical semiquinone, and two-electron fully reduced hydroquinone. Semiquinone and hydroquinone have pKa values of 8.3 and 6.7, respectively,40 and can be present in their neutral or anionic forms under physiological conditions. Among the five redox forms, two redox pairs, oxidized flavin/anionic semiquinone (FAD/FAD•−) and neutral semiquinone/anionic hydroquinone (FADH•/FADH−), are often involved in intermolecular ET reactions. Because of flavin’s chemical versatility, flavoproteins are ubiquitous and participate in a broad spectrum of biological activities,42,46–51 including light-driven DNA repair in photolyase1,18,27,32–34 and signal transduction in blue-light photoreceptors.52–59
The steady-state spectroscopic properties of these redox flavins have been extensively studied, especially their absorption spectra in different proteins and solution.41,42,60–64 On the other side, the emission spectra of flavoproteins or flavins in solvent have not been well characterized until we reported recently.28,65 Fig. 3 shows the steady-state absorption and emission spectra of flavin cofactors in various redox states and environments. Oxidized FAD in solution exhibits two broad absorption bands with peaks at 450 nm for S0←S1 and at 375 nm for S0←S266 while the oxidized FMN enclosed in the flavodoxin mutant shows some peak features at 476, 454, and 432 nm for S0←S1 and with some red shift in wavelength for S0←S2. The emission peak of oxidized flavin is around 530 nm in solution and shifts to the blue side in a hydrophobic protein environment such as in flavodoxin (Fig. 3A).67,68 It has been reported that the fluorescence intensity of excited FAD in solution is about 10-fold weaker than that of excited riboflavin or FMN,69–71 owing to intramolecular ET quenching with a stacked conformation between the isoalloxazine ring and adenine moiety. Numerous experimental and theoretical studies69–78 suggested that two FAD conformations, open and stacked, exist in solution (Fig. 3A), while this cofactor takes a folding configuration in both photolyase and cryptochrome.8–13,79,80
Fig. 3.
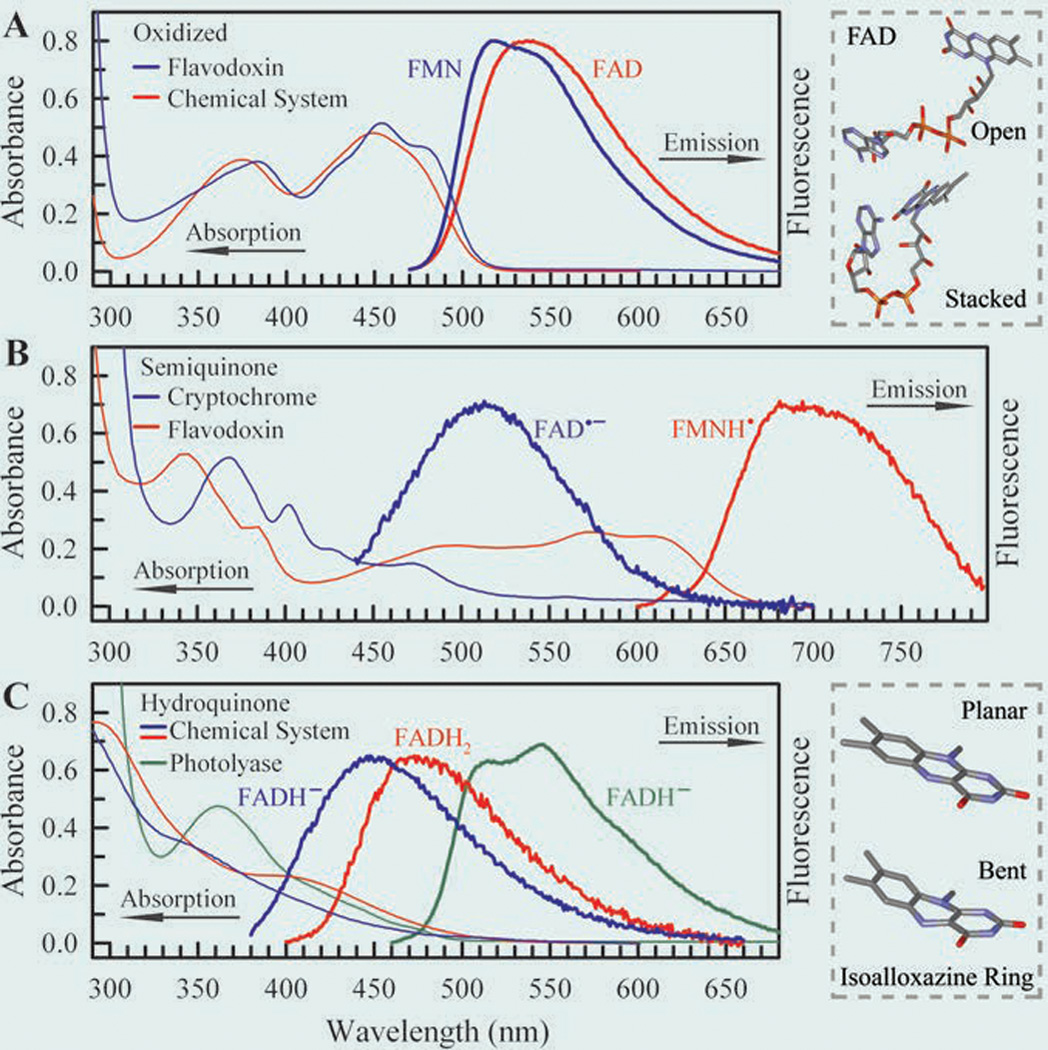
Steady-state absorption and emission spectra of flavins in five redox states.28 (A) Oxidized state, (B) semiquinone radical state, and (C) fully reduced hydroquinone state. The excitation wavelengths are 400 nm for oxidized FAD and FMN, 420 nm for anionic radical FAD•−, 580 nm for neutral radical FMNH•, and 360 nm for fully reduced FADH− and FADH2. The fluorescence emissions of FAD•− in the cryptochrome and FADH− and FADH2 in solution are very weak. Upper right frame: Schematic representation of FAD in open and stacked conformations. The stacked conformation was adopted from U-shaped FAD in photolyase. Lower right frame: Schematic representation of the isoalloxazine ring in planar and “butterfly” bent conformations.
Fig. 3B shows the absorption and emission spectra of stable radical semiquinones in two flavoproteins. The radicals are not stable in solution. Absorption of the neutral radical FMNH• in flavodoxin mutant extends until 700 nm, and the previous study has assigned the absorption between 650–555 nm as D0←D1 and the absorption between 525–410 nm as the D0←D2 transitions in neutral semiquinoid flavin of photolyase.81 A weak, broad emission spectrum peaking at 700 nm was recently observed (Fig. 3B). We also observed a similar fluorescence profile for FADH• in photolyase with a peak at 715 nm. The anionic radical FAD•− in insect type 1 cryptochrome also gives a weak emission spectrum with a peak at 513 nm upon 420-nm excitation (Fig. 3B), and it was shown that the emission peak depends on excitation wavelength.65 These emission spectra of two radicals have not been reported before and were observed for the first time,28 enabling the fluorescence studies feasible.
Fig. 3C shows the absorption and emission spectra of fully reduced anionic and neutral flavins (FADH− and FADH2) in solution and in photolyase. The absorption spectra in solution are consistent with those previously reported for reduced FMNH− and FMNH2.61 As previously reported, the 350–440 nm absorption can be assigned as two transitions of S0←S1 peaked around 420 nm and S0←S2 peaked around 350 nm.82 Upon 360-nm excitation, we observed weak fluorescence emission peaking at 455 nm for anionic hydroquinone (FADH−) and at 480 nm for neutral hydroquinone (FADH2). However, in photolyase, the anionic hydroquinone (FADH−) clearly shows an absorption band with a peak at 360 nm, and its emission spectrum shows a structured fluorescence profile peaking at 515 and 545 nm (Fig. 3C). The fluorescence intensity of fully reduced flavins in solution is much weaker than that obtained in the protein, suggesting very different dynamic behaviors.28 The huge red shift of the emission in photolyase is directly related to the efficient repair of CPD in the enzyme complex (see below). All the excited-state dynamics of these five redox states in solution and proteins were reported in ref. 28.
3. Ultrafast active-site solvation dynamics in photolyases
Dynamic solvation in binding and active sites plays a critical role in protein recognition and enzyme reaction and such local motions optimize spatial configurations and minimize energetic pathways.18,29–34,83–90 These dynamics involve local constrained protein and trapped-water motions within Å distance and occur on ultrafast time scales. Specifically, the active-site solvation can modulate ultrafast charge-transfer processes and significantly influence the dynamics and efficiencies of ET processes through occurring at nonequilibrium states.91–94 Unlike the typical techniques which use external labeling of bulky dye molecule or synthetic amino acids to observe the relaxation of active sites in proteins,95–97 direct characterization with intrinsic chromophores or amino acids in proteins eliminates significant local perturbations and reveals intact environment responses,98–103 as recently also examined in green fluorescence proteins.104 We have recently studied a series of flavoproteins using the intrinsic flavin molecule as an optical probe and especially found the important functional role of local solvation in photolyase.29–34 Fig. 4 shows the X-ray structures of E. coli CPD photolyase wild type (EcCPD) and A. thaliana (6–4) photolyase wild type (At(6–4)) with chromophore molecules.31 Both the X-ray structures and molecular dynamics (MD) simulations (Fig. 4) show certain water molecules trapped at the active sites besides charged and polar amino acids surrounding the functional chromophore of FADH−. Thus, upon excitation the local polar environments at the active sites would proceed to a series of relaxations.
Fig. 4.

Active-site structures of photolyases and solvation dynamics probed by intrinsic flavin cofactor FADH−.31 (A-B) Top panel: X-ray structures of E. coli CPD photolyase and A. thaliana (6–4) photolyase with the catalytic cofactor (FADH−) in the center. Middle panel: Close-up view of the flavin and the active site of CPD photolyase (Left) and of (6–4) photolyase (Right) with the neighboring polar/charged residues and trapped-water molecules within 8 Å from one snapshot of 1-ns MD simulations. Bottom panel: Corresponding surface maps of the MD snapshots, showing the local topography, chemical property (red, negative charged residue; blue, positive charged residue), and trapped-water molecules at these active sites. (C-D) Upper panel: 3D representation of fs-resolved emission spectra of EcCPD (C) and At(6–4) (D) relative to time (ps) and emission energy (cm−1). The intensity was scaled by a color code. Lower panel: Snapshots of fs-resolved spectra at three typical delay times for the two sites with their corresponding steady-state emission spectra. For comparison and clarity, the steady-state emission peaks were marked by the gray dotted lines as references to show spectral peak and shape evolution.
