

Background: RGS7 plays an essential role in regulating neuronal G protein signaling.
Results: Elimination of GPR158 in mice reduces RGS7 expression and membrane localization. Unique domains in GPR158 control RGS7 catalytic activity.
Conclusion: The function of RGS7 in the brain is critically regulated by binding to GPR158.
Significance: This introduces a new player and its mechanism for regulating RGS activity in the nervous system.
Keywords: G Protein-coupled Receptor (GPCR), GTPase, Heterotrimeric G Protein, Neurobiology, Regulator of G Protein Signaling (RGS), Signal Transduction
Abstract
Regulators of G protein signaling control the duration and extent of signaling via G protein-coupled receptor (GPCR) pathways by accelerating the GTP hydrolysis on G protein α subunits thereby promoting termination of GPCR signaling. A member of this family, RGS7, plays a critical role in the nervous system where it regulates multiple neurotransmitter GPCRs that mediate vision, memory, and the action of addictive drugs. Previous studies have established that in vivo RGS7 forms mutually exclusive complexes with the membrane protein RGS7-binding protein or the orphan receptor GPR158. In this study, we examine the impact of GPR158 on RGS7 in the brain. We report that knock-out of GPR158 in mice results in marked post-transcriptional destabilization of RGS7 and substantial loss of its association with membranes in several brain regions. We further identified the RGS7-binding site in the C terminus of GPR158 and found that it shares significant homology with the RGS7-binding protein. The proximal portion of the GPR158 C terminus additionally contained a conserved sequence that was capable of enhancing RGS7 GTPase-activating protein activity in solution by an allosteric mechanism acting in conjunction with the regulators of the G protein signaling-binding domain. The distal portion of the GPR158 C terminus contained several phosphodiesterase E γ-like motifs and selectively recruited G proteins in their activated state. The results of this study establish GPR158 as an essential regulator of RGS7 in the native nervous system with a critical role in controlling its expression, membrane localization, and catalytic activity.
Introduction
G protein-coupled receptors (GPCRs)2 form a large family of transmembrane signaling molecules that mediate many vital physiological processes. In the nervous system, they play essential role in sensory reception, neurotransmitter signaling, differentiation, and regulation of neuronal excitability (1). GPCRs transduce their signals via activation of the heterotrimeric G proteins (2). In the prototypic sequence of events, binding of a specific ligand induces a conformational change in the receptor that leads it to catalyze the exchange of GDP to GTP on the Gα subunit of the G protein. Upon nucleotide exchange, Gα-GTP dissociates from the Gβγ subunits, and in this state both entities activate a variety of effectors that are responsible for the generation of cellular response. The signaling continues until Gα hydrolyzes GTP and re-associates with Gβγ subunits. The signaling termination is a highly regulated process as appropriate duration of signaling is essential for normal physiological function. Termination of G protein signaling relies on the action of the regulators of G protein signaling (RGS) proteins, a family encoded by more than 30 genes in mammals (3–5). RGS proteins act as GTPase-activating proteins (GAP) that stimulate the GTP hydrolysis on the Gα subunits catalyzing their timely inactivation and thus naturally opposing stimulatory action of GPCRs.
Among the many RGS proteins expressed in the nervous system, a particularly important role belongs to the R7 subfamily (R7 RGS) that includes RGS6, RGS7, RGS9, and RGS11. Collectively, R7 RGS proteins play key roles in the regulation of several fundamental physiological processes such as vision, memory, motor control, reward behavior, and nociception (6, 7). The distinguishing feature of the R7 RGS proteins is the complexity of their macromolecular organization; they contain four structurally distinct domains and form stable heteromers with several proteins. These interactions determine GAP activity, proteolytic stability, and subcellular localization of the R7 RGS complexes (8–11). The C terminus of the R7 RGS proteins is composed of the catalytic RGS domain that binds to Gα-GTP and stimulates its GTPase activity (12–14). The RGS domain is preceded by a central Gγ-like domain, which forms a nondissociable complex with the type 5 G protein β subunit (Gβ5) (15–17). This binding is obligatory and is required for folding and proteolytic stability of the R7 RGS complexes. The N terminus of R7 RGS proteins is formed by the DEP (Disheveled, egl-10, pleckstrin) and DHEX (DEP helical extension) domains that are intertwined together to form a single module (9, 18). The DEP/DHEX module is crucial for binding to small SNARE-like membrane proteins as follows: RGS9 anchor protein (R9AP) and R7-binding protein (R7BP) (19–21). The specificity of these interactions varies among individual R7 RGS proteins. Although R9AP can bind only to RGS9 and RGS11, R7BP interacts with all R7 RGS members (9, 21). Association with R9AP and/or R7BP was shown to result in membrane recruitment of R7 RGS proteins (20, 22, 23), potentiation of their catalytic activity (19, 24, 25), and influencing Gα selectivity (26).
The consequence of disrupting R7 RGS association with R7BP and R9AP anchors was examined in vivo using mouse knock-out models. R9AP expression is limited to the retina where it is present only in photoreceptors and ON-bipolar neurons (20, 27, 28). Accordingly, knock-out of R9AP resulted in elimination of RGS9-1 and RGS11 (27, 29, 30) that are expressed in these neurons, respectively. When transgenically expressed in the photoreceptors, the mutant of RGS9 incapable of binding to R9AP also failed to appropriately localize to the outer segment, a membranous compartment of the cell (31). Similarly, knock-out of R7BP, which is broadly expressed in the nervous system, resulted in proteolytic destabilization of RGS9-2 in the striatum, a region of the brain where RGS9-2 is preferentially expressed (8). Furthermore, RGS9-2 was markedly mislocalized from the plasma membrane of striatal neurons in R7BP knockouts (32). Together, these observations confirm the essential role of membrane anchoring subunits R9AP and R7BP in dictating localization, expression, and the ability of R7 RGS complexes to regulate G protein signaling in vivo. Curiously, despite robust localization of RGS7 on the membranes of native neuronal tissues (33, 34) and the demonstrated ability of R7BP to recruit it there in transfected cells (22), knock-out of R7BP or R9AP in mice has very minor effects on RGS7 localization and does not impact its expression (33, 35–37). These observations suggest the possible involvement of alternative mechanisms that regulate RGS7 in the nervous system.
Using an unbiased proteomic approach, we have recently identified a new type of membrane anchors for R7 RGS proteins, GPR158 and GPR179 that belong to a group of orphan GPCRs (38). GPR158/179 compete with R7BP for binding to R7 RGS proteins and can recruit them on the plasma membrane in transfected cells (38). Biochemically, GPR179 can bind to all R7 RGS members, but its expression is restricted to the ON-bipolar neurons in the retina (38). Loss of GPR179 results in a mislocalization of RGS7 and RGS11 in these neurons and leads to night blindness (38–40). GPR158 is broadly expressed in the nervous system and preferentially binds to RGS7 (38). However, the role of GPR158 in influencing RGS7 homeostasis and function in the nervous system is unknown.
In this study, we examined the impact of GPR158 on the expression, localization, and catalytic activity of RGS7 using a combination of approaches involving in vivo mouse genetics and in vitro enzyme kinetics and protein-protein interaction assays. We report that knock-out of GPR158 in mice decreases RGS7 expression across the brain and results in substantial loss of its membrane localization. We identified the binding site for RGS7 in GPR158, and we show that it acts in combination with other regulatory elements to enhance RGS7 GAP activity toward Gαo by an allosteric mechanism. Together, our results indicate that GPR158 is an essential regulator of RGS7 function in the nervous system.
Experimental Procedures
Mice, Antibodies, and Genetic Constructs
The generation of R7BP knock-out mice has been described (8). A line of GPR158 knock-out mice was created from ES cell clone 10108A-A5, generated by Regeneron Pharmaceuticals, Inc., and made into live mice by the KOMP Repository and the Mouse Biology Program at the University of California at Davis. In these mice, the first two exons encoding ∼⅓ of the entire GPR158 sequence were replaced with the LacZ cassette containing a stop codon. All procedures involving mice were reviewed and approved by the IACUC committee at the Scripps Research Institute.
We generated rabbit antibodies against the intracellular C terminus of mouse GPR158 (aa 665–1200; GPR158CT). Two GST-tagged proteins encoding the GPR158 sequences 665–961 and 962–1200 were purified by affinity chromatography on glutathione-Sepharose high performance beads (GE Healthcare), mixed, and used for the rabbit immunization. Antibodies from the immune sera were then affinity-purified against the same peptides used for the immunization. Polyclonal RGS7 antibodies (RGS7NT) were affinity-purified from rabbit sera after immunization with synthetic peptides (Pocono Rabbit Farm & Laboratory, Inc.). Briefly, synthetic peptide from the N terminus of mouse RGS7 (GNNYGQTSNGVADESPC) was covalently immobilized to beaded agarose using SulfoLink immobilization kit (Pierce). Antibodies against RGS7 were then purified by affinity chromatography from immune sera. Generation of sheep anti-RGS9-2 and sheep anti-RGS6 antibodies was described previously (21). Rabbit anti-Gβ1 was a kind gift from Dr. Barry Willardson (Brigham Young University, Provo, UT). Rabbit anti-Gβ5, rabbit anti-RGS7 (7RC-1), and rabbit anti-R7BP were gifts from Dr. William Simonds (NIDDK, National Institutes of Health, Bethesda). Mouse anti-GAPDH (Millipore), mouse anti-HA (Millipore), and rabbit anti-Gαo (Cell Signaling) were purchased. Chicken anti-RGS7 antibodies (Pierce) were used for immunodetection of RGS7 after GST pulldown assays; rabbit anti-RGS7 antibodies (7RC-1) were used for immunoprecipitation and Western blotting in transfected cells, and rabbit RGS7NT antibodies were used for the detection of RGS7 in native tissues. Rabbit anti-GPR158 (Assay Biotech) antibodies against GPR158 N terminus were used for live staining immunocytochemistry and immunoprecipitation, and rabbit GPR158CT antibodies were used in the Western blot experiments.
