

Background: Several nucleoside antibiotics contain a uridine-5′-carboxamide core of unclear origin.
Results: The A-102395 biosynthetic gene cluster was cloned, a genetic system was developed, and three enzymes were characterized in vivo and in vitro.
Conclusion: Uridine-5′-carboxamide originates from UMP and l-Thr by sequential reactions catalyzed by a dioxygenase and transaldolase.
Significance: The results provide the first opportunity to methodically interrogate the biosynthesis of these unusual antibiotics.
Keywords: antibiotic resistance, antibiotics, biosynthesis, dioxygenase, natural product biosynthesis
Abstract
A-500359s, A-503083s, and A-102395 are capuramycin-type nucleoside antibiotics that were discovered using a screen to identify inhibitors of bacterial translocase I, an essential enzyme in peptidoglycan cell wall biosynthesis. Like the parent capuramycin, A-500359s and A-503083s consist of three structural components: a uridine-5′-carboxamide (CarU), a rare unsaturated hexuronic acid, and an aminocaprolactam, the last of which is substituted by an unusual arylamine-containing polyamide in A-102395. The biosynthetic gene clusters for A-500359s and A-503083s have been reported, and two genes encoding a putative non-heme Fe(II)-dependent α-ketoglutarate:UMP dioxygenase and an l-Thr:uridine-5′-aldehyde transaldolase were uncovered, suggesting that C–C bond formation during assembly of the high carbon (C6) sugar backbone of CarU proceeds from the precursors UMP and l-Thr to form 5′-C-glycyluridine (C7) as a biosynthetic intermediate. Here, isotopic enrichment studies with the producer of A-503083s were used to indeed establish l-Thr as the direct source of the carboxamide of CarU. With this knowledge, the A-102395 gene cluster was subsequently cloned and characterized. A genetic system in the A-102395-producing strain was developed, permitting the inactivation of several genes, including those encoding the dioxygenase (cpr19) and transaldolase (cpr25), which abolished the production of A-102395, thus confirming their role in biosynthesis. Heterologous production of recombinant Cpr19 and CapK, the transaldolase homolog involved in A-503083 biosynthesis, confirmed their expected function. Finally, a phosphotransferase (Cpr17) conferring self-resistance was functionally characterized. The results provide the opportunity to use comparative genomics along with in vivo and in vitro approaches to probe the biosynthetic mechanism of these intriguing structures.
Introduction
Capuramycin-type antibiotics include A-500359s from Streptomyces griseus SANK 60196 (1–4), A-503083s from Streptomyces sp. SANK 62799 (5), and A-102395 from Amycolatopsis sp. SANK 60206 (6). They were discovered using an activity-based screen to identify inhibitors of bacterial phospho-N-acetylmuramyl-pentapeptide translocase 1 (TL12; annotated as MraY), a potential, clinically unexploited antibiotic target that initiates the lipid cycle of peptidoglycan cell wall biosynthesis and is essential for bacterial survival (7, 8). Studies into the biological activity of A-500359 A, the major congener isolated from S. griseus SANK 60196, and several semisynthetic analogues have revealed a potential utility of these natural products as anti-tuberculosis antibiotics (9, 10). For example, SQ641 and SQ922, two leads under preclinical development by Sequella (Rockville, MD), have been shown to have several clinically desirable attributes, including in vitro activity against multiple-drug-resistant strains of Mycobacterium tuberculosis (the primary causative agent of tuberculosis); high efficacy in a murine model of tuberculosis; rapid kill time in vitro and in vivo; and no toxicity to mice (11–17). Given the widespread documentation of extensively drug-resistant M. tuberculosis and the recent reality of totally drug-resistant M. tuberculosis (18), the development of drugs with novel targets, such as the capuramycin-type antibiotics, makes them attractive leads for tuberculosis chemotherapy (19, 20).
The namesake capuramycin, initially discovered in 1986 from an antibacterial screening program (21), consists of three structurally distinct, modular components: a uridine-5′-carboxamide (CarU), an unsaturated α-d-mannopyranuronate, and an l-α-amino-ϵ-caprolactam (l-ACL) (Fig. 1A) (22). Structural elucidation of the A-500359s revealed that A-500359 B 1a (IC50 = 18 nm against TL1) is identical to the original capuramycin, and A-500359 A 1b contains a 6‴-C-methylated l-ACL, a modification that has no effect on TL1 inhibition (IC50 = 17 nm) (1, 2). Two additional structural variants of note include the deaminocaprolactam (de-ACL) congeners that contain a free carboxylic acid (A-500359 F, 1c) or a methyl ester (A-500359 E, 1d), the former of which has a significantly decreased TL1 inhibitory activity (IC50 = 1.1 μm), whereas the latter has a slight decrease (IC50 = 27 nm) (3). The A-503083s 2a-2d have structures identical to those of A-500359s with one exception; in all cases, CarU is modified with a 2′-O-carbamoyl group that has minimal effect on TL1 inhibition and antibacterial activity (5). A-102395 3 significantly diverges from the other capuramycins in both structure, wherein the l-ACL is substituted with an unusual arylamine-containing polyamide, and activity, with 3 being the most potent inhibitor of TL1 (IC50 = 11 nm) yet completely inactive against Mycobacteria sp. and other bacterial strains (6).
FIGURE 1.
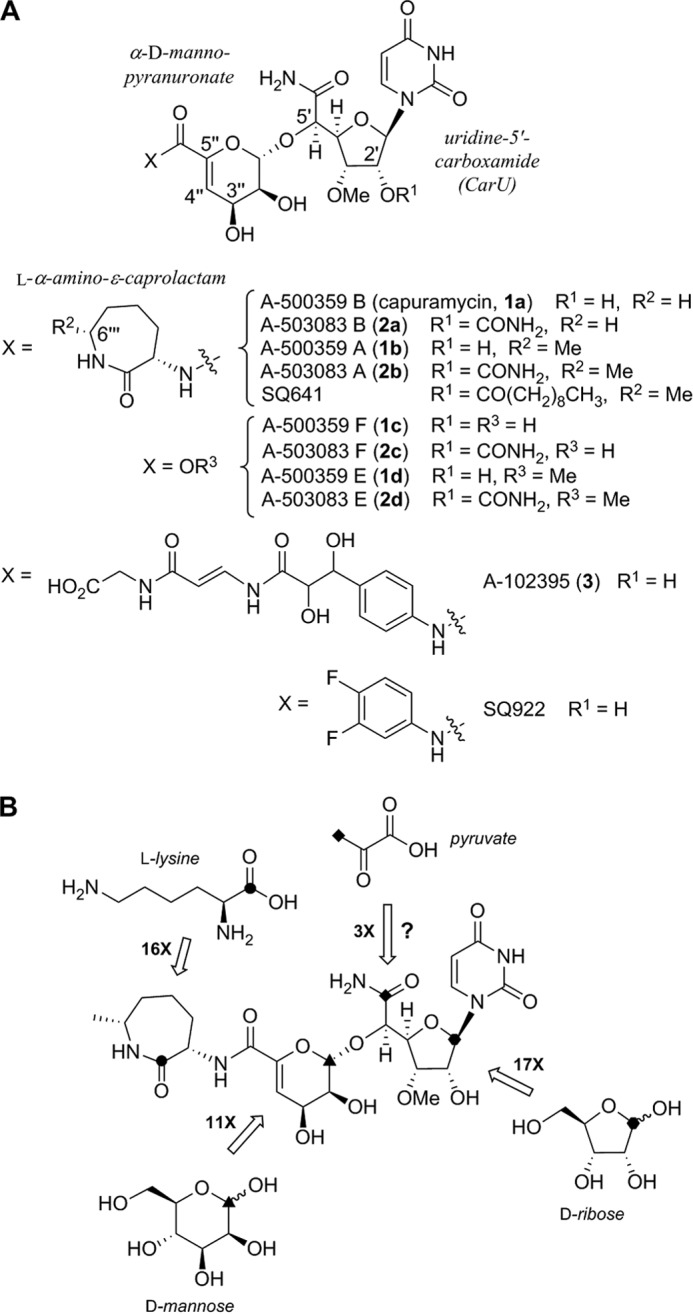
Structure and biosynthesis of representative capuramycin-type inhibitors of TL1. A, the CarU and α-d-mannopyranuronate components are found in all capuramycin-type antibiotics. SQ641 and SQ922 are semisynthetic leads prepared from 2b and 1c, respectively. B, prior results from isotopic enrichment studies using the indicated 13C-labeled precursors. The numerical value represents the -fold enrichment at the indicated site relative to a reference carbon.
Shortly after the rediscovery of capuramycin, the biosynthesis of each component was interrogated using feeding experiments with the producing strain of 1 (4). As expected, high incorporation was observed for l-ACL using l-[1-13C]Lys (16-fold enrichment of C-1‴); α-d-mannopyranuronate using d-[1-13C]mannose (11-fold enrichment of C-1″); and CarU using d-[1-13C]ribose (17-fold enrichment of C-1′) (Fig. 1B). The metabolic origin of the 5′-carboxamide of CarU was less clear, although a modest 3-fold enrichment of C-6′ using [3-13C]pyruvate led to a proposal that the biosynthesis involves an aldol reaction between phosphoenolpyruvate and uridine-5′-aldehyde (UA). This mechanism of ribose chain extension is analogous to that previously proposed for the biosynthesis of polyoxin and nikkomycin, two nucleoside antibiotics that also contain a high carbon sugar albeit with distinct chemical features (23). However, the specific enzymatic transformations required for the assembly of the high carbon sugar nucleoside of either the capuramycins or polyoxin and nikkomycin have not been biochemically established.