To systematically study the solvation dynamics in the active sites of photolyases, we need to obtain their solvation correlation functions. Traditionally, we can construct fs-resolved emission spectra using wavelength-resolved fluorescence transients to derive solvation correlation functions.84,105 We usually need to assume the emission spectra following a log-normal distribution. However, the emission spectra of FADH−* (Figs. 4C–D bottom panels) show an irregular distribution with double peaks at 515 and 545 nm for EcCPD and 505 and 540 nm for At(6–4). Thus, given the heterogeneous electrostatic distributions and different structural flexibilities of the functional sites, to evaluate the complete local relaxation, we need to directly measure the fs-resolved emission spectra and examine how the emission spectra change with local relaxation.
Figs. 4C–D show the three-dimension representation of the emission evolution with time (top) and a few snapshots at different delay times (bottom) for both active sites. For FADH−* at the active site of EcCPD, we observed only a small spectral shift but instead an obvious shape change at the blue side of the emission. The central distribution of the emission spectrum at 0.5 ps is already around the steady-state peaks. The X-ray structure shows 4 water molecules buried at the bottom of the active site and 8 water molecules around the entrance of the active site within 8 Å from the isoalloxazine ring. Our MD simulations (1 ns) found 22 water molecules around the moiety ring (Fig. 4A). There are at least four polar/charged amino acid sidechains in direct van der Waals contact with the moiety ring. Thus, the active site is a highly polar environment. On the contrary to the recent calculations,106 the dipole-moment change between the ground and excited states is estimated to be around 1–2 D.29,107 Therefore, the observed small Stokes shift indicates a relatively rigid active site despite the polar environment. The observed continuous solvation dynamics from a few to five hundreds of picoseconds reflect the local collective motions of trapped water molecules and charged/polar sidechains. These constrained motions induce only a small Stokes shift and stabilization energy, but strongly perturb the excited state, resulting in the obvious spectral shape changes.108
The fs-resolved emission spectra of FADH−* at the active site of At(6–4) are drastically different from those of EcCPD. We not only observed a significant spectral shape change but also a large spectral shift. At 0.5 ps, the spectrum peaks around 500 nm and at a later time of 1 ns, the peak moves to 540 nm. Clearly, the solvation relaxation is much larger than EcCPD. The X-ray structure shows only 7 water molecules around the ring moiety within 8 Å with six buried inside the active site and one near the entrance. Our MD simulations (1 ns) show the least 14 water molecules near the ring moiety at the active site, consistent with the smaller hole size at the entrance shown in the X-ray structures for two photolyases (Figs. 4A–B). However, there are four more polar/charged amino acid residues in direct van der Waals contact with the isoalloxazine ring (Fig. 4A–B). Clearly, the active site is also a highly polar environment with dominant polar/charged sidechains. Thus, the significant relaxation reflects a relatively flexible active site with large plasticity, similar to the observation for sequence-homology cryptochrome.109 The observed large solvation in the transients results from the substantial motions of the local trapped-water and flexible polar/charged sidechains, leading to a large Stokes shift. Meanwhile, such heterogeneous local motions greatly modulate the excited state, resulting in the significant spectral shape changes. The flexible active site is also supported by another observation. We observed a fast component of 30 ps with 5–7% amplitude in the transient gated at the emission peak of At(6–4), a deactivation process through the butterfly and/or twist motions of the isoalloxazine ring as also shown in our recent studies of its homologous cryptochromes.28,65 The different absorption and emission spectra31 and distinct time-zero emission spectra in the two active sites (Fig.s 4C–D) also reflect the chromophore’s different structural and electrostatic environments. It would be erroneous if we conclude from the steady-state emission spectra that the FADH−* in EcCPD would have a larger dynamic Stokes shift and in this case it has a smaller one.
To extract the correlation functions for the active sites, the conventional method using the Stokes shift changes with time would not be applicable here. Alternatively, we used the change of average frequencies with time. For the two active sites, the average frequency shift of At(6–4) is about three times larger than that of EcCPD, reaching to 798 cm−1. It is striking that both sites have all three similar relaxation timescales with comparable first and third components of the stabilization energies, but very different second components of the stabilization energies. The first ultrafast relaxation in a few picoseconds mostly results from the local reorientation motions of trapped water and/or neighboring unscreened polar/charged sidechains. Thus, the comparable time scale, stabilization energy and solvation speed indicate that both active sites have the similar initial ultrafast response. The second relaxation in tens of picoseconds reflects the coupled water-protein motions, a collective rearrangement of the local configuration.102,103,110 Since the active site of At(6–4) has more polar/charged sidechains and is more flexible, the second relaxation mostly comes from those coupled water-sidechains large motions in proximity with the chromophore. This result is significant and reflects larger mobility of protein sidechains in the At(6–4) active site. On the other hand, the protein sidechains at the active site of EcCPD are much less mobile. These distinct local dynamics in the two active sites well correlate with their functions. For EcCPD, since the anionic CPD lesion has very low reaction barrier and splits within tens of picoseconds, a relatively rigid, optimized electrostatic environment would be ideal to avoid any futile side reactions. On the other side, to optimize both electron and proton transfers in (6–4) photolyase, especially the sensitive proton transfer from the neighboring sidechain,10,21,27 the extra flexibility of sidechains would be necessary to search for a favorable configuration for proton tunneling. Finally, the third relaxation with the similar time scales in subnanoseconds, stabilization energies in tens of wavenumbers and solvation speeds represents the intrinsic collective active-site protein motions and is the same for both photolyases. As a result, all charge-transfer steps in CPD and 6–4PP repair are modulated by these active-site solvation dynamics that occur on the similar time scales from a few to hundreds picoseconds, and all these ET processes follow a stretched-exponential behavior.111 Therefore, the relaxation in local active sites modulates the enzymatic reaction dynamics through a synergy of all elementary steps and enhances the repair efficiency, especially by restraining the unproductive charge recombination step in EcCPD.
4. Dynamics and mechanism of CPD repair by photolyase
The understanding of the complete CPD repair dynamics have been challenging. Michel-Beyerle and colleagues observed electron injection from the excited cofactor to the substrate in 1997 using 100-ps temporal resolution.16 MacFarlane & Stanley showed the formation of the repaired thymine product with 800-fs temporal resolution in 2003.17 However, the overall repair dynamics remained unknown, and the entire mechanism and photocycle still were not determined. With femtosecond temporal resolution, we recently uncovered the veil of the entire repair dynamics, molecular mechanism and repair photocycle with unprecedented details.
4.1 Sequential splitting mechanism of the cyclobutane ring
The kinetic scheme of CPD repair can be basically summarized in Fig. 5. In previous studies, we performed fs-resolved fluorescence and absorption measurements and proved that photolyase repairs CPD with a cyclic ET mechanism.18 The initial charge separation occurs in 250 ps while the charge recombination after complete repair finishes in 700 ps. Both ET processes follow the stretched-exponential dynamical behavior due to the active-site solvation.
Fig. 5.

A proposed sequential CPD repair scheme. After the forward electron transfer (FET, reaction rate kFET) from FADH−* to thymine dimer upon light excitation, the repair channel includes splitting of two bonds of C5-C5’ (ksp1) and C6-C6’ (ksp2) in thymine dimer accompanied with unproductive back electron transfer (kBET) without repair. The subsequent electron return (kER) after complete ring splitting finishes the reaction photocycle. ktotal is the overall decay rate of intermediate state FADH• after the initial charge separation.
To resolve the detailed mechanism of CPD splitting after electron injection and to probe the intermediates and repaired products (thymine), the detection wavelengths were extended from visible to deep UV light. Fig. 6 shows a striking pattern of the transient-absorption signals of the complex of photolyase with substrate T<>T, probed at fifteen wavelengths. At 430 nm, the signal is the summation of all three flavin species (FADH−*, FADH• and FADH−) and returns to zero with a time constant of 700 ps. At 335 nm in the UV region, we captured the signals of thymine-related intermediates and the long-component formation of final repaired thymine molecules (insets B-E in Fig. 6). By knowing the dynamics of FADH−* and the absorption coefficients of FADH• and FADH− (inset A in Fig. 6), all the absorption transients only can be systematically fit with the sequential model shown in Fig. 5, but not any other concerted schemes of thymine splitting and electron return at the same time. The deconvolution of various species is shown in insets B-E and the resulting absorption coefficients are given in inset A of Fig. 6. Thus, we obtained the entire dynamics of thymine dimer splitting.
Fig. 6.

Femtosecond-resolved transient-absorption dynamics of reactants, various intermediates and final products involved in repair of thymine dimer.32 The repair dynamics are probed systematically from 800 to 260 nm. Several typical results in the UV region are shown with a distinct pattern. Inset A: The absorption coefficients of all species involved in repair. Insets B-E: Transient-absorption signals probed at 335, 300, 270 and 266 nm. These dynamics are systematically fitted by total flavin-related species (FADH−* + FADH• + FADH−, dashed red), thymine dimer intermediate T-T− (dashed cyan), thymine anion T− (dashed dark red) and thymine products T (dashed dark yellow).