Cloning of full-length mouse GPR158, R7BP, Gβ5, RGS7, and DEPless RGS7 into the pcDNA3.1/V5-His-TOPO were described previously (9, 13, 21, 38, 41, 42). Full-length human RGS7 (GenBankTM accession number AY587875) was subcloned into pcDNA3.1/V5-His-TOPO with N-terminal HA tag and an RGS7 original stop codon. For expression in Escherichia coli, full-length mouse R7BP was subcloned into pET-28a (+). Plasmids encoding mouse GPR158 with an internal deletion of aa 671–961 (GPR158Δ-CD1/2/3) and a C- terminal deletion of aa 962–1200 (GPR158-ΔCD4) were generated by reverse PCR using as template the plasmid encoding the full-length GPR158. Plasmids encoding GST-fused proteins were generated using In-Fusion HD cloning system (Clontech) in the pGEX-2T vector. All these constructs contained GST sequence fused in-frame with sequences encoding the following regions derived from mouse GPR158: CD1 (aa 665–775); CD2 (aa 777–830); CD3 (aa 866–944); CD4 aa (962–1200); CD1/2 (aa 665–830); CD2/3 (aa 777–944); CD1/2/3 (aa 665–961); and the full-length GPR158 C terminus (CT, aa 665–1200). All constructs were verified by sequencing.
Cell Culture, Transfection, Western Blotting, and Immunoprecipitation
COS1 cells were used for immunocytochemistry experiments, and HEK293T/17 cells were used for Western blotting and immunoprecipitation studies. Both cell lines were cultured at 37 °C and 5% CO2 in DMEM supplemented with 10% FBS, minimum Eagle's medium nonessential amino acids, 1 mm sodium pyruvate, and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin). Cells were transfected using Lipofectamine LTX (Invitrogen) and Plus reagent (Invitrogen) and used 24 h later. For Western blotting analysis, cells were harvested and lysed in ice-cold immunoprecipitation buffer (300 mm NaCl, 50 mm Tris-HCl, pH 7.4, 1% Triton X-100, and complete protease inhibitor mixture) by sonication. For immunoprecipitation, lysates were cleared by centrifugation at 14,000 rpm for 15 min, and the supernatants were incubated with 20 μl of Dynabeads (Invitrogen) and 2 μg of antibodies on a rocker at 4 °C for 1 h. After three washes with immunoprecipitation buffer, proteins were eluted with 50 μl of 2× SDS sample buffer and analyzed by SDS-PAGE.
Immunocytochemistry
The staining of GPR158 has been performed on live cells before permeabilization, and S7 staining was performed after permeabilization of cells. Live transfected COS1 cells were incubated overnight at 4 °C with rabbit anti-GPR158 antibody (Assay Biotech) in PBS containing 2% donkey serum. After a brief wash with PBS, cells were fixed for 15 min with 4% paraformaldehyde, permeabilized for 5 min with 0.1% Triton X-100/PBS, blocked with 10% donkey serum in PBS for 1 h, and incubated with mouse anti-HA tag (Millipore) primary antibody in 2% donkey serum in PBS for 1 h. After three washes, sections were incubated with Alexa Fluor 488 anti-rabbit and Alexa Fluor 546 anti-mouse secondary antibodies for 1 h. Cells were stained 5 min with DAPI before mounting in Fluoromount G (SouthernBiotech). Cells were imaged using confocal microscopy (LSM 780; Carl Zeiss; Plan Neofluar 63×/1.3 NA Korr differential interference contrast M27 objective in water) at room temperature. Image acquisition and processing were accomplished using ZEN 2011 (64 bit) software (Carl Zeiss) with only minor manipulations of the images setting the fluorescence intensity in nonsaturating conditions and maintaining similar parameters for each acquired image.
Subcellular Fractionation
Brains were quickly removed from euthanized mice, and different brain regions (cortex, striatum, and hippocampus) were dissected on ice. Tissues were homogenized in ice-cold lysis buffer (150 mm NaCl, 50 mm Tris-HCl, pH 7.4, 1 mm EDTA, 2.5 mm MgCl2, and complete protease inhibitor mixture (Roche Applied Science)) by sonication. Lysates were adjusted to the same protein concentration by diluting with the lysis buffer, and equal amounts were subjected to ultracentrifugation (200,000 × g for 30 min at 4 °C). The supernatant was recovered and designated as the cytosolic fraction. The pellet was washed with the lysis buffer and re-sedimented by centrifugation (200,000 × g for 30 min at 4 °C). The pellet was then resuspended in immunoprecipitation buffer (300 mm NaCl, 50 mm Tris-HCl, pH 7.4, 1% Triton X-100 and complete protease inhibitor mixture), incubated on a rocker for 30 min at 4 °C, and cleared by centrifugation at 14,000 rpm for 15 min. The supernatant was saved and designated as membrane fraction.
Immunogold Electron Microscopy
Immunohistochemical reactions were carried out using the pre-embedding immunogold method as described earlier (43). Briefly, after blocking with 10% serum for 1 h at room temperature, free-floating sections were incubated for 48 h with rabbit anti-RGS7 antibodies (1–2 μg/ml). Sections were washed and incubated for 3 h with goat anti-rabbit IgG coupled to 1.4 nm gold (Nanoprobes Inc) at 1:100 dilution. Sections were washed, postfixed in 1% glutaraldehyde, and processed for silver enhancement of the gold particles with an HQ silver kit (Nanoprobes Inc.). The reacted sections were treated with osmium tetroxide (1% in 0.1 m phosphate buffer), block-stained with uranyl acetate, dehydrated in graded series of ethanol, and flat-embedded on glass slides in Durcupan (Fluka) resin. Regions of interest were cut at 70–90 nm on an ultramicrotome (Reichert Ultracut E; Leica). Staining was performed on drops of 1% aqueous uranyl acetate followed by Reynolds's lead citrate. Ultrastructural analyses were performed in a Jeol-1010 electron microscope. Quantification of immunogold labeling was carried out in reference areas totaling ∼ 2000 μm2 for each sample as described previously (36). Immunoparticles identified in each reference area and present in different subcellular compartments (dendritic spines and dendritic shafts) were counted. We measured the radial distance of each immunoparticle to the plasma membrane, being 0 for those just located in the plasma membrane. The data were expressed as percentage of immunoparticles along the radial distance from the plasma membrane expressed in nanometers.
Quantitative Real Time PCR
Total RNA from the whole brain was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. The RNA in the aqueous phase was further purified using the RNeasy spin column (Qiagen). RNA quantification and quality controls were performed using spectrophotometric analysis and the Bioanalyzer 2100 lab-on-a-chip technology (Agilent Technologies). Reverse transcription was carried out using Superscript III first-strand synthesis supermix for quantitative RT-PCR (Invitrogen) according to manufacturer's instructions starting from 0.5 μg of total RNA. To analyze the RNA expression pattern of the target genes, we used the 7900HT Fast Real Time PCR System (Applied Biosystems) with TaqMan probes under the following conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 1 min. Three biological replicates and three technical replicates for each sample were used. 10 ng of each sample were used in each real time PCR (TaqMan Gene Expression Assay ID probes: RGS7, Mm01317058_m1; RGS9, Mm01250425_m1; GPR158, Mm00558878_m1; R7BP, Mm00712799_m1, from Applied Biosystems). The expression ratio of the target genes was calculated using the β-actin (ID 4352341E) as reference using the 2−ΔΔCT method (44). Data are shown as mean ± S.E.
Purification of Recombinant Proteins and GST Pulldown Assays
Recombinant RGS7 was co-expressed with Gβ5S using the Sf9 baculovirus system as described previously (21). Protein complexes were purified by nickel-nitrilotriacetic acid chromatography taking advantage of the His tag engineered at the N terminus of RGS7 as described previously (13). Recombinant Gαo purification was described (41). Recombinant His-tagged R7BP was expressed in BL21 (DE3) E. coli upon induction with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 28 °C for 24 h of culturing. Cell lysate was loaded on a His TALON column, and the proteins were eluted by increasing imidazole concentrations. Recombinant GST fusion proteins were expressed in BL21 E. coli cells after induction with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and purified by affinity chromatography on glutathione-Sepharose high performance beads (GE Healthcare). The purity of the recombinant proteins was assessed by Coomassie staining after gel separation and found to be at least 80%. GST pulldown assays were performed as described previously (21). Briefly, 100 μl of purified recombinant GST fusion proteins (4 μm) were attached to 20 μl of glutathione-agarose beads (GE Healthcare) by incubating in binding buffer (20 mm Tris-HCl, pH 7.8, 300 mm NaCl, 0.25% N-dodecanoyl sucrose, 2 mm MgCl2, 50 μg/ml bovine serum albumin) for 1 h on ice. The beads were washed twice with binding buffer and incubated with 50 μl of 2 μm purified RGS7-Gβ5 complexes or Gαo for 15 min, followed by three washes. Gαo was loaded with GDP, GTPγS, or GDP in the presence of freshly prepared AlF4 for 30 min at 30 °C. The proteins were eluted in 50 mm Tris-HCl, 250 mm NaCl, 15 mm glutathione at pH 8.0 and analyzed by Western blotting with specific antibodies.
GTPase Activity Assays
Single-turnover GTPase assays using recombinant proteins were conducted as described previously (45). Purified Gαo (3.5 μm) was pre-loaded with 12 μm [γ-32P]GTP (25 Ci/mmol, MP Biomedicals) in 100 μl of charging buffer (50 mm HEPES, pH 7.5, 100 mm NaCl, 1 mm DTT, 5 μg/ml BSA, 0.05% polyoxyethylene 10 lauryl ether, and 10 mm EDTA) for 20 min at room temperature. The mixture was then placed on ice. Unbound [γ-32P]GTP was removed by protein desalting spin column (Pierce), and the buffer of the [γ-32P]GTP-bound Gαo was exchanged to GTPase buffer (10 mm Tris-HCl, pH 7.8, 100 mm NaCl, 8 mm MgCl2, and 1 mm DTT). The reactions were started by combining 10 μl of 1.2 μm of [γ-32P]GTP-bound Gαo with 20 μl of GTPase buffer solution supplemented with RGS7-Gβ5 with or without additional proteins. Final concentration in the reaction was 0.4 μm for Gαo, 0.25 μm for RGS7, and 0.5 μm for tested proteins (R7BP and purified GPR158 C-terminal fragments). The reaction was stopped by the addition of 100 μl of 6% perchloric acid at the desired time points. 10 μl of the [γ-32P]GTP-bound Gαo solution was removed (time 0) directly to 100 μl of 6% perchloric acid. The 32Pi formation released from hydrolyzed GTP was measured in the supernatant by activated charcoal assay (46). The amount of radioactivity was determined by scintillation counting. Complete (100%) GTP hydrolysis was performed in the presence of excess RGS7 (1.2 μm) for at least 30 min. Percentage of GTP hydrolysis was calculated as (time point − time 0)/(100% GTP hydrolysis − time 0) × 100%. To determine values for the rate of GTP hydrolysis (1/τ), the data were fitted to the exponential equation using nonlinear regression. To emphasize the modulation of catalytic activity of RGS7, the parameter of kGAP was calculated, defined as the difference between the rates of GTP hydrolysis in the presence of RGS7 and the Gαo basal GTP hydrolysis rate.