The biosynthetic gene cluster for 1 and 2 have been identified, and evidence for the involvement of 21 open reading frames (ORFs) was provided by gene expression profiling of the wild-type, 1-producing strain and several null mutants created by random chemical mutagenesis (24, 25). Additional evidence that the correct gene cluster was identified was provided by functional assignment of genes involved in 2 biosynthesis: one (capP) encoding an ATP-dependent phosphotransferase that modifies the 3″-OH of the unsaturated α-d-mannopyranuronate as a mechanism of self-resistance (26) and the other (capW) encoding an l-ACL:2d transacylase (25). Bioinformatic analysis of the gene clusters unexpectedly revealed two shared ORFs encoding putative proteins with sequence similarity to LipL: a non-heme Fe(II)-dependent α-ketoglutarate (αKG):uridine-5′-monophosphate (UMP) dioxygenase (27) and LipK, a pyridoxal-5-phosphate-dependent l-Thr:UA transaldolase (28). These two enzymes sequentially convert UMP to (5′S,6′S)-5′-C-glycyluridine (GlyU) during the biosynthesis of A-90289, a member of a distinct group of TL1 inhibitors that contain a GlyU component in the final product (Fig. 2) (29, 30). Additional members of the GlyU-containing nucleoside antibiotics with identified biosynthetic gene clusters include liposidomycins (31), caprazamycins (32), muraymycin (33), muraminomicin (34), and the recently discovered sphaerimicins (35). Thus, the uncovering of these two genes within the clusters for 1 and 2 suggested that the biosynthesis of CarU proceeds via condensation of l-Thr with UA to generate GlyU as a cryptic pathway intermediate. Herein we provide evidence through newly designed feeding experiments that this is indeed the case. The probability for the involvement of an l-Thr:UA transaldolase in CarU biosynthesis was subsequently exploited to clone and characterize the 3 gene cluster. The first genetic system for a producer of the capuramycin-type antibiotics was developed using the 3-producing strain, which, in addition to in vitro characterization of enzymes with sequence similarity to CapP (Cpr17), LipL (Cpr19), and LipK (CapH), has provided new insight into the resistance and biosynthetic strategies for the capuramycin-type antibiotics.
FIGURE 2.
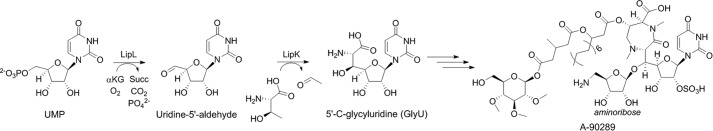
Structure and biosynthesis of A-90289, a GlyU-containing nucleoside antibiotic. The pathway starts with UMP that is converted to GlyU by the sequential reactions catalyzed by LipL and LipK.
Experimental Procedures
Standards
UA and GlyU were prepared following previously described procedures (27, 28). 2a and 2c were isolated from Streptomyces sp. SANK 62799, and 3 was isolated from Amycolatopsis sp. SANK 60206 following the procedures described (5, 6). Mass, 1H, and 13C NMR spectroscopic analyses of 3 were identical with the prior report (Fig. 3).
FIGURE 3.

1H and 13C NMR spectra of 3 in methanol-d4.
Isotopic Enrichment
Fermentation media and growth conditions for Streptomyces sp. SANK 62799 were as described previously (5). A seed culture was incubated at 28 °C for 48 h, when 1.5 ml was used to inoculate fresh media (50 ml of liquid medium in a 250-ml flask). After fermentation for 70 h, 25 mg of filter-sterilized [1-13C]Gly, [2-13C]Gly, or l-[13C4,15N]Thr was added to each flask. Fermentation was continued an additional 72 h. An equal volume of methanol was added directly to the culture and mixed vigorously prior to centrifugation to remove cell debris. The supernatant was lyophilized, and the dried powder was resuspended in water (100 mg/ml) for HPLC purification using a C-18 reverse phase semipreparative column. A series of linear gradients was developed from A (0.1% TFA, 2.5% acetonitrile) to B (0.1% TFA, 90% acetonitrile) in the following manner (beginning time and ending time with linear increase to percentage of B): 0–6 min, 0% B; 6–26 min, 100% B; 26–30 min, 100% B; 30–34 min, 0% B; and 34–35 min, 0% B. The flow rate was kept constant at 3.5 ml/min, and elution was monitored at 260 nm. Purified 2a and 2c were analyzed by NMR, and relative peak intensities were assigned based on C-2″ and C-3″ from the natural abundance 13C NMR spectra.
Cloning of the 3 Gene Cluster
Amycolatopsis sp. SANK 60206 genomic DNA was partially digested with Sau3AI to give ∼40-kb DNA fragments that were dephosphorylated with bacterial alkaline phosphatase and ligated into BamHI-digested cosmid vector SuperCos1 (Stratagene, Cedar Creek, TX), which was dephosphorylated by bacterial alkaline phosphatase after XbaI digestion. The ligation products were packaged with Gigapack III Gold packaging extract as described by the manufacturer (Stratagene), and the resulting recombinant phage was used to transfect Escherichia coli XL-1 Blue MR. Approximately 8,000 colonies from the obtained genomic library were screened by colony hybridization using a digoxigenin-labeled lipK/capH homologous DNA fragment obtained by PCR using degenerate primers (supplemental Table 1) and genomic DNA from Amycolatopsis sp. SANK 60206. Hybridization was carried out using DIG Easy Hyb (Roche Applied Science) at 42 °C, and the resulting filter was washed under high stringency conditions (0.1× SSC including 0.1% SDS, 68 °C). Detection was performed using CDP-Star (Roche Applied Science) according to the manufacturer's procedures.
Based on restriction digest analysis, three positive cosmids, pNCap01, pNCap02, and pNCap03, were isolated and sequenced using a Roche Applied Science GS FLX system (Operon Biotechnologies). The terminal region of pNCap03 was used as a probe to identify a 10-kb DNA fragment following BamHI digestion of genomic DNA, which was cloned into pUC19 to yield pUC/B10k. This insert was subsequently used as a probe to rescreen the library to identify a fourth cosmid, pNCap04, which was also sequenced to complete the genetic locus. Potential open reading frames were defined using Frameplot version 4.0, and database comparison for sequence homology was performed with BLAST search tools using the National Center for Biotechnology Information (Bethesda, MD).
Gene Inactivation and Production Analysis
Genes were inactivated using REDIRECT technology (36). In short, pNCap03 was introduced into E. coli BW25141/pKD78, and the linear PCR fragment obtained with template pIJ773 was introduced by electroporation. Following confirmation of the genotype using PCR, the modified cosmids were introduced into Amycolatopsis sp. SANK 60206 by conjugation using E. coli ET12567(pUZ8002). Apramycin resistance was used to select for a double-crossover event, the genotype of which was subsequently confirmed by both PCR and Southern blot analysis.
For comparative analysis of 3 production in the wild-type and mutant strains, 2 liters of culture broth was centrifuged at 2,600 × g for 20 min to remove mycelia. The pH of the supernatant was adjusted to 3.0 with concentrated HCl followed by the addition of an equal volume of methanol. After vigorous mixing for 1 h, the insoluble material was removed by centrifugation (6,000 × g for 40 min), and the solvent from the recovered supernatant was removed by rotary evaporation and lyophilization. Dried bacterial extracts (250–400 mg) were dissolved in 1.6 ml of 1:1 water/methanol, and the pH was adjusted to 3–4 with concentrated HCl if necessary prior to the addition of 6.4 ml of cold acetonitrile. The resulting mixture was vortexed for 5 min, followed by sonication for 1 min, and the insoluble material was removed by centrifugation (2,600 × g for 10 min). The supernatant was recovered and dried under a stream of nitrogen. The dried sample was resuspended in 50 μl of methanol for analysis by LC-MS-MS.
LC-MS-MS analysis of 3 was carried out using a Shimadzu UFLC equipped with an Apollo C18 column (250 × 4.6 mm, 5 μm) coupled to an AB Sciex 4000-Qtrap hybrid linear ion trap triple quadrupole mass spectrometer in multiple-reaction-monitoring mode. The mobile phase consisted of 10 mm ammonium formate with 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). A series of linear gradients was developed from A to B in the following manner (beginning time and ending time with linear increase to percentage of B): 0 min, 2% B; 0–8 min, 2% B; 8–27 min, 90% B; 27–30 min, 90% B; and 30–32 min, 2% B. The flow rate was kept constant at 0.5 ml/min with a column temperature of 30 °C, and elution was monitored at 260 nm. The mass spectrometer was operated in the negative electrospray ionization mode with optimal ion source settings determined with 3 standard with a declustering potential of −135 V, entrance potential of −10 V, collision energy of −60 V, collision cell exit potential of −7 V, curtain gas of 20 p.s.i., ion spray voltage of −4,200 V, ion source gas 1/gas 2 of 40 p.s.i., and temperature of 550 °C. Multiple-reaction-monitoring transitions monitored were as follows: 763.316/189.9 and 763.316/120. Negative electrospray ionization mode was used to analyze samples because it was more sensitive compared with positive electrospray ionization mode for 3.
Reverse Transcriptase-PCR Analysis
All procedures were performed according to the provided protocol. A TRIzol Max bacterial RNA isolation kit (Ambion) was used to extract total RNA from wild-type and mutant strains. The SuperScript III first-strand synthesis system for RT-PCR (Invitrogen) was used to synthesize the total cDNA from total RNA. Takara Taq was used to amplify target genes using total cDNA as template.
Characterization of Recombinant Enzymes
Procedures for cloning of genes, heterologous production and purification of proteins, and enzyme assays used standard protocols (26–28). The primers used for cloning were as follows: cpr17, 5′-GGTATTGAGGGTCGCATGACCGAGACCGAGATCAC-3′ (forward)/5′-AGAGGAGAGTTAGAGCCCTATCCGTGGAATCGGGCCAG-3′ (reverse); cpr19, 5′-GGTATTGAGGGTCGCATGCAGCAGCTGCAAGCCG-3′ (forward)/5′-AGAGGAGAGTTAGAGCCTCAATTGGAGGCGCGGGG-3′ (reverse); capH, 5′-GGTATTGAGGGTCGCATGACTGATATCAGGGAGCTCCGCAAG-3′ (forward)/5′-AGAGGAGAGTTAGAGCCTCAGGAGAACTCGACGAAATAGAAGGGAG-3′ (reverse). DNA templates for PCR cloning were pNCap02 for cpr17 and cpr19 and cosmid N-4 for capH (25). The capH gene was subcloned into pUWL20pw and expressed as described (25).
Results
GlyU Is a Biosynthetic Intermediate for CarU
An ORF encoding a protein with high amino acid sequence similarity to the transaldolase LipK is located within the two gene clusters that have been identified for the CarU-containing nucleoside antibiotics: 1 (ORF14, 55% identity/81% similarity) and 2 (CapH, 47% identity/79% similarity) (35). Similarly, an ORF encoding a protein with moderate sequence similarity to the dioxygenase LipL is found within the 1 and 2 gene clusters (Table 1). Both LipK and LipL were previously biochemically characterized to establish that C-6′ and C-7′ of GlyU originates from C-2 and C-1, respectively, of l-Thr through a transaldol-like reaction using UA as the aldol acceptor (Fig. 2) (27, 28). LipK, the catalyst for the transaldol-like reaction, was completely inactive when l-Thr was replaced with Gly or l-Ser despite having the closest sequence homology with proteins annotated as serine hydroxymethyltransferases that utilize tetrahydrofolic acid to interconvert Gly and l-Ser (28). With this information in hand, the metabolic origin of the 5′-carboxamide of CarU was re-investigated by undertaking feeding experiments with isotopically enriched Gly and l-Thr using the 2-producing strain.