Our data indicate that in contrast to the computational reaction models112 the thymine dimer splits by a sequential pathway. The first-bond C5-C5’ breakage (1/ksp1) finishes in much less than 10 ps, consistent with the theoretical prediction of a nearly barrierless process.113–120 Also, the driving force of direct back ET between T<>T− and FADH• is quite large (~2.0 eV),121, 122 which falls in the Marcus inverted ET region and should occur in hundreds of picoseconds. Thus, the slow formation (kFET) and ultrafast decay (ksp1) of T<>T− result in negligible accumulation of T<>T− population and a close-to-unity efficiency of C5-C5’ splitting to T-T−. Indeed, we observed the formation and decay of T-T− intermediate after the first-bond C5-C5’ breakage (Fig. 5 and insets B, D and E in Fig. 6). The decay dynamics in 87 ps mainly represents the second-bond C6-C6’ splitting. Given the total repair quantum yield as 0.82,15,37,123,124 the forward ET branching as 0.85 and the yield of C5-C5’ breaking as 1.0, the branching of C6-C6’ splitting is 0.96. Therefore, the second-bond breakage finishes in 90 ps (1/ksp2), much longer than that of theoretical calculations, and the back ET without the second-bond splitting occurs in 2.4 ns (1/kBET), much slower than the reported value of a recent time-resolved study with subnanosecond resolution.125 After the second-bond C6-C6’ cleavage, the signal of T− forms and then decays in 700 ps (1/kER and inset C of Fig. 6), reflecting the electron return from T− to FADH• that is completely decoupled from the second-bond breaking. This is confirmed by the observation of the final products of two repaired thymine molecules, formed in two sequential steps in 90 and 700 ps upon the initial electron injection (see insets D and E). The complete repair photocycle is summarized in Fig. 7.
Fig. 7.

Complete photocycle of CPD repair by photolyase.32 All resolved elementary steps of CPD (thymine dimer) repair on the ultrafast time scales and the elucidated molecular mechanism.
4.2 Electron tunneling pathways and role of adenine in repair of CPD
The FADH− cofactor in photolyase has an unusual bent U-shaped conformation with the isoalloxazine and adenine rings in close proximity (Fig. 2A). The crystal structure of A. nidulans photolyase with CPD complex shows that the adenine moiety of FADH− is at van der Waals distances with both base moieties of CPD, 3.1 Å to the 5’ side and 3.2 Å to 3’, and the first carbon atom linked to the isoalloxazine ring at 3.6 Å.9 It was thought that the repair reaction by photolyase involves electron tunneling directly106, 126 or through the adenine moiety.127, 128 However, the cyclic electron tunneling pathways, forward (kFET), backward (kBET) and return (kER), are a matter of some debate. One view is that the electron tunneling is mediated by the intervening adenine with a total distance of about 8 Å.127,128 An alternative model suggests that electron tunneling occurs directly from the o-xylene ring of FADH− to the 3’ side of CPD with a short distance of 4.3 Å.106, 126 However, intramolecular electron hopping from the isoalloxazine ring to adenine moiety has the unfavorable redox potentials (ΔG0~0-+0.1 eV)122, 129 and we also did not observe any ultrafast quenching of FADH−* fluorescence without substrate.18,32 To examine the electron tunneling directionality, we used a series of substrates, U<>U, U<>T, T<>U and T<>T (chemical structures in Fig. 8A), as electron acceptors to follow electron tunneling pathways.
Fig. 8.
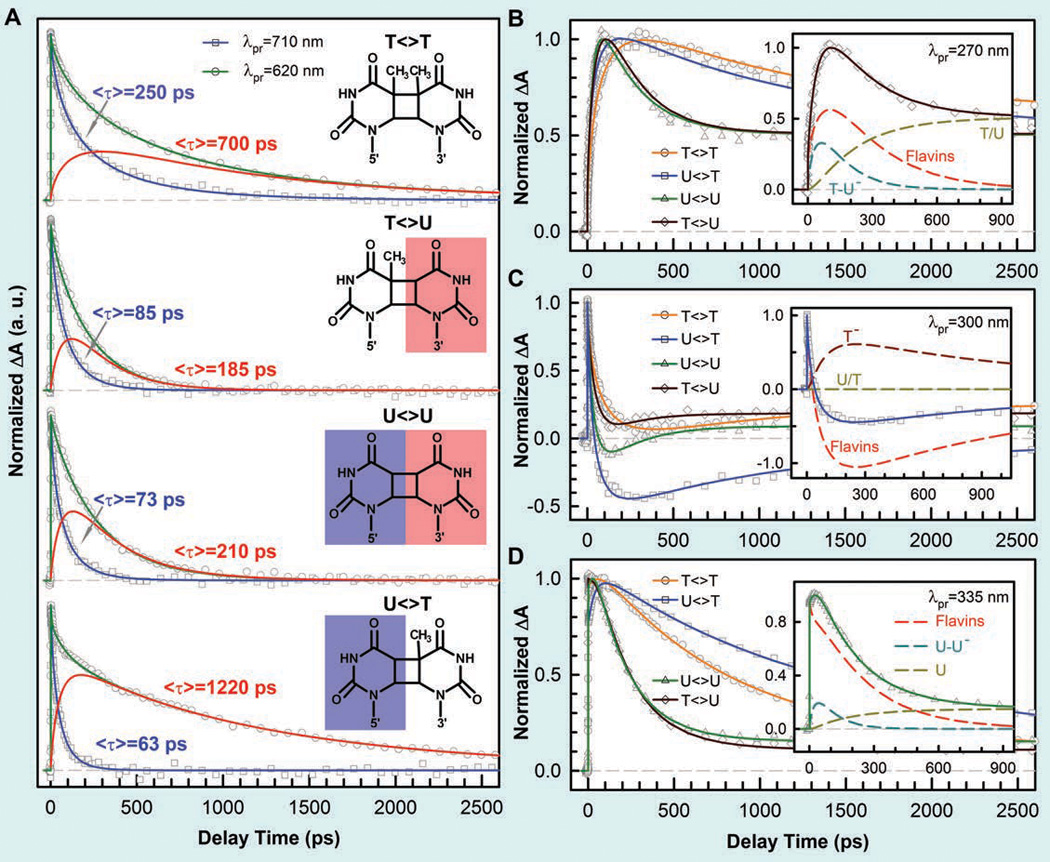
Femtosecond-resolved transient-absorption dynamics of DNA repairs with different combination of bases.32 (A) Transient-absorption signals of repair with T<>T, T<>U, U<>U, and U<>T probed at 710 and 620 nm. The dynamics of FADH− (blue line) was probed at 710 nm. The signal at 620 nm is the combination of FADH− and intermediate FADH• (red line) contributions. The chemical structures of various CPD substrates are also shown with highlight at uracil sides. The blue shading of U indicates the forward electron tunneling to the 5′ side of DNA and the red shading for electron return starting at the 3′ side after the complete two-bonds splitting. (B–D) Repair dynamics of T<>T (orange), U<>T (blue), U<>U (green), and T<>U (dark red) probed at 270 nm (B), 300 nm (C) and 335 nm (D). Insets show the deconvolution of total flavin-related species (dashed red), CPD intermediate anions T−-U/U−-U (dashed cyan), and T− (dashed dark red), and the products of T/U (dashed dark yellow) of repair with T<>U, U<>T, and U<>U in (B), (C), and (D), respectively.
At 710 nm, only signal of FADH−* was probed and we observed the dynamics of forward ET for T<>U, U<>T and U<>U in 85, 63, and 73 ps, much faster than that of T<>T in 250 ps (Fig. 8A). Thus, the ET dynamics (rates) from the excited FADH− to four CPD substrates vary in a decreasing order of 5’-U<>T-3’ > U<>U > T<>U > T<>T. This observation is significant and indicates that the electron tunnels toward ending at the 5’ side, not the 3’ side, of the dimers. U<>T and T<>U have the similar reduction potentials, but the ET time lengthens by more than 35% from 63 ps of U<>T to 85 ps of T<>U, i.e., the rate decreases to 74%. Theoretical studies showed the similar binding configurations for U<>T and T<>U and thus the observed differences in ET rates should not come from the electronic couplings because of the similar donor-acceptor separation.9,127
The uracil base has a reduction potential more positive than thymine by ~0.11 V.129 Although the electronic interactions between T and U in the covalent CPD species delocalize in the CPD states and result in the similar redox properties of U<>T and T<>U, the U moiety without a methyl group at the C5 position at the 5’ side should have a larger electron affinity than the T moiety. Thus, if the adenine moiety of FADH− is involved in electron tunneling, our results indicate that the local configuration and orientation favors the electron tunneling to the 5’ side, rather than the 3’ side (Fig. 2A), although the distances from the adenine to the two sides are nearly equal, 3.1 and 3.2 Å, respectively. The larger ET rate of U<>T vs. T<>U also excludes the electron tunneling pathway directly from the o-xylene ring of the flavin to the 3’ side of the dimer (Fig. 2A).106, 126 Such a hybrid tunneling pathway mediated by the adenine moiety is also observed in 6–4 photoproduct repair by 6–4 photolyase.27,33
With the measured forward ET dynamics, we can determine the electronic coupling constant (J) and related free energy for different substrates. We can use the semi-classical ET expression.91
| (1) |
Where h and kB are Planck and Boltzmann constants, respectively, and T is temperature in Kelvin. For the four different substrates, the coupling constant (J) and the reorganization energy (λ) could be considered as constants. Considering the free energy ΔG0 of forward ET between FADH−* and T<>T as −0.44 eV and the reported reorganization energy of λ to be 1.2–1.4 eV in flavoprotein ETs130–132 as a fitting reference value, and also taking into account a larger J and a smaller λ in the forward ET than in the electron return, we obtained the free energy for ET with U<>U to be −0.59 eV, λ=1.21 eV and J=3.0 meV.33 The coupling constant of 3.0 meV is in a good agreement with the theoretical calculation that assumed electron tunneling through adenine instead of space.127 Using the obtained λ and J values, we calculated free energy (ΔG0) of −0.61 eV for U<>T and −0.57 eV for T<>U. The difference of 40 meV for U<>T and T<>U probably results from the preferred electron tunneling directionality to the 5’ side rather than 3’ side of the dimer, assuming the same electronic coupling constants for both ETs.