Bioinformatics Analysis
The amino acid sequences of GPR158 from 82 different species were identified by homology using BLAST. Multiple sequence alignment of these sequences was performed using the software COBALT (47) and setting the parameters for conservation analysis to “identity.” Using this approach, we were able to identify amino acids that were conserved across species and therefore good candidates for a functional analysis (48). Coiled-coil motifs have been predicted using the software COILS that compares a sequence to a database of known parallel two-stranded coiled coils and derives a similarity score (49). The secondary structure of GPR158 CD1/2/3 sequence has been analyzed using the on-line available program XtalPred-RF. The PGL domains in PDEγ, RGS9-2, GPR158, and GPR179 have been aligned using the multiple sequence alignment software Clustal Omega. The consensus motif has then been identified using the software ESPript 3.0.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (Prism 6.0, GraphPad, San Diego). Groups were compared using Student's t test. GTPase assay results were analyzed using one-way ANOVA followed by post hoc Tukey's test.
Results
GPR158, but Not R7BP, Is Required for the Stabilization of RGS7 in Vivo
We previously showed that GPR158 forms a stable complex with RGS7-Gβ5 in vitro and in vivo (38). To test the involvement of GPR158 in the regulation of RGS7 function in vivo, we generated a line of GPR158 knock-out mice (GPR158 KO). In these mice ∼⅓ of the entire coding sequence of GPR158 is replaced with the LacZ cassette containing the stop codon, making the expression of any remaining parts of the GPR158 gene impossible (Fig. 1A). Indeed, Western blot analysis confirms the elimination of GPR158 from the brain at the protein level (Fig. 1B). Next, we examined possible changes in the levels of R7 RGS proteins expressed in the brain comparing whole brain lysates from GPR158 KO mice to their wild-type (WT) littermates. In parallel, we performed a similar analysis using mice lacking R7BP (R7BP KO). The results revealed that the protein levels of RGS7 in GPR158 KO were reduced by more than 2-fold as compared with the wild-type littermates (44 ± 8%, n = 3, p < 0.01, t test) (Fig. 1C). This effect was selective for RGS7 as we did not detect any changes in the levels of RGS6 and RGS9-2, the other two R7 RGS proteins highly expressed in the brain. In contrast, knock-out of R7BP selectively reduced the expression of RGS9-2 but not RGS7, consistent with previous observations (8). To assess whether the lack of one membrane anchor for RGS7 leads to a compensatory regulation in the levels of the other, we evaluated the expression levels of GPR158 in R7BP KO mice and the levels of R7BP in GPR158 KO mice. We found that the lack of GPR158 did not affect the expression levels of R7BP (Fig. 1C), and vice versa, R7BP KO mice did not show any change in GPR158 protein levels (Fig. 1D).
FIGURE 1.

Generation and biochemical characterization of GPR158 knock-out mice. A, targeting strategy for the disruption of the GPR158 gene. Boxes indicate coding exons; angled arrow denotes the position of the transcription start site. B, representative Western blot analysis of the levels of indicated proteins in whole brain lysates from GPR158 knock-out mice (GPR158 KO), R7BP knock-out mice (R7BP KO), and their respective wild-type littermates (WT). An equal amount of total protein was loaded in each lane. C, analysis of changes in protein expression by quantification of band densities in Western blot experiments for indicated proteins in wild-type and GPR158 KO brain samples. Each band has been normalized to the density of the GAPDH-reactive band. Results are reported as percentage of expression in wild-type samples (mean ± S.E.; n = 3; **, p < 0.01; t test). D, analysis of changes in protein expression by quantification of band densities in Western blot experiments for indicated proteins in wild-type and R7BP KO brain samples (mean ± S.E.; n = 3; ***, p < 0.001; t test). Protein levels have been normalized to GAPDH expression. E, analysis of mRNA expression levels in the brain of GPR158 KO compared with wild-type littermates. mRNA levels were measured by quantitative real time PCR using TaqMan probes and β-actin for the normalization. Results are reported as percentage of wild-type expression levels (mean ± S.E.; n = 3; ***, p < 0.001; t test).
Knock-out of R7BP is known to result in a post-translational destabilization of RGS9-2 increasing its susceptibility to proteolytic degradation. Therefore, we analyzed the RGS7 mRNA levels in GPR158 KO brains to assess whether the change observed at the protein level could indeed be explained by post-transcriptional destabilization as opposed to transcriptional regulation of the gene. We extracted total RNA from the brains of WT and GPR158 KO mice and measured levels of several transcripts by quantitative real time PCR. We found no significant changes in RGS7 mRNA levels nor in the levels of RGS9-2 and R7BP. The only mRNA we found to be dramatically down-regulated in GPR158 KO mice was that of GPR158, entirely consistent with the expectations based on the disruption of its gene (Fig. 1E). In summary, these results indicate that knock-out of GPR158 leads to selective down-regulation of RGS7 expression in the brain likely via affecting its post-translational stability.
GPR158 Is Essential for Membrane Localization of RGS7 in Several Brain Regions
Previous studies identified a relatively minor role of R7BP in dictating RGS7 localization on the membrane with the majority of RGS7 remaining associated with the membranes upon R7BP elimination (33, 35–37). We previously reported that GPR158, just like R7BP, was able to localize RGS7 to the plasma membrane in transfected cells (38). We therefore hypothesized that GPR158 may control the majority of RGS7 membrane localization in the nervous system. To test this hypothesis, we isolated several brain regions where RGS7 is prominently expressed (50), namely cortex (Fig. 2A), striatum (Fig. 2B), and hippocampus (Fig. 2C), and we used them to analyze the distribution of RGS7 between cytosolic and membrane fractions in WT versus GPR158 KO mice by sedimentation following cell lysis. We observed a significant decrease in the total levels of RGS7 in GPR158 KO mice as compared with wild-type littermates in all brain regions examined (cortex, 73 ± 2%, p < 0.001; striatum, 55 ± 9%, p < 0.01; hippocampus, 47 ± 6%, p < 0.001). Subcellular fractionation experiments revealed exacerbation of RGS7 reduction in the membrane fraction of GPR158 KO tissues relative to WT with no significant difference in the RGS7 content in the cytosol.
FIGURE 2.

Subcellular fractionation and quantification of RGS7 levels in different brain regions of wild-type and GPR158 knock-out mice. Representative Western blots detecting RGS7 before and after subcellular fractionation of cortex (A), striatum (B), and hippocampus (C). GAPDH and Gβ1 were used as markers for cytosolic and membrane fractions, respectively. RGS7 levels have been normalized to the levels of GAPDH in total lysates and cytosol fraction or Gβ1 in the membrane fraction. The quantification of RGS7 protein band density is represented as percentage of RGS7 levels in wild type (mean ± S.E.; n = 6. **, p < 0.01; ***, p < 0.001; Student's paired t test).
Because elimination of GPR158 influences total expression level of RGS7, we next performed a relative analysis of RGS7 distribution across membrane versus cytosol compartments in each brain region, normalizing it to total RGS7 levels in the sample. When comparing this distribution between WT and GPR158 KO, we found a substantial loss of RGS7 from the membrane fraction in cortex (Fig. 3A; from 68.4 ± 2.3% in WT to 46.0 ± 4.7% in KO, p < 0.001), striatum (Fig. 3B; 42.1 ± 2.6% in WT to 16.8% ± 2.9% in KO, p < 0.001), and hippocampus (Fig. 3C; 44.0 ± 3.6% in WT to 25.2 ± 3.8% in KO, p < 0.001). This was accompanied by the corresponding gain of RGS7 content in the cytoplasm of the cortex (Fig. 3A; from 31.6 ± 3.6% in WT to 54.0 ± 2.5% in KO, p < 0.05), striatum (Fig. 3B; 57.9 ± 2.6% in WT to 83.2 ± 2.4% in KO, p < 0.05), and hippocampus (Fig. 3C; 56.0 ± 4.2% in WT to 74.8 ± 5.6% in KO, p < 0.05).
FIGURE 3.
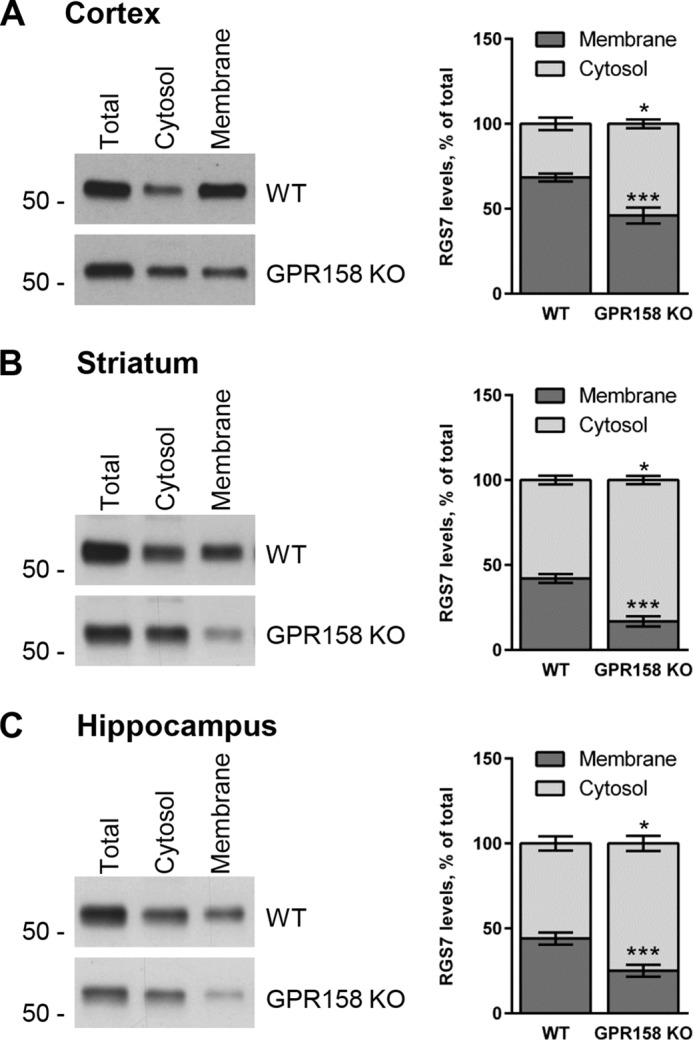
Impact of GPR158 deletion on relative distribution of RGS7 between cytosol and membrane in different brain regions. Fractionation of tissues from cortex (A), striatum (B), and hippocampus (C) was performed as described in Fig. 2. Total, cytosolic, and membrane fractions were loaded on the gel side by side, and RGS7 content was determined by Western blotting. The levels of RGS7 in each fraction have been normalized on total RGS7 levels. Results are expressed as percentage of RGS7 in cytosol versus membrane (mean ± S.E.; n = 4. *, p < 0.05; ***, p < 0.001; Student's paired t test).