TABLE 1.
Sequence comparison in percentage identity/percentage similarity among putative αKG:UMP dioxygenases
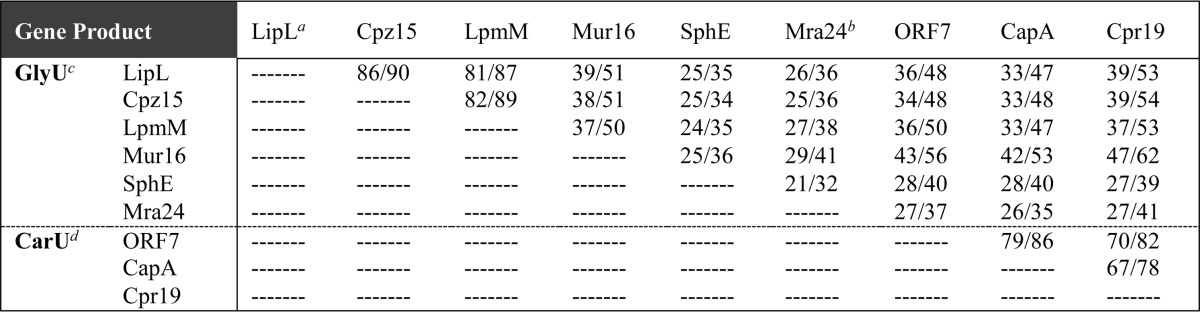
a LipL is encoded within the biosynthetic gene cluster for A-90289, Cpz15 within the cluster for caprazamycin, LpmM within the cluster for liposidomycin, Mur16 within the cluster for muraymycin, SphE within the cluster for sphaerimicin, Mra24 within the cluster for muraminomicin, ORF7 within the cluster for A-500359, CapA within the cluster for A-503083, and Cpr19 within the cluster for A-102395.
b Mra24 appears truncated and inactive, missing 57 amino acids at the C terminus when aligned with LipL.
c GlyU, 5′-C-glycyluridine-containing nucleoside antibiotics.
d CarU, uridine-5′-carboxamide-containing nucleoside antibiotics.
To initially probe the metabolic origin of the 5′-carboxamide, either [1-13C]Gly or [2-13C]Gly was added to the culture both of the 2-producing strain. Under the growth conditions employed, 2a was the major congener produced and, as predicted from the bioinformatics analysis, no significant enrichment at C-6′ was observed based on comparative analysis of the 13C NMR spectra (data not shown). However, a 6% enrichment of the 3′-O-methyl group was observed using [2-13C]Gly, which is similar to incorporation results that have been reported for other actinomycete-derived O-methylated metabolites (37). In contrast to the Gly enrichment experiments, the de-ACL derivative 2c was the major congener produced when fed with l-[13C4,15N]Thr. Comparative analysis of the 13C NMR spectra of purified 2c revealed a 15% enrichment at C-6′ (Fig. 4), which is substantially greater than that previously reported with pyruvate (4). This high level of enrichment is comparable with the levels observed for enrichment of l-ACL with l-Lys, which has been supported by biochemical studies (25). Thus, the data are consistent with direct incorporation of l-Thr via a transaldol-like mechanism. Additionally, a 13C-15N heteronuclear spin coupling at C-6′ of the 13C-NMR spectrum (JCN = 18 Hz) was detected that, along with analysis of the LC-MS spectrum (data not shown), is consistent with concomitant 15N incorporation from l-Thr during the biosynthesis of CarU.
FIGURE 4.
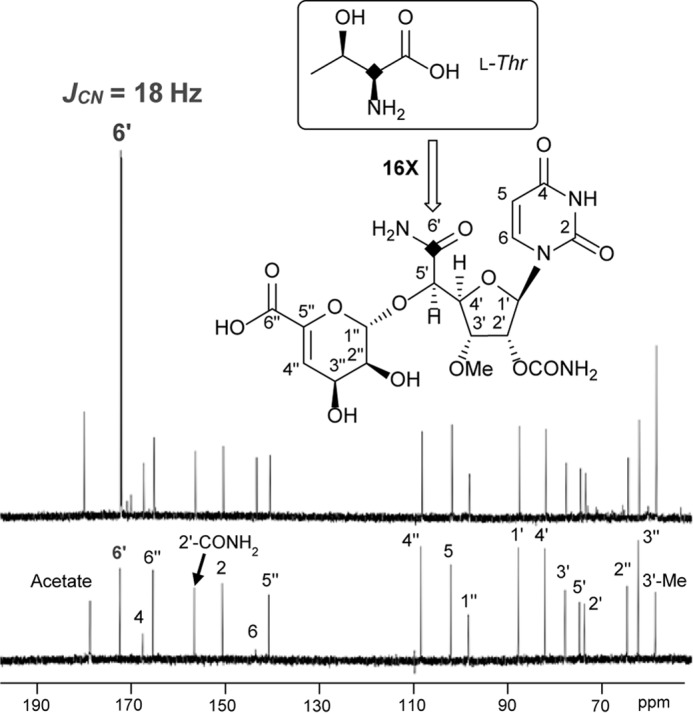
Isotopic enrichment using l-[13C4,15N]Thr. A, 13C NMR spectrum of 2c obtained with l-[13C4,15N]Thr feeding. B, 13C NMR spectrum of 2c obtained without feeding (natural abundance 13C).
Transaldolase as a Genetic Fingerprint for Identifying the 3 Gene Cluster
After providing evidence that CarU biosynthesis proceeds with direct incorporation of l-Thr, the probable involvement of an l-Thr:UA transaldolase was exploited as a genetic fingerprint to identify the biosynthetic gene cluster for 3. We have previously designed degenerate primers based on distinctive sequence blocks found within the functionally assigned l-Thr:UA transaldolases (35), and these primer sets were utilized to amplify DNA fragments of the expected size from the genomic DNA of the producing strain of 3 (Fig. 5). The pattern and size of the PCR product were similar to those previously reported using template DNA from the producing strains of 1 and 2, and, as expected, no PCR product was obtained using genomic DNA isolated from Streptomyces lividans TK21, whose whole genome has been sequenced and does not harbor the homologous gene (Fig. 5).
FIGURE 5.
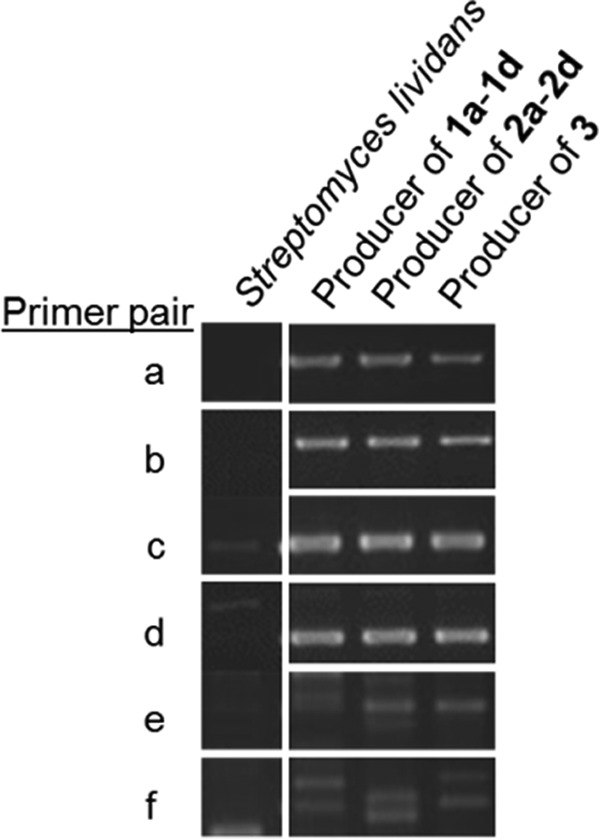
PCR amplification of the probable l-Thr:uridine-5′-aldehyde transaldolase gene. Template genomic DNA was isolated from S. lividans TK21 (negative control) and the producing strains of the indicated CarU-containing nucleoside antibiotics. The primers were designed according to the conserved sequence as described previously (35), and primer pairs that were utilized are as follows: A-F1/C-R1 (a); A-F1/D-R1 (b); A-F2/C-R1 (c); A-F2/D-R1 (d); B-F1/C-R1 (e); and B-F1/D-R (f).
The PCR products amplified using the genomic DNA from the producing strain of 3 were subsequently utilized as probes to identify three overlapping cosmids (pNCap01–03) encompassing ∼57-kb DNA, which were sequenced and analyzed to reveal 49 complete ORFs (Fig. 6A). One of the ORFs (cpr25) encoded the expected l-Thr:UA transaldolase with moderate (43–50%) to high (70–79%) amino acid sequence identity to transaldolases encoded in gene clusters for GlyU-containing and CarU-containing nucleoside antibiotics, respectively. Due to the novel chemical structure of the polyamide component of 3 and hence uncertainty regarding its mechanism of biosynthesis and incorporation, the cluster was extended by first cloning a BamHI-digested, 10-kb DNA fragment that overlapped with pNCap03. The cloned DNA fragment was subsequently used as a probe to identify a fourth cosmid, pNCap04. Following sequencing and bioinformatic analysis, a total of ∼85 kb of contiguous DNA was identified, including 69 complete ORFs and an additional ORF (cpr51) in pNCap04 that has a homolog within the 1 and 2 biosynthetic gene cluster, thus bringing the total shared ORFs for CarU-containing nucleoside antibiotics to 16 (Fig. 6, A and B). The closest homologs and putative functions of the ORFs are listed in supplemental Table 1; ORFs predicted to be involved in the biosynthesis of 3 were annotated as cpr17-cpr57, for capuramycin-related nucleoside.
FIGURE 6.

Characterization of the 3 biosynthetic gene cluster. A, four overlapping cosmids were identified and sequenced to define the genetic organization of the 3 biosynthetic gene cluster. The cpr25 gene encodes the putative l-Thr:uridine-5′-aldehyde transaldolase that was targeted to identify the genetic locus. B, comparative analysis of the sequenced region of chromosomal DNA from the 2- and 3-producing strains. Highlighted in blue are the minimal ORFs predicted to be essential for 3 biosynthesis, whereas the two ORFs in black, cpr19 and cpr25, are predicted to encode for a non-heme Fe(II)-dependent αKG:UMP dioxygenase and l-Thr:uridine-5′-aldehyde transaldolase, respectively. C, shared biosynthetic pathway leading to the core structure of 2 and 3 and divergence upon amide bond formation.