With the understanding of the forward ET dynamics probed at 710 nm, we tuned the probe wavelength to 620 nm to detect the flavin intermediate FADH•.18 To detect the substrate-related intermediates and products, we extended the probe wavelengths to UV region from 360 to 266 nm and observed a series of striking features of transients.33 At wavelengths shorter than 360 nm, we observed the anionic intermediates of T−-U, U−-U and U−-T (Figs. 8B–D). Following the C5-C5’ bond breaking within a few picoseconds, the dynamics of T−-U, U−-U and U−-T decay in 64, 31 and 30 ps, representing the total dynamics (rates) of both the C6-C6’ bond breakage of anionic CPD intermediates and futile back ET after the C5-C5’ splitting. Given the splitting branching of T−-U, U−-U and U−-T as 0.94, 0.88 and 0.90, respectively,33 the T−-U takes the similar bond splitting time (1/ksp2) of 75 ps as T−-T in 90 ps, but the C6-C6’ bond in U−-U and U−-T splits on a much faster time scale of 35 ps (Fig. 9A). Clearly, the second C6-C6’ bond breaking has a longer time with T and a shorter time with U at the 5’ side. Thus, after the C5-C5’ bond breakage, the excess electron mainly remains at the 5’ side because the T moiety at the 5’ side with a methyl group at the C5 position can significantly stabilize the anionic radical and thus has a longer time of the C6-C6’ splitting. This observation further supports the electron tunneling toward the 5’ side of the dimer, which also must pass through the adenine moiety. According to the transition-state theory, we estimated the activation energy of ~0.174 eV (4.0 kcal/mol) for T−-T bond breaking, ~0.170 eV (3.9 kcal/mol) for T−-U bond splitting, and ~0.152 eV (3.5 kcal/mol) for U− -T and U−-U bond cleavage (Fig. 9B). Thus, the stabilization in T−-T by the methyl group(s) at the C5 (and C5’) position leads to ~0.022 eV (0.5 kcal/mol) more for the activation of the bond splitting than U−-U. From the measured quantum yields, we obtained the back ET time scales (1/kBET) of 1175, 315 and 260 ps for T−-U, U−-T and U−-U, respectively, much shorter than that for T−-T in 2.4 ns (Fig. 9A). Thus, the back ET dynamics change in a decreasing order of 5’-U−-U-3’ > U−-T > T−-U > T−-T. Using the same λ and J values of 1.21 eV and 3 meV as forward ET, respectively, we obtained the free energy of back ET to be −0.22 eV for T−-T, −0.29 eV for T−-U, −0.42 eV for U−-T, and −0.44 eV for U−-U (Fig. 9B) and these BET reactions are all in the Marcus normal region.33
Fig. 9.

Reaction times and free energy diagrams of the elementary steps in splitting of CPD substrates.33 (A) Reaction times of each elementary step observed in repair of various substrates (T<>T, T<>U, U<>U, and U<>T), including the forward ET, the C6−C6′ bond splitting, futile back ET and final electron return to complete the photocycle. Note the order in reaction times of three ET processes for four different substrates and also the faster bond splitting than the back ET and electron return. (B) The free energy profiles along the reaction coordinate after the forward ET in repair of CPD substrates with the time scales of the dynamics shown at the top. On the anionic surface, the solid curve represents the splitting of T<>T− while the dashed curve for U<>U−. On the neutral surface, the bond-breaking activation barrier (dashed curve) is very high according to theoretical calculations. Note the different regions, normal or inverted, of three ET processes and the ring reclosure after the futile back ET.
After a sequential breakage of both C-C bonds, the cyclobutane ring is repaired but the electron must return semiquinone FADH• to complete the photocycle and restore the catalytic state of FADH−. In UV region, we did observe the signal of T− and U− intermediates around 300 nm and the products of T and U in various substrates. The (T+U)− and (U+U)− have faster return dynamics (1/kER) of 185 and 210 ps than (T+T)− with 700 ps, while (U+T)− has the longest electron return time of 1220 ps (Fig. 9A). The electron-return dynamics are much slower than the ring splitting and thus these two processes are decoupled. Also, the electron-return dynamics for the four substrates vary in a decreasing order of 5’-T+U−-3’ > U+U− > T+T− > U+T−. The back electron tunneling from T− passing the adenine-mediated pathway again to final FADH• has a large driving force of −2.26 eV122,129 and such tunneling is in the Marcus inverted region. Using eq. (1) for the electron-return dynamics of T+T− and U+U− (ΔG0 around −2.15 eV) and by consideration of a smaller J and a larger λ in the electron return than in the forward ET, we obtained the electronic coupling and reorganization energy of 2.6 meV and 1.37 eV for return tunneling, and the free energy of −2.12 eV for U+U−. Using the obtained J and λ values, we obtained a smaller free energy of −2.10 eV for T+U− and a larger free energy of −2.32 eV for U+T− (Fig. 9B). Since the thymine has lower reduction potential than uracil, the fact that the driving force for U+T− is larger than T+U− suggests that the electron stays at the 3’ side of DNA after ring splitting. From the X-ray structure,9,11,13 several water molecules, polar/charged residues, and the highly reducing FADH• are all around the 3’ side, which probably stabilizes and solvates the electron after the complete bond breaking.
4.3 Active-site mutation and repair efficiency modulation
The repair efficiency of thymine dimer by photolyase is higher than those of all chemical model systems (0.004–0.41) synthesized so far,133–135 indicating that the amino acids in the active site must significantly contribute to the repair efficiency by modulating the redox properties and dynamics of the flavin/CPD pair. To examine how the protein active site controls the higher repair efficiency, we mutated a series of residues (E274A, R226A, R342A, N378C and M345A) at the active site of EcCPD and here showed two typical mutants, N378C near the cofactor side and E274A near the substrate side, which makes critical contributions to the repair efficiency (Fig. 10A). We systematically studied these two mutants by probing all involved species from visible to UV. Four typical results are shown in Figs. 10B–D at 800, 620, 270 and 266 nm. The final results of forward ET, back ET, second-bond splitting, and electron return are shown in Fig. 11B with the measured total repair quantum yields in Fig. 11A.
Fig. 10.

Effect of active-site mutations on repair dynamics.32 (A) X-ray structure of the active site of A. nidulans photolyase with two critical residues of N386 (N378 in E. coli) and E283 (E274 in E. coli). The hydrogen-bonding distances of the two residues with FADH− and CPD are also shown, respectively. (B–D) Femtosecond-resolved absorption transients of the repair of damaged CPD by the wild type and two mutants (N378C and E274A) probed at 800 and 620 nm (B), 270 nm (C), and 266 nm (D). Insets in (B) and (C) show the deconvolution of various species’ contributions of N378C mutant probed at 620 and 270 nm, respectively, while the inset in (D) for E274A mutant probed at 260 nm.
Fig. 11.

Quantum yields and various reactions times of four elementary steps for wild type (EcCPD) and the two mutants.32 (A) The overall repair quantum yields (QY) of the two mutants N378C and E274A were measured, relative to the known wild-type one (0.82), by monitoring the formation of thymine bases at 266-nm absorption with certain visible-light irradiation of the enzyme-substrate solution. (B) The reaction times of each elementary step in CPD repair by the wild type and the two mutants with our measured total QY from (A). The vertical dashed lines represent the two-step repair efficiency of the FET to lifetime emission (LT) and the second-bond splitting (SP2) to BET. The ER is decoupled from the CPD splitting.
Both mutants exhibit the lower quantum yields of 0.69 for N378C and 0.40 for E274A, resulting from a combination of two-step quantum yields, forward ET relative to lifetime emission (kLT) and second-bond splitting relative to back ET (two pairs of dashed lines in Fig. 11B). Both mutants modulate the ET redox potentials, N378C for FADH− at the cofactor side and E274A for T<>T at the 5’ side, leading to the longer forward ET times as 1300 ps for N378C and 580 ps for E274A (Fig. 11B), respectively, and thus resulting in the lower first-step branching yields to 0.73 for N378C and to 0.65 for E274A. After the forward ET, the C5-C5' bonds in anionic CPD break ultrafast in both mutants, similar to the wild type. For the second-bond C6-C6’ cleavage, we obtained the same splitting time in 90 ps as the wild type for N378C, consistent with the fact that the mutation only affects the cofactor. For E274A, we observed a faster second-bond splitting time of 30 ps, probably due to the destabilization of the splitting transition state by the mutation of E274A that abolishes two hydrogen bonds with T<>T at the 5’ side (Fig. 11A). The observation of thymine dimer repair by mutant E274A also excludes any possibility of proposed proton transfer(s) between E274 with T<>T during repair that has been suggested based on theoretical consideration.112,136 However, the mutation near the cofactor side or the substrate side also increases the rate of unproductive back ET, 1300 ps for N378C and 50 ps for E274A (Fig. 11B). Thus, the second-step branchings drop to 0.94 for N378C and to 0.62 for E274A. Finally, the decrease of the total repair quantum yield for these two mutants is an overall effect from both branching ratios.
5. Dynamics and mechanism of CPD repair in a biomimetic flavin−thymine dimer adduct
Numerous biomimetic systems have been synthesized to mimic the repair function by photolyase, but all have shown low repair efficiency, for example, 0.016−0.062 for flavin−thymine dimer systems137,138 and 0.06−0.40 for indole−thymine dimer systems.133,135 The molecular mechanisms of these low-efficiency processes have not been understood.113,114 Here, we report our direct mapping of the repair processes in a biomimetic flavin−thymine dimer adduct by following the entire temporal evolution of reactants and intermediates. By capturing the complete repair photocycle and comparing with CPD photolyase, we can understand how CPD photolyase achieves its high repair efficiency (0.8–0.9).
Instead of simply using a solution mixture of thymine dimer and flavin, we employed a covalent linkage between the lumiflavin (LF) and thymine dimer (T<>T) to hold the flavin photosensitizer and dimer together (Fig. 12A). The molecular structures of LFH−−T<>T and LFH− are highlighted in pink and blue, respectively, in the left inset of Fig. 12A. The absorption and emission spectra of fully reduced FMNH− in solution are shown in the right inset of Fig. 12A. The absorption spectrum is consistent with the results of previous studies,61,137 and the emission spectrum is similar to that in our earlier report on FADH−* emission.28 Upon 360 nm excitation, we observed weak fluorescence emission peaked at 435 nm for fully reduced FMNH−. More importantly, we observed the excitation-wavelength dependence of the emission spectra.28 Also, as mentioned in section 2, the fluorescence intensity of fully reduced flavin in solution is much weaker than that observed in photolyase, suggesting very different dynamic behaviors of fully reduced flavin in the two environments.
Fig. 12.