We further analyzed the consequence of GPR158 elimination on RGS7 subcellular localization by immunogold electron microscopy (EM) that provides high resolution information on protein localization within cellular compartments. In the prefrontal cortex, we detected most of the RGS7-positive immunoparticles associated with the plasma membrane (PM) compartment in both dendritic spines and shafts (Fig. 4, A–E). A significant fraction of RGS7 immunoreactivity was also detected in the immediate vicinity of the PM. This distribution was changed in GPR158 knock-out brains, where we found less immunoparticles associated with the PM and more in the cytoplasm. Indeed, quantitative analysis of distribution of 4293 immunoparticles in the WT and 4175 in GPR158 KO sections revealed a significant shift of RGS7 distribution from the PM to the intracellular compartments (Fig. 4F). Together, these data indicate that GPR158 is responsible for anchoring a significant fraction of RGS7 to the membranes across the brain.
FIGURE 4.

Impact of GPR158 deletion on subcellular localization of RGS7 in prefrontal cortex analyzed by immunogold EM. A–E, electron micrographs of the medial prefrontal cortex showing immunoparticles for RGS7, as detected using a pre-embedding immunogold method in wild-type (WT) and GPR158 knock-out (GPR158KO) brains. Dendritic spines (s) and axon terminals (at) are marked. Arrows indicate locations of immunoparticles at the plasma membrane, and arrowheads identify RGS7 immunoparticles found just below the membrane or intracellularly. Scale bars, 0.2 μm. F, quantitative analysis of RGS7-positive immunoparticle distribution as a function of the distance from the PM. Error bars are mean ± S.E.; n = 3 mice. **, p < 0.01; ***, p < 0.001; one-way ANOVA followed by Bonferroni's post hoc test). Den, dendrite.
RGS7 Requires Gβ5 for Forming Complexes with GPR158
We have previously established that binding to both R7BP and GPR158 required the DEP domain of RGS7 (9, 38). However, in the case of the RGS9-R7BP interaction, the DEP domain was found to be insufficient, and binding further required the presence of Gβ5 in the complex (51). The competition between R7BP and GPR158 for binding to RGS7 suggests that these membrane anchors also bind to the same determinants on RGS7. Thus, we tested whether binding of RGS7 to GPR158 is also dependent on Gβ5. First, we confirmed that Gβ5 was indeed present in the ternary complexes and that the interaction was sensitive to the presence of the DEP domain while comparing GPR158 and R7BP binding to RGS7 side-by-side (Fig. 5B). As expected, RGS7 effectively co-immunoprecipitated with R7BP or GPR158 following their co-expression in HEK293 cells. However, this interaction was completely eliminated when truncated RGS7 lacking the DEP domain (DEPless RGS7) was used. Both complexes contained Gβ5, the binding of which did not require the presence of GPR158 or R7BP (Fig. 5B).
FIGURE 5.

RGS7 requires an intact DEP domain and Gβ5 to interact with GPR158. A, schematic representation of key structural elements in RGS7, Gβ5, and GPR158 and their spatial organization. B, analysis of RGS7 interaction with GPR158, R7BP, and Gβ5 by co-immunoprecipitation (IP). Either full-length RGS7 or its truncated mutant lacking the DEP domain (DEPless RGS7) was co-transfected in HEK293 cells with or without GPR158 or R7BP. Left panels show total expression levels of the indicated proteins as analyzed by Western blotting. Panels on the right examine protein content in the eluates after immunoprecipitation. C, requirement of Gβ5 for the interaction of RGS7 with GPR158. The constructs encoding GPR158 were co-transfected into HEK293 cells with or without RGS7 and Gβ5 as indicated. To obtain comparable levels of RGS7 expression in the absence of Gβ5, nine times more plasmid encoding RGS7 was used in the transfection (9×). NT, N terminus; CT, C terminus.
Next, we performed a reciprocal experiment and determined the requirement for Gβ5 in GPR158-RGS7 complex formation by performing immunoprecipitation experiments in HEK293 cells transfected with GPR158 and RGS7 with or without Gβ5. Because the expression levels of RGS7 are dramatically reduced when Gβ5 is absent (10), we changed the ratio of transfected plasmids to match RGS7 levels between the conditions (Fig. 4C). Following RGS7 immunoprecipitation, we detected the robust presence of GPR158 in the eluate only when Gβ5 was present (Fig. 4C). Together, these results indicate that binding to GPR158 requires both the DEP domain of RGS7 and Gβ5.
C Terminus of GPR158 Contains Several Conserved Regions That Share Similarity with Known G Protein Regulators
Having established the requirements for GPR158 binding in the RGS7-Gβ5 complex, we next aimed at identifying determinants in GPR158 for binding to RGS7-Gβ5. Reasoning that such interaction must involve an intracellular portion of GPR158, our attention was brought to the C terminus of the molecule that contains 535 amino acids accounting for almost half of the GPR158 molecule. Comparison of GPR158 amino acid sequences from 82 different species revealed the presence of several highly conserved regions at the C terminus of the molecule (Fig. 6A). The sequence identity in the first half of the C terminus (aa 665–961) was particularly high and contained three distinct isles of conservation, which we named CD1 (conserved domain 1), CD2, and CD3. The second half of the C terminus was less conserved, and we refer to it as CD4. Closer analysis of the CD1/2/3 region revealed that several of its features made it similar to known membrane anchors for RGS7, R7BP and R9AP. First, the size of this domain is comparable with the size of R7BP/R9AP. Second, it contained a predicted coiled-coil motif, which plays an essential role in RGS binding in R7BP/R9AP. Third, the coiled-coil domain was positioned at the same distance from the site of the membrane attachment and showed ∼35% amino acid identity with the corresponding coiled-coil regions of R7BP. Fourth, all three proteins were predicted to contain three α-helices connected by unstructured loops (Fig. 6B). Based on these observations, we hypothesized that CD1/2/3 domain of GPR158 may contain the RGS7-binding site. It should be noted, however, that we did not detect any appreciable sequence homology of CD1/2/3 with R9AP or R7BP at the amino acid level when using unbiased sequence comparison algorithms.
FIGURE 6.

Analyses of conserved features present in GPR158 C terminus. A, alignment of the amino acid sequence of mouse GPR158 intracellular C terminus (aa 665–1200) across 82 species. Regions that show 100% amino acid identity are highlighted in gray. These evolutionarily conserved sequences are referred to as conserved domains (CDs), and their boundaries are denoted by the indicated amino acid numbers starting from the beginning of the full-length GPR158 sequence. CD4 domain contains several PGL sequences shown in detail in C. B, scheme of the predicted secondary structural elements present in the GPR158-CD1/2/3 domain compared with the secondary structure of R9AP and R7BP. Blue cylinders represent predicted coiled-coil regions. White cylinders are predicted α-helices. N (N) and C terminus (C) of each protein are indicated, and the structures are aligned using the position of the plasma membrane as a reference. The sequence, including the conserved domains CD1/2/3 of GPR158, has been chosen for the alignment because of the comparable length with R9AP and R7BP. C, alignment of the sequence surrounding the PGL motifs from PDEγ, RGS9-2, GPR158, and one representative repeat in GPR179. The consensus motif shown has been identified with the software ESPript 3.0. D, schematic representation of the GPR158 structural organization indicating the position of the conserved domains in the intracellular C terminus.
Interestingly, sequence analysis of the CD4 region revealed the presence of a short motif repeated three times, two of which show high conservation across species. A BLAST search across the mouse proteome with this repeated region as a bait revealed the presence of highly similar sequence in three other proteins as follows: the retinal homolog of GPR158 and GPR179, where the motif has been found repeated 21 times, the γ subunit of the retinal cGMP phosphodiesterase (PDEγ), and the striatal enriched RGS protein RGS9-2 (Fig. 6C). The crystal structure of this sequence in PDEγ was solved, and it was shown to bind to the switch II region of the Gα subunit of the G protein transducin (14). This binding requires conserved cysteine and tryptophan residues and plays an essential role in enhancing the interaction of Gα with RGS9-1 thus potentiating its GAP activity (52–54). A similar role was also documented for the homologous region found in RGS9-2 (41). We therefore named this conserved sequence the “PDEγ-like” (PGL) domain. Thus, bioinformatics analysis identified four distinct regions/domains in the C terminus of GPR158 schematically depicted in Fig. 6D.