The genetic architecture of the clusters for 1 and 2 is virtually identical, excluding a single ORF encoding a truncated 2′-O-carbamoyltransferase within the 1 gene cluster that leads to the sole structural variation between these two groups of capuramycin-type antibiotics. Conversely, the 16 conserved ORFs within the 3 gene cluster have a genetic organization different from that found in 1 and 2 (Fig. 6B). Nonetheless, individual comparison of the gene products revealed a relatively high amino acid sequence identity, which includes the aforementioned transaldolase (Cpr 25; 79% identity with CapH) and non-heme Fe(II)-dependent αKG:UMP dioxygenase (Cpr19; 76% identity with CapA). Additional shared gene products of note include a phosphotransferase Cpr17, probably involved in self-resistance (53% identity with CapP), a carboxymethyltransferase (Cpr27; 57% identity with CapS), and a putative amide bond-forming N-transacylase (Cpr51; 65% identity with CapW). CapS and CapW have been shown to function sequentially to activate the carboxylic acid of 2c in the form of the methyl ester 2d, followed by transacylation to incorporate the l-ACL to generate 2a (Fig. 6C). The uncovering of genes for Cpr27 and Cpr51 suggests that the unusual arylamine-containing polyamide of 3 is incorporated using a comparable mechanism (i.e. carboxylic acid 1c → methyl ester 1d → amide 1a) (Fig. 6C), which was unexpected given the unique chemical nature of the acyl acceptors.
Phosphorylation by Cpr17 as a Mechanism of Self-resistance
Heterologous expression of cpr17 in Streptomyces albus yielded a recombinant strain that was, in contrast to the strain harboring the empty vector, resistant to 1a at 500 μg/ml (Fig. 7A). The cpr17 gene was subsequently cloned and expressed in E. coli to yield soluble protein for activity assessment. CapP, which was previously shown to confer resistance by catalyzing regiospecific phosphorylation at the 3″-OH of 2a and 2d (26), was used as a control. Activity of Cpr17 was initially assessed by HPLC using 2a, 2c, and 2d as potential substrates, and new peaks appeared with 2a and 2d. The retention time of the new peak was identical when Cpr17 was substituted with CapP (Fig. 7, B and C), consistent with the formation of 3″-phospho-2a and -2d, respectively. CapP utilized 2c with a very low turnover of ≤0.2 min−1 (26), and accordingly no appreciable activity was detected using Cpr17 or CapP with 2c under the reaction conditions employed here (data not shown). The activity of Cpr17 and CapP was tested with 3, and HPLC analysis revealed a new peak in both cases with an (M − H)− ion at m/z = 843.0 (Fig. 7, D–F), consistent with the molecular formula for 3″-phospho-3 (C31H37N6O20P; expected (M − H)− ion at m/z = 843.2). Although the limited availability of 3 precluded structural elucidation and product bioactivity analysis at the current time, the results suggest that Cpr17, like CapP, functions as capuramycin-type antibiotic phosphotransferase involved in self-resistance.
FIGURE 7.

Characterization of the phosphotransferase Cpr17. A, resistance to 1a conferred to S. albus upon heterologous expression of cpr17 using variable amounts of 1a (0 μg/ml (column 1), 100 μg/ml (column 2), and 500 μg/ml (column 3)). The top two rows (pWHM3 (row 1) and pWHM3-ermEp-cpr17 (row 2)) consist of mycelia diluted 1:10 following homogenization of a liquid culture of S. albus, whereas the bottom two rows (pWHM3 (row 3) and pWHM3-ermEp-cpr17 (row 4)) consist of mycelia diluted 1:100 prior to spotting on ISP2 agar plates. B–D, HPLC traces with the indicated substrate incubated without enzyme (i), with Cpr17 (ii), and with CapP (iii). ▾, 3″-phospho-2a; ♦, 3″-phospho-2d; ●, 3″-phospho-3. A260, absorbance at 260 nm. *, unidentified contamination peak. E, mass spectrum of the peak at retention time 7.7 min (D, trace i) corresponding to authentic 3. F, representative mass spectrum of the peak at retention time 7.6 min (D, trace ii or iii) corresponding to 3″-phospho-3. All mass data are reported in negative ion mode. G, plots for single-substrate kinetic analysis of Cpr17.
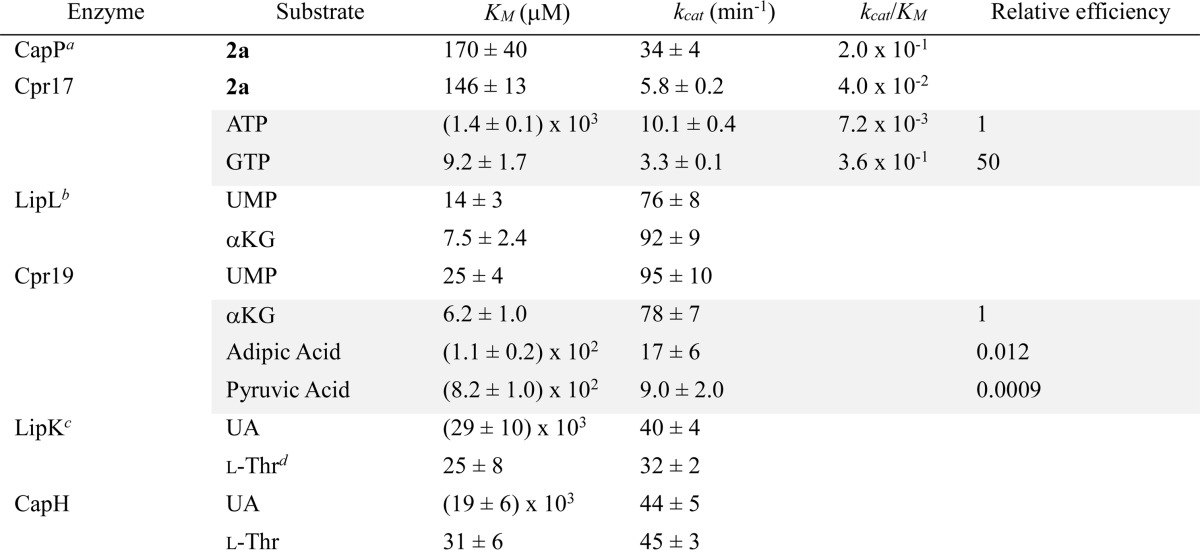
After identifying the function of Cpr17, single-substrate kinetic experiments were performed using 2a as a substrate. Using nearly saturating ATP and variable 2a, standard Michaelis-Menten kinetics were observed, yielding a Km = 146 ± 13 μm and kcat = 5.8 ± 0.2 min−1 (Fig. 6G), the former constant similar to and the latter 5-fold lower than that reported for CapP (Table 2). Single-substrate kinetic experiments with saturating 2a and variable ATP yielded a Km = 1.4 ± 0.1 mm and kcat = 10.1 ± 0.4 min−1 (Fig. 7G). The high observed Km for ATP prompted us to test GTP as an alternative phosphate donor, which served as an excellent substrate for Cpr17, yielding a Km = 9.2 ± 1.7 μm and kcat = 3.3 ± 0.1 min−1 (Fig. 7G), a 50-fold improvement in catalytic efficiency, suggesting that GTP is the in vivo substrate for Cpr17. A similar nucleotide preference has been reported recently for certain aminoglycoside phosphotransferases (38–40).
TABLE 2.
Michaelis-Menten kinetic parameters
Development of a Genetic System and Biochemical Interrogation of CarU Biosynthesis
Unlike the producing strains for 1 and 2, which have not been amenable to genetic manipulation, gene inactivation was possible with the 3-producing strain, thus enabling the first in vivo analysis of the gene clusters for the capuramycin-type antibiotics. Both cpr19 and cpr25 were individually targeted for inactivation by double crossover homologous recombination, and successful inactivation of each was confirmed by PCR and Southern blot analysis (Fig. 8, A–C). The resulting Δcpr19 and Δcpr25 mutant strains were unable to produce 3 (Fig. 8D), consistent with an essential role in the biosynthesis of 3. Gene complementation has yet to be successful in this strain, and thus to exclude possible polar effects, the expression levels of the upstream and downstream genes were analyzed by RT-PCR. Using mRNA extracted from the Δcpr25 mutant strain at the onset of 3 production, expression of cpr25 was not detected, whereas genes flanking cpr25 were clearly expressed (Fig. 8E). In contrast, all three genes were detected within the wild-type strain, suggesting that there is no polar effect due to cpr25 inactivation. Ambiguous results were obtained with expression analysis within the Δcpr19 mutant strain, and hence we turned our attention to in vitro characterization for direct evidence of the involvement of cpr19 in the biosynthesis of 3.
FIGURE 8.

Gene inactivation in Amycolatopsis sp. SANK 60206. A, strategy utilized for gene inactivation demonstrated with cpr25. B, PCR analysis of genomic DNA isolated from the wild type (WT; expected 1.2 kb) and Δcpr25 mutant (Δ; expected 1.8 kb) strains. C, Southern blot analysis of ApaI-digested genomic DNA from WT and Δcpr25 mutant strains using the indicated probe. D, LC-MS-MS analysis of authentic 3 (i) and production from wild-type strain (ii), Δcpr19 (iii), Δcpr25 (iv), and Δcpr51 (v). E, RT-PCR analysis of the indicated genes using mRNA isolated from the Δcpr25 mutant strain or wild-type strain. F, RT-PCR analysis of the indicated genes using mRNA isolated from the Δcpr51 mutant strain or wild-type strain. M, DNA ladder.
The gene encoding cpr19 was heterologously expressed in E. coli to obtain pure, recombinant protein (Fig. 9A). Based on prior characterization of LipL, Cpr19 was expected to convert UMP to UA in a reaction that is dependent upon Fe(II), O2, and αKG. HPLC analysis of reactions with all of these components revealed a new peak that co-eluted with authentic UA, and as expected, the formation of the product was absolutely dependent on the inclusion of O2 and αKG (Fig. 9B). Inductively coupled plasma MS revealed a fraction (4%) of Cpr19 co-purified with Fe(II), thus explaining the residual activity in the absence of additional Fe(II). Optimal activity for Cpr19 was observed between 100 and 1000 μm FeCl2 (Fig. 9C). Similar to other enzymes of this dioxygenase superfamily, including E. coli taurine dioxygenase (41), ascorbic acid enhanced the activity, reaching an optimum between 1 and 5 mm (Fig. 9D). No tested divalent or transition metals were able to substitute for Fe(II), and similarly to LipL, Zn2+ inhibited the reaction (Fig. 9E). Finally, single-substrate kinetic analysis yielded a Km = 25 ± 4 μm and kcat = 95 ± 10 min−1 with respect to UMP and Km = 6.2 ± 1.0 μm and kcat = 78 ± 7 min−1 with respect to αKG (Fig. 9F) (Table 2).