(A) Femtosecond-resolved fluorescence transients of reduced FMNH− and LFH− −T<>T gated at 450 nm upon 325 nm excitation.36 The molecular structures of LFH−−T<>T and LFH− are highlighted in pink and blue, respectively, in the left inset. The right inset shows the absorption and emission spectra (360 nm excitation) of reduced FMNH−. The two arrows indicate the pump wavelength (λpu) and gated fluorescence emission wavelength (λfl). (B) Repair scheme with forward ET (τET) after light excitation, ultrafast first C5−C5’ bond splitting (τSP1), back ET (τBET) and ring reclosure without repair, and the repair channel including C6−C6’ bond splitting (τSP2) and electron return (τER). (C) Femtosecond-resolved absorption signal of LFH−*−T<>T probed at 580 nm upon 325 nm excitation, with detection of both LFH−*−T<>T (green curve; mainly probed at 710 nm) and LFH• (blue curve). The total LFH• signal is from the one dominant contribution of the initially formed LFH• (dashed cyan curve) and two minor contributions of the branched LFH• in the repair channel (≥85%, dashed purple; <15%, dashed lime-green). A minor plateau from the deactivation channels in the 710 nm transient was removed for clarity. The inset shows the dynamics of the two fitted minor channels.
We first studied the excited FMNH−* in solution and the fluorescence transient of FMNH−* in the absence of dimer exhibited multiple decay dynamics with lifetimes (1/kd) of 5.8 ps (82%), 35 ps (16%), and 1.5 ns (2%) (Fig. 12A).28,36 Such multiple-decay dynamics reflect the ultrafast deactivation of the excited FMNH−* with a butterfly bending and/or distortion motions of the isoalloxazine ring.28 With LFH−-T<>T, the transient completely decays to zero in less than 150 ps (Fig. 12A). By considering the ET reaction in each deactivation process of excited LFH−*, we obtained an ET dynamics (1/kFET) in 79 ps. Thus, the first-step ET quantum branching drops to 0.124 considering all three deactivation processes and is much lower than that in photolyase (~0.85).
According to recent quantum-chemical calculations,113 the splitting of the C5−C5′ bond of the anionic thymine dimer (T<>T−) is a downhill reaction and occurs in less than 1 ps. Therefore, there is trace amount of T<>T− accumulation. After the C5−C5′ ultrafast splitting, the reaction can evolve along one of two pathways (Fig. 12B): (1) the nonproductive pathway including back ET (τBET) and ring reclosure without repair, or (2) the repair channel including C6−C6′ bond splitting (τSP2) and then electron return after repair (τER). By observing the absorption of various intermediates (Fig. 12C), we obtained that the C6-C6’ bond splits in 435 ps (1/kSP2) with a concurrent unproductive back ET in 95 ps (1/kBET). The fast back ET of 95 ps in solution indicates a relatively unstable charge-separated intermediate (LFH•−T-T−), leading to a significant competition between ring splitting (435 ps) and futile back ET. The branching of the dimer ring splitting to electron return is 0.179, much smaller than that in photolyase (0.96). Considering the two-step branching ratios together, we obtained the total repair efficiency of 0.022, a similar order as the steady-state measurement (0.062) but more than one order smaller than that in photolyase. After both C-C bonds breaking, the electron is delocalized at both thymine bases and the electron at the linked T1 thymine base (85%) returns to flavin within about 23 ps (inset in Fig. 12A).36 Such ultrafast dynamics of electron return in the Marcus inverted region must be due to the low activation energy, probably facilitated through hot vibrational modes of the products. On the other side, the negative charge could also stay at the distal base T2 with a much longer time scale, leading to a long-lived LFH• (15%).
Our observations reveal that the underlying molecular mechanism for the low repair quantum yield of all flavin−thymine dimer adducts is the short-lived excited flavin and the ultrafast dynamics of futile back ET. In contrast, in CPD photolyase, the enzyme can utilize geometric restriction and electrostatic interactions to confine the flavin cofactor and lengthen its excited-state lifetime (1.3 ns). The excited cofactor could exist long enough to inject electron into the CPD to form a charge-separated intermediate and thus reach a high forward ET quantum yield. Moreover, the active site stabilizes the charge-separated intermediate in photolyase (2.4 ns) and speeds up the ring splitting (90 ps). Such modulation of the dynamics leaves enough time to cleave the ring, resulting in a high splitting efficiency. These two high-efficiency processes lead to perfect repair of damaged DNA by photolyase.
6. Dynamics and mechanism of 6–4PP repair by photolyase
For 6–4PP repair, various hypothetical repair models,10,19–26 including formation of an oxetane intermediate in the ground state before the photochemical reaction19,21 and, more recently, a model that includes formation of a water molecule following the primary photochemical reaction,10 have been proposed to rationalize the bond breakages and group arrangements for complete repair. Most models invoke proton transfer from a neighbouring histidine residue, leading to a plausible scheme as shown in Fig. 13.
Fig. 13.

A proposed 6–4PP repair scheme includes forward electron transfer (FET, reaction rate k1) after light excitation, back electron transfer (BET, reaction rate k2) without repair, and the repair channel, comprising of initial proton transfer (PT, reaction rate k3) and late proton and electron return (PR and ER, reaction rate k4) after repair.
6.1 Mechanism of proton and electron transfer
To examine the proposed repair models of 6–4PP by At(6–4) and to identify the related reaction intermediates, we followed the similar strategy as we studied for CPD repair. We first characterized the dynamics of forward ET.27 By capturing the fluorescence emission from excited FADH− at 550 nm (Fig. 14A), the fs-resolved transient of the complex can be best represented by a stretched-single-exponential decay with a time constant of 225 ps and β=0.8 (Fig. 14B). Considering the lifetime of FADH−* in 3.0 ns, the forward ET takes 280 ps. When the potential active-site proton donor, His 364, was replaced by charged (Lys and Asp), polar (Asn and Tyr) and hydrophobic (Ala and Met) residues, we observed the similar electron-transfer dynamics with time constants of 165–379 ps. This finding, along with the observation that the 6–4PP is in its standard form in the structure of enzyme-substrate complex,10,23 excludes the early repair model that requires a mixture of oxetane precursor and ground state 6–4PP before photoexcitation.19,21 We conclude that after one electron injection the 6–4PP repair takes place completely in the anionic ground state of 6–4PP.
Fig. 14.

Femtosecond-resolved dynamics of flavin species involved in the repair of damaged DNA by (6–4) photolyase enzyme.27 (A) Absorption spectra and coefficients of purified protein with FADH• (red), converted active form FADH− (blue) and damaged 6–4PP (yellow), and derived FADH−* (dashed green) and a 6–4PP related intermediate (dashed purple) from this study. Inset shows the fluorescence emission of FADH−* with an arrow indicating the gated wavelength. (B) Normalized signals, detected by both fluorescence (gated around the emission peak of 550 nm) and absorption (probed at 800 nm) methods without (red) and with (blue) the substrate in the active site, show the same lifetime and forward electron-transfer decays. λfl, wavelength of fluorescence; λpr, probing wavelength; a.u., arbitrary units. (C) Transient absorption signal probed at 640 nm with both FADH−* (blue) and FADH• (green) contributions. The total FADH• signal is from the two contributions of the initially formed FADH• and the branched FADH• in the repair channel. Inset shows a flat signal in tens of picoseconds, reflecting an apparent fast rising signal. ΔA, absorption change. (D) Transient-absorption signals of the His364Asn mutant (green line) and wild-type (WT) enzymes in D2O (dark-red line) and in H2O (light-red line) probed at 640 nm. The corresponding relative steady-state quantum yield measurements are shown in inset.
After the photoinduced charge separation (FADH•+6–4PP•−), the reaction can evolve along two pathways: back ET (reaction rate constant k2) or 6–4PP repair (k3) (Fig. 13). Knowing the forward ET dynamics of FADH−*, we can map out the temporal evolution of FADH• by probing at wavelengths from 500 to 700 nm (Fig. 14A) to follow 6–4PP repair. In Fig. 14C, the transient probed at 640 nm (red curve) shows drastically different behavior from that probed at 800 nm (blue curve), owing to the capture of the radical FADH• (green curve). We observed an apparent rise signal of FADH• in 51 ps (initial flat part of Fig. 14C inset) and a long plateau, indicating that complete 6–4PP repair takes longer than several nanoseconds. Because the forward ET takes about 280 ps, the 51 ps is the overall decay time of the initially formed FADH• (dashed purple curve in Fig. 14C), but appears as an apparent rise. Slower formation and faster decay result in apparent reverse kinetics and less FADH• accumulation. The dynamics of the branched FADH• in the repair channel (dashed light-blue curve in Fig. 14C) exhibits a complex formation (largely determined by the k1 process) and a slow decay (k4), but the amplitude is mainly determined by the k3 rate. By deconvolution, we obtained the back ET in 57 ps (k2) and the repair channel in 481 ps (k3) to form a 6–4PP•− related intermediate. From these rate constants, we obtained a repair branching constant of 0.097, a value that is in excellent agreement with the reported steady-state repair quantum yield of 0.1, suggesting that after the k3 step all subsequent reactions proceed to the final 6–4PP repair without any back ET that would lead to a futile cycle.
These findings reveal that the underlying reason for the low repair quantum yield of (6–4) photolyase (0.1), compared with cyclobutane pyrimidine dimer photolyase (0.82), is the ultrafast rate of back ET (k2 = 57 ps) from 6–4PP•− to FADH•, relative to the parallel repair channel (k3 = 481 ps), although the branching of the forward ET is quite high (0.8). For the series of mutants (His364Asn/Met/Tyr/Ala/Asp/Lys) designed to examine the proposed reaction mechanism, we observed that all transients probed in the 500–700 nm region show similar back-electron transfer dynamics in the range of 80–295 ps,27 but decay to zero without any long plateaus (shown in Fig. 14D for the His 364 Asn mutant). This observation is critical to the proposed reaction scheme. Even though the ET from FADH−* to 6–4PP is essentially normal, the repair channel is completely shut off and all FADH• formed by initial charge separation follows a futile ET cycle back to FADH•− by charge recombination without repair (Fig. 13). These results are also consistent with our steady-state quantum yield measurements, which revealed a total lack of repair in any of these mutants (inset in Fig. 14D). Collectively, our data, in agreement with an earlier report,21 indicate that His 364 in the active site is a functional residue that is irreplaceable in the repair channel, and a proton transfer from His 364 to 6–4PP is conceivably the rate-limiting step (k3) in our reaction scheme (Fig. 13).