Identification of the RGS7-binding Site in GPR158 and Regions Involved in Association with Gα
Because GPR158 is essential for membrane localization of otherwise soluble RGS7, we used plasma membrane recruitment as a convenient readout of the RGS7 interaction with GPR158. Live staining of transfected COS1 cells showed that GPR158 is localized on the plasma membrane (Fig. 7A), consistent with our earlier observations (38). Instead, when transfected alone, the RGS7-Gβ5 complex showed a preferential localization in cytoplasm and nuclei (Fig. 7B). When RGS7-Gβ5 was co-expressed with full-length GPR158 (GPR158-FL), nearly all RGS7 was found on the plasma membrane co-localizing with GPR158 (Fig. 7C). Thus, the ability of GPR158 to change localization of RGS7 in transfected cells can be used as an indication of their interaction. Using this strategy, we assessed the behavior of two truncated versions of GPR158. The first construct encoded GPR158 with a deletion of the conserved domain CD4 (GPR158-ΔCD4), and the second construct encoded GPR158 with an internal deletion in the first half of the C terminus but contained CD4 (GPR158-ΔCD1/2/3). Just like full-length GPR158, the GPR158-ΔCD4 mutant was efficiently targeted to the plasma membrane and was able to localize the entire pool of RGS7 to the plasma membrane (Fig. 7D). In contrast, although the GPR158-ΔCD1/2/3 construct was still efficiently targeted to the plasma membrane, it completely failed to alter the subcellular localization of RGS7 that remained cytoplasmic and nuclear (Fig. 7E). These results suggest that the first half of the GPR158 C terminus containing CD1, CD2, and CD3 domains is necessary for the interaction with RGS7. We confirmed this conclusion by examining GPR158-RGS7 binding using co-immunoprecipitation assay. Full-length GPR158 and its truncated versions were co-expressed with RGS7 in HEK293 cells followed by immunoprecipitation of GPR158 using specific antibodies. All GPR158 constructs were expressed at similar levels and were equivalently precipitated. We were able to detect robust RGS7 co-immunoprecipitation with GPR158-FL and GPR158-ΔCD4 but not with GPR158-ΔCD1/2/3 (Fig. 8A). This co-immunoprecipitation was specific, because no RGS7 was pulled down by GPR158 antibodies in the absence of GPR158. We further conducted a reciprocal experiment, immunoprecipitating RGS7 and probing the eluates for the presence of GPR158. Again, both GPR158-FL and GPR158-ΔCD4 were specifically co-immunoprecipitating with RGS7 but not GPR158-ΔCD1/2/3 (Fig. 8B). Together, these results indicate that the first half of the GPR158 C terminus is necessary for RGS7 binding.
FIGURE 7.

C terminus of GPR158 is required for the recruitment of RGS7 to the plasma membrane in transfected cells. COS1 cells have been co-transfected with the indicated constructs, and localization of RGS7 and GPR158 was determined by immunocytochemistry. RGS7 was always co-transfected with the construct expressing Gβ5. GPR158 (green) was stained in live cells under nonpermeabilizing conditions using an antibody against its extracellular domain. RGS7 (red) was immunostained after fixation and permeabilization using an antibody against an HA tag present in its C terminus. DAPI staining (blue) was used to visualize the cell nuclei. A, GPR158 is localized on the plasma membrane in the absence of RGS7 co-transfection. B, RGS7 is localized in the cytoplasm and nuclei of COS1 cells when GPR158 is not co-transfected. C, co-expression of the full-length GPR158 (GPR158-FL) localizes RGS7 to the plasma membrane. D, GPR158 construct lacking CD4 domain (GPR158-ΔCD4) transfected in COS1 cells is localized to the plasma membrane and is capable of recruiting RGS7 to the plasma membrane. E, GPR158 construct with internal deletion of CD1/2/3 domains but containing CD4 (GPR158-ΔCD1/2/3) is localized to the plasma membrane but does not recruit RGS7 whose localization appears restricted to cytoplasm and nuclei.
FIGURE 8.

Mapping of RGS7 binding domain in GPR158. A and B, HEK293 cells were transfected with the indicated constructs for the expression of RGS7, Gβ5, and either full-length GPR158 (GPR158-FL) or truncated versions lacking either CD4 domain (GPR158-ΔCD4) or CD1/2/3 domains (GPR158-ΔCD1/2/3). A, co-immunoprecipitation of GPR158 constructs with RGS7 using antibodies against the N terminus of GPR158. Proteins were detected by Western blotting before (left panels) and after (right panels) immunoprecipitation (IP). B, reverse immunoprecipitation using RGS7 antibodies. Proteins were detected by Western blotting before (left panels) and after (right panels) immunoprecipitation. C, pulldown of the purified recombinant RGS7-Gβ5 complex by GST-purified GPR158 fragments immobilized on the beads. Upper panel shows Western blotting performed using a chicken anti-RGS7 antibody, and lower panel depicts Ponceau S staining that serves as a control for loading and purity of the bait proteins. D, pulldown of Gαo in different activation states (inactive, transition state, and active) by purified GST-CD1/2/3 and GST-CD4 as baits. Ponceau S staining (lower panel) was used as a control for the protein input. GST protein alone was used as control for nonspecific binding.
Next, we probed the sufficiency of the GPR158 C terminus for RGS7 binding. We purified the GST-tagged C-terminal region of GPR158 containing its entire sequence (aa 665–1200) and used it as bait in the pulldown assay with the purified recombinant RGS7-Gβ5 complex. As evident from the results presented in Fig. 8C, this construct effectively pulled down the RGS7 complex. This interaction was specific as bait beads containing GST alone failed to pull-down RGS7-Gβ5. We further used this assay to narrow down the region that mediates RGS7 binding. We generated a series of constructs with various combinations of the CD1 through CD4 regions and tested them in the pulldown assay. Only constructs containing the CD1 domain, including CD1 alone, were able to bind RGS7. These results indicate the first 110 amino acids (CD1) of the GPR158 C terminus constitute the binding site for RGS7.
The C termini of GPCRs are involved in activation of G proteins and have been shown to engage in interactions with G proteins (55–57). In addition, we found that the C terminus of GPR158 contains PGL domains whose homologs are known to interact with the Gα subunits of the Gi/o family. Therefore, we tested whether both the proximal (CD1/2/3) and distal (CD4) portions of the C-terminal GPR158 are capable of binding Gαo. For this, we performed the pulldown assay using purified GST-tagged CD1/2/3, CD4, or GST alone as baits analyzing their ability to retain purified Gαo in its inactive state (GDP-bound), transition state (GDP + AlF4), and active state (GTPγS-bound). We found that CD1/2/3 was able to bind Gαo irrespective of its activation state (Fig. 8D). However, the CD4 domain that contained three PGL repeats was able to bind selectively to Gαo-GTPγS but not to the Gαo-GDP or AlF4-induced transition state (Fig. 8D). This binding was specific, as GST protein alone failed to retain Gαo across different conformational states.
C Terminus of GPR158 Potentiates the GAP Activity of RGS7 toward Gαo in Solution
We next sought to determine functional consequences of RGS7 binding to GPR158. The RGS7-Gβ5 complex shows potent GAP activity toward Gαo that serves as its physiological substrate in native neurons. Therefore, we used in vitro single turnover GTPase assay to measure the ability of recombinant RGS7-Gβ5 complex to stimulate GTP hydrolysis catalyzed by purified Gαo in solution. As expected from previous observations, introduction of RGS7 resulted in a dramatic acceleration of the GTP hydrolysis rate, reflecting its GAP activity toward Gαo (Fig. 9A). The addition of recombinant C terminus of GPR158 (GPR158CT), but not R7BP, resulted in pronounced potentiation of this effect. Neither GPR158CT nor R7BP had any effect on intrinsic GTP hydrolysis by Gαo in the absence of RGS7-Gβ5, indicating that GPR158CT acts by potentiating the function of RGS7 rather than by regulating the activity of Gαo directly.
FIGURE 9.

GPR158-CT potentiates catalytic activity of RGS7 toward Gαo in solution. In vitro single turnover GTPase assay measuring the rate of GTP hydrolysis by the Gαo. A, comparison of the GTP hydrolysis rate by Gαo alone (control) and in the presence of a combination of the indicated purified proteins: R7BP and GPR158-CT with and without RGS7-Gβ5. Data were fit with nonlinear single exponential functions, and the representative traces are shown. Right panel provides quantification of the GTP hydrolysis rates (1/τ). B and C, impact of individual purified C-terminal domains of GPR158 on the catalytic activity of RGS7 toward Gαo. The Gαo and purified complex RGS7-Gβ5 were present in all the experimental conditions. Control experiment contains 0.5 μm GST recombinant proteins. The rate of intrinsic GTP hydrolysis by Gαo was subtracted to obtain the catalytic activity (kGAP) of RGS7. The results were from three independent experiments (*, p < 0.05; ***, p < 0.001; comparing with control experiment with RGS7-Gβ5; one-way ANOVA followed by post hoc Tukey's test).
Next, we determined which elements in GPR158CT are responsible for the effect. We found that proximal part (CD1/2/3) but not distal part (CD4) of the C terminus potently stimulated GAP activity of RGS7, enhancing it by nearly 4-fold (from 0.11 ± 0.01 to 0.37 ± 0.03 s−1) (Fig. 9B). Interestingly, the minimal RGS7 binding domain (CD1) was not sufficient for the GAP activity potentiation. Neither did other elements of the proximal portion (CD2 or CD3) potentiate RGS7 activity when added in isolation. In an effort to identify the minimal region responsible for the potentiating effect, we tested the activity of purified proteins containing combinations of the domains, CD1/2 and CD2/3 (Fig. 9C). Although we found the CD2/3 construct completely inactive, CD1/2 had a partial effect on RGS7-mediated acceleration of Gαo GTPase activity. Together, these results indicate that the GAP activity of RGS7 is potently regulated by the C terminus of GPR158 that requires a synergistic contribution of its RGS7-binding regions (CD1) and allosterically acting elements located in CD2 and CD3 domains.
Discussion
The results of this study establish GPR158 as an essential modulator of RGS7 function in vivo. Using genetically engineered mice, we found that elimination of GPR158 in the brain substantially reduced expression of RGS7 and its association with the membranes. Components of the GPCR signaling pathway are intimately associated with the plasma membranes, hence targeting otherwise cytoplasmic RGS proteins to the membranes is expected to enhance their ability to regulate G protein signaling. Determining localization of R7 RGS proteins in neurons appears to be a complex process that relies on their association with auxiliary membrane proteins. Previous studies have examined the role of two such proteins, R9AP and R7BP, in localizing RGS proteins in vivo (58). R9AP and R7BP were found to be indispensable for membrane localization of the entire fraction of RGS9 in the retina and brain, respectively (8, 31, 59). However, localization of RGS7 in the native neurons was affected only modestly by R7BP knock-out and only in some brain regions (33, 35, 36). We now show that association of RGS7 with the plasma membrane across the brain is determined by GPR158 to a great extent. We found that GPR158 contains the R7BP-like domain in its intracellular C terminus and that this region mediates its binding to RGS7. The structural similarity between the C terminus of GPR158 and R7BP provides an explanation for the mutually exclusive nature of their complex formation with RGS7, as both proteins compete for the same binding determinants (38). These observations further suggest that in the nervous system RGS7 is likely present in two alternative configurations, where it is bound to either R7BP or GPR158.