FIGURE 9.

In vitro characterization of the non-heme Fe(II)-dependent αKG:UMP dioxygenase Cpr19. A, SDS-PAGE of purified His6-Cpr19 (expected 35.3 kDa). The engineered N-terminal His6 tag contributes ∼5 kDa to the native molecular mass. B, HPLC analysis of the reaction catalyzed by Cpr19 using UMP as the prime substrate with all components (i) or the omission of ascorbic acid (ii), ferrous iron (iii), αKG (iv), or O2 (v). Similarly to LipL (27), Cpr19 co-purified with iron (4 ± 0.5%), explaining the formation of trace amounts of product without exogenous ferrous iron. ●, uridine-5′-aldehyde; A260, absorbance at 260 nm. C, optimal activity of Cpr19 with respect to varied Fe(II). D, optimal activity of Cpr19 with respect to varied ascorbic acid. E, activity with the addition of Zn2+ using the optimized reaction conditions. For comparison, the activity of LipL with Zn2+ is shown (from Ref. 27). F, plots for single-substrate kinetic analysis of CapH using optimized activity conditions. G, uncoupled oxidation of α-ketoglutarate to succinate in the absence of prime substrate (UMP), which for Cpr19 yielded a kcat = 1.6 min−1, corresponding to a relative turnover of 2.5% compared with the UMP-containing reaction. 14×, a 14-fold increase in enzyme utilized compared with data shown for 1× Cpr19. Error bars, S.D.
The overall biochemical and kinetic properties of Cpr19 were comparable with those previously reported for LipL excluding two of note (27). First, although LipL is able to catalyze uncoupled oxidative decarboxylation of αKG, the rate of formation of succinate in the absence of UMP was moderately greater for Cpr19 (kcat = 1.6 min−1 compared with kcat = 0.4 min−1), corresponding to a relative turnover of 2.5% to the UMP-containing reaction (Fig. 9G). Second, in contrast to the high specificity of LipL, Cpr19 catalyzed UA formation with alternative α-keto acids pyruvate and α-ketoadipate. Single substrate kinetic analysis yielded constants of Km = 8.2 × 102 ± 90 mm and kcat = 9.0 ± 2.0 min−1 with respect to pyruvate and Km = 1.1 × 102 ± 16 mm and kcat = 17 ± 6 min−1 with respect to α-ketoadipate (Table 2), a relative efficiency of 8.7 × 10−4 and 1.2 × 10−2, respectively, compared to the preferred substrate αKG. Similarly to LipL, however, Cpr19 was inactive with α-ketobutyrate, α-ketovalerate, and oxaloacetate.
After demonstrating that UA is a biosynthetic intermediate of CarU-containing nucleosides, the probable l-Thr:UA transaldolase, Cpr25, was targeted for characterization; however, soluble protein could not be obtained using multiple hosts and a variety of expression conditions. Instead, Cpr25 was substituted with CapH that is encoded within the 2 biosynthetic gene cluster (5). Similarly to LipK (28), soluble CapH was only produced in S. lividans TK64 (Fig. 10A), and UV-visible spectroscopic analysis revealed trace amounts of PLP co-purified with CapH (Fig. 10B). Enzyme assays monitored by HPLC revealed identical traces to LipK (Fig. 10C), and the product peak was characterized by spectroscopic analysis to demonstrate that CapH is an l-Thr:UA transaldolase. The biochemical properties of CapH were similar to those of LipK, and kinetic analysis following acetaldehyde production (28) yielded kinetic constants of Km = 19 ± 6 mm and kcat = 44 ± 5 min−1 with respect to UA and Km = 25 ± 8 μm and kcat = 32 ± 2 min−1 with respect to l-Thr (Fig. 10D), values comparable with those for LipK (Table 2).
FIGURE 10.

In vitro characterization of the transaldolase CapH. A, SDS-PAGE analysis of partially purified His6-CapH (lane 2, expected molecular mass of 50.4 kDa). Lane 1, molecular mass standards from Bio-Rad; the engineered N-terminal His6-tag contributes 5 kDa to the predicted native molecular mass. B, UV-visible spectrum of CapH isolated from S. lividans TK64. C, HPLC analysis of the reaction without enzyme (i) or with CapH (ii) or LipK (iii). ●, (5′S,6′S)-5′-C-glycyluridine; A260, absorbance at 260 nm. D, plots for single-substrate kinetic analysis of CapH. Error bars, S.E.
Cpr51 Is Essential for 3 Biosynthesis
To determine whether cpr51, encoding a putative N-transacylase with a proposed functional role comparable with that of CapW, was involved in the biosynthesis of 3, the gene was inactivated by double-crossover homologous recombination, which was confirmed by PCR and Southern blot analysis (data not shown). Utilizing mRNA extracted from the Δcpr51 mutant strain at the onset of 3 production, expression of cpr51 was not detected, whereas genes flanking cpr51 were clearly expressed (Fig. 8F). As expected, the resulting Δcpr51 mutant strain was unable to produce 3 (Fig. 8D), consistent with an essential role in 3 biosynthesis.
Discussion
We have performed isotope enrichment experiments, identified the 3 gene cluster, characterized a phosphotransferase involved in resistance, and characterized two biosynthetic enzymes to support a shared biosynthetic paradigm for the CarU-containing nucleoside antibiotics 1-3. Interestingly, the biosynthesis of CarU occurs through the morphing of the primary metabolites UMP and l-Thr and not uridine and phosphoenolpyruvate as previously suggested. The pathway to CarU proceeds through the unusual β-hydroxy-l-amino acid GlyU as an intermediate following sequential catalysis by a non-heme Fe(II), αKG:UMP dioxygenase, and an l-Thr:UA transaldolase. The function of the dioxygenase Cpr19/LipL, the generation of UA from the mononucleotide, is novel for enzymes of the non-heme Fe(II), αKG-dependent dioxygenases and thus mechanistically intriguing (42–45). Based on the mechanism for taurine dioxygenase, which catalyzes the transformation of taurine to aminoacetaldehyde and sulfite and is perhaps the best characterized enzyme of the superfamily (46–48), Cpr19/LipL probably catalyze insertion of one atom of O2 into αKG and the other at C-5′ of UMP, leading to a geminal hydroxyl-phosphoester intermediate. This labile intermediate is proposed to undergo phosphate elimination to yield UA.
In addition to being mechanistically interesting, the identification of the new activity of Cpr19/LipL has biosynthetic implications for multiple chemical entities. Not only is this dioxygenase activity required to initiate the biosynthesis of the high carbon sugar nucleosides CarU and GlyU, but it is also required for biosynthesis of the aminoribosyl moiety found in the GlyU-containing nucleoside antibiotics (Fig. 2) (49). During the assembly of this unusual sugar, an l-Met:UA aminotransferase uses the Cpr19/LipL product to form 5-amino-5-deoxyuridine. The discovery of the dioxygenase and aminotransferase activities involved in aminoribose biosynthesis has subsequently led to the uncovering of the biosynthetic gene cluster for jawsamycin, whose structure contains a 5-amino-5-deoxyuridine (50). Finally, a survey of the whole genomes of diverse actinomycetes reveals a high prevalence of genes encoding proteins of the dioxygenase superfamily within other putative natural product biosynthetic operons, suggesting the potential for discovery of comparable dioxygenase activities.
Following the reaction by Cpr19, the transaldolase Cpr25 generates a new β-hydroxyamino acid that is ultimately converted to the carboxamide functionality of CarU. Carboxamides in primary metabolites are made by one of four enzymatic processes: an ATP-dependent enzymatic process from a carboxylic acid using an NH3/Gln as the amine source, a hydrolytic process, lyase chemistry, or (in the unusual case of l-Arg catabolism in certain microorganisms) an oxidative decarboxylation catalyzed by a flavin-dependent l-Arg 2-monooxygenase (51). Carboxamides are found in some secondary metabolites, such as phenazines (52), oxytetracycline (53), AB-400 (54, 55), and sarubicin A (56), and genetic or biochemical evidence suggests that they are produced from the carboxylic acid by an ATP-dependent process. A gene encoding a protein with similarity to those catalyzing ATP-dependent carboxamide formation or any alternative mechanism is not apparent within the 1–3 gene clusters. The revelation of a 13C-15N heteronuclear spin coupling in the 13C NMR spectrum upon feeding labeled l-Thr suggests that the C–N bond remains intact during CarU biosynthesis (57), providing additional support for a different, potentially novel mechanism of carboxamide formation. Because the conversion of GlyU to CarU is likely to involve decarboxylation, hydroxylation, and/or oxidation, it is possible that the PLP-dependent enzyme Cpr23, a second non-heme Fe(II)-dependent dioxygenase Cpr18, and/or the putative oxidoreductase Cpr20 are involved.
The formation of the unsaturated α-d-mannopyranuronate that is attached to the CarU core is not obvious from bioinformatics analysis, excluding the attachment of the sugar by the glycosyltransferase, Cpr24. GDP-d-mannuronic acid, expected to be an intermediate based on prior isotopic enrichment studies, is a building block of alginate, a major anionic polysaccharide found in some strains of bacteria (58). During alginate biosynthesis, GDP-d-mannuronic acid is formed from GDP-d-mannose by a GDP-mannose 6-dehygrogenase, yet the two remaining shared proteins encoded in the 1–3 gene clusters with similarity to sugar biosynthetic enzymes are Cpr21, a putative NDP-hexose 2,3-dehydratase, and Cpr22, a putative NDP-hexose 4-epimerase/dehydratase. Although Cpr21 and Cpr22 are very different in sequence and domain architecture from GDP-mannose 6-dehygrogenase, it is possible that one or both may perform this chemistry. Finally, the 4,5-unsaturation found in alginate is a result of lyase (β-elimination) chemistry (58), yet it appears that the double bond is introduced by a dehydratase reaction, possibly by Cpr20, -21, or -22. Finally, Cpr29 is proposed to catalyze 3′-O-methylation, a modification that is found in all capuramycin-type antibiotics, although the timing of methylation remains unclear. The three remaining shared genes in the 1–3 gene cluster (cpr28, -30, and -31) are predicted to encode three subunits of a functional carbon monooxide dehydrogenase complex with an unclear role in biosynthesis. The genetic system developed here should help in defining their role along with the other aforementioned gene products in 1c biosynthesis.