To test the proton transfer from His 364 during repair, the enzymatic reaction was carried out in D2O. As shown in Fig. 14D for wild-type photolyase, we observed a different transient with an obviously lower plateau, reflecting a slower repair process (1.25 ns) but with the similar forward (240 ps) and back (68 ps) electron transfer dynamics as observed in H2O. The X-ray structure shows a hydrogen-bond distance of 2.7 Å between the amine group of His 364 and the hydroxyl group at the C5 position of the 5' base (Fig. 2); hence, a proton transfer from His 364 to the hydroxyl group is quite feasible. The lower plateau of the transient in D2O is about half of that in H2O (Fig. 14D), and thus corresponds to a halving in the repair branching, in good agreement with the steady-state measured quantum yield ratio of 1:2 for D2O:H2O (Fig. 14D inset). We also studied the repair dynamics and the steady-state enzyme activity over a pH range from 7 to 9 and did not observe any changes, consistent with the observation that His 364 remains protonated over even a wide basic pH range. All these results are consistent with the proton transfer from His 364 to 6–4PP to generate a protonated neutral radical 6–4PPH• as a key step in the repair pathway (Fig. 13). This critical proton transfer, facilitated by the initial photoinduced electron transfer, completely blocks the futile back ET from 6–4PP•− and allows the reaction to proceed to repair with 100% efficiency after this step.
6.2 Reaction photocycle and repair quantum yield
On the basis of these findings and previous data, including the crystal structure of the enzyme–substrate complex, a catalytic photocycle for the repair of thymine (6–4) photoproduct was proposed (Fig. 15). In this scheme, the primary reactions are the initial electron transfer (I to II in Fig. 15) and the subsequent proton transfers (II to III in Fig. 15). The ET-induced transfer of a proton from a His residue in photolyase to the 6–4PP substrate is a key step in the repair photocycle, like the ‘dividing line’ in the transition state, and makes the subsequent reactions ‘downhill’ without the possibility of back reaction. This critical step competes with the back electron transfer, resulting in an overall repair quantum yield of about 0.1, which is probably the maximum value that could be achieved for such a structurally and chemically challenging reaction. The successive elementary steps naturally proceed to an intramolecular proton transfer from the hydroxyl group on the C5 of the 5' base to the N3 at the 3' base to form a transient zwitterion, and then an oxygen-atom attack of the C4 position at the 3' base to form a transient oxetane-type structure (III in Fig. 15). The transient-water-molecule-formation model, which proposes direct breakage of the C–O bond at the 5’ base after the initial proton transfer, seems unlikely because it necessitates a series of proton-transfer reactions, including the protonation of the carbonyl group at the 3’ base, and there are no potential proton donors in proximity to this carbonyl group. In addition, any interruption in such a complicated scheme proposed by this water model would be expected to give rise to permanent damaged DNA; this is not observed in the repair reaction by (6–4) photolyase. Our scheme, in which a simple transient-oxetane formation facilitates the oxygen-atom transfer from the 5’ to 3’ base followed by C6–C4 bond split (IV in Fig. 15), would be less prone to mutagenic side reactions because, after oxygen-atom transfer and C–C bond cleavage, the proton returns to the essential His 364 residue and the electron returns to FADH•, restoring the enzyme to its active form and the 6–4PP to two thymine bases (V in Fig. 15).
Fig. 15.

Repair photocycle of (6–4) thymine photoproduct by (6–4) photolyase.27 The resolved elementary steps include a forward electron transfer in 225 ps upon excitation (I to II), a back electron transfer in 50 ps without repair (II to I) and a parallel, catalytic proton transfer between the enzyme (His364) and the substrate (II to III), induced by the initial electron transfer, in 425 ps. This proton transfer is a critical step in repair and determines the overall repair quantum yield. The subsequent repair reactions involve a series of atom arrangements with bond breaking and making (III to IV), and final proton and electron returns (to His364 residue and flavin cofactor) to convert the 6–4PP to two thymine bases on time scales of longer than ten nanoseconds (IV to V).
7. Conclusions and outlook
In this perspective, we have summarized the recent advances on understanding UV-damaged DNA repair by photolyases. We first reported our systematic characterization of the steady-state spectra of flavin in various redox states and observed the new fluorescence emission of semiquinone states, providing a novel fluorescence tool for probing the radical semiquinone species in flavoproteins. Using the intrinsic flavin chromophore as an optical probe, we characterized the active-site solvation in CPD and (6–4) photolyases and determined the timescales of their dynamics in a few to hundreds of picoseconds. The active-site motions are critical to the understanding of the repair dynamics in photolyases. With femtosecond resolution and site-directed mutagenesis, we followed the entire dynamic evolution of the repair processes by monitoring the initial reactants, capturing various intermediates, and detecting the final product formation. We observed six elementary steps all on the ultrafast timescales including excited-state decay, three intermolecular electron-transfer reactions, and two bond-breaking and -making processes. We further identified the key functional residues at the active sites and explained the molecular origin of high repair efficiency. By examining a biomimetic system with much low repair efficiency, we conclude that the active sites of the photolyases provide a favorable electrostatic environment and local flexibility to eliminate the ultrafast deactivation processes and give enough time to inject one electron to the substrate, and balance the two competing bifurcations with all the dynamics in synergy by speeding up the bond splitting and slowing down the charge recombination. For 6–4 photolyase, we reported a sequential ET-induced proton transfer repair mechanism with a photon, an electron and a proton to collectively rearrange atoms and break chemical bonds. Thus, we obtained two complete repair photocycles by photolyases and elucidated the unique electron-tunneling pathways mediated by the intervening adenine of the folded flavin configuration and the molecular origin of high repair efficiency.
The future studies will be extended to examine the newly discovered class II, class III and single-strand DNA CPD photolyases and to compare their structures, dynamics and functional efficiencies with the class I photolyase. Such studies can provide the important correlations between structures, dynamics and functions. For 6–4 photolyase, more detailed studies are needed to capture intermediates and complete the characterization of all involved elementary steps. Related to 6–4PP repair, the investigation of the UV-damaged Dewar repair is also required. All these studies will provide not only the fundamental knowledge of a significant enzyme reaction but also a molecular basis for practical applications such as rational drug design for curing skin cancer.
Supplementary Material
{kind=link}
Acknowledgement
The authors thank Prof. Aziz Sancar for a longtime collaboration and helpful discussion (Univ. of North Carolina) and all group members involved in this DNA repair project, especially to Drs. Jiang Li, Chih-Wei Chang, Ya-Ting Kao, Chaitanya Saxena, and Chuang Tan. The work is supported in part by the National Institute of Health (Grant GM074813), the Packard Fellowship, the Camille Dreyfus Teacher-Scholar and the Guggenheim Fellowship (to D.Z.).
References
- 1.Sancar A. Chem. Rev. 2003;103:2203. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 2.Taylor JS. Acc. Chem. Res. 1994;27:76. [Google Scholar]
- 3.Daya-Grosjean L, Dumaz N, Sarasin A. J. Photochem. Photobiol. B. 1995;28:115. doi: 10.1016/1011-1344(95)07130-t. [DOI] [PubMed] [Google Scholar]
- 4.Lima-Bessa KM, Menck CFM. Curr. Biol. 2005;15:R58. doi: 10.1016/j.cub.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 5.Dulbecco R. Nature. 1949;163:949. doi: 10.1038/163949b0. [DOI] [PubMed] [Google Scholar]
- 6.Husain I, Carrier WL, Regan JD, Sancar A. Photochem. Photobiol. 1988;48:233. doi: 10.1111/j.1751-1097.1988.tb02814.x. [DOI] [PubMed] [Google Scholar]
- 7.Todo T, Takemori H, Ryo H, Ihara M, Matsunaga T, Nikaido O, Sato K, Nomura T. Nature. 1993;361:371. doi: 10.1038/361371a0. [DOI] [PubMed] [Google Scholar]