The alternating nature of RGS7 complexing with either R7BP or GPR158 also raises a question about redundancy of membrane anchoring mechanisms for RGS7. In transfected cells, localizing RGS7 on the plasma membrane by either R7BP or GPR158 was shown to augment the ability of RGS7 to accelerate termination of G protein signaling (26, 38). Thus, the consequences of RGS7 binding to R7BP or GPR158 may be functionally identical. Alternatively, interactions with R7BP and GPR158 may provide a selectivity filter directing RGS7 action to a particular molecular environment, e.g. through scaffolding with other elements of GPCR pathways. Such a possibility seems plausible considering recent findings on GPR179, a close homolog of GPR158. GPR179 is selectively expressed by retina ON-bipolar neurons, and its loss results in synaptic transmission deficits leading to night blindness (39, 40). Just like GPR158, GPR179 associates with R7 RGS proteins and was shown to play an essential role for their synaptic targeting (38). Interestingly, in addition to RGS proteins, GPR179 also forms macromolecular complexes with the mGluR6 receptor and the TRPM1 effector channel (60–62). Elimination of either GPR179 or RGS proteins produces similar deficits in synaptic transmission associated with the increase in tonic inhibition of the TRPM1 channel by the active G protein (62). This suggests that placing RGS proteins in an appropriate molecular environment allows them to adjust mGluR6 signaling, thereby enabling regulation of the synaptic signaling cascade in the bipolar neurons. Determining whether GPR158 can similarly participate in selective recruitment of RGS7 to regulation of specific GPCR signaling pathways in the brain or whether its function is to simply recruit RGS7 to the plasma membrane appears to be an interesting future direction.
In this study we show that in addition to regulating membrane localization, GPR158 plays a role in setting expression levels of RGS7. We observed that the loss of GPR158 diminishes total RGS7 levels in the brain without affecting expression of other R7 RGS proteins. This regulation occurs at the post-transcriptional level and likely involves proteolytic stabilization of RGS7, although the exact mechanisms involved in control of RGS7 expression by GPR158 remain to be established. Several RGS proteins show modulation of their levels in response to a variety of stimuli e.g. stress, drug exposure, changes in neurotransmitter signaling, etc. (50, 63–65). These changes are thought to reflect homeostatic adaptations in the extent of GPCR signaling as abundance of RGS proteins has a direct effect on the lifetime of activated G proteins. One of the best documented examples of this is provided by RGS9, a close homolog of RGS7. The expression levels of RGS9 are differentially affected by exposure to cocaine, morphine, amphetamine, or upon changes in neuronal excitability and oxygenation (32, 50, 66–68). The stability of RGS9 is modulated post-translationally via its association with R7BP; binding to R7BP increases its expression, and dissociation makes it susceptible to degradation by cellular cysteine proteases decreasing the expression levels (8). Accordingly, the current model suggests that stimuli that affect RGS9 expression at the post-translational level do so by modulating the extent of its association with R7BP (66, 69). Interestingly, in the case of RGS7, the primary role in regulating post-translational stability and expression levels belongs to GPR158 instead of R7BP. Similarly to RGS9, the levels of RGS7 have been reported to be modulated by exposure to inflammatory mediators (70, 71) and antidepressant drugs (72). Although the mechanisms of this regulation remain to be elucidated, by analogy with RGS9, it is tempting to speculate that the modulation of RGS7 association with GPR158 may serve as one such critical regulatory point.
One of the major findings of this work is the demonstration that the activity of RGS7 is modulated allosterically by its auxiliary binding partner. We show that binding of RGS7 to the cytoplasmic C-terminal domain of GPR158 substantially increases its GAP activity toward Gαo in solution. Activity of R7 RGS proteins has been shown to be augmented by R7BP and R9AP (6). The action of these proteins requires membrane recruitment of RGS proteins as minimal RGS-binding elements of R9AP (73, 74) and R7BP (Fig. 9A), devoid of membrane anchoring elements, are unable to potentiate the GAP activity of R7 RGS proteins in solution. Similarly, we find that the CD1 domain of GPR158, which harbors minimal binding site for RGS7 and shares structural similarity with R7BP/R9AP, cannot potentiate its activity. However, longer fragment of GPR158 C terminus containing CD2 and CD3 domains in addition to CD1 potently stimulated RGS7 GAP activity in solution. These results indicate the following: 1) mere binding of the R9AP/R7BP/CD1 module is insufficient for altering RGS activity; 2) CD2/3 domains of GPR158 contain unique elements that allosterically potentiate the activity of RGS7 while it is bound to CD1. We have found that the CD1/2/3 module of GPR158 can also interact with Gαo in a state-independent manner. This evidence allows us to hypothesize that CD1/2/3 can modulate the activity of RGS7 by two possible mechanisms. By simultaneously binding to both RGS7 and its substrate Gαo, CD1/2/3 may enhance the probability of their interaction thereby speeding up the reaction. Alternatively, CD1/2/3 may induce a conformational change in RGS7 by direct protein-protein interaction. To the best of our knowledge, this is the first time an allosteric potentiation of RGS GAP activity by a direct interaction with a protein partner outside of the membrane context is documented.
Another interesting observation made in this study is that GPR158 and related orphan receptor GPR179 contain a short amino acid sequence repeated several times in their C-terminal regions. This sequence was first described in the γ subunit of rod cGMP PDEγ, thus prompting us to name it the PDEγ-like (PGL) domain. A similar sequence has also been found in the C terminus of RGS9-2 (41). In both PDEγ and RGS9-2, these PGL sequences bind to the active state of the Gα subunit and serve to enhance interaction between RGS proteins and their Gα substrates. Remarkably, we found that the C-terminal portion of GPR158 bearing three PGL domains also selectively interacts with activated Gαo-GTP but not with inactive Gαo-GDP or transition states. These PGL domains in GPR158 may further increase the efficiency of G protein regulation by RGS7 when in complex with GPR158. Interestingly, the C terminus of mouse GPR179 contains 21 PGL domains, prompting us to speculate that perhaps the action of this module might be to scaffold the activated Gα proteins presenting them for RGS7 to be inactivated, thus increasing temporal resolution of GPCR signaling.
Our studies primarily focused on the role of GPR158 from the perspective of RGS function. It remains possible that GPR158 serves as a bona fide signaling GPCR, a possibility that so far has not been addressed. Both GPR158 and GPR179 are distant members of the class C family of GPCRs and share some features that are necessary for G protein activation by these GPCRs. Whether GPR158 or GPR179 can bind and activate heterotrimeric G proteins is unknown at this point. These studies are largely hindered by the lack of knowledge on any extracellular ligands that might interact with these orphan receptors. The possibility that GPR158/179 might be signaling GPCRs is exemplified by recent studies on the muscarinic M3 receptor (M3R), which has been shown to form complexes with the RGS7 complex (75). Unlike binding to GPR158/179 or R9AP/R7BP, association with M3R does not require Gβ5 and is mediated by the 3rd intracellular loop and the 8th α-helix in the M3R C-terminal tail, which bear neither amino acid nor structural homology to CD1/R9AP/R7BP (76). This interaction appears to regulate the efficiency of M3R signaling by a non-GAP mechanism, but it is unclear whether M3R-RGS7 interaction occurs in vivo and if so how much impact it has on RGS7 in the brain. It is also possible that GPR158 might have an additional G protein-independent mode in regulating cellular signaling. For example, a C-terminal fragment of GPR158 was shown to contain a nuclear localization sequence and play an important role in directing GPR158 in the nucleus to regulate cellular proliferation (77). However, regardless of whether or not GPR158/179 serve as GPCRs and/or affect signaling via an alternative mechanism, it is clear that they can impact cellular homeostasis by affecting the activity, localization, and expression of RGS7.
In conclusion, we established that GPR158 is a critical modulator of RGS7 in the central nervous system. We showed that GPR158 is required to achieve a high level of RGS7 expression in the brain and controls localization of a large fraction of RGS7 on the membranes. In addition, we demonstrated that distinct intracellular domains of GPR158 act allosterically to regulate the GAP activity of RGS7. These findings establish GPR158 as an essential player in regulating RGS7 function in the nervous system.
Acknowledgments
We thank Dr. Dipak Patil for help with protein structural analysis and Natalia Martemyanova for breeding and maintaining the mouse colony. We gratefully acknowledge William Simonds (NIDDK, National Institutes of Health, Bethesda) for the rabbit anti-Gβ5, rabbit anti-RGS7 (7RC-1), and rabbit anti-R7BP antibodies and Dr. Barry Willardson (Brigham Young University, Provo, UT) for anti-Gβ1 antibodies. The Gpr158 mouse strain used for this research project was created from ES cell clone 10108A-A5, generated by Regeneron Pharmaceuticals, Inc., and made into live mice by the KOMP Repository and the Mouse Biology Program at the University of California Davis.
This work was supported, in whole or in part, by National Institutes of Health Grants DA036082 and DA036596 (to K. A. M.). This work was also supported by Spanish Ministry of Education and Science Grant BFU-2012-38348 and Junta de Comunidades de Castilla-La Mancha Grant PPII11-0284-9301 (to R. L.).
This article was selected as a Paper of the Week.
- GPCR
- G protein-coupled receptor
- R7BP
- RGS7-binding protein
- RGS
- regulator of G protein signaling
- GAP
- GTPase-activating protein
- ANOVA
- analysis of variance
- PGL
- phosphodiesterase Eγ-like
- aa
- amino acid
- CT
- C terminus
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- PDE
- phosphodiesterase
- PM
- plasma membrane.