Similar to the disaccharadyl core, the arylamine-containing polyamide moiety of 3 contains highly unusual chemical features. The 3-(4-aminophenyl)-2,3-dihydroxypropanoic acid component of the polyamide is predicted to be assembled from chorismic acid and malonyl-CoA by minimally eight ORFs, cpr12 and cpr32–cpr38 (Fig. 11). The pathway initially proceeds by the conversion of chorismic acid to p-aminobenzoic acid, a transformation that occurs during the de novo synthesis of essential folates in plants, fungi, and bacteria and requires three enzyme activities: an l-Gln amidotransferase, a 4-amino-4-deoxychorismate (ADC) synthase, and ADC lyase (59). In E. coli, these three activities, which have been biochemically confirmed, are found on proteins encoded by three distinct genes (pabA, pabB, and pabC, respectively) located at remote locations within the chromosome. A survey of 100 Streptomyces genomes revealed that pabB and pabC are located in a conserved, probable three-gene operon (the third gene encodes a protein with sequence similarity to acetyltransferases of the GNAT family), and pabA is located elsewhere within the chromosome. Twenty of these 100 genomes contain at least one additional gene encoding a PabB but, contrastingly, as part of a bidomain protein containing an N-terminal PabA domain. Although not yet biochemically confirmed, a protein with identical bidomain architecture has been uncovered for the biosynthesis of amicetin (60), pristinamycin (61), aureothin (62), candicidin/FR-008 (63), chloramphenicol (64–66), the recently identified albicidin (67), and now 3. Thus, cpr38 encoding this bidomain protein is proposed to catalyze a two-step reaction involving amidohydrolysis of l-Gln with ammonia channeled and incorporated into chorismic acid to generate ADC. Cpr12, encoded by a gene that was initially believed to be outside the gene cluster, has similarity to proteins annotated as ADC lyase or aminotransferase class IV, of which ADC lyase belongs (59), suggesting that this protein catalyzes the elimination of pyruvate to form PABA.
FIGURE 11.
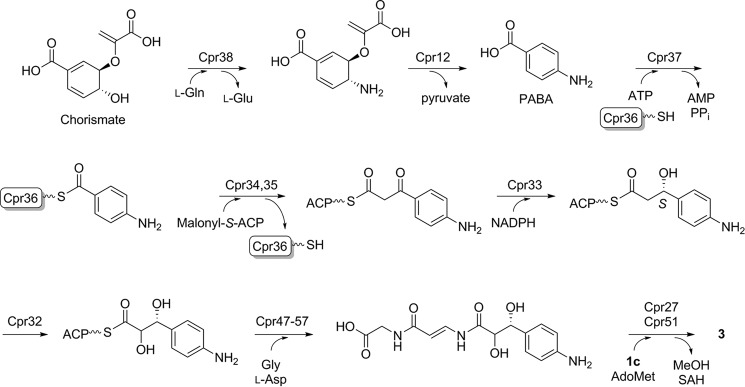
Proposed biosynthetic pathway of the unique arylamine-containing polyamide of 3.
The remaining steps require C–C bond formation between the carboxylic acid of PABA and a C-2 extender unit. This is putatively initiated by Cpr37-catalyzed activation of PABA as the acyl-adenylate with loading to the free-standing carrier protein Cpr36 to form the thioester-linked PABA. Homologous genes are found within the actinomycin gene cluster (68), wherein biosynthesis is proposed to be initiated by activation and loading of a 4-methyl-3-hydroxyanthranilic acid starter unit to a free-standing carrier protein. Adjacent to cpr36 are two genes encoding proteins with similarity to a ketosynthase and chain length factor (Cpr34 and Cpr35, respectively) that work in concert to catalyze decarboxylative condensation between a malonyl-S-acyl carrier protein (ACP) and recipient thioester during aromatic polyketide biosynthesis catalyzed by type II polyketide synthases (69). In several instances, type II polyketide synthase systems recruit fatty acid biosynthetic machinery during polyketide assembly, and it is probably the same scenario here, wherein an ACP and malonyl-CoA:ACP transacylase from fatty acid biosynthesis generate the co-substrate for decarboxylative condensation by the Cpr34/35 heterodimer. A comparable mechanism of condensation has previously been proposed during hygromycin B biosynthesis upon identification of its gene cluster (70).
Following formation of the β-ketothioester, a putative 3-oxoacyl-ACP reductase, Cpr33, catalyzes reduction to the β-hydroxythioester intermediate, and the luciferase-like monoxygenase, Cpr32, catalyzes α-hydroxylation to give 3-(4-aminophenyl)-2,3-dihydroxypropanoic acid. These last two enzymatic steps to generate the vicinal diol of 3-(4-aminophenyl)-2,3-dihydroxypropanoic acid are of particular interest because the stereochemistry in 3 has not been established. Sequence analysis of Cpr33 suggests the closest similarity to 3-oxoacyl-ACP reductase (FabG) from bacterial type II fatty acid synthases, which catalyze hydride addition to the si face to generate the R configuration (71). It is thus reasonable to assume the same relative orientation of acyl-ACP binding and reduction, in this case hydride addition to the re face to generate the S configuration (the PABA starter unit changes priorities of the carbonyl carbon; Fig. 11). Stereoselective reduction of other members of the short chain dehydrogenase family, to which 3-oxoacyl-ACP reductases belong, has been the subject of several studies, culminating in sequence signatures to predict the stereochemical outcome, which is consistent with that proposed for Cpr33 (72, 73). Introduction of the C-2 hydroxyl is probably catalyzed by Cpr32 that has sequence similarity to proteins annotated as FMN-dependent monooxygenases of the bacterial luciferase family, which includes a variety of FMN- or coenzyme F420-dependent oxidoreductases that act on structurally diverse substrates. It is not possible to predict the stereochemistry of α-hydroxylation based solely on this information, but assuming that catalysis does not involve epimerization at C-3, C-2 hydroxylation leads to a change in substituent priority at C-3 so that the ultimate product is proposed to have the 3R configuration.
The remaining steps in polyamide biosynthesis involve formation of two amide bonds, and as expected, there are putative proteins encoded within ORFs cpr47–cpr57 that are associated with amide bond-forming events, including an adenylation-like domain protein with predicted specificity to a hydrophilic amino acid (Cpr54), two carrier proteins (Cpr48 and Cpr55), a condensation domain protein (Cpr47), and three transglutaminase-like proteins (Cpr49, Cpr50, and Cpr57). Based on the functional assignment of an unrelated transglutaminase-like protein as a condensation catalyst that uses two substrate thioesters during andrimid biosynthesis (74), the putative transglutaminases, all of which have similarity to uncharacterized MitI in the mitomycin C gene cluster (75), potentially catalyze amide bond formation. It is likely that this component is derived from a Gly and l-Asp (or l-Asn) unit, although a detailed proposal for polyamide biosynthesis at this stage would be highly speculative without isotopic enrichment or genetic evidence. Finally, components of Cpr41–Cpr46 have similarity to a pyruvate dehydrogenase complex or acetolactate synthase, and their role in 3 biosynthesis, if any, is unclear, but possibly they are utilized for pyruvate and by-product recycling that is generated by Cpr12.
The proposed final step in 3 biosynthesis is the coupling of the arylamine-containing polyamide to the capuramycin core via amide bond formation. Of the aforementioned natural products containing PABA, only the aromatic amine of PABA from amecitin is modified to an amide as also observed in 3. In the case of amicetin, gene inactivation of aviN encoding a putative malonyl-CoA:ACP transacylase revealed it as the catalyst for amide bond formation (60). Contrastingly, the formation of amide bond for 2a is orchestrated by an unconventional mechanism featuring sequential catalysis by CapS and CapW (25). Despite the stark structural contrast of the amine acceptor (aliphatic amine of aminocaprolactam versus the arylamine of the polyamide), genes encoding putative CapS and CapW homologues (cpr27 and cpr51, respectively) were identified in the 3 gene cluster, and gene inactivation demonstrated that cpr51 is essential for 3 biosynthesis. Although no pathway intermediates could be identified, Cpr51 is proposed to catalyze the coupling reaction, and future studies will be aimed at defining the function of Cpr51 and its role and catalytic properties relative to CapW.
In summary, we have established that UMP and, unexpectedly, l-Thr serve as the metabolic precursors of the CarU nucleoside found in the capuramycin-type antibiotics. Using this information, the biosynthetic gene cluster for 3 was identified to enable a thorough, comparative genomic approach to propose a biosynthetic pathway for the capuramycin-type antibiotics. A mechanism of self-resistance to 3 was also defined, and the phosphotransferase Cpr17 conferring this resistance was shown to prefer GTP as the phosphate donor. Finally, the first genetic system for the capuramycin-type antibiotic producers was developed, a critical advancement for interrogating the biosynthesis of the unusual chemical components of this family of antibiotics.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health, NIAID, Grant AI087849 (to S. V. L.). This work was also supported by National Center for Advancing Translational Sciences Grant UL1TR000117, NSF CAREER award MCB-1149427 (to S. G.-T.), and the Kentucky Science and Education Foundation (to S. V. L.).
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) KP995196.

This article contains supplemental Table 1.
- TL1
- translocase 1
- CarU
- uridine-5′-carboxamide
- l-ACL
- l-α-amino-ϵ-caprolactam
- αKG
- α-ketoglutarate
- GlyU
- 5′-C-glycyluridine
- UA
- uridine-5′-aldehyde
- PABA
- para-aminobenzoic acid
- ADC
- 4-amino-4-deoxychorismate
- ACP
- acyl carrier protein.