- 8.Park HW, Kim ST, Sancar A, Deisenhofer J. Science. 1995;268:1866. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 9.Mees A, Klar T, Gnau P, Hennecke U, Eker APM, Carell T, Essen LO. Science. 2004;306:1789. doi: 10.1126/science.1101598. [DOI] [PubMed] [Google Scholar]
- 10.Maul MJ, Barends TRM, Glas AF, Cryle MJ, Domratcheva T, Schneider S, Schlichting I, Carell T. Angew. Chem. Int. Ed. Engl. 2008;47:10076. doi: 10.1002/anie.200804268. [DOI] [PubMed] [Google Scholar]
- 11.Pokorny R, Klar T, Hennecke U, Carell T, Batschauer A, Essen LO. Proc. Nat. Acad. Sci. U.S.A. 2008;105:21023. doi: 10.1073/pnas.0805830106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hitomi K, DiTacchio L, Arvai AS, Yamamoto J, Kim ST, Todo T, Tainer JA, Iwai S, Panda S, Getzoff ED. Proc. Nat. Acad. Sci. U.S.A. 2009;106:6962. doi: 10.1073/pnas.0809180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiontke S, Geisselbrecht Y, Pokorny R, Carell T, Batschauer A, Essen LO. EMBO J. 2011;30:4437. doi: 10.1038/emboj.2011.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brettel K, Byrdin M. Curr. Opin. Struc. Biol. 2010;20:693. doi: 10.1016/j.sbi.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Kim ST, Sancar A. Biochemistry. 1991;30:8623. doi: 10.1021/bi00099a019. [DOI] [PubMed] [Google Scholar]
- 16.Langenbacher T, Zhao XD, Bieser G, Heelis PF, Sancar A, Michel-Beyerle ME. J. Am. Chem. Soc. 1997;119:10532. [Google Scholar]
- 17.MacFarlane AW, Stanley RJ. Biochemistry. 2003;42:8558. doi: 10.1021/bi034015w. [DOI] [PubMed] [Google Scholar]
- 18.Kao Y-T, Saxena C, Wang L, Sancar A, Zhong D. Proc. Nat. Acad. Sci. U.S.A. 2005;102:16128. doi: 10.1073/pnas.0506586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao XD, Liu JQ, Hsu DS, Zhao SY, Taylor JS, Sancar A. J. Biol. Chem. 1997;272:32580. doi: 10.1074/jbc.272.51.32580. [DOI] [PubMed] [Google Scholar]
- 20.Joseph A, Prakash G, Falvey DE. J. Am. Chem. Soc. 2000;122:11219. [Google Scholar]
- 21.Hitomi K, Nakamura H, Kim ST, Mizukoshi T, Ishikawa T, Iwai S, Todo T. J. Biol. Chem. 2001;276:10103. doi: 10.1074/jbc.M008828200. [DOI] [PubMed] [Google Scholar]
- 22.Borg OA, Eriksson LA, Durbeej B. J. Phys. Chem. A. 2007;111:2351. doi: 10.1021/jp0676383. [DOI] [PubMed] [Google Scholar]
- 23.Glas AF, Schneider S, Maul MJ, Hennecke U, Carell T. Chem. Eur. J. 2009;15:10387. doi: 10.1002/chem.200901004. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto J, Hitomi K, Hayashi R, Getzoff ED, Iwai S. Biochemistry. 2009;48:9306. doi: 10.1021/bi900956p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domratcheva T, Schlichting I. J. Am. Chem. Soc. 2009;131:17793. doi: 10.1021/ja904550d. [DOI] [PubMed] [Google Scholar]
- 26.Sadeghian K, Bocola M, Merz T, Schutz M. J. Am. Chem. Soc. 2010;132:16285. doi: 10.1021/ja108336t. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Liu Z, Tan C, Guo X, Wang L, Sancar A, Zhong D. Nature. 2010;466:887. doi: 10.1038/nature09192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kao Y-T, Saxena C, He T-F, Guo L, Wang L, Sancar A, Zhong D. J. Am. Chem. Soc. 2008;130:13132. doi: 10.1021/ja8045469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang C-W, He T-F, Guo L, Stevens JA, Li T, Wang L, Zhong D. J. Am. Chem. Soc. 2010;132:12741. doi: 10.1021/ja1050154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He T-F, Guo L, Guo X, Chang C-W, Wang L, Zhong D. Biochemistry. 2013;52:9120. doi: 10.1021/bi401137u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang C-W, Guo L, Kao Y-T, Li J, Tan C, Li T, Saxena C, Liu Z, Wang L, Sancar A, Zhong D. Proc. Nat. Acad. Sci. U.S.A. 2010;107:2914. doi: 10.1073/pnas.1000001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z, Tan C, Guo X, Kao YT, Li J, Wang L, Sancar A, Zhong D. Proc. Natl. Acad. Sci. U.S.A. 2011;108:14831. doi: 10.1073/pnas.1110927108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Guo X, Tan C, Li J, Kao Y-T, Wang L, Sancar A, Zhong D. J. Am. Chem. Soc. 2012;134:8104. doi: 10.1021/ja2105009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Zhang M, Guo X, Tan C, Li J, Wang L, Sancar A, Zhong D. Proc. Natl. Acad. Sci. USA. 2013;110:12972. doi: 10.1073/pnas.1311077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan C, Liu Z, Li J, Guo X, Wang L, Sancar A, Zhong D. 2014 in review. [Google Scholar]
- 36.Kao Y-T, Song QH, Saxena C, Wang L, Zhong D. J. Am. Chem. Soc. 2012;134:1501. doi: 10.1021/ja2112788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong D, Sancar A, Stuchebrukhov A. Proc. Natl. Acad. Sci. USA. 2012;109:E1463. [Google Scholar]
- 38.Walsh C. Acc. Chem. Res. 1980;13:148. [Google Scholar]
- 39.Massey V, Hemmerich P. Biochem. Soc. Trans. 1980;8:246. doi: 10.1042/bst0080246. [DOI] [PubMed] [Google Scholar]
- 40.Müller F. In: The Flavin Redox-System and its Biological Function. In Topics in Current Chemistry. Boschke FL, editor. Vol. 108. Berlin: Springer-Verlag; 1983. pp. 1971–1107. [DOI] [PubMed] [Google Scholar]
- 41.Müller F, editor. Chemistry and Biochemistry of Flavoenzymes. I–III. Boca Raton, FL: CRC Press; 1990–1991. [Google Scholar]
- 42.Fraaije MW, Mattevi A. Trends Biol. Sci. 2000;25:126. doi: 10.1016/s0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- 43.Massey V. Biochem. Soc. Trans. 2000;28:283. [PubMed] [Google Scholar]
- 44.Bornemann S. Nat. Prod. Rep. 2002;19:761. doi: 10.1039/b108916c. [DOI] [PubMed] [Google Scholar]
- 45.Joosten V, van Berkel WJH. Curr. Opin. Chem. Biol. 2007;11:195. doi: 10.1016/j.cbpa.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 46.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Nature. 1999;397:441. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 47.Senda T, Senda M, Kimura S, Ishida T. Antioxid. Redox Signal. 2009;11:1741. doi: 10.1089/ars.2008.2348. [DOI] [PubMed] [Google Scholar]
- 48.Fitzpatrick PF. Acc. Chem. Res. 2001;34:299. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- 49.Dagley S. Annu. Rev. Microbiol. 1987;41:1. doi: 10.1146/annurev.mi.41.100187.000245. [DOI] [PubMed] [Google Scholar]
- 50.Massey V. J. Biol. Chem. 1994;269:22459. [PubMed] [Google Scholar]
- 51.Dong CJ, Flecks S, Unversucht S, Haupt C, van Pee KH, Naismith JH. Science. 2005;309:2216. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozturk N, Selby CP, Annayev Y, Zhong D, Sancar A. Proc. Nat. Acad. Sci. U.S.A. 2011;108:516. doi: 10.1073/pnas.1017093108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin CT, Shalitin D. Annu. Rev. Plant Biol. 2003;54:469. doi: 10.1146/annurev.arplant.54.110901.160901. [DOI] [PubMed] [Google Scholar]
- 54.Lin CT, Todo T. Genome Biol. 2005;6:220. doi: 10.1186/gb-2005-6-5-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Partch CL, Sancar A. Photochem. Photobiol. 2005;81:1291. doi: 10.1562/2005-07-08-IR-607. [DOI] [PubMed] [Google Scholar]
- 56.Song SH, Ozturk N, Denaro TR, Arat NO, Kao YT, Zhu H, Zhong D, Reppert SM, Sancar A. J. Biol. Chem. 2007;282:17608. doi: 10.1074/jbc.M702874200. [DOI] [PubMed] [Google Scholar]
- 57.Ozturk N, Song SH, Selby CP, Sancar A. J. Biol. Chem. 2008;283:3256. doi: 10.1074/jbc.M708612200. [DOI] [PubMed] [Google Scholar]
- 58.Kennis JTM, Groot ML. Curr. Opin. Struct. Biol. 2007;17:623. doi: 10.1016/j.sbi.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 59.Christie JM. Annu. Rev. Plant Biol. 2007;58:21. doi: 10.1146/annurev.arplant.58.032806.103951. [DOI] [PubMed] [Google Scholar]
- 60.Heelis PF. Chem. Soc. Rev. 1982;11:15. [Google Scholar]
- 61.Ghisla S, Massey V, Lhoste JM, Mayhew SG. Biochemistry. 1974;13:589. doi: 10.1021/bi00700a029. [DOI] [PubMed] [Google Scholar]
- 62.Land EJ, Swallow AJ. Biochemistry. 1969;8:2117. doi: 10.1021/bi00833a050. [DOI] [PubMed] [Google Scholar]
- 63.Sakai M, Takahashi H. J. Mol. Struct. 1996;379:9. [Google Scholar]
- 64.Massey V, Palmer G. Biochemistry. 1966;5:3181. doi: 10.1021/bi00874a016. [DOI] [PubMed] [Google Scholar]
- 65.Kao Y-T, Tan C, Song SH, Ozturk N, Li J, Wang L, Sancar A, Zhong D. J. Am. Chem. Soc. 2008;130:7695. doi: 10.1021/ja801152h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stanley RJ, MacFarlane AW. J. Phys. Chem. A. 2000;104:6899. [Google Scholar]
- 67.Malicka J, Groth M, Karolczak J, Czaplewski C, Liwo A, Wiczk W. Biopolymers. 2001;58:447. doi: 10.1002/1097-0282(20010405)58:4<447::AID-BIP1020>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 68.Reichardt C. Solvents and Solvent Effects in Organic Chemistry. 3rd ed. Weinheim: Wiley-VCH; 2003. [Google Scholar]
- 69.Weber G. Biochem. J. 1950;47:114. doi: 10.1042/bj0470114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barrio JR, Tolman GL, Leonard NJ, Spencer RD, Weber G. Proc. Nat. Acad. Sci. U.S.A. 1973;70:941. doi: 10.1073/pnas.70.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van den Berg PAW, Feenstra KA, Mark AE, Berendsen HJC, Visser AJWG. J. Phys. Chem. B. 2002;106:8858. [Google Scholar]