References
- 1. Wettschureck N., Offermanns S. (2005) Mammalian G proteins and their cell type-specific functions. Physiol. Rev. 85, 1159–1204 [DOI] [PubMed] [Google Scholar]
- 2. Neer E. J. (1995) Heterotrimeric G proteins: organizers of transmembrane signals. Cell 80, 249–257 [DOI] [PubMed] [Google Scholar]
- 3. Hollinger S., Hepler J. R. (2002) Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol. Rev. 54, 527–559 [DOI] [PubMed] [Google Scholar]
- 4. Ross E. M., Wilkie T. M. (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69, 795–827 [DOI] [PubMed] [Google Scholar]
- 5. Kimple A. J., Bosch D. E., Giguère P. M., Siderovski D. P. (2011) Regulators of G-protein signaling and their Gα substrates: promises and challenges in their use as drug discovery targets. Pharmacol. Rev. 63, 728–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson G. R., Posokhova E., Martemyanov K. A. (2009) The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochem. Biophys. 54, 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cowan C. W., He W., Wensel T. G. (2001) RGS proteins: lessons from the RGS9 subfamily. Prog. Nucleic Acid Res. Mol. Biol. 65, 341–359 [DOI] [PubMed] [Google Scholar]
- 8. Anderson G. R., Lujan R., Semenov A., Pravetoni M., Posokhova E. N., Song J. H., Uversky V., Chen C. K., Wickman K., Martemyanov K. A. (2007) Expression and localization of RGS9-2/G 5/R7BP complex in vivo is set by dynamic control of its constitutive degradation by cellular cysteine proteases. J. Neurosci. 27, 14117–14127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anderson G. R., Semenov A., Song J. H., Martemyanov K. A. (2007) The membrane anchor R7BP controls the proteolytic stability of the striatal specific RGS protein, RGS9-2. J. Biol. Chem. 282, 4772–4781 [DOI] [PubMed] [Google Scholar]
- 10. Chen C. K., Eversole-Cire P., Zhang H., Mancino V., Chen Y. J., He W., Wensel T. G., Simon M. I. (2003) Instability of GGL domain-containing RGS proteins in mice lacking the G protein β-subunit Gβ5. Proc. Natl. Acad. Sci. U.S.A. 100, 6604–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Witherow D. S., Wang Q., Levay K., Cabrera J. L., Chen J., Willars G. B., Slepak V. Z. (2000) Complexes of the G protein subunit Gβ5 with the regulators of G protein signaling RGS7 and RGS9. Characterization in native tissues and in transfected cells. J. Biol. Chem. 275, 24872–24880 [DOI] [PubMed] [Google Scholar]
- 12. He W., Lu L., Zhang X., El-Hodiri H. M., Chen C. K., Slep K. C., Simon M. I., Jamrich M., Wensel T. G. (2000) Modules in the photoreceptor RGS9–1.Gβ 5L GTPase-accelerating protein complex control effector coupling, GTPase acceleration, protein folding, and stability. J. Biol. Chem. 275, 37093–37100 [DOI] [PubMed] [Google Scholar]
- 13. Skiba N. P., Martemyanov K. A., Elfenbein A., Hopp J. A., Bohm A., Simonds W. F., Arshavsky V. Y. (2001) RGS9-G β 5 substrate selectivity in photoreceptors. Opposing effects of constituent domains yield high affinity of RGS interaction with the G protein-effector complex. J. Biol. Chem. 276, 37365–37372 [DOI] [PubMed] [Google Scholar]
- 14. Slep K. C., Kercher M. A., He W., Cowan C. W., Wensel T. G., Sigler P. B. (2001) Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature 409, 1071–1077 [DOI] [PubMed] [Google Scholar]
- 15. Snow B. E., Krumins A. M., Brothers G. M., Lee S. F., Wall M. A., Chung S., Mangion J., Arya S., Gilman A. G., Siderovski D. P. (1998) A G protein γ subunit-like domain shared between RGS11 and other RGS proteins specifies binding to Gβ5 subunits. Proc. Natl. Acad. Sci. U.S.A. 95, 13307–13312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cabrera J. L., de Freitas F., Satpaev D. K., Slepak V. Z. (1998) Identification of the Gβ5-RGS7 protein complex in the retina. Biochem. Biophys. Res. Commun. 249, 898–902 [DOI] [PubMed] [Google Scholar]
- 17. Makino E. R., Handy J. W., Li T., Arshavsky V. Y. (1999) The GTPase activating factor for transducin in rod photoreceptors is the complex between RGS9 and type 5 G protein β subunit. Proc. Natl. Acad. Sci. U.S.A. 96, 1947–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheever M. L., Snyder J. T., Gershburg S., Siderovski D. P., Harden T. K., Sondek J. (2008) Crystal structure of the multifunctional Gβ5-RGS9 complex. Nat. Struct. Mol. Biol. 15, 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drenan R. M., Doupnik C. A., Jayaraman M., Buchwalter A. L., Kaltenbronn K. M., Huettner J. E., Linder M. E., Blumer K. J. (2006) R7BP augments the function of RGS7/Gβ5 complexes by a plasma membrane-targeting mechanism. J. Biol. Chem. 281, 28222–28231 [DOI] [PubMed] [Google Scholar]
- 20. Hu G., Wensel T. G. (2002) R9AP, a membrane anchor for the photoreceptor GTPase accelerating protein, RGS9–1. Proc. Natl. Acad. Sci. U.S.A. 99, 9755–9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martemyanov K. A., Yoo P. J., Skiba N. P., Arshavsky V. Y. (2005) R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J. Biol. Chem. 280, 5133–5136 [DOI] [PubMed] [Google Scholar]
- 22. Drenan R. M., Doupnik C. A., Boyle M. P., Muglia L. J., Huettner J. E., Linder M. E., Blumer K. J. (2005) Palmitoylation regulates plasma membrane-nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family. J. Cell Biol. 169, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song J. H., Waataja J. J., Martemyanov K. A. (2006) Subcellular targeting of RGS9-2 is controlled by multiple molecular determinants on its membrane anchor, R7BP. J. Biol. Chem. 281, 15361–15369 [DOI] [PubMed] [Google Scholar]
- 24. Hu G., Zhang Z., Wensel T. G. (2003) Activation of RGS9–1GTPase acceleration by its membrane anchor, R9AP. J. Biol. Chem. 278, 14550–14554 [DOI] [PubMed] [Google Scholar]
- 25. Lishko P. V., Martemyanov K. A., Hopp J. A., Arshavsky V. Y. (2002) Specific binding of RGS9-Gβ 5L to protein anchor in photoreceptor membranes greatly enhances its catalytic activity. J. Biol. Chem. 277, 24376–24381 [DOI] [PubMed] [Google Scholar]
- 26. Masuho I., Xie K., Martemyanov K. A. (2013) Macromolecular composition dictates receptor and G protein selectivity of regulator of G protein signaling (RGS) 7 and 9–2 protein complexes in living cells. J. Biol. Chem. 288, 25129–25142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cao Y., Masuho I., Okawa H., Xie K., Asami J., Kammermeier P. J., Maddox D. M., Furukawa T., Inoue T., Sampath A. P., Martemyanov K. A. (2009) Retina-specific GTPase accelerator RGS11/G β 5S/R9AP is a constitutive heterotrimer selectively targeted to mGluR6 in ON-bipolar neurons. J. Neurosci. 29, 9301–9313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang J., Jeffrey B. G., Morgans C. W., Burke N. S., Haley T. L., Duvoisin R. M., Brown R. L. (2010) RGS7 and -11 complexes accelerate the ON-bipolar cell light response. Invest. Ophthalmol. Vis. Sci. 51, 1121–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeffrey B. G., Morgans C. W., Puthussery T., Wensel T. G., Burke N. S., Brown R. L., Duvoisin R. M. (2010) R9AP stabilizes RGS11-G β5 and accelerates the early light response of ON-bipolar cells. Vis. Neurosci. 27, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keresztes G., Martemyanov K. A., Krispel C. M., Mutai H., Yoo P. J., Maison S. F., Burns M. E., Arshavsky V. Y., Heller S. (2004) Absence of the RGS9·Gβ5 GTPase-activating complex in photoreceptors of the R9AP knockout mouse. J. Biol. Chem. 279, 1581–1584 [DOI] [PubMed] [Google Scholar]
- 31. Martemyanov K. A., Lishko P. V., Calero N., Keresztes G., Sokolov M., Strissel K. J., Leskov I. B., Hopp J. A., Kolesnikov A. V., Chen C. K., Lem J., Heller S., Burns M. E., Arshavsky V. Y. (2003) The DEP domain determines subcellular targeting of the GTPase activating protein RGS9 in vivo. J. Neurosci. 23, 10175–10181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anderson G. R., Lujan R., Martemyanov K. A. (2009) Changes in striatal signaling induce remodeling of RGS complexes containing Gβ5 and R7BP subunits. Mol. Cell. Biol. 29, 3033–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Panicker L. M., Zhang J. H., Posokhova E., Gastinger M. J., Martemyanov K. A., Simonds W. F. (2010) Nuclear localization of the G protein β 5/R7-regulator of G protein signaling protein complex is dependent on R7 binding protein. J. Neurochem. 113, 1101–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grabowska D., Jayaraman M., Kaltenbronn K. M., Sandiford S. L., Wang Q., Jenkins S., Slepak V. Z., Smith Y., Blumer K. J. (2008) Postnatal induction and localization of R7BP, a membrane-anchoring protein for regulator of G protein signaling 7 family-Gβ5 complexes in brain. Neuroscience 151, 969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cao Y., Song H., Okawa H., Sampath A. P., Sokolov M., Martemyanov K. A. (2008) Targeting of RGS7/Gβ5 to the dendritic tips of ON-bipolar cells is independent of its association with membrane anchor R7BP. J. Neurosci. 28, 10443–10449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ostrovskaya O., Xie K., Masuho I., Fajardo-Serrano A., Lujan R., Wickman K., Martemyanov K. A. (2014) RGS7/Gβ5/R7BP complex regulates synaptic plasticity and memory by modulating hippocampal GABABR-GIRK signaling. eLife 3, e02053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou H., Chisari M., Raehal K. M., Kaltenbronn K. M., Bohn L. M., Mennerick S. J., Blumer K. J. (2012) GIRK channel modulation by assembly with allosterically regulated RGS proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 19977–19982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Orlandi C., Posokhova E., Masuho I., Ray T. A., Hasan N., Gregg R. G., Martemyanov K. A. (2012) GPR158/179 regulate G protein signaling by controlling localization and activity of the RGS7 complexes. J. Cell Biol. 197, 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peachey N. S., Ray T. A., Florijn R., Rowe L. B., Sjoerdsma T., Contreras-Alcantara S., Baba K., Tosini G., Pozdeyev N., Iuvone P. M., Bojang P., Jr., Pearring J. N., Simonsz H. J., van Genderen M., Birch D. G., et al. (2012) GPR179 is required for depolarizing bipolar cell function and is mutated in autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 90, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Audo I., Bujakowska K., Orhan E., Poloschek C. M., Defoort-Dhellemmes S., Drumare I., Kohl S., Luu T. D., Lecompte O., Zrenner E., Lancelot M. E., Antonio A., Germain A., Michiels C., Audier C., et al. (2012) Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 90, 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martemyanov K. A., Hopp J. A., Arshavsky V. Y. (2003) Specificity of G protein-RGS protein recognition is regulated by affinity adapters. Neuron 38, 857–862 [DOI] [PubMed] [Google Scholar]