References
- 1. Muramatsu Y., Muramatsu A., Ohnuki T., Ishii M. M., Kizuka M., Enokita R., Tsutsumi S., Arai M., Ogawa Y., Suzuki T., Takatsu T., Inukai M. (2003) Studies on novel bacterial translocase I inhibitors, A-500359s: I. Taxonomy, fermentation, isolation, physico-chemical properties and structure elucidation of A-500359 A, C, D, and G. J. Antibiot. 56, 243–252 [DOI] [PubMed] [Google Scholar]
- 2. Muramatsu Y., Ishii M. M., Inukai M. (2003) Studies on novel bacterial translocase I inhibitors, A-500359s: II. Biological activities of A-500359 A, C, D and G. J. Antibiot. 56, 253–258 [DOI] [PubMed] [Google Scholar]
- 3. Muramatsu Y., Miyakoshi S., Ogawa Y., Ohnuki T., Ishii M. M., Arai M., Takatsu T., Inukai M. (2003) Studies on novel bacterial translocase I inhibitors, A-500359s: III. Deaminocaprolactam derivatives of capuramycin: A-500359 E, F, H, M-1 and M-2. J. Antibiot. 56, 259–267 [DOI] [PubMed] [Google Scholar]
- 4. Ohnuki T., Muramatsu Y., Miyakoshi S., Takatsu T., Inukai M. (2003) Studies on novel bacterial translocase I inhibitors, A-500359s: IV. Biosynthesis of A-500359s. J. Antibiot. 56, 268–279 [DOI] [PubMed] [Google Scholar]
- 5. Muramatsu Y., Ohnuki T., Ishii M. M., Kizuka M., Enokita R., Miyakoshi S., Takatsu T., Inukai M. (2004) A-503083 A, B, E and F, novel inhibitors of bacterial translocase I, produced by Streptomyces sp. SANK 62799. J. Antibiot. 57, 639–646 [DOI] [PubMed] [Google Scholar]
- 6. Murakami R., Fujita Y., Kizuka M., Kagawa T., Muramatsu Y., Miyakoshi S., Takatsu T., Inukai M. (2007) A-102395, a new inhibitor of bacterial translocase I, produced by Amycolatopsis sp. SANK 60206. J. Antibiot. 60, 690–695 [DOI] [PubMed] [Google Scholar]
- 7. Ikeda M., Wachi M., Jung H. K., Ishino F., Matsuhashi M. (1991) The Escherichia coli mraY gene encoding UDP-N-acetylmuramoyl-pentapeptide:undecaprenyl-phosphate phospho-N-acetylmuramoyl-pentapeptide transferase. J. Bacteriol. 173, 1021–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boyle D. S., Donachie W. D. (1998) mraY is an essential gene for cell growth in Escherichia coli. J. Bacteriol. 180, 6429–6432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hotoda H., Furukawa M., Daigo M., Murayama K., Kaneko M., Muramatsu Y., Ishii M. M., Miyakoshi S., Takatsu T., Inukai M., Kakuta M., Abe T., Harasaki T., Fukuoka T., Utsui Y., Ohya S. (2003) Synthesis and antimycobacterial activity of capuramycin analogs: Part 1. Substitution of the azepan-2-one moiety of capuramycin. Bioorg. Med. Chem. Lett. 13, 2829–2832 [DOI] [PubMed] [Google Scholar]
- 10. Hotoda H., Daigo M., Furukawa M., Murayama K., Hasegawa C. A., Kaneko M., Muramatsu Y., Ishii M. M., Miyakoshi S., Takatsu T., Inukai M., Kakuta M., Abe T., Fukuoka T., Utsui Y., Ohya S. (2003) Synthesis and antimycobacterial activity of capuramycin analogs. Part 2: Acylated derivatives of capuramycin-related compounds. Bioorg. Med. Chem. Lett. 13, 2833–2836 [DOI] [PubMed] [Google Scholar]
- 11. Reddy V. M., Einck L., Nacy C. A. (2008) In vitro antimycobacterial activities of capuramycin analogues. Antimicrob. Agents Chemother. 52, 719–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nikonenko B. V., Reddy V. M., Protopopova M., Bogatcheva E., Einck L., Nacy C. A. (2009) Activity of SQ641, a capuramycin analog, in a murine model of tuberculosis. Antimicrob. Agents Chemother. 53, 3138–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reddy V. M., Bogatcheva E., Einck L., Nacy C. A. (2011) Nanoemulsion formulation enhances intracellular activity of capuramycin analogues against Mycobacterium tuberculosis. Drug Delivery Lett. 10.2174/2210304x11101020150 [DOI] [Google Scholar]
- 14. Reddy V. M., Dubuisson T., Einck L., Wallis R. S., Jakubiec W., Ladukto L., Campbell S., Nacy C. A. (2012) SQ109 and PNU-100480 interact to kill Mycobacterium tuberculosis in vitro. J. Antimicrob. Chemother. 67, 1163–1166 [DOI] [PubMed] [Google Scholar]
- 15. Bogatcheva E., Hanrahan C., Nikonenko B., de los Santos G., Reddy V., Chen P., Barbosa F., Einck L., Nacy C., Protopopova M. (2011) Identification of SQ609 as a lead compound from a library of dipiperidines. Bioorg. Med. Chem. Lett. 21, 5353–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nikonenko B. V., Einck L., Nacy C. A. (2010) Anti-tuberculosis drug therapy in mice of different inbred strains. Infect. Genet. Evol. 10, 1151–1154 [DOI] [PubMed] [Google Scholar]
- 17. Reddy V. M., Einck L., Andries K., Nacy C. A. (2010) In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrob. Agents Chemother. 54, 2840–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shah N. S., Wright A., Bai G.-H., Barrera L., Boulahbal F., Martín-Casabona N., Drobniewski F., Gilpin C., Havelková M., Lepe R., Lumb R., Metchock B., Portaels F., Rodrigues M. F., Rüsch-Gerdes S., Van Deun A., Vincent V., Laserson K., Wells C., Cegielski J. P. (2007) Worldwide emergence of extensively drug-resistant tuberculosis. Emerging Infect. Dis. 13, 380–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wong E. B., Cohen K. A., Bishai W. R. (2013) Rising to the challenge: new therapies for tuberculosis. Trends Microbiol. 21, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson M., McNeil M. R., Brennan P. J. (2013) Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol. 8, 855–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamaguchi H., Sato S., Yoshida S., Takada K., Itoh M., Seto H., Otake N. (1986) Capuramycin, a new nucleoside antibiotic: taxonomy, fermentation, isolation and characterization. J. Antibiot. 39, 1047–1053 [DOI] [PubMed] [Google Scholar]
- 22. Seto H., Otake N., Sato S., Yamaguchi H., Takada K., Itoh M., Lu H. S. M., Clardy J. (1988) The structure of a new nucleoside antibiotic, capuramycin. Tetrahedron Lett. 10.1016/S0040-4039(00)86055-4 [DOI] [Google Scholar]
- 23. Isono K., Sato T., Hirasawa K., Funayama S., Suzuki S. (1978) Biosynthesis of the nucleoside skeleton of polyoxins. J. Am. Chem. Soc. 10.1021/ja00480a052 [DOI] [Google Scholar]
- 24. Funabashi M., Nonaka K., Yada C., Hosobuchi M., Masuda N., Shibata T., Van Lanen S. G. (2009) Identification of the biosynthetic gene cluster of A-500359s in Streptomyces griseus SANK60196. J. Antibiot. 62, 325–332 [DOI] [PubMed] [Google Scholar]
- 25. Funabashi M., Yang Z., Nonaka K., Hosobuchi M., Fujita Y., Shibata T., Chi X., Van Lanen S. G. (2010) An ATP-independent strategy for amide bond formation in antibiotic biosynthesis. Nat. Chem. Biol. 6, 581–586 [DOI] [PubMed] [Google Scholar]
- 26. Yang Z., Funabashi M., Nonaka K., Hosobuchi M., Shibata T., Pahari P., Van Lanen S. G. (2010) Functional and kinetic analysis of the phosphotransferase CapP conferring selective self-resistance to capuramycin antibiotics. J. Biol. Chem. 285, 12899–12905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Z., Chi X., Funabashi M., Baba S., Nonaka K., Pahari P., Unrine J., Jacobsen J. M., Elliott G. I., Rohr J., Van Lanen S. G. (2011) Characterization of LipL as a non-heme, Fe(II)-dependent α-ketoglutarate:UMP dioxygenase that generates uridine-5′-aldehyde during A-90289 biosynthesis. J. Biol. Chem. 286, 7885–7892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barnard-Britson S., Chi X., Nonaka K., Spork A. P., Tibrewal N., Goswami A., Pahari P., Ducho C., Rohr J., Van Lanen S. G. (2012) Amalgamation of nucleosides and amino acids in antibiotic biosynthesis: discovery of an l-threonine:uridine-5′-aldehyde transaldolase. J. Am. Chem. Soc. 134, 18514–18517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Funabashi M., Baba S., Nonaka K., Hosobuchi M., Fujita Y., Shibata T., Van Lanen S. G. (2010) The biosynthesis of liposidomycin-like A-90289 antibiotics featuring a new type of sulfotransferase. ChemBioChem 11, 184–190 [DOI] [PubMed] [Google Scholar]
- 30. Fujita Y., Kizuka M., Funabashi M., Ogawa Y., Ishikawa T., Nonaka K., Takatsu T. (2011) A-90289 A and B, new inhibitors of bacterial translocase I, produced by Streptomyces sp. SANK 60405. J. Antibiot. 64, 495–501 [DOI] [PubMed] [Google Scholar]
- 31. Kaysser L., Siebenberg S., Kammerer B., Gust B. (2010) Analysis of the liposidomycin gene cluster leads to the identification of new caprazamycin derivatives. ChemBioChem 11, 191–196 [DOI] [PubMed] [Google Scholar]
- 32. Kaysser L., Lutsch L., Siebenberg S., Wemakor E., Kammerer B., Gust B. (2009) Identification and manipulation of the caprazamycin gene cluster lead to new simplified liponucleoside antibiotics and give insights into the biosynthetic pathway. J. Biol. Chem. 284, 14987–14996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng L., Chen W., Zhai L., Xu D., Huang T., Lin S., Zhou X., Deng Z. (2011) Identification of the gene cluster involved in muraymycin biosynthesis from Streptomyces sp. NRRL 30471. Mol. BioSyst. 7, 920–927 [DOI] [PubMed] [Google Scholar]
- 34. Chi X., Baba S., Tibrewal N., Funabashi M., Nonaka K., Van Lanen S. G. (2013) The muraminomicin biosynthetic gene cluster and enzymatic formation of the 2-deoxyaminoribosyl appendage. MedChemComm 4, 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Funabashi M., Baba S., Takatsu T., Kizuka M., Ohata Y., Tanaka M., Nonaka K., Spork A. P., Ducho C., Chen W. C., Van Lanen S. G. (2013) Structure-based gene targeting discovery of sphaerimicin, a bacterial translocase I inhibitor. Angew. Chem. Int. Ed. Engl. 52, 11607–11611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gust B., Chandra G., Jakimowicz D., Yuqing T., Bruton C. J., Chater K. F. (2004) λ Red-mediated genetic manipulation of antibiotic-producing Streptomyces. Adv. Appl. Microbiol. 54, 107–128 [DOI] [PubMed] [Google Scholar]