- 72.Visser AJWG. Photochem. Photobiol. 1984;40:703. doi: 10.1111/j.1751-1097.1984.tb04640.x. [DOI] [PubMed] [Google Scholar]
- 73.Kotowycz G, Teng N, Klein MP, Calvin M. J. Biol. Chem. 1969;244:5656. [PubMed] [Google Scholar]
- 74.Kainosho M, Kyogoku Y. Biochemistry. 1972;11:741. doi: 10.1021/bi00755a011. [DOI] [PubMed] [Google Scholar]
- 75.Copeland RA, Spiro TG. J. Phys. Chem. 1986;90:6648. [Google Scholar]
- 76.Weber G, Tanaka F, Okamoto BY, Drickame HG. Proc. Nat. Acad. Sci. U.S.A. 1974;71:1264. doi: 10.1073/pnas.71.4.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hahn J, Michel-Beyerle ME, Rosch N. J. Mol. Model. 1998;4:73. [Google Scholar]
- 78.Islam SDM, Susdorf T, Penzkofer A, Hegemann P. Chem. Phys. 2003;295:137. [Google Scholar]
- 79.Brautigam CA, Smith BS, Ma ZQ, Palnitkar M, Tomchick DR, Machius M, Deisenhofer J. Proc. Natl. Acad. Sci. USA. 2004;101:12142. doi: 10.1073/pnas.0404851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zoltowski BD, Vaidya AT, Top D, Widom J, Young MW, Crane BR. Nature. 2013;496:252. doi: 10.1038/nature10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kodali G, Siddiqui SU, Stanley RJ. J. Am. Chem. Soc. 2009;131:4795. doi: 10.1021/ja809214r. [DOI] [PubMed] [Google Scholar]
- 82.Siddiqui SU, Kodali G, Stanley RJ. J. Phys. Chem. B. 2008;112:119. doi: 10.1021/jp075830e. [DOI] [PubMed] [Google Scholar]
- 83.Pal SK, Zewail AH. Chem. Rev. 2004;104:2099. doi: 10.1021/cr020689l. [DOI] [PubMed] [Google Scholar]
- 84.Zhong D. Adv. Chem. Phys. 2009;143:83. [Google Scholar]
- 85.Levy Y, Onuchic JN. Annu. Rev. Biophys. Biomol. Struct. 2006;35:389. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 86.Zhong D, Pal SK, Zewail AH. ChemPhysChem. 2001;2:219. doi: 10.1002/1439-7641(20010417)2:4<219::AID-CPHC219>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 87.Mattos C. Trends Biochem. Sci. 2002;27:203. doi: 10.1016/s0968-0004(02)02067-4. [DOI] [PubMed] [Google Scholar]
- 88.Qiu W, Wang L, Lu W, Boechler A, Sanders DAR, Zhong D. Proc. Nat. Acad. Sci. U.S.A. 2007;104:5366. doi: 10.1073/pnas.0608498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pocker Y. Cell. Mol. Life Sci. 2000;57:1008. doi: 10.1007/PL00000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Helms V. ChemPhysChem. 2007;8:23. doi: 10.1002/cphc.200600298. [DOI] [PubMed] [Google Scholar]
- 91.Sumi H, Marcus RA. J. Chem. Phys. 1986;84:4894. [Google Scholar]
- 92.Nagasawa Y, Yartsev AP, Tominaga K, Bisht PB, Johnson AE, Yoshihara K. J. Phys. Chem. 1995;99:653. [Google Scholar]
- 93.Wang HY, Lin S, Allen JP, Williams JC, Blankert S, Laser C, Woodbury NW. Science. 2007;316:747. doi: 10.1126/science.1140030. [DOI] [PubMed] [Google Scholar]
- 94.LeBard DN, Kapko V, Matyushov DV. J. Phys. Chem. B. 2008;112:10322. doi: 10.1021/jp8016503. [DOI] [PubMed] [Google Scholar]
- 95.Pierce DW, Boxer SG. J. Phys. Chem. 1992;96:5560. [Google Scholar]
- 96.Cohen BE, McAnaney TB, Park ES, Jan YN, Boxer SG, Jan LY. Science. 2002;296:1700. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]
- 97.Jesenská A, Sykora J, Olzynska A, Brezovsky J, Zdrahal Z, Damborsky J, Hof M. J. Am. Chem. Soc. 2009;131:494. doi: 10.1021/ja804020q. [DOI] [PubMed] [Google Scholar]
- 98.Homoelle BJ, Edington MD, Diffey WM, Beck WF. J. Phys. Chem. B. 1998;102:3044. [Google Scholar]
- 99.Pal SK, Peon J, Zewail AH. Proc. Nat. Acad. Sci. U.S.A. 2002;99:1763. doi: 10.1073/pnas.042697899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qiu W, Zhang L, Kao Y-T, Lu W, Li T, Kim J, Sollenberger GM, Wang L, Zhong D. J. Phys. Chem. B. 2005;109:16901. doi: 10.1021/jp0511754. [DOI] [PubMed] [Google Scholar]
- 101.Qiu W, Kao Y-T, Zhang L, Yang Y, Wang L, Stites WE, Zhong D, Zewail AH. Proc. Nat. Acad. Sci. U.S.A. 2006;103:13979. doi: 10.1073/pnas.0606235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang L, Wang L, Kao Y-T, Qiu W, Yang Y, Okobiah O, Zhong D. Proc. Nat. Acad. Sci. U.S.A. 2007;104:18461. doi: 10.1073/pnas.0707647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang L, Yang Y, Kao Y-T, Wang L, Zhong D. J. Am. Chem. Soc. 2009;131:10677. doi: 10.1021/ja902918p. [DOI] [PubMed] [Google Scholar]
- 104.Abbyad P, Childs W, Shi XH, Boxer SG. Proc. Nat. Acad. Sci. U.S.A. 2007;104:20189. doi: 10.1073/pnas.0706185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu W, Kim J, Qiu W, Zhong D. Chem. Phys. Lett. 2004;388:120. [Google Scholar]
- 106.Prytkova TR, Beratan DN, Skourtis SS. Proc. Nat. Acad. Sci. U.S.A. 2007;104:802. doi: 10.1073/pnas.0605319104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kodali G. Excited state electronic properties of DNA photolyase and fluorescent nucleobase analogues (FBA): An experimental and theoretical study. UMI Thesis Publishing; 2009. [Google Scholar]
- 108.Philip AF, Nome RA, Papadantonakis GA, Scherer NF, Hoff WD. Proc. Nat. Acad. Sci. U.S.A. 2010;107:5821. doi: 10.1073/pnas.0903092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Partch CL, Clarkson MW, Ozgur S, Lee AL, Sancar A. Biochemistry. 2005;44:3795. doi: 10.1021/bi047545g. [DOI] [PubMed] [Google Scholar]
- 110.Li T, Hassanali AA, Kao Y-T, Zhong D, Singer SJ. J. Am. Chem. Soc. 2007;129:3376. doi: 10.1021/ja0685957. [DOI] [PubMed] [Google Scholar]
- 111.Kao Y-T, Saxena C, Wang L, Sancar A, Zhong D. Cell Biochem. Biophys. 2007;48:32. doi: 10.1007/s12013-007-0034-5. [DOI] [PubMed] [Google Scholar]
- 112.Masson F, Laino T, Rothlisberger U, Hutter J. ChemPhysChem. 2009;10:400. doi: 10.1002/cphc.200800624. [DOI] [PubMed] [Google Scholar]
- 113.Hassanali AA, Zhong D, Singer SJ. J. Phys. Chem. B. 2011;115:3848. doi: 10.1021/jp107722z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hassanali AA, Zhong D, Singer SJ. J. Phys. Chem. B. 2011;115:3860. doi: 10.1021/jp107723w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Voityuk AA, Rosch N. J. Phys. Chem. A. 1997;101:8335. [Google Scholar]
- 116.Durbeej B, Eriksson LA. J. Am. Chem. Soc. 2000;122:10126. [Google Scholar]
- 117.Saettel NJ, Wiest O. J. Am. Chem. Soc. 2001;123:2693. doi: 10.1021/ja005775m. [DOI] [PubMed] [Google Scholar]
- 118.Harrison CB, O'Neil LL, Wiest O. J. Phys. Chem. A. 2005;109:7001. doi: 10.1021/jp051075y. [DOI] [PubMed] [Google Scholar]
- 119.Tachikawa H, Kawabata H. J. phys. Chem. B. 2008;112:7315. doi: 10.1021/jp801564t. [DOI] [PubMed] [Google Scholar]
- 120.Masson F, Laino T, Tavernelli I, Rothlisberger U, Hutter J. J. Am. Chem. Soc. 2008;130:3443. doi: 10.1021/ja076081h. [DOI] [PubMed] [Google Scholar]
- 121.Boussicault F, Kruger O, Robert M, Wille U. Org. Biomol. Chem. 2004;2:2742. doi: 10.1039/B406923D. [DOI] [PubMed] [Google Scholar]
- 122.Gindt YM, Schelvis JPM, Thoren KL, Huang TH. J. Am. Chem. Soc. 2005;127:10472. doi: 10.1021/ja051441r. [DOI] [PubMed] [Google Scholar]
- 123.Payne G, Sancar A. Biochemistry. 1990;29:7715. doi: 10.1021/bi00485a021. [DOI] [PubMed] [Google Scholar]
- 124.Ramsey AJ, Alderfer JL, Jorns MS. Biochemistry. 1992;31:7134. doi: 10.1021/bi00146a015. [DOI] [PubMed] [Google Scholar]
- 125.Thiagarajan V, Byrdin M, Eker APM, Muller P, Brettel K. Proc. Natl. Acad. Sci. U.S.A. 2011;108:9402. doi: 10.1073/pnas.1101026108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Acocella A, Jones GA, Zerbetto F. J. Phys. Chem. B. 2010;114:4101. doi: 10.1021/jp101093z. [DOI] [PubMed] [Google Scholar]
- 127.Antony J, Medvedev DM, Stuchebrukhov AA. J. Am. Chem. Soc. 2000;122:1057. [Google Scholar]
- 128.Wang H, Chen X, Fang W. Phys. Chem. Chem. Phys. 2014;165:25432. doi: 10.1039/c4cp04130e. [DOI] [PubMed] [Google Scholar]
- 129.Seidel CAM, Schulz A, Sauer MHM. Phys. Chem. 1996;100:5541. [Google Scholar]
- 130.Wilson EK, Huang LX, Sutcliffe MJ, Mathews FS, Hille R, Scrutton NS. Biochemistry. 1997;36:41. doi: 10.1021/bi961224q. [DOI] [PubMed] [Google Scholar]
- 131.Twitchett MB, Ferrer JC, Siddarth P, Mauk AG. J. Am. Chem. Soc. 1997;119:435. [Google Scholar]
- 132.Roth JP, Wincek R, Nodet G, Edmondson DE, McIntire WS, Klinman JP. J. Am. Chem. Soc. 2004;126:15120. doi: 10.1021/ja047050e. [DOI] [PubMed] [Google Scholar]
- 133.Kim ST, Hartman RF, Rose SD. Photochem. Photobiol. 1990;52:789. doi: 10.1111/j.1751-1097.1990.tb08683.x. [DOI] [PubMed] [Google Scholar]
- 134.Carell T, Epple R. Eur. J. Org. Chem. 1998:1245. [Google Scholar]
- 135.Song QH, Tang WM, Hei XM, Wang HB, Guo QX, Yu SQ. Eur. J. Org. Chem. 2005:1097. doi: 10.1021/jo048006k. [DOI] [PubMed] [Google Scholar]
- 136.Essen LO, Klar T. Cell. Mol. Life Sci. 2006;63:1266. doi: 10.1007/s00018-005-5447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Epple R, Wallenborn EU, Carell T. J. Am. Chem. Soc. 1997;119:7440. [Google Scholar]
- 138.Song QH, Tang WJ, Ji XB, Wang HB, Guo QX. Chem. Eur. J. 2007;13:7762. doi: 10.1002/chem.200700251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.