- 42. Porter M. Y., Xie K., Pozharski E., Koelle M. R., Martemyanov K. A. (2010) A conserved protein interaction interface on the type 5 G protein β subunit controls proteolytic stability and activity of R7 family regulator of G protein signaling proteins. J. Biol. Chem. 285, 41100–41112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lujan R., Nusser Z., Roberts J. D., Shigemoto R., Somogyi P. (1996) Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur. J. Neurosci. 8, 1488–1500 [DOI] [PubMed] [Google Scholar]
- 44. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 45. Self A. J., Hall A. (1995) Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol. 256, 67–76 [DOI] [PubMed] [Google Scholar]
- 46. Cowan C. W., Wensel T. G., Arshavsky V. Y. (2000) Enzymology of GTPase acceleration in phototransduction. Methods Enzymol. 315, 524–538 [DOI] [PubMed] [Google Scholar]
- 47. Papadopoulos J. S., Agarwala R. (2007) COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073–1079 [DOI] [PubMed] [Google Scholar]
- 48. Marchler-Bauer A., Zheng C., Chitsaz F., Derbyshire M. K., Geer L. Y., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Lanczycki C. J., Lu F., Lu S., Marchler G. H., Song J. S., Thanki N., et al. (2013) CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41, D348–D352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lupas A., Van Dyke M., Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 50. Gold S. J., Ni Y. G., Dohlman H. G., Nestler E. J. (1997) Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J. Neurosci. 17, 8024–8037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Masuho I., Wakasugi-Masuho H., Posokhova E. N., Patton J. R., Martemyanov K. A. (2011) Type 5 G protein β subunit (Gβ5) controls the interaction of regulator of G protein signaling 9 (RGS9) with membrane anchors. J. Biol. Chem. 286, 21806–21813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Grant J. E., Guo L. W., Vestling M. M., Martemyanov K. A., Arshavsky V. Y., Ruoho A. E. (2006) The N terminus of GTPγS-activated transducin α-subunit interacts with the C terminus of the cGMP phosphodiesterase γ-subunit. J. Biol. Chem. 281, 6194–6202 [DOI] [PubMed] [Google Scholar]
- 53. Skiba N. P., Hopp J. A., Arshavsky V. Y. (2000) The effector enzyme regulates the duration of G protein signaling in vertebrate photoreceptors by increasing the affinity between transducin and RGS protein. J. Biol. Chem. 275, 32716–32720 [DOI] [PubMed] [Google Scholar]
- 54. Slepak V. Z., Artemyev N. O., Zhu Y., Dumke C. L., Sabacan L., Sondek J., Hamm H. E., Bownds M. D., Arshavsky V. Y. (1995) An effector site that stimulates G-protein GTPase in photoreceptors. J. Biol. Chem. 270, 14319–14324 [DOI] [PubMed] [Google Scholar]
- 55. Dohlman H. G., Thorner J., Caron M. G., Lefkowitz R. J. (1991) Model systems for the study of seven-transmembrane-segment receptors. Annu. Rev. Biochem. 60, 653–688 [DOI] [PubMed] [Google Scholar]
- 56. Oldham W. M., Hamm H. E. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71 [DOI] [PubMed] [Google Scholar]
- 57. König B., Arendt A., McDowell J. H., Kahlert M., Hargrave P. A., Hofmann K. P. (1989) Three cytoplasmic loops of rhodopsin interact with transducin. Proc. Natl. Acad. Sci. U.S.A. 86, 6878–6882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jayaraman M., Zhou H., Jia L., Cain M. D., Blumer K. J. (2009) R9AP and R7BP: traffic cops for the RGS7 family in phototransduction and neuronal GPCR signaling. Trends Pharmacol. Sci. 30, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cao Y., Kolesnikov A. V., Masuho I., Kefalov V. J., Martemyanov K. A. (2010) Membrane anchoring subunits specify selective regulation of RGS9·Gβ5 GAP complex in photoreceptor neurons. J. Neurosci. 30, 13784–13793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Orlandi C., Cao Y., Martemyanov K. A. (2013) Orphan receptor GPR179 forms macromolecular complexes with components of metabotropic signaling cascade in retina ON-bipolar neurons. Invest. Ophthalmol. Vis. Sci. 54, 7153–7161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cao Y., Pahlberg J., Sarria I., Kamasawa N., Sampath A. P., Martemyanov K. A. (2012) Regulators of G protein signaling RGS7 and RGS11 determine the onset of the light response in ON bipolar neurons. Proc. Natl. Acad. Sci. U.S.A. 109, 7905–7910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ray T. A., Heath K. M., Hasan N., Noel J. M., Samuels I. S., Martemyanov K. A., Peachey N. S., McCall M. A., Gregg R. G. (2014) GPR179 is required for high sensitivity of the mGluR6 signaling cascade in depolarizing bipolar cells. J. Neurosci. 34, 6334–6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Holden N. S., George T., Rider C. F., Chandrasekhar A., Shah S., Kaur M., Johnson M., Siderovski D. P., Leigh R., Giembycz M. A., Newton R. (2014) Induction of regulator of G-protein signaling 2 expression by long-acting β2-adrenoceptor agonists and glucocorticoids in human airway epithelial cells. J. Pharmacol. Exp. Ther. 348, 12–24 [DOI] [PubMed] [Google Scholar]
- 64. Kach J., Sethakorn N., Dulin N. O. (2012) A finer tuning of G-protein signaling through regulated control of RGS proteins. Am. J. Physiol. Heart Circ. Physiol. 303, H19–H35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ingi T., Krumins A. M., Chidiac P., Brothers G. M., Chung S., Snow B. E., Barnes C. A., Lanahan A. A., Siderovski D. P., Ross E. M., Gilman A. G., Worley P. F. (1998) Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. J. Neurosci. 18, 7178–7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Anderson G. R., Cao Y., Davidson S., Truong H. V., Pravetoni M., Thomas M. J., Wickman K., Giesler G. J., Jr., Martemyanov K. A. (2010) R7BP complexes with RGS9-2 and RGS7 in the striatum differentially control motor learning and locomotor responses to cocaine. Neuropsychopharmacology 35, 1040–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Seeman P., Ko F., Jack E., Greenstein R., Dean B. (2007) Consistent with dopamine supersensitivity, RGS9 expression is diminished in the amphetamine-treated animal model of schizophrenia and in postmortem schizophrenia brain. Synapse 61, 303–309 [DOI] [PubMed] [Google Scholar]
- 68. Zachariou V., Georgescu D., Sanchez N., Rahman Z., DiLeone R., Berton O., Neve R. L., Sim-Selley L. J., Selley D. E., Gold S. J., Nestler E. J. (2003) Essential role for RGS9 in opiate action. Proc. Natl. Acad. Sci. U.S.A. 100, 13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Terzi D., Cao Y., Agrimaki I., Martemyanov K. A., Zachariou V. (2012) R7BP modulates opiate analgesia and tolerance but not withdrawal. Neuropsychopharmacology 37, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hausmann O. N., Hu W. H., Keren-Raifman T., Witherow D. S., Wang Q., Levay K., Frydel B., Z Slepak V., R Bethea J. (2002) Spinal cord injury induces expression of RGS7 in microglia/macrophages in rats. Eur. J. Neurosci. 15, 602–612 [DOI] [PubMed] [Google Scholar]
- 71. Benzing T., Brandes R., Sellin L., Schermer B., Lecker S., Walz G., Kim E. (1999) Upregulation of RGS7 may contribute to tumor necrosis factor-induced changes in central nervous function. Nat. Med. 5, 913–918 [DOI] [PubMed] [Google Scholar]
- 72. Singh R. K., Shi J., Zemaitaitis B. W., Muma N. A. (2007) Olanzapine increases RGS7 protein expression via stimulation of the Janus tyrosine kinase-signal transducer and activator of transcription signaling cascade. J. Pharmacol. Exp. Ther. 322, 133–140 [DOI] [PubMed] [Google Scholar]
- 73. Baker S. A., Martemyanov K. A., Shavkunov A. S., Arshavsky V. Y. (2006) Kinetic mechanism of RGS9–1 potentiation by R9AP. Biochemistry 45, 10690–10697 [DOI] [PubMed] [Google Scholar]
- 74. Masuho I., Celver J., Kovoor A., Martemyanov K. A. (2010) Membrane anchor R9AP potentiates GTPase-accelerating protein activity of RGS11 x Gβ5 complex and accelerates inactivation of the mGluR6-G(o) signaling. J. Biol. Chem. 285, 4781–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sandiford S. L., Slepak V. Z. (2009) The Gβ5-RGS7 complex selectively inhibits muscarinic M3 receptor signaling via the interaction between the third intracellular loop of the receptor and the DEP domain of RGS7. Biochemistry 48, 2282–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Karpinsky-Semper D., Tayou J., Levay K., Schuchardt B. J., Bhat V., Volmar C. H., Farooq A., Slepak V. Z. (2015) Helix 8 and the i3 loop of the muscarinic M3 receptor are crucial sites for its regulation by the Gβ5-RGS7 complex. Biochemistry 54, 1077–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Patel N., Itakura T., Gonzalez J. M., Jr., Schwartz S. G., Fini M. E. (2013) GPR158, an orphan member of G protein-coupled receptor Family C: glucocorticoid-stimulated expression and novel nuclear role. PLoS One 8, e57843. [DOI] [PMC free article] [PubMed] [Google Scholar]