- 37. Chang Z., Sitachitta N., Rossi J. V., Roberts M. A., Flatt P. M., Jia J., Sherman D. H., Gerwick W. H. (2004) Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine Cyanobacterium lyngbya majuscula. J. Nat. Prod. 67, 1356–1367 [DOI] [PubMed] [Google Scholar]
- 38. Smith C. A., Toth M., Frase H., Byrnes L. J., Vakulenko S. B. (2012) Aminoglycoside 2″-phosphotransferase IIIa (APH(2″)-IIIa) prefers GTP over ATP: structural templates for nucleotide recognition in the bacterial aminoglycoside-2″ kinases. J. Biol. Chem. 287, 12893–12903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shi K., Berghuis A. M. (2012) Structural basis for dual nucleotide selectivity of aminoglycoside 2"-phosphotransferase IVa provides insight on determinants of nucleotide specificity of aminoglycoside kinases. J. Biol. Chem. 287, 13094–13102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Toth M., Chow J. W., Mobashery S., Vakulenko S. B. (2009) Source of phosphate in the enzymic reaction as a point of distinction among aminoglycoside 2"-phosphotransferases. J. Biol. Chem. 284, 6690–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eichhorn E., Van Der Ploeg J. R., Kertesz M. A., Leisinger T. (1997) Characterization of α-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J. Biol. Chem. 272, 23031–23036 [DOI] [PubMed] [Google Scholar]
- 42. Bollinger J. M., Jr., Krebs C. (2006) Stalking intermediates in oxygen activation by iron enzymes: motivation and method. J. Inorg. Biochem. 100, 586–605 [DOI] [PubMed] [Google Scholar]
- 43. Straganz G. D., Nidetzky B. (2006) Variations of the 2-His-1-carboxylate theme in mononuclear non-heme FeII oxygenases. ChemBioChem 7, 1536–1548 [DOI] [PubMed] [Google Scholar]
- 44. Mantri M., Zhang Z., McDonough M. A., Schofield C. J. (2012) Autocatalysed oxidative modifications to 2-oxoglutarate dependent oxygenases. FEBS J. 279, 1563–1575 [DOI] [PubMed] [Google Scholar]
- 45. Hausinger R. P. (2004) Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 21–68 [DOI] [PubMed] [Google Scholar]
- 46. Price J. C., Barr E. W., Tirupati B., Bollinger J. M., Jr., Krebs C. (2003) The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508 [DOI] [PubMed] [Google Scholar]
- 47. Price J. C., Barr E. W., Hoffart L. M., Krebs C., Bollinger J. M., Jr. (2005) Kinetic dissection of the catalytic mechanism of taurine:α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 44, 8138–8147 [DOI] [PubMed] [Google Scholar]
- 48. Grzyska P. K., Ryle M. J., Monterosso G. R., Liu J., Ballou D. P., Hausinger R. P. (2005) Steady-state and transient kinetic analyses of taurine/α-ketoglutarate dioxygenase: effects of oxygen concentration, alternative sulfonates, and active-site variants on the FeIV-oxo intermediate. Biochemistry 44, 3845–3855 [DOI] [PubMed] [Google Scholar]
- 49. Chi X., Pahari P., Nonaka K., Van Lanen S. G. (2011) Biosynthetic origin and mechanism of formation of the aminoribosyl moiety of peptidyl nucleoside antibiotics. J. Am. Chem. Soc. 133, 14452–14459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hiratsuka T., Suzuki H., Kariya R., Seo T., Minami A., Oikawa H. (2014) Biosynthesis of the structurally unique polycyclopropanated polyketide-nucleoside hybrid jawsamycin (FR-900848). Angew. Chem. Int. Ed. Engl. 53, 5423–5426 [DOI] [PubMed] [Google Scholar]
- 51. Kaneoke M., Shiota K., Kusunose M., Shimizu E., Yorifuji T. (1993) Function of the arginine oxygenase pathway in utilization of l-arginine-related compounds in Arthrobacter globiformis and Brevibacterium helvolum. Biosci. Biotechnol. Biochem. 10.1271/bbb.57.814 [DOI] [Google Scholar]
- 52. Mavrodi D. V., Bonsall R. F., Delaney S. M., Soule M. J., Phillips G., Thomashow L. S. (2001) Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 183, 6454–6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang W., Ames B. D., Tsai S.-C., Tang Y. (2006) Engineered biosynthesis of a novel amidated polyketide, using the malonamyl-specific initiation module from the oxytetracycline polyketide synthase. Appl. Environ. Microbiol. 72, 2573–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Miranzo D., Seco E. M., Cuesta T., Malpartida F. (2010) Isolation and characterization of pcsB, the gene for a polyene carboxamide synthase that tailors pimaricin into AB-400. Appl. Microbiol. Biotechnol. 85, 1809–1819 [DOI] [PubMed] [Google Scholar]
- 55. Seco E. M., Miranzo D., Nieto C., Malpartida F. (2010) The pcsA gene from Streptomyces diastaticus var. 108 encodes a polyene carboxamide synthase with broad substrate specificity for polyene amides biosynthesis. Appl. Microbiol. Biotechnol. 85, 1797–1807 [DOI] [PubMed] [Google Scholar]
- 56. Gould S. J., Eisenberg R. (1991) Biosynthesis of sarubicin A: synthesis and incorporation of 6-hydroxy [13CO15NH2] anthranilamide. J. Org. Chem. 56, 6666–6671 [Google Scholar]
- 57. Gould S. J., Chang C. C., Darling D. S., Roberts J. D., Squillacote M. (1980) Streptonigrin biosynthesis: 4. Details of the tryptophan metabolism. J. Am. Chem. Soc. 102, 1707–1712 [Google Scholar]
- 58. Remminghorst U., Rehm B. H. A. (2006) Bacterial alginates: from biosynthesis to applications. Biotechnol. Lett. 28, 1701–1712 [DOI] [PubMed] [Google Scholar]
- 59. Ye Q. Z., Liu J., Walsh C. T. (1990) p-Aminobenzoate synthesis in Escherichia coli: purification and characterization of PabB as aminodeoxychorismate synthase and enzyme X as aminodeoxychorismate lyase. Proc. Natl. Acad. Sci. U.S.A. 87, 9391–9395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang G., Zhang H., Li S., Xiao J., Zhang G., Zhu Y., Niu S., Ju J., Zhang C. (2012) Characterization of the amicetin biosynthesis gene cluster from Streptomyces vinaceusdrappus NRRL 2363 implicates two alternative strategies for amide bond formation. Appl. Environ. Microbiol. 78, 2393–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Blanc V., Gil P., Bamas-Jacques N., Lorenzon S., Zagorec M., Schleuniger J., Bisch D., Blanche F., Debussche L., Crouzet J., Thibaut D. (1997) Identification and analysis of genes from Streptomyces pristinaespiralis encoding enzymes involved in the biosynthesis of the 4-dimethylamino-l-phenylalanine precursor of pristinamycin I. Mol. Microbiol. 23, 191–202 [DOI] [PubMed] [Google Scholar]
- 62. He J., Hertweck C. (2003) Iteration as programmed event during polyketide assembly: molecular analysis of the aureothin biosynthesis gene cluster. Chem. Biol. 10, 1225–1232 [DOI] [PubMed] [Google Scholar]
- 63. Campelo A. B., Gil J. A. (2002) The candicidin gene cluster from Streptomyces griseus IMRU 3570. Microbiology 148, 51–59 [DOI] [PubMed] [Google Scholar]
- 64. Brown M. P., Aidoo K. A., Vining L. C. (1996) A role of pabAB, a p-aminobenzoate synthase gene of Streptomyces venezuelae ISP5230, in chloroamphenicol biosynthesis. Microbiology 142, 1345–1355 [DOI] [PubMed] [Google Scholar]
- 65. He J., Magarvey N., Piraee M., Vining L. C. (2001) The gene cluster for chloramphenicol biosynthesis in Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular non-ribosomal peptide synthetase gene. Microbiology 147, 2817–2829 [DOI] [PubMed] [Google Scholar]
- 66. Yanai K., Sumida N., Okakura K., Moriya T., Watanabe M., Murakami T. (2004) Para-position derivatives of fungal anthelmintic cyclodepsipeptides engineered with Streptomyces venezuelae antibiotic biosynthetic genes. Nat. Biotechnol. 22, 848–855 [DOI] [PubMed] [Google Scholar]
- 67. Cociancich S., Pesic A., Petras D., Uhlmann S., Kretz J., Schubert V., Vieweg L., Duplan S., Marguerettaz M., Noëll J., Pieretti I., Hügelland M., Kemper S., Mainz A., Rott P., Royer M., Süssmuth R. D. (2015) The gyrase inhibitor albicidin consists of p-aminobenzoic acids and cyanoalanine. Nat. Chem. Biol. 11, 195–197 [DOI] [PubMed] [Google Scholar]
- 68. Keller U., Lang M., Crnovcic I., Pfennig F., Schauwecker F. (2010) The actinomycin biosynthetic gene cluster of Streptomyces chrysomallus: a genetic hall of mirrors for synthesis of a molecule with mirror symmetry. J. Bacteriol. 192, 2583–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shen B. (2000) Aromatic polyketide biosynthesis. in Topics in Current Chemistry: Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Vol. II, Book 209, (Leeper F. J., Vederas J. C., ed), pp. 1–51, Springer-Verlag, Berlin [Google Scholar]
- 70. Palaniappan N., Ayers S., Gupta S., Habib E.-S., Reynolds K. A. (2006) Production of hygromycin A analogs in Streptomyces hygroscopicus NRRL 2388 through identification and manipulation of the biosynthetic gene cluster. Chem. Biol. 13, 753–764 [DOI] [PubMed] [Google Scholar]
- 71. Toomey R. E., Wakil S. J. (1966) Studies on the mechanism of fatty acid synthesis: XV. Preparation and general properties of β-ketoacyl acyl carrier protein reductase from Escherichia coli. Biochim. Biophys. Acta 116, 189–197 [DOI] [PubMed] [Google Scholar]
- 72. Reid R., Piagentini M., Rodriguez E., Ashley G., Viswanathan N., Carney J., Santi D. V., Hutchinson C. R., McDaniel R. (2003) A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry 42, 72–79 [DOI] [PubMed] [Google Scholar]
- 73. Caffrey P. (2003) Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. ChemBioChem 4, 654–657 [DOI] [PubMed] [Google Scholar]
- 74. Fortin P. D., Walsh C. T., Magarvey N. A. (2007) A transglutaminase homologue as a condensation catalyst in antibiotic assembly lines. Nature 448, 824–827 [DOI] [PubMed] [Google Scholar]
- 75. Mao Y., Varoglu M., Sherman D. H. (1999) Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem. Biol. 6, 251–263 